
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical investigation of fluorine-containing Li-salts as slurry cathode additives for tunable rheology in super high solid content NMP slurries†
Francesco
Colombo
 *,
Marcus
Müller
,
Andreas
Weber
,
Noah
Keim
,
Fabian
Jeschull
,
Werner
Bauer
and
Helmut
Ehrenberg
*,
Marcus
Müller
,
Andreas
Weber
,
Noah
Keim
,
Fabian
Jeschull
,
Werner
Bauer
and
Helmut
Ehrenberg
Karlsruhe Institute of Technology – Institute for Applied Materials e Energy Storage Systems (IAM-ESS), P.O. Box 3640, 76021, Karlsruhe, Germany. E-mail: sonofaciledaricordare@gmail.com
First published on 2nd November 2023
Abstract
Slurries with high solid contents are attractive because they can minimize usage and recycling of toxic and expensive organic solvents, but have been, so far, very challenging to realize due to their high viscosities, strong slurry gelation and poor coating results. Herein, we demonstrate the application of well-known Li electrolyte salts, namely LiTFA, LiTFSI or LiODFB, as slurry additives, which allow the achievement of an outstanding high solid content of 75.5 wt% for a NMC622-NMP slurry. These kinds of additives are chosen in order to neutralize and chemically complex the NMC622 basic surface and because of their well-known interaction within a battery system when used as electrolyte salts or additives. The investigation shows how high solid content induced slurry gelation can be tuned and controlled depending on the type of the additive and on its affinity towards the NMC622 surface. LiTFA shows the best slurry gelation controlling capabilities and LiTFSI has enhanced long-term capacity retention among the additives, rivalling the best performing reference electrode. EIS performed on fatigued cathodes after 1000 cycles shows how the contact impedance between the electrode composite and the Al current collector rises when Li-salts are used in comparison to the reference. Post-mortem SEM images show cathodic delamination for the additive-containing electrodes. Incremental capacity curves and post-mortem EDX investigations suggest Li plating on graphite anodes as a supplementary cell degradation mechanism when additives are employed.
Introduction
A basic solvent born slurry for cathodes consists of the active material (AM), carbon black (CB) as the electronically conductive agent and polyvinylidene difluoride (PVdF) as a binder. For electrode slurries N-methyl-2-pyrrolidone (NMP), despite its low vapor pressure (making NMP energy consuming to remove during drying) and toxicity, is the most commonly used dispersing medium owing to its potential for dissolving PVdF. Slurry optimization plays an important role in manufacturing electrodes and has to be tuned to match the requirements of each different coating line. Slurry viscosity has to be controlled throughout dwell times in storage tanks in order to prevent sedimentation, clogging of tubing, filters and feeds along the coating line that may arise from flocculation and precipitation of solid components. For production processes such as coating, spraying, and extruding, the elastic gel-like behaviour of the sample is often more important than the simple viscous response to shear.1 In battery technology PVdF emerged as the standard binder for cathode coatings, and electrode processing with PVdF is a key step in battery manufacturing.2–4 Most cathode materials, like LiNi0.80Co0.15Al0.05O2 (NCA) or the varieties of Li(Ni,Mn,Co)O2 (NMC), have a mean particle size in the range from 5 to 10 μm. Such large AM particles allow the production of slurries with higher solid contents (SCs), thanks to their reduced specific surface area.5 Indeed, low solid content (SC) slurries have a more critical economic and ecologic impact as the consumption of solvents and energy increases during manufacturing, since NMP is evaporated and recovered (recycled) in the electrode drying step. Therefore, new and greener production solutions like super high SC slurries are needed in order to reduce the application of solvents. Nevertheless, Li(Ni0.6Mn0.2Co0.2)O2 (NMC622) high SCs are typically associated with strong gelation, especially beyond practical limits6,7 of 70.0 wt% or higher. Gelation can compromise the slurry rheology, thus hindering processability and yielding only poor-quality coatings. Upon mixing PVdF and CB in NMP, CB can readily aggregate in network structures due to its high surface area and sub μm primary particle dimensions, where attractive van der Waals forces become relevant. The PVdF binder can indeed easily adsorb on CB surfaces thus accommodating the high surface tension between CB and solvent and providing stabilization (CB immobilization) to the colloidal system.3,8,9 When large 5–10 μm NMC particles are mixed with NMP, PVdF and CB to produce a slurry, gel formation can be caused by the interaction of the AM particles with the entangled polymer chains trapped in the CB-PVdF colloidal domains. Indeed, whenever a polymer is transported to a solvent wetted solid surface through diffusion or convection, attachment can take place (its kinetics depends on different potential barriers, like, for instance, repulsion of the polymer from pre-adsorbed solvent molecules). The adsorption speed is ruled by the rate of the re-conformation process of the flexible freshly adsorbed polymer layer.10 The conformation of the polymer adsorbed on a solid interface encompasses surface adherent trains as well as solution protruding parts called loops or tails.11,12 A polymer density profile can be defined so that a high monomeric unit density can be found in close contact to the AM surface in train conformation, while a more rarefied density of tails and loops is detected heading into the solution. The extension of the polymer tail from the AM into the solution is directly proportional to the molecular weight of the polymer employed. For low adsorbing polymers extended loops or tails prevail, whereas for high adsorption energy a larger portion of trains in direct contact with the surface is expected.13 Whereas fully polymer covered surfaces in good solvents tend to repel each other at all distances and have the potential to sterically stabilize a suspension against agglomeration; unsaturated layers in good solvents can produce bridging flocculation if this process is fast enough to compete with the flattening and compaction of the polymer sheath around the ceramic AM surface.14 Therefore, extended polymer layers can interact with one another and, when segments of one polymer chain adsorb on different particles, aggregation by bridging flocculation takes place. In this latter case, physical gelation is reported to occur due to an interplay of the type of high-molecular weight PVdF binder used (Mw = 370![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 – 1300000 g mol−1) and of the moderately high slurry SC employed (20 vol% or 60 wt%).3 Gel formation is beneficial for this standard SC slurries3 whenever the strength of its aggregated state is high enough to prevent segregation of components and sedimentation of AM particles by effectively immobilizing particles. Nevertheless, coating problems can readily occur when the strength of the slurry aggregated state is too high. If the average AM inter-particle distance gets much higher compared to the radius of gyration of the polymer, the probability of gel formation is expected to decrease considerably, which is generally the case for low SCs. Gelation has also been reported as being even more likely for smaller sub-μm particles,15 so that, in this case, the slurry SC threshold at which gelation occurs is expected to be lower. Moreover, PVdF chemical instability towards basic surfaces, such as the ones of NMC, was reported as another possible source of gelation in PVdF-based slurries.16 Indeed, apart from bridging flocculation triggered physical gelation, chemical slurry gelation can act on layered cathode materials due to the presence of Li2O, LiOH and Li2CO3 residual Li compounds on their surface.16,17 Residual Li basic compounds are abundant at the surface of higher Ni content NMC16 and, particularly, in overlithiated NMC materials (e.g. the NMC622 used in this study) where LiOH excess is directly employed in their synthesis to compensate for the lithium loss during the calcination process.18 Moreover, if not stored in a controlled environment, NMC surface's Li2O can readily react with ambient H2O and CO2 to further produce additional LiOH and Li2CO3 basic impurities.19 Beside causing gas evolution problems due to their reaction with carbonate solvents20 of liquid electrolyte in the final cell, residual Li compounds can actively catalyse PVdF binder dehydrofluorination21–24 in the cathode slurry state thereby producing C
000 – 1300000 g mol−1) and of the moderately high slurry SC employed (20 vol% or 60 wt%).3 Gel formation is beneficial for this standard SC slurries3 whenever the strength of its aggregated state is high enough to prevent segregation of components and sedimentation of AM particles by effectively immobilizing particles. Nevertheless, coating problems can readily occur when the strength of the slurry aggregated state is too high. If the average AM inter-particle distance gets much higher compared to the radius of gyration of the polymer, the probability of gel formation is expected to decrease considerably, which is generally the case for low SCs. Gelation has also been reported as being even more likely for smaller sub-μm particles,15 so that, in this case, the slurry SC threshold at which gelation occurs is expected to be lower. Moreover, PVdF chemical instability towards basic surfaces, such as the ones of NMC, was reported as another possible source of gelation in PVdF-based slurries.16 Indeed, apart from bridging flocculation triggered physical gelation, chemical slurry gelation can act on layered cathode materials due to the presence of Li2O, LiOH and Li2CO3 residual Li compounds on their surface.16,17 Residual Li basic compounds are abundant at the surface of higher Ni content NMC16 and, particularly, in overlithiated NMC materials (e.g. the NMC622 used in this study) where LiOH excess is directly employed in their synthesis to compensate for the lithium loss during the calcination process.18 Moreover, if not stored in a controlled environment, NMC surface's Li2O can readily react with ambient H2O and CO2 to further produce additional LiOH and Li2CO3 basic impurities.19 Beside causing gas evolution problems due to their reaction with carbonate solvents20 of liquid electrolyte in the final cell, residual Li compounds can actively catalyse PVdF binder dehydrofluorination21–24 in the cathode slurry state thereby producing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unsaturated bonds in the polymer chain. PVdF binder thus actively produce HF through dehydrofluorination of its chain that, upon back reaction with basic Li compounds, ultimately leads to H2O. Whereas polymer CC double bonds are capable of polymerizing to crosslink PVdF chains, water is a non-solvent for PVdF polymer which can induce chemical slurry gelation. In this respect, NMC surface basic compounds were previously reported being removed by water25,26 or by acidic solutions27,28 washings or by direct LiPF6 addition into the cathode slurry for lab-scale formulations.16 Herein, a new approach is presented which tries to encompass the benefits of both slurry and electrolyte additives at the pilot plant scale. Electrolyte additives/salts, widely recognized in the literature for enhancing battery performance when dissolved in the electrolyte, are employed and they are used one step ahead, directly in the slurry mixing, to ease its processability. Indeed, established Li electrolyte salts, namely lithium trifluoroacetate (LiTFA), lithium bis(trifluoromethane)sulfonimide (LiTFSI) and lithium difluoro(oxalato)borate (LiODFB), are employed as slurry additives as a way to create a unique AM surface chemistry that, by complexation of the NMC622 and, therefore, by modification of its interaction with PVdF, could tune the rheology and suppress the gelation of high SCs slurries. LiTFSI is a thermally29 (melting temperature of 236 °C and not decomposing until 360 °C) and electrochemically stable (oxidation potential at 4.3 V vs. Li/Li+ in EC/DEC 1:1 (v/v) binary solution30 at 25 °C) electrolyte salt. It has a high dissociation constant (good even in low dielectric constant solvents) and high ionic conductivity owing to its bulky anion guarantying enhanced electron delocalization.31 This salt has been described in rechargeable Li-ion batteries since 1984,32 nevertheless its commercial use is not established due to its corrosion issue against the Al current collector.29 LiODFB is a new generation electrolyte Li-salt that is thermally stable29 up to 240 °C and displays extended
electrochemical stability (oxidation potential33 at 5.57 V vs. Li/Li+ in 1:1:1 wt EC/DMC/EMC electrolytes at 25 °C). When dissolved, LiODFB has as good an ionic conductivity as LiPF6 at room temperature and it promotes a stable Al passivation.29 LiODFB has an improved solubility in alkyl carbonate solvents compared to its parent LiBOB molecule34–36 and it promotes the formation of solid–electrolyte interfaces29 both on the anode33,37 and cathode38,39 side. Thanks to its strong passivation capabilities LiODFB is not only used as an electrolyte salt but also as an electrolyte additive capable of guaranteeing improved high temperature34/high voltage39 cell stability and superior battery cycle life.40 LiTFA is an additive reported for use in Li metal batteries41 to regulate the composition of the Li+ solvation sheath in the electrolyte and to promote stable and uniform SEI formation. This can significantly suppress Li dendrite formation for improved full-cell Coulombic efficiencies. Indeed, LiTFA preferential reduction compared to all the remaining electrolyte components can produce a stable Li metal passivation with uniform LiF and Li2O distribution.41 These kinds of additives were chosen with the intent to chemically modify the basic surface of NMC622 by exploiting the acidic nature29 of LiTFA42 and LiTFSI43 and the passivation capability of LiODFB. Moreover, these Li-salts were selected because of their well-known interaction within a cell when used as electrolyte salts or additives. The study provides a holistic view on the impact of slurry additives on parameters such as rheological properties, processability, particle distribution, as well as electrochemical performance indicators from coin-cell based NMC half-cell configurations and pouch-cell type graphite-NMC full cells.
C unsaturated bonds in the polymer chain. PVdF binder thus actively produce HF through dehydrofluorination of its chain that, upon back reaction with basic Li compounds, ultimately leads to H2O. Whereas polymer CC double bonds are capable of polymerizing to crosslink PVdF chains, water is a non-solvent for PVdF polymer which can induce chemical slurry gelation. In this respect, NMC surface basic compounds were previously reported being removed by water25,26 or by acidic solutions27,28 washings or by direct LiPF6 addition into the cathode slurry for lab-scale formulations.16 Herein, a new approach is presented which tries to encompass the benefits of both slurry and electrolyte additives at the pilot plant scale. Electrolyte additives/salts, widely recognized in the literature for enhancing battery performance when dissolved in the electrolyte, are employed and they are used one step ahead, directly in the slurry mixing, to ease its processability. Indeed, established Li electrolyte salts, namely lithium trifluoroacetate (LiTFA), lithium bis(trifluoromethane)sulfonimide (LiTFSI) and lithium difluoro(oxalato)borate (LiODFB), are employed as slurry additives as a way to create a unique AM surface chemistry that, by complexation of the NMC622 and, therefore, by modification of its interaction with PVdF, could tune the rheology and suppress the gelation of high SCs slurries. LiTFSI is a thermally29 (melting temperature of 236 °C and not decomposing until 360 °C) and electrochemically stable (oxidation potential at 4.3 V vs. Li/Li+ in EC/DEC 1:1 (v/v) binary solution30 at 25 °C) electrolyte salt. It has a high dissociation constant (good even in low dielectric constant solvents) and high ionic conductivity owing to its bulky anion guarantying enhanced electron delocalization.31 This salt has been described in rechargeable Li-ion batteries since 1984,32 nevertheless its commercial use is not established due to its corrosion issue against the Al current collector.29 LiODFB is a new generation electrolyte Li-salt that is thermally stable29 up to 240 °C and displays extended
electrochemical stability (oxidation potential33 at 5.57 V vs. Li/Li+ in 1:1:1 wt EC/DMC/EMC electrolytes at 25 °C). When dissolved, LiODFB has as good an ionic conductivity as LiPF6 at room temperature and it promotes a stable Al passivation.29 LiODFB has an improved solubility in alkyl carbonate solvents compared to its parent LiBOB molecule34–36 and it promotes the formation of solid–electrolyte interfaces29 both on the anode33,37 and cathode38,39 side. Thanks to its strong passivation capabilities LiODFB is not only used as an electrolyte salt but also as an electrolyte additive capable of guaranteeing improved high temperature34/high voltage39 cell stability and superior battery cycle life.40 LiTFA is an additive reported for use in Li metal batteries41 to regulate the composition of the Li+ solvation sheath in the electrolyte and to promote stable and uniform SEI formation. This can significantly suppress Li dendrite formation for improved full-cell Coulombic efficiencies. Indeed, LiTFA preferential reduction compared to all the remaining electrolyte components can produce a stable Li metal passivation with uniform LiF and Li2O distribution.41 These kinds of additives were chosen with the intent to chemically modify the basic surface of NMC622 by exploiting the acidic nature29 of LiTFA42 and LiTFSI43 and the passivation capability of LiODFB. Moreover, these Li-salts were selected because of their well-known interaction within a cell when used as electrolyte salts or additives. The study provides a holistic view on the impact of slurry additives on parameters such as rheological properties, processability, particle distribution, as well as electrochemical performance indicators from coin-cell based NMC half-cell configurations and pouch-cell type graphite-NMC full cells.
Experimental
Electrode materials
Slurries for the manufacturing of cathode coatings were prepared by mixing NMC622 (BASF, Germany) as an active material, CB (C-NERGY Super C65, Imerys Graphite & Carbon, Bodio, Switzerland) as a conductive additive, high Mw PVdF binder (Kynar® HSV 900, homopolymer, Mw = 900.000–1.300.000 g mol−1, Arkema Inc., France) and NMP (1-methyl-2-pyrrolidone, Emplura, Sigma-Aldrich, USA) as the solvent. NMC622 particle size distribution was measured with a laser scattering particle size analyser (LA-950V2, Horiba, Japan) as D10 = 6.12 μm, D50 = 9.46 μm and D90 = 14.17 μm. The specific BET surface of the NMC622 0.3334 m2 g−1 and C65 58.7 m2 g−1 was measured with a surface area analyser (Gemini VII, Micromeritics, USA). An NMC622 powder density of 4.63 g cm−3 was measured with a helium pycnometer (Pycnomatic ATC, Porotec, Germany). LiTFSI (99.95% trace metals basis, Sigma-Aldrich, USA), LiTFA (97%, Alfa Aesar, USA) and LiODFB (99.5%, Sigma-Aldrich, USA) were used as slurry additives.Mixing
First of all, the binder was dissolved in NMP to produce binder stock solutions with 9 wt% of PVdF with a dissolver mixer (Dispermat, VMA Getzmann GmbH, Germany) at 700 rpm for 1 h; subsequently, 15 min mixing at 700 rpm under vacuum was applied for degassing of stock solutions. The required amount of additive was already pre-dissolved in the NMP to obtain the correct additive concentration in the final slurry (Table 1). The additive concentration for each NMP-PVdF stock solution is related to the amount of PVdF. Corresponding slurry concentrations can be found in Table 1. For cathode slurries production an ARV 310 Thinky Planetary Mixer (Thinky Corporation, Japan) was employed. In the first step C65 and PVdF stock solution are mixed 3 times, each time for 2 min at 2000 rpm. Subsequently NMC622 is added and mixed 3 times for 1 min at 2000 rpm. Finally, the required amount of pure NMP to reach the target SC is added and mixed 14 times, each time for 1 min at 2000 rpm. The general composition of the slurries and the SCs employed are shown in Tables 1 and 3, respectively.| Sample name | Li-salt ADDITIVE (wt%) | NMC622 (wt%) | Carbon black C65 (wt%) | PVdF (wt%) | Anode balancing (%) |
|---|---|---|---|---|---|
| Additive-free | 0 | 97.00 | 1.50 | 1.50 | 117 |
| LiTFA 0.5 wt%PVdF | 0.007 | 96.99 | 1.50 | 1.50 | |
| LiTFA 1 wt%PVdF | 0.015 | 96.99 | 1.50 | 1.50 | |
| LiTFA 2 wt%PVdF | 0.030 | 96.97 | 1.50 | 1.50 | 113 |
| LiODFB 2 wt%PVdF | 0.030 | 96.97 | 1.50 | 1.50 | 113 |
| LiTFSI 2 wt%PVdF | 0.030 | 96.97 | 1.50 | 1.50 | 117 |
| LiTFA 8 wt%PVdF | 0.120 | 96.88 | 1.50 | 1.50 |
Rheology
Sedimentation of a non-transparent slurry can hardly be detected by optical means; therefore, rheology is the preferred characterization method to measure the flowability of a dispersion as well as its stability. A full characterization of a viscoelastic sample like an NMP-based slurry (NMP + NMC + PVdF + CB) starts with a steady-state flow test (shear stress against shear strain) to measure the viscosity of the system and it continues with dynamic oscillatory shear and frequency sweep measurements that can, respectively, probe the strength of the slurry aggregated state and its time dependent structure stability.1 Rheological characterization of the viscoelastic samples is carried out using a rheometer (MCR 302, Paar Physica, Austria) in stress-controlled mode with a plate-plate geometry (PP50, gap 500 μm) at 25 °C constant temperature. Steady-state flow, amplitude and frequency sweep tests are performed one after the other soon after finishing the mixing process. Five minutes resting time was applied to keep the residual normal force at an acceptable level at 25 °C until thermalization conditions are fulfilled before starting rheological measurements. Steady-state flow viscosities were measured applying shear stresses from 0 Pa to 1000 Pa to access the viscosity and shear thinning behaviour. Amplitude sweeps are performed at fixed frequency ω = 10 Hz within the strain region γ = (0.01–10000%). Frequency sweeps are measured within the linear viscoelastic range (LVR) of the sample, if existing, at fixed strain γ = 0.1% within the frequency window ω = (0.01–1000 Hz). From the critical strain value limiting the LVR (γcrit value beyond which the material response is increasingly non-linear viscoelastic) and the modulus of G′ within this region (where the equality 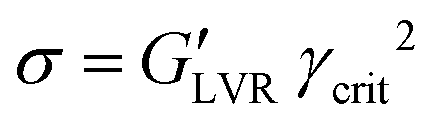 holds) it's possible to calculate the cohesive energy:44
holds) it's possible to calculate the cohesive energy:44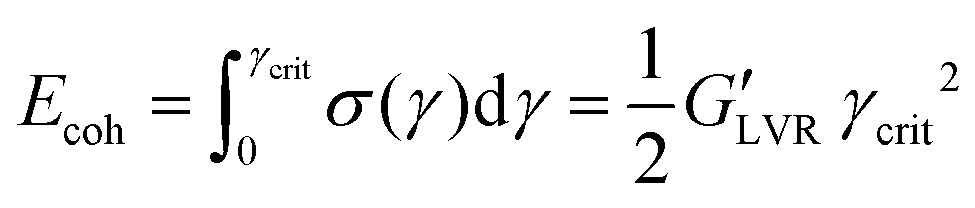 | (1) |
Electrode manufacturing
Electrode production was carried out by casting the NMP based slurries on a 20 μm Al foil employing a roll-to-roll coater (KTFS, Mathis AG, Switzerland) with doctor blade and two drying chambers set to 80 °C and 120 °C and a speed of 0.2 m min−1.After coating, a calendar (Saueressig GLK 200) was used at 50 °C to reduce the porosity of all the sheets to 33%. The adhesion strength of the coating before and after calendaring is measured with a zwikiLine Z2.5/TN with 10N load cell (Zwick-Roell) via 90° peel test (DIN EN 28510-1). 80 mm × 17 mm electrode stripes are fixed with double-sided adhesive tape gluing the NMC622 coating downwards to a metallic holder and pressed with a load of 200 kg for 2 s. The aluminium foil was stripped off from the electrode layer at a constant speed of 600 mm min−1 and the adhesion strength in N m−1 is evaluated at a 30 mm distance. A post-coating heat treatment of 10 min at 167 °C is applied to restore optimal adhesion when employing LiTFA and LiTFSI as slurry additives. After documenting weight and thickness, the through-plane resistance of the coating is measured with a milliohm meter in DC mode (RM3544, Hioki E.E. Corp., Japan) punching out 14 mm electrode disks and placing them between 14 mm copper cylinders under 6.5 kPa pressure. All cathode manufacturing data (composition, mass loading, adhesion, porosity, electrical resistance) are reported in Tables 1 and 2.
| Electrode sample | Mass loading (mg cm−2) | Thickness (μm) | Resistance (Ω) – cal | Adhesion (N m−1) – uncal | Adhesion (N m−1) – cal | Adhesion (N m−1) – after heat treatment |
|---|---|---|---|---|---|---|
| Additive free | 11.28 | 38.8 | 108 | 56.1 | 44.3 | — |
| LiTFA 2 wt%PVdF | 11.64 | 39.8 | 76 | 7.0 | 14.1 | 28.7 |
| LiODFB 2 wt%PVdF | 11.73 | 41.1 | 88 | 45.4 | 63.4 | — |
| LiTFSI 2 wt%PVdF | 11.21 | 38.4 | 73 | 13.1 | 10.3 | 32.4 |
Cell assembly and electrochemical characterization
:1 EC/DMC, 1 M LiPF6, BASF, Germany) as electrolyte.
Anode composition and balancing versus cathode is reported in Table 1. Two pouch cells are built and tested for each coating.
EIS spectra were recorded in regular intervals of ΔSOC = 10%, starting from SOC = 0% (SOC 0) to SOC = 100% (SOC 1) both during charging and discharging in the subsequent C/10 (charge/discharge) cycle; 2.5 V and 4.2 V are used as cut-off voltages. The electrochemical measurements are performed at room temperature using a multi-channel potentiostat (MPG2, BioLogic, France) system within 50 mHz to 20 kHz frequency range and applying a 10 mV AC voltage perturbation. After an initial galvanostatic formation cycle at 0.1C (1C = 175 mA h g−1). Each EIS measurement comprised of a forward scan (from high to low frequency) and a backward scan (from low to high frequencies), thus yielding two EIS spectra per SOC. Two EIS cells and spectra are investigated for each sample. The same procedure is applied with the 16 mm fatigued cathodes. The fatigued electrodes are retrieved from the cycled pouch cells (after 1000 cycles) and washed in DMC before EC-Ref cell assembly.
Results
Rheology
Coating of a 75.5 wt% SC reference slurry without additives was not possible as a result of very high slurry viscosity that also impeded rheological characterization with the used device. Nevertheless, coating of the additive-free cathode sample employing a lower 70.0 wt% SC slurry was possible owing to reduced viscosity, although only the very first part of the coating was usable to produce good quality electrodes. Indeed, a continuous casting process with the 70.0 wt% SC slurry was hindered by the fast slurry gelation (Fig. S2, ESI†). Li-salts are employed in small amounts and herein expressed in wt%PVdF, i.e. the amount of additive with respect to the weight of PVdF used in the slurry formulation.Rheological data of the additive-free 70.0 wt% SC slurry are reported in Table 3 for comparison. LiODFB, LiTFSI and LiTFA are tested as cathode slurry additives to investigate their effect on slurry stability and they were tested at 2 wt%PVdF. Flow curves can help predicting slurry stability by showing the existence of a yield point and viscosity changes upon days of storage can reflect the slurry segregation tendency (e.g. sedimentation). Steady-state viscosity measurements are performed keeping 50 s−1 as a suitable shear rate reference value lying well within the processability window of the coating device; this point can be optically localized following the grey vertical reference line in the flow curves plots. From the flow curves measured soon after mixing (Fig. 1(A)) for all the 2 wt%PVdF Li-salts slurries only minor changes can be measured in viscosity values and fall within the uncertainty of the measurement.
| Slurry sample | SC (wt%) | η (Pa s) at 50 s−1 – 0 h | η (Pa s) at 50 s−1 – 3 days | E coh (mJ m−3) – 0 h | E coh (mJ m−3) – 3 days |
|---|---|---|---|---|---|
| Additive-free | 70.0 | 0.52 | 4.1 | 40.6 | 97.6 |
| LiTFA 0.5 wt%PVdF | 75.5 | 3.5 | 12.4 | 25.0 | 104.7 |
| LiTFA 1 wt%PVdF | 75.5 | 2.9 | 6.9 | — | 35.1 |
| LiTFA 2 wt%PVdF | 75.5 | 2.5 | 3.9 | — | 27.4 |
| LiODFB 2 wt%PVdF | 75.5 | 2.7 | 6.0 | — | 39.0 |
| LiTFSI 2 wt%PVdF | 75.5 | 2.7 | 7.0 | — | 42.2 |
| LiTFA 8 wt%PVdF | 75.5 | 2.1 | 2.7 | — | 15.8 |
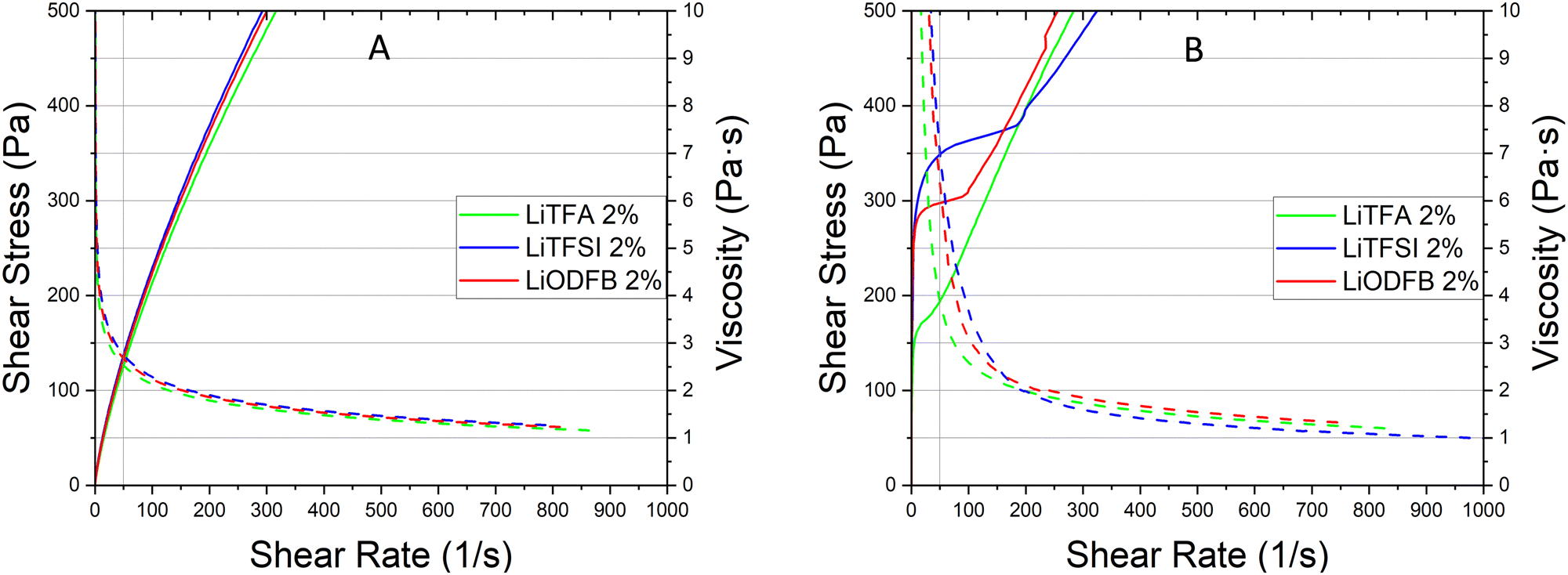 | ||
| Fig. 1 (A) Flow and viscosity (full and dotted lines) curves soon after mixing of NMP slurries at SC 75.5 wt% with NMC622 powder, high Mw PVdF, carbon black and 2 wt%PVdF LiTFA (green), 2 wt%PVdF LiODFB (red) and 2 wt%PVdF LiTFSI (blue). (B) Flow and viscosity curves of the same slurries after 3 days storage. | ||
In Fig. 1(B) the flow curve of the top solution for all the 2 wt%PVdF slurries after 3 days of resting time are plotted; the rise of the viscosity at every shear rate compared to the value measured directly after mixing as well as higher yield points with their extended vertical branches in the low shear rate domain are disclosing a stiff gel formation. A lower +55% slurry viscosity increase is detected after 3 days (Fig. 1(B)) for LiTFA (green curve), while LiODFB (red curve) and LiTFSI (blue curve) score much higher viscosity surges of +125% and +155%, respectively (Table 3). 2 wt%PVdF LiTFA proved itself to be a more effective gelation retarding additive; nevertheless 2 wt%PVdF LiODFB and 2 wt%PVdF LiTFSI were also capable of guaranteeing a smooth coating process thanks to slurries with a predominant flowing-like character soon after mixing.
Since a precise determination of the yield point from flow curves is not so easy, a better characterization of the slurry aggregated state can be provided by oscillatory shear measurements in the linear visco-elastic regions (LVR) of the samples. In amplitude sweeps, whenever slurry gelation sets in (e.g. high SC slurries), a large increase of the cohesive energy is normally measured. Amplitude sweeps measured soon after mixing (Fig. 2(A)) for all the salts at 2 wt%PVdF can, again, confirm that all the slurries have a predominant liquid-like character just after production, with LiTFA being the most liquid-like viscoelastic sample (green curve). Indeed, the frequency sweep measured soon after mixing, as reported in Fig. S1 (ESI†), also corroborates the dominant flowing-like character for all the slurry samples. From the amplitude sweeps measured after 3 days of storage (Fig. 2(B)) gelation driven effects become apparent with G′ > G′′ in the LVR region for each 2 wt%PVdF slurry sample, at variance with the soon after mixing measurements and thus confirming the existence of a dominant gel state. A higher Ecoh = 42.2 mJ m−3 gel strength upon storage is measured for 2 wt%PVdF LiTFSI (blue curve), while weaker cohesion states are calculated for 2 wt%PVdF LiODFB (red curve) with Ecoh = 39.0 mJ m−3 and for 2 wt%PVdF LiTFA (green curve) with Ecoh = 27.4 mJ m−3 (Table 3). All the slurries showed a favourable predominant liquid-like character coupled with low viscosities soon after mixing and, very importantly, gelation effects were actively tuned and delayed, so that they become visible only after 3 days of storage. In this latter respect, LiTFA and LiTFSI are, respectively, the best and the worst gelation controlling additive tested. An extended rheological investigation is then performed for LiTFA, the best gelation controlling additive. This Li-salt allowed casting when used at concentrations lower than 2 wt%PVdF. The slurry study was performed for the addition of various amounts of LiTFA in the range between 0.5 to 8 wt%PVdF. From the flow curves measured soon after mixing for the LiTFA additive (Fig. 3(A)) a gradual decrease of the gelation related slurry viscosity with increasing LiTFA concentration is detected (Table 3). All the flow curves of the freshly prepared NMP slurries display a pseudo-plastic behaviour with viscosity decreasing at increased shear rate; moreover, the very steep vertical slope of all the flow curves approaching the lower shear rate limit hint at the presence of a yield point. The existence of a yield point becomes apparent in the flow curves measured upon 3 days resting time for all LiTFA concentrations (Fig. 3(B)) that also show a neat viscosity rise at every shear rate compared to the viscosities, measured soon after mixing. Small slurry viscosity increases are detected after 3 days (Fig. 3(B)) for the two highest concentrations (+24% for 8 wt%PVdF (blue) and +55% for 2 wt%PVdF (green)), while a high gelation related viscosity surge is detected for the two lowest concentrations with +139% and +250% for 1 wt%PVdF (black) and 0.5 wt%PVdF (red), respectively (Table 3). Amplitude sweeps were measured, soon after slurry mixing, for all LiTFA-containing samples as in Fig. 4(A). It's shown in the figure that higher additive amounts above 0.5 wt%PVDF of additive yield slurries with a predominant liquid-like character, as demonstrated by the loss modulus G′′ being higher than the storage modulus G′ over the entire shear strain domain. The lowest additive concentration of 0.5 wt%PVDF shows an opposite behaviour with G′ dominating over G′′ in the low shear strain domain (Fig. 4(A), red curve). This is a clear indication that a predominant gel-like slurry has already formed upon mixing. From the amplitude sweep measurements after 3 days of slurry storage (Fig. 4(B)) it's apparent that all the samples created a gel network, which is represented by G′ > G′′ in the LVR region. By calculating the energy of the aggregated state upon 3 days of storage with eqn (1) for each LiTFA treated slurry (Table 3) there is clearly a trend which sees a gradual decrease of the gel strength going from 0.5 wt%PVdF with the highest cohesion energy down to 8 wt%PVdF with the lowest. Interestingly, the lowest concentration of 0.5 wt%PVdF is the only one having a G′ > G′′ LVR region already upon mixing; its cohesion energy is Ecoh = 25.0 mJ m−3. Finally, the time dependent processes of the viscoelastic system can be measured in the LVR by applying a fixed deformation at different frequencies in a frequency sweep measurement. This measurement is like a unique fingerprint of the slurry structure that can be very sensitive to formulation changes and reveals its time related properties like shelf-life stability (long-time storage), idle consistence or segregation tendency3. In the long-time scale limit (ω ≈ 0), whenever the loss modulus G′′ is dominating over the storage modulus G′, the liquid-like character of the visco-elastic system prevails (more energy will be dissipated through heat production than stored elastically) and long shelf-life stability cannot be expected. From the frequency sweep soon after mixing (Fig. 5) for all the LiTFA additive amounts, the time response of the visco-elastic slurry system is explored: all the measurements show the presence of a low ω (ω ≈ 0) G′ > G′′ inversion point which is shifting towards much higher frequencies for the minimum amount 0.5 wt%PVdF of LiTFA used (red curve). The minimum amount of 0.5 wt%PVdF LiTFA already has a predominant solid gel-like character all through the frequency domain. This feature indicates that the slurry could be stable upon prolonged storage and, if the strength of its aggregated state is high enough, sedimentation of active material is not expected. Nevertheless, coating problems can readily occur when the strength of the slurry aggregated state is too high. Hence, in conclusion, LiTFA, LiTFSI and LiODFB employed as slurry additives enabled the casting of regular coatings without gelation artefacts (at variance with the additive-free electrode, Fig. S2, ESI†) in a continuous roll-to-roll process by favourably tuning the rheology at the challenging 75.5 wt% SC.
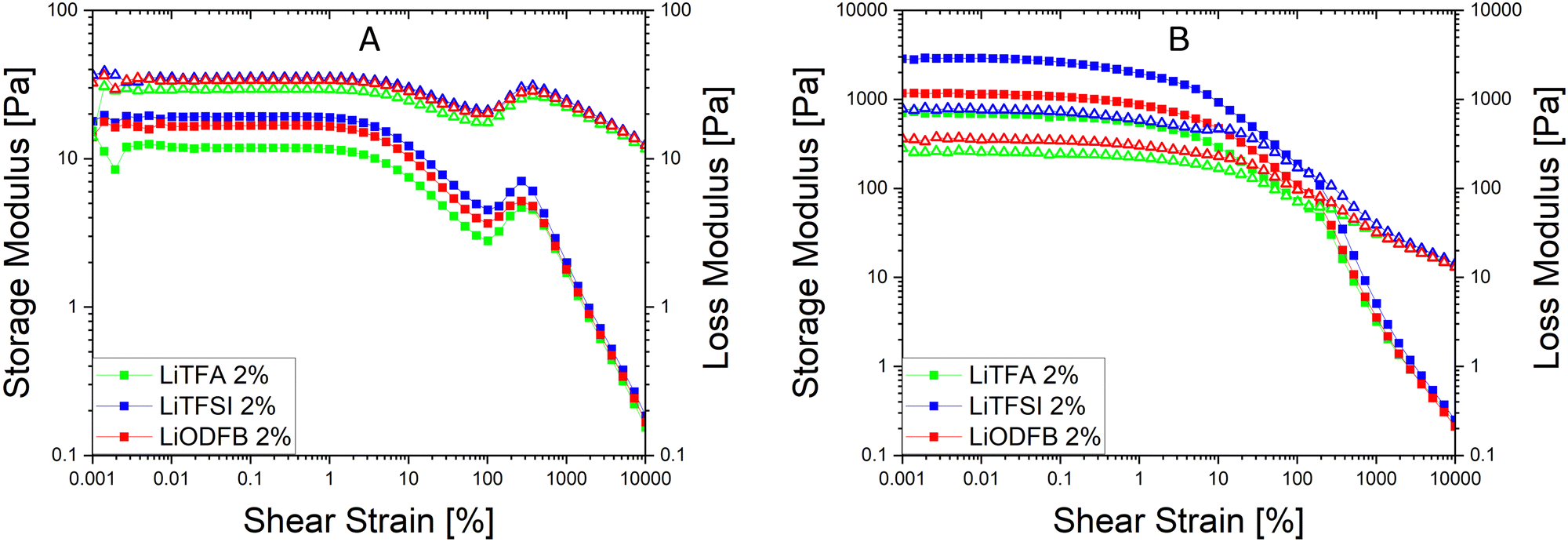 | ||
| Fig. 2 (A) Amplitude sweeps (squares = G′, triangles = G′′, angular frequency ω = 10 s−1) soon after mixing of NMP slurries at SC 75.5 wt% with NMC622 powder, high Mw PVdF, carbon black and 2 wt%PVdF LiTFA (green), 2 wt%PVdF LiODFB (red) and 2 wt%PVdF LiTFSI (blue). (B) Amplitude sweeps of the same slurries after 3 days of storage. | ||
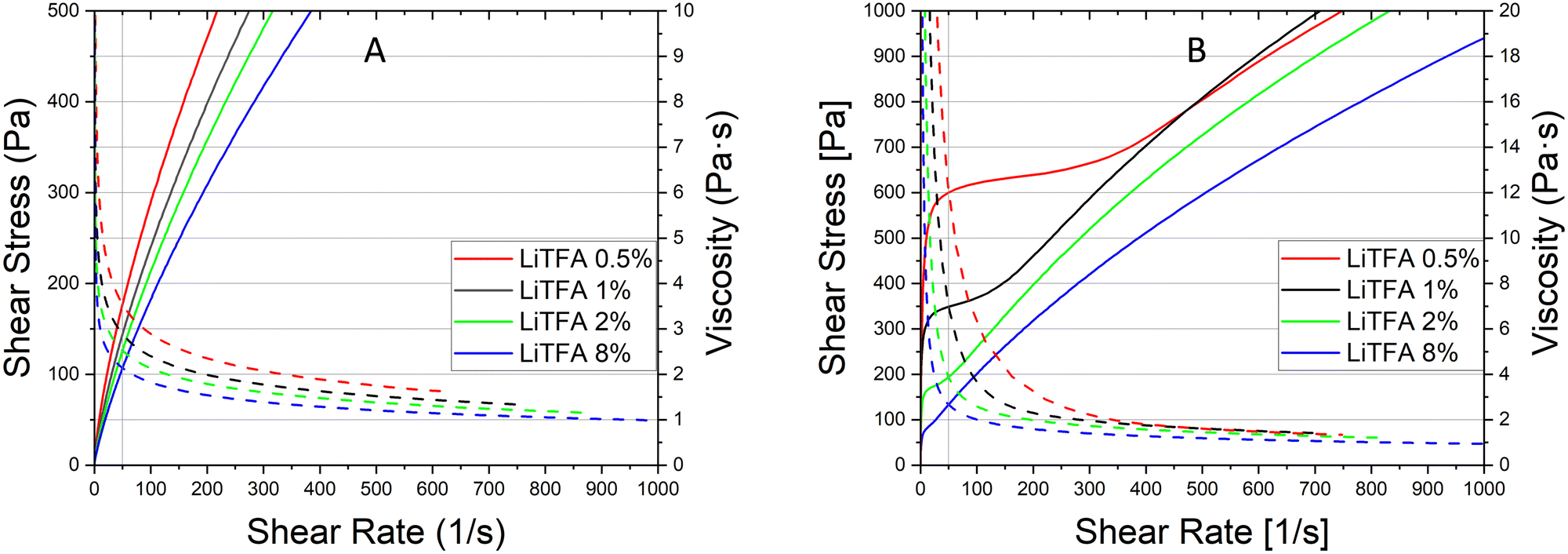 | ||
| Fig. 3 (A) Flow and viscosity (full and dotted lines) curves soon after mixing of NMP slurries at SC 75.5 wt% with NMC622 powder, high Mw PVdF, carbon black and LiTFA at 8 wt%PVdF (blue), 2 wt%PVdF (green), 1 wt%PVdF (black), and 0.5 wt%PVdF (red). (B) Flow and viscosity curves of the same slurries after 3 days storage. | ||
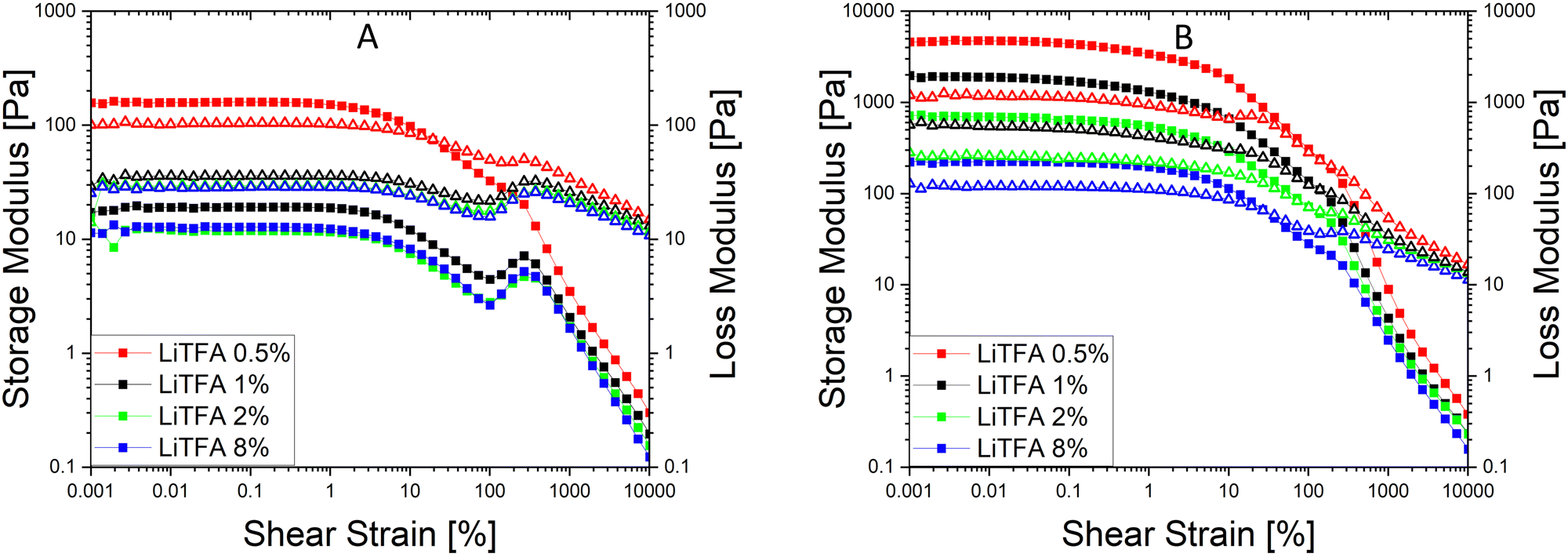 | ||
| Fig. 4 (A) Amplitude sweeps (squares = G′, triangles = G′′, angular frequency ω = 10 s−1) soon after mixing of NMP slurries at SC 75.5 wt% with NMC622 powder, high Mw PVdF, carbon black and LiTFA at 8 wt%PVdF (blue), 2 wt%PVdF (green), 1 wt%PVdF (black), and 0.5 wt%PVdF (red). (B) Amplitude sweeps of the same slurries after 3 days of storage. | ||
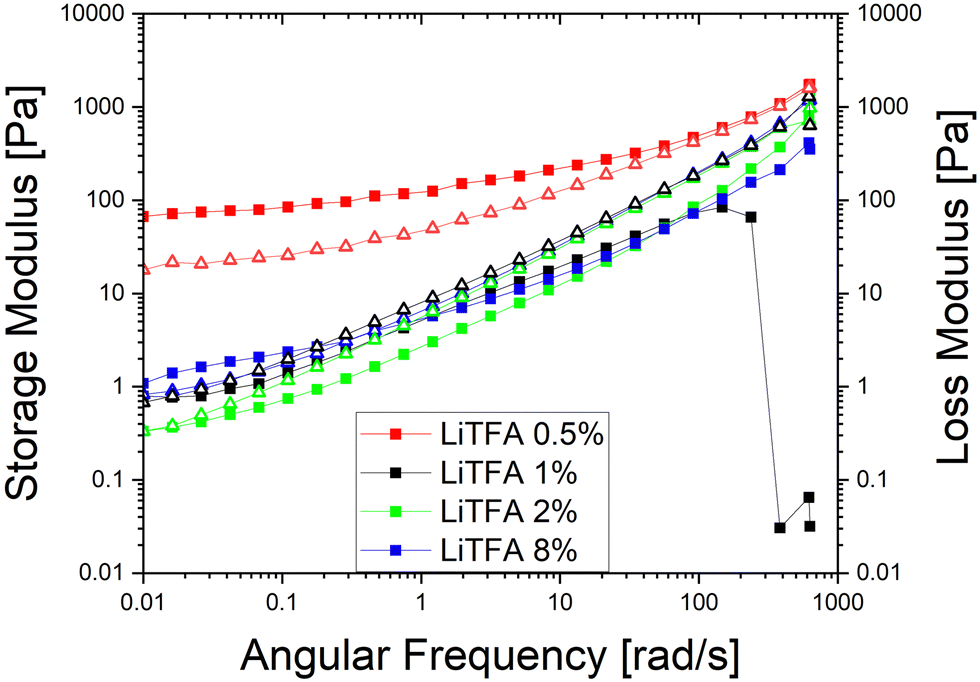 | ||
| Fig. 5 Frequency sweeps (squares = G′, triangles = G′′, strain γ = 0.1%) measured soon after slurry mixing of slurries at SC 75.5 wt% with NMC622 powder, high Mw PVdF, carbon black and LiTFA at 8 wt%PVdF (blue), 2 wt%PVdF (green), 1 wt%PVdF (black), and 0.5 wt%PVdF (red). | ||
Electrochemistry
Cycling experiments were carried out on NMC-graphite full cells (pouch-cell format) as described in the Experimental section. The electrochemical performance of 2 wt%PVdF Li-salts containing cathodes was tested against an additive-free electrode as a reference sample (cathode and cell manufacturing data are reported in Tables 1 and 2). During the formation cycles at a low C-rate of C/20, the cells achieved a mean discharge capacity of 165.6 ± 1.9 mA h g−1 with a narrow spread amongst the different samples. The rate-capability test (Fig. 6(A)) between C/2 and 5C (after 2 C/20 formation cycles) shows comparable rate stability for all coatings, including the reference (black curve). Slight capacity retention differences at the highest rates of 3C and 5C fall within the uncertainty of the measurement (control experiment in Fig. S6, ESI†). The long-term cycling (Fig. 6(B)) further demonstrates faster degradation for all additive-containing cells compared to the reference. The cathode coating containing LiTFSI performs close to the additive-free sample in terms of capacity retention. To gain deeper insights on high C-rate and long cycling behaviour, differential capacity dQ/dV curves were analysed, as shown in Fig. 7. The first symmetric 0.05C/0.05C (charge/discharge) formation step is plotted as a black curve, while asymmetric 1C/3C curves are plotted from cycle 40 (dark red) until cycle 1000 (lightest red shade) for selected cycles. The high discharge rates (3C) caused a broadening of the signals. Two main oxidation peaks have been reported for NMC622 half cells at 3.64 V and 3.74 V vs. Li+/Li associated with the oxidation of Ni2+/3+ to Ni4+ and a unique 3.66 V vs. Li+/Li reduction peak; having Ni a formal mixed valence state of +2 and +3.49 Whenever NMC622 is cycled in a full cell the aforementioned oxidation peaks split into multiple peaks due to the coupling with graphite anode multi stage insertion reactions;50–52 the area under each peak is proportional to the capacity delivered by each correspondent electrochemical process.47 Changes in the first upper oxidation peak with an onset at 3.5 V (black curve) are reported being mainly caused by reactions at the graphite anode side.47,53,54 The second main peak at around 3.6 V (black curve) with its left (3.57 V) and right (3.71 V) satellite peaks are dominated by the sequential Ni redox reactions of NMC622. With increasing cycle number, their intensity/peak area decreased, which is a sign of loss of active material that can be caused by either structural changes while cycling, dissolution of transition metal ions or surface film modification.47 In particular the depression of the peak at 3.71 V while cycling is associated with changes of the lithium inventory due to SEI formation and other electrolyte decomposition driven side reactions.50,53 Looking at the anodic upper portion of dQ/dV plots and moving from the formation step (black curve) to 1C charging (red shaded curves) during battery cycling/aging all peaks are broadening and moving to higher potentials. The main peak at around 3.6 V (black curve) remains visible together with the peak at 3.5 V (black curve) and the peak at 3.71 V (black curve), which are still detectable as left and right shoulders of the main peak. Two out of three cells containing Li-salt additives show a pronounced main peak area decline, which correlates with an increased loss of active material through cycling compared to the reference. Specifically, the 2 wt%PVdF LiTFSI sample and the reference behaved largely alike, showing noticeable smaller degrees of degradation as compared to the 2 wt%PVdF LiODFB and 2 wt%PVdF LiTFA samples. Interestingly, an additional very first, small anodic peak (green arrows) that was not present in the formation step (black curve) develops through long-cycling for LiTFA and LiODFB and, less pronouncedly for LiTFSI.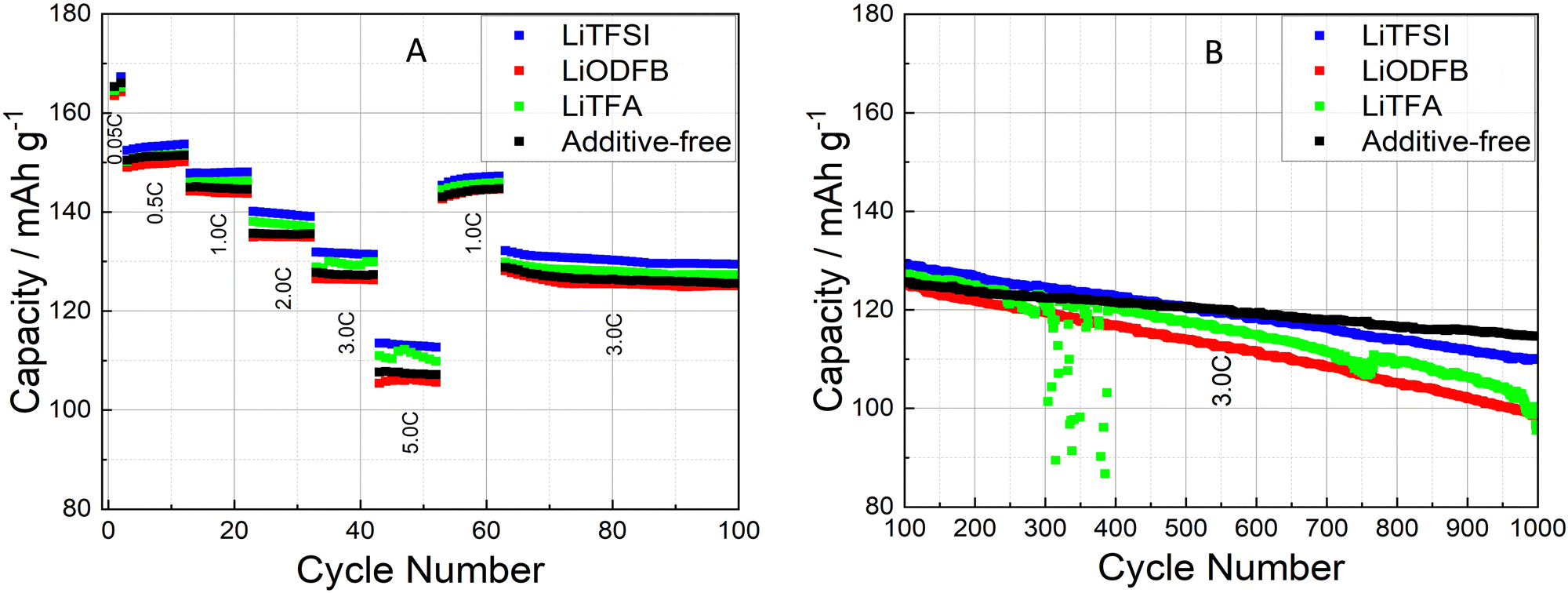 | ||
| Fig. 6 High C-rate performance of graphite-NMC full cells (A) and long-term cycling (B) for additive-free (black), 2 wt%PVdF LiTFA (green), 2 wt%PVdF LiODFB (red) and 2 wt%PVdF LiTFSI (blue) cathodes. Green outliers due to temporary potentiostat failure. | ||
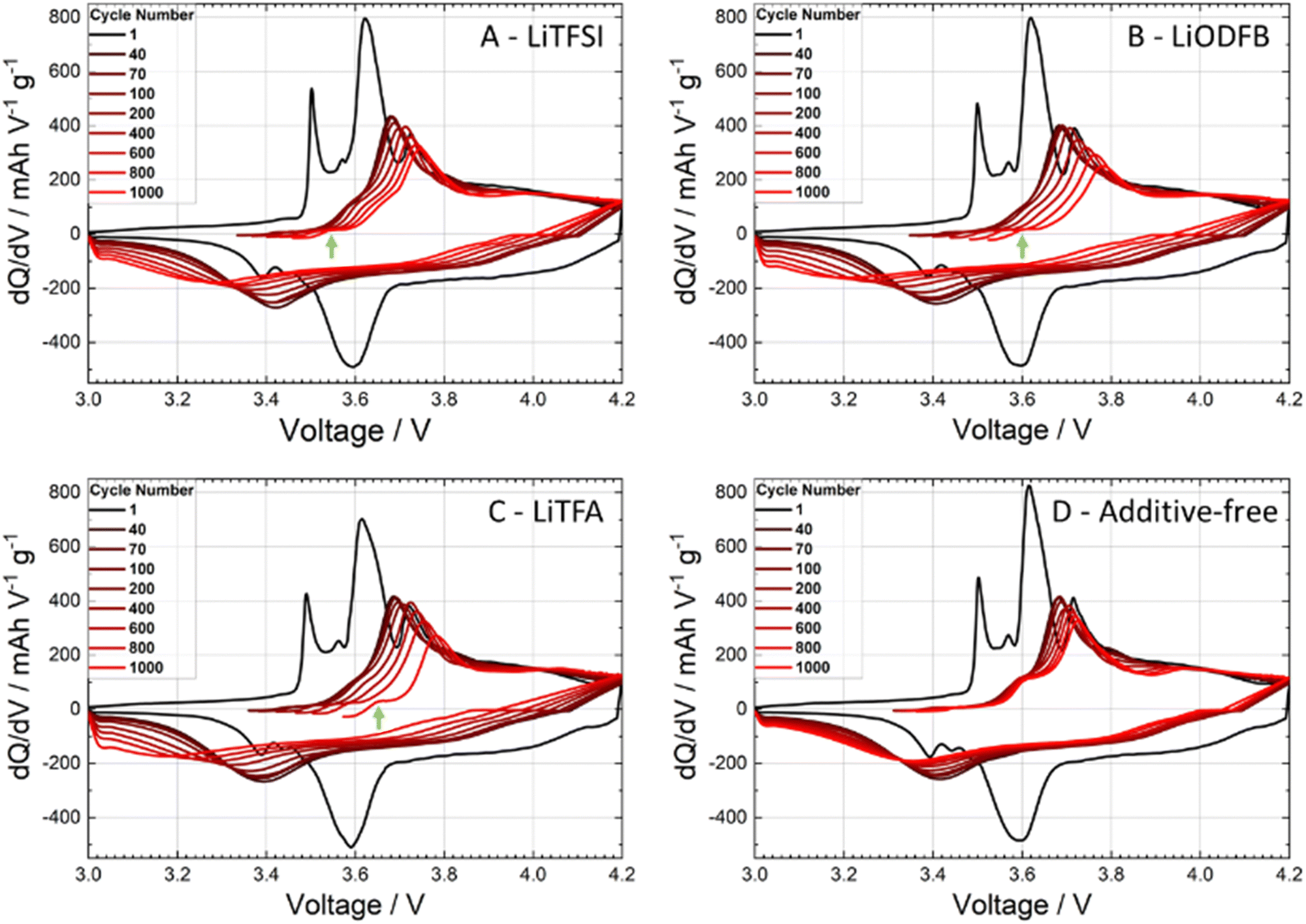 | ||
| Fig. 7 dQ/dV curves at the 0.05C formation step (black) and at 1C/3C (charge/discharge – red shades) during cell cycling for (A) 2 wt%PVdF LiTFSI, (B) 2 wt%PVdF LiODFB, (C) 2 wt%PVdF LiTFA and (D) additive-free cells at cycles 1 (black), 40, 70, 100, 200, 400, 600, 800, and 1000 (lightest red). | ||
This peak, which is almost not present for the reference cell, could probably be caused by anodic Li plating.19 From the voltage difference between the main upper and lower anodic/cathodic redox peaks (at around 3.6 V for the black curve) cell polarization effects can be followed through cycling (Table S1, ESI†); a higher polarization (ΔV) of the cell is associated with a higher overall cell impedance and lower cycling performances. As shown in Table S1 (ESI†), all cells have similar ΔV up to cycle 100, while from cycle 200 onwards, all additive-containing cells developed higher overpotentials. In this respect 2 wt%PVdF LiTFSI is, again, closest to the reference performance, while the 2 wt%PVdF LiODFB and 2 wt%PVdF LiTFA samples developed notably higher polarization, in accordance with their faster capacity fading.
In order to gain a more detailed understanding of possible differences in the electrode processes, electrochemical impedance spectroscopy (EIS) is employed as a way to gauge the influence of the slurry additives on the electrochemical behaviour and on the electrode–electrolyte interface formed for different cathode formulations. EIS measurements were performed in a first series of experiments on fresh cathodes and, subsequently, on fatigued cathodes after retrieval from cycled pouch cells after 1000 cycles, using a 3-electrode setup with Li-metal as the counter and pseudo-reference electrode. After one conditioning cycle EIS was performed during OCV sequences between constant current intervals of ΔSOC = 0.1 on a full charge–discharge cycle. In Fig. 8 the EIS data for all four fresh cathodes is shown at SOC = 0.8. The spectra are representative for the impedance at SOC > 0.5. Although minor changes in the Nyquist plot are still observed in the EIS spectra at high SOC, they are notably smaller than in the early stages of the charge process (SOC < 0.5). The complete dataset for each sample from SOC = 0.1 to SOC = 1 is presented in Fig. S7–S10 (ESI†). It can be readily seen that the electrode impedance at SOC = 0.1 and SOC = 0.2 deviates strongly in the low frequency range. This effect is commonly ascribed to diffusion processes. The lower frequency charge transfer arc becomes apparent for electrodes at SOC = 0.2 or higher and, as also reported by others,55–58 its impedance rapidly decreases with increasing SOC. The curves have been fitted with the SCRF48 model (inset in Fig. 8) in which Rel is the uncompensated resistance between working and reference electrodes. R0 and QCPE0 are ascribed to the contact impedance between the metal current collector and the active electrode composite. Specifically, R0 is the resistance of transferring electrons from the metallic substrate to the percolated conductive network of the electrode composite (CB and/or AM contacting particles) and QCPE0 is the constant phase element which accounts for the double layer that is built up at the current collector metal interface (i.e. Al2O3 or any passivation layer on the Al foil). Rf and QCPEf are linked to surface film formation on the carbon-black and active material particles at high potentials originating from electrolyte degradation (cathode–electrolyte interphase, CEI, formation) or changes at the particle interface, e.g. by oxygen evolution reactions. The use of the SCRF model taking into account passivation or surface layers appeared to be the most suitable option for the analysis of the coatings due to the well-known reactivity of NMC622 with any moisture or air during processing and storage.17R1 and QCPE1 represents the charge transfer resistance for the insertion reaction and the corresponding double layer capacitance. In series to R1 an open Warburg element Wo is used in order to express the electrolyte diffusion resistance.
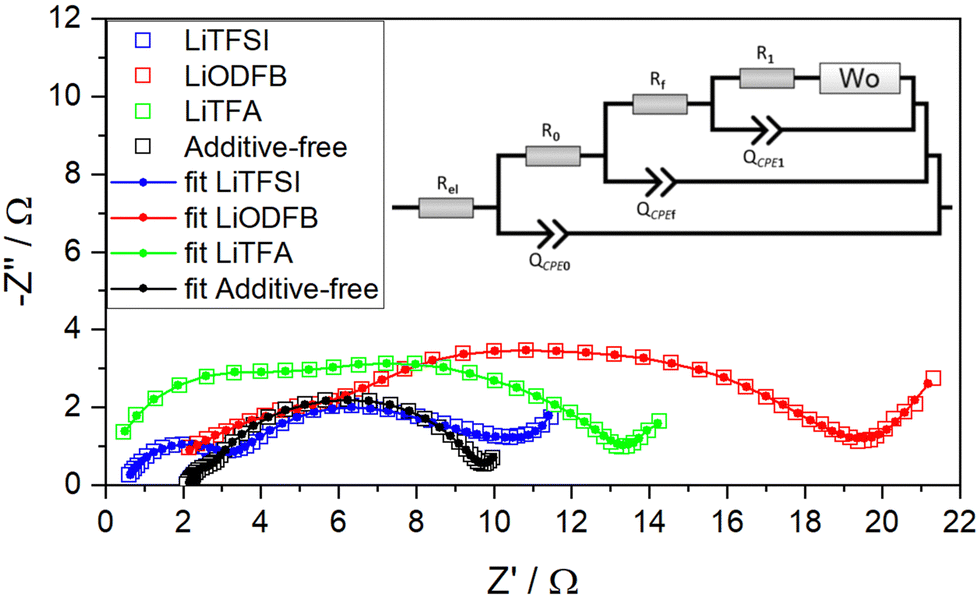 | ||
| Fig. 8 Measured Nyquist plot (empty squares) and simulated (solid lines and dots) impedance spectra of fresh NMC622 electrodes at 0.8 SOC for additive-free (black), 2 wt%PVdF LiTFA (green), 2 wt%PVdF LiODFB (red) and 2 wt%PVdF LiTFSI (blue) cathodes. Frequency range from 50 mHz (right) to 20 kHz (left). The model used for simulation is reported as tinset. The corresponding Bode representation is plotted in Fig. S11–S14 (ESI†). | ||
Note that during the equivalent circuit fitting, the CPE exponent α was limited to a value of 0.75 (for an ideal capacitor α would be 1). The fact that α approached the lower set limit indicated a strong deviation that is typically explained by combinations of several non-ideal capacitive contributions such as surface roughness, distribution of reaction rates and varying thicknesses of surface layers.7 The Nyquist plot for the additive-free sample is depicted as a black curve in Fig. 8, where it shows a small high frequency arc due to the contact impedance together with a broadened semicircle extending from medium to low frequencies. This latter semicircle encompasses both surface film impedance and charge transfer impedance, respectively. It is apparent how the introduction of additives in the coatings caused an overall increase in the charge-transfer and interfacial impedance compared to the additive-free reference. In particular, as can be seen from the LiODFB- (red), LiTFSI- (blue) and LiTFA-containing (green) samples in Fig. 8, every additive addition causes a notable increase in the high frequency arc, while the medium-low frequency arc expands notably only when employing LiODFB and LiTFA, respectively. Interestingly, if LiTFSI is considered, the medium to low frequency response remains very close to the reference (additive-free) sample. The model parameters obtained from the equivalent circuit fits are shown in Table 4 and indicate that the major impedance increases with respect to the additive-free sample originated from the contact resistance R0. R0 increases in magnitude in the order additive-free < LiTFSI < LiODFB < LiTFA. Also, the active material surface film resistance Rf is increased, especially for LiODFB. For LiTFA- and LiTFSI-containing samples the values are notably smaller, but higher than for the additive-free sample. The charge transfer resistance R1 showed slightly higher values for all additive-containing cathodes. Finally, the uncompensated resistance Rel in the additive-containing samples show lower values than the additive-free formulation. According to the SEM images of ion-milled cross-sections of pristine calendered electrodes shown in Fig. S3–S5 (ESI†), the addition of additives did not alter the electrode morphology in the dry state.
| Sample | R el (Ω) | R 0 (Ω) | Q CPE0 (S × sα) | α CPE0 | R 0 Q CPE0 (sα) | R f (Ω) | Q CPEf (S × sα) | α CPEf | R f Q CPEf (sα) | R 1 (Ω) | Q CPE1 (S × sα) | α CPE1 | R 1 Q CPE1 (sα) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Additive-free | 1.3 | 0.5 | 9.8 × 10−5 | 1 | 4.9 × 10−5 | 3.9 | 2.4 × 10−3 | 0.84 | 9.4 × 10−3 | 2.1 | 2.7 × 10−2 | 0.75 | 5.7 × 10−2 |
| LiTFA | 0.3 | 5.7 | 2.2 × 10−4 | 0.89 | 1.3 × 10−3 | 6.2 | 4.3 × 10−3 | 0.78 | 2.7 × 10−2 | 3.3 | 4.9 × 10−2 | 0.86 | 1.6 × 10−1 |
| LiODFB | 0.4 | 3.2 | 3.9 × 10−4 | 0.75 | 1.2 × 10−3 | 10.0 | 2.6 × 10−3 | 0.75 | 2.6 × 10−2 | 4.7 | 4.2 × 10−2 | 0.75 | 2.0 × 10−1 |
| LiTFSI | 0.5 | 2.0 | 4.5 × 10−4 | 0.78 | 9.0 × 10−4 | 4.6 | 8.0 × 10−3 | 0.75 | 3.7 × 10−2 | 3.6 | 1.2 × 10−1 | 0.75 | 4.2 × 10−1 |
Post-mortem SEM images of ion-milled cross-sections (Fig. 9) are also taken for fatigued cathodes, which were retrieved from their respective cycled pouch cells after 1000 cycles. As clearly illustrated by the SEM images, electrode delamination after long-cycling is present for all the 2 wt%PVdF additive-containing electrodes, whilst intact Al/composite contacting is retained only by the additive-free electrode. Moreover, cracking of AM after long cycling is detected only for the additive-free sample in Fig. 9(D). EIS measurements were also performed on fatigued cathodes to shed more light towards cathodic capacity fading mechanisms after 1000 cycles, as shown in Fig. 9. For each electrode formulation two different electrode samples were transferred into the 3-electrode setup. Compared to the previous measurements the variance between electrodes of the same type (and from the same electrode), has considerably increased in the mid- to low-frequency regions of the additive-containing electrode formulations. The deviation is particularly large in the case of the LiODFB-containing samples. Therefore, a qualitative discussion of EIS is pursued. The EIS results show consistent changes in the high- and mid-frequency region that was previously ascribed to the contact impedance, which expands notably in fatigued samples and, in particular, when Li-salts are used as additives. Indeed, the breadth of the high frequency arc, to which the contact resistance is most likely a major contributer, is notably lower for the fatigued additive-free cathode, when compared to the other samples. Among the additive-containing samples, 2 wt%PVdF LiTFSI shows the lowest contact resistance, which is consistent with the measurements conducted after one cycle (Fig. 8), followed by 2 wt%PVdF LiTFA, and by the 2 wt%PVdF LiODFB sample that suffered a significant increase in cell impedance. Apparently, higher contact resistances when Li-salts are used correlate with the loss of contact at the Al/composite interface, as shown in the post-mortem SEM cross-sections in Fig. 9 and Fig. S20 (ESI†). When considering the medium to low frequencies semicircle due to surface film and charge transfer impedance, the arc widths for 2 wt%PVdF LiTFSI, 2 wt%PVdF LiTFA and the additive-free cathode are similar and their differences fall within the uncertainty of the measurements.
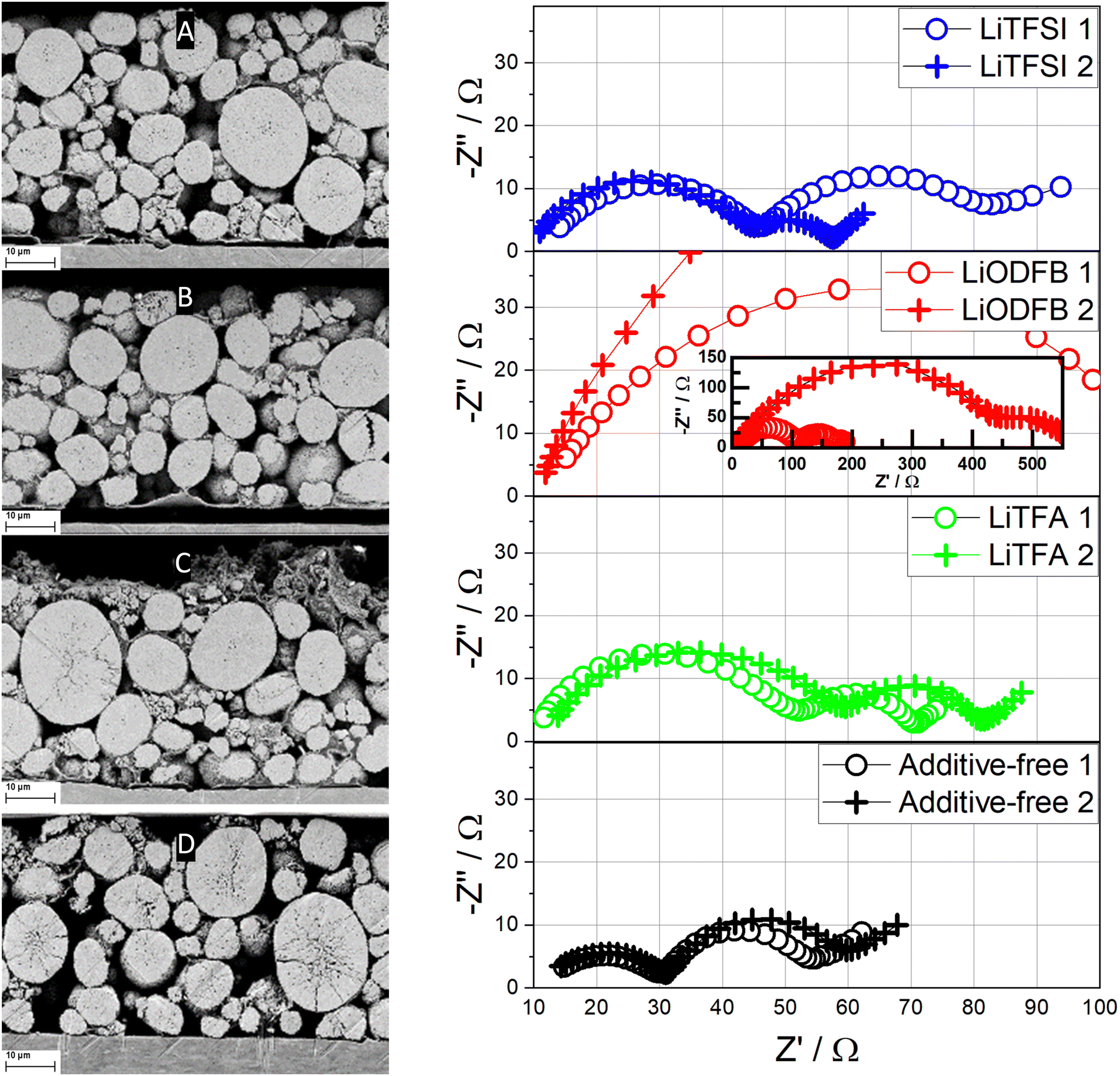 | ||
| Fig. 9 Left: Post-mortem SEM images of ion-milled cross-sections of fatigued electrodes (after 1000 cycles) of 2 wt%PVdF LiTFSI (A), 2 wt%PVdF LiODFB (B), 2 wt%PVdF LiTFA (C) and additive-free (D). Right: Nyquist plots (two different cells) of fatigued (after 1000 cycles) NMC622 electrodes at 0.8 SOC for the 2 wt%PVdF LiTFSI, 2 wt%PVdF LiODFB, 2 wt%PVdF LiTFA and the additive-free cathodes. Frequency range from 50 mHz (right) to 20 kHz (left). The corresponding Bode representations are plotted in Fig. S15–S18 (ESI†). | ||
Discussion
In the first section of this work, it was shown that Li-salts can actively delay the gelation and reduce the viscosity of PVdF-based slurries, thus allowing slurries with higher solid content (SC). Based on molecule adsorption experiments reported by others,59 the complexation of basic sites on the NMC622 surface by chemical coordination with Li-salts is envisaged as the main mechanism behind the observed slurry rheology changes. Li-salts in the slurry provide Li+ ions to the basic oxygen sites on the NMC622 surface. Hereby, the electron-accepting Lewis-acidic Li+ ions can neutralize the electron-donating Lewis-basic oxygen sites on the NMC622 surface by means of electrostatic acid–base interactions. PVdF is known to be a Lewis-acidic polymer60 that, owing to its dipole moment between neighbouring CF2 and CH2 units, typically adsorbs only onto Lewis-basic oxide surfaces.61 Hence, Li+ ions can change the net AM surface charge by turning negatively charged Lewis-basic oxygen sites into positively charged electron-accepting Lewis-acidic sites. As a result, PVdF adsorption and therefore bonding to the AM surface can be partially prevented owing to the Li+ ions in the slurry. Moreover, it should be considered that surface adsorbed Li+ ions on the NMC622 surface may also be coordinated by their respective anions, thus resulting in different PVdF–surface interactions depending on the chemical nature of the anion. The binding strength and surface coverage may depend on the specific Li-salt anion used and on its peculiar properties such as electron-donating (basic) character or polarizability. Each Li-salt thus creates a unique surface chemistry, which is summarized in Fig. 10.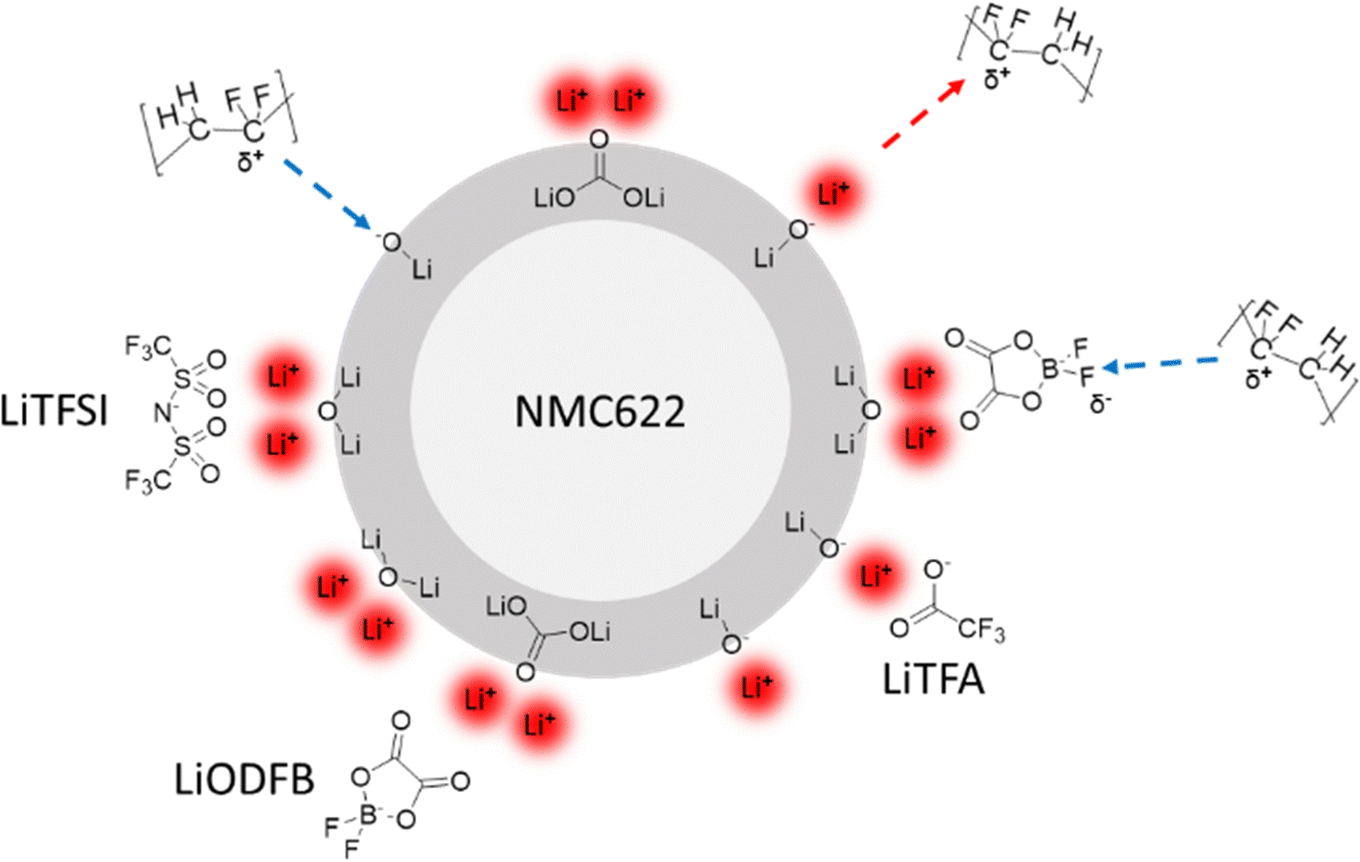 | ||
| Fig. 10 Scheme of Li+ ions neutralizing the basic surface sites (dark grey region – not in scale with the particle size) of NMC622 in the slurry. Adsorbed Li+ ions are complexed by Li-salts’ anions. PVdF attractive and repulsive acid–base interactions with surface species are represented by blue and red dotted arrows, respectively. | ||
Therefore, Li-salts can actively hinder chemical slurry gelation by neutralization of the NMC622 surface basic groups and, at the same time, they can also reduce physical slurry gelation by partially preventing PVdF binder adsorption.
From an electrochemical perspective, the impedance and capacity retention improve in the order LiODFB < LiTFA < LiTFSI < additive-free. When comparing the uncompensated resistance Rel, it is the additive-containing samples that show a lower Rel than the additive-free formulations. This could be due to additional Li-salts in in the electrode pores, which locally improve the ionic conductivity. In terms of contact-resistance, R0, the lowest value was found for the additive-free formulations. Interestingly, it is the LiTFSI and LiTFA sample that showed a slightly improved rate-capability over the additive-free and LiODFB-containing sample, despite the fact that R0 was highest for LiTFA. From the EIS data it is apparent how the introduction of additives in the coatings caused an overall increase in the charge-transfer (R1) and interfacial impedance (Rf) compared to the additive-free reference sample. As shown in Table 4, the surface film resistance Rf is more than doubled in the case of LiODFB and significantly increased for LiTFA, whereas with LiTFSI as slurry additive, Rf is close to the value observed for an additive-free electrode. We hypothesize that different NMC622 surface adsorption of the three Li-salt anions could correlate with diverse surface film resistance Rf values, so that, in particular, the enhanced adsorption of ODFB− at the NMC622 surface may be related with the highest Rf for this additive. Moreover, ODFB− is less likely to form a passivation layer (CEI) owing to its better electrochemical stability and thus not decomposing while cycling. Generally, the use of 8 fitting parameters can easily lead to an over-parametrization, especially when the relaxation frequencies of processes in the mid to low frequency range are similar. For instance, the Nyquist plot for the additive-free sample, depicted as a black curve in Fig. 8, shows a small high frequency semicircle due to the contact impedance followed by a broadened semicircle extending from medium to low frequencies. In samples containing slurry additives, the expanded mid to low frequency arc is even more clearly visible. With exception of the LiODFB sample, where the two RQ circuit elements in the mid to low frequency domain can be distinguished, it is challenging to separate individual contributions unambiguously for the other formulations and thus larger uncertainties in the parameter values are to be expected when comparing their values and interpreting their impact on the overall electrochemistry. Specifically, this can be expressed by the magnitude of the relaxation frequencies, which only applies strictly when capacitors are used (not CPEs). However, for α-values > 0.75, the product of R and Q should yield an estimate of the relaxation frequency according to eqn (2).
| R∙Q = τα = (RC)α | (2) |
As can be seen in Table 4 the relaxation frequencies of the individual RQ circuit couples in the equivalent circuit are about an order of magnitude apart from each other (R0 one order apart from Rf and Rf one order of magnitude apart from R1, respectively). Only in the additive-free sample RfQCPEf and R1QCPE1 are separated by only half an order of magnitude, while the R0QCPE0 element is more clearly separated from the RfQCPEf element by two orders of magnitude. It should be highlighted that values found for Rf and R1 are in same range between 2 to 10 Ω, thus making it challenging to derive clear trends. In more general terms it can be said that the sum of Rf and R1(Rf + R1) correlate to some extent with the long-term capacity retention.
Similar to the NMC surface, the Al current collector exhibits a native Al2O3 surface that is basic by nature62 and thus possesses a population of unsaturated Lewis oxygen sites that can be coordinated in the same way as shown in Fig. 10 for the NMC surface. Augmented Al surface passivation due to Li-salts adsorption could explain higher R0 values compared to the additive-free reference and, in particular, LiODFB and LiTFA-containing samples that show the highest R0. We hypothesize that the surface adsorbed Li+ ions onto the native alumina layer at the current collector are still strongly interacting with the ODFB− anions, due to their pronounced polar nature. As a result, adsorption of PVdF is facilitated, owing to the overall negative surface charge generated by the adsorbed anions. In this case PVdF might coordinate adsorb ODFB− anions, as depicted in Fig. 10 for the NMC case. Whereas for LiTFSI and LiTFA, PVdF adsorption is more pronouncedly hindered due to the non-neutralized and positively charged Al surface, which is most likely complexed by adsorbed anions only to a lesser extent. This speculated mechanism agrees with the much better adhesion values of the LiODFB coating (as good as for the additive-free electrode) and with the much lower values measured for LiTFSI and LiTFA (Table 2). Moreover, replacement of adsorbed Li-salts’ anions at the Al current collector surface (i.e. at the Al/composite electrode interface) with species of the electrolyte (carbonate solvents/LiPF6) while cycling can explain the delamination tendency observed in the post-mortem SEM cross-sections (Fig. 9) of Li-salts containing electrodes. EIS measurements were repeated on fatigued cathodes after 1000 cycles (Fig. 9) to shed more light on cathodic degradation mechanisms. EIS clearly shows how the contact resistance at the Al current collector is the main source of impedance growth upon long cycling for Li-salt containing cathodes compared to the additive-free reference. Higher contact resistances correlate with poorer coating adhesion, which is most likely a result of the altered adhesion properties between PVdF and the Lewis-basic oxide surface of the Al current collector. It is striking that, the best adhesion value measured for LiODFB corresponds to the highest contact resistance among all samples in the post-mortem EIS. Indeed, despite the good coating adhesion with LiODFB as a slurry additive in the dry state, the EIS and SEM results of the fatigued electrodes suggest adhesion failure. These observations could be explained by anion exchange processes, once the electrode is in contact with the liquid, LiPF6-based, electrolyte, leading to changes in the coating adhesion properties. In particular, we hypothesize a superior ODFB− adsorption onto the Al current collector while casting the electrodes compared to the TFSI− and TFA− anions, that would promote, at variance with the other additives, a good adhesion in the dry state and, at the same time, an increased anion exchange rate with electrolyte species while cycling. This latter process can hasten the contact loss of the coating from the current collector upon long cycling and produces delamination, which is then correlated to an increased contact resistance due to the rise in Al passivation generated by LiPF6. One important cause behind faster capacity fading, higher overpotentials and active material losses for all the additive-containing cells, as detected in the dQ/dV curves in comparison to the additive-free cell, has to be therefore assigned to increased contact resistance when employing slurry additives and to the concomitant growth of insulating layers at the Al/composite interface upon long cycling. Instead, the additive-free cell shows notably reduced active material loss through cycling and slower overpotential increase in the dQ/dV curves, which can be linked to comparatively low R0 and, as clearly visible in the post-mortem SEM cross-sections of Fig. 9, to crack formation on the NMC622 particles. Interestingly, the post-mortem SEM cross-sections of Li-salts containing electrodes (Fig. 9) show almost no cracking on NMC622 particles. It is not excluded that slight cathodic delamination and replacement of adsorbed species at the current collector surface upon long cycling triggered the dissolution of tiny amounts of transition metal ions that could, eventually, migrate and incorporate into the SEI. The post-mortem EDX analysis of all the cycled anodes revealed an increased oxygen atomic fraction for all the additive containing cells compared to the additive-free sample, especially for the LiTFA sample (Table S3, ESI†). The increased oxygen content is attributed to an augmented SEI formation with proceeding cycling. Although EDX-analysis could not prove the presence of transition metal migration from the cathode to the anode, it cannot be excluded that traces below the detection limit of EDX cause an accelerated SEI growth. The dissolution of transition metals, even in very small amounts, can pose severe threats to graphite anodes by decomposition of the SEI layers. Ni, Co, and Mn can chemically react with pre-existing SEI components, they can catalyse solvent reduction into Li2CO3 inside the SEI layers damaging its mechanical integrity through crack formation, thus consuming mobile Li-ions and forming more inorganic components.63–66 It has been reported67 that one single Mn2+ ion integrated into SEI could cause a loss of ∼100 additional lithium ions, thereby hastening the depletion of cyclable active lithium. Moreover, an increased anodic side reaction was already hinted by the dQ/dV curves analysis, where a very first, small anodic peak that was not present in the formation step (green arrows in Fig. 7), developed along cycling for LiTFA and LiODFB and, less pronouncedly for LiTFSI. This peak, which is almost absent for the reference cell, could be attributed to Li plating19 at the graphite anode side and its effects are consistent with augmented SEI formation for all the additive-containing cells, as elucidated in the EDX analysis of cycled anodes (Table S3, ESI†). Particularly, the highest oxygen atomic fraction detected for LiTFA is in perfect correlation with the most pronounced anodic peak development through cycling measured for this additive. Last but not least, it cannot be ruled out that long-term capacity fading effects could stem from TFA− or any of the other anions damaging the graphite. Indeed, co-intercalation of anions68 into the layered carbonaceous anodic structure and their subsequent decomposition may lead to graphite exfoliation or premature disintegration through gas formation.
Conclusions
Three Li-salts, namely LiTFA, LiTFSI and LiODFB have been herein investigated as different slurry additives. Their potential as multi-purpose additives capable of encompassing the benefits of both processing and electrolyte additives have been carefully explored at the pilot scale. They were selected because they do not represent fundamentally new battery components, by virtue of being, already, well-known electrolyte salts or additives. Their use as slurry additives is therefore more suitable than that of new substances, that could, potentially, lead to unknown interactions within the complex battery system. It was shown how Li-salts can, even in very low concentrations, actively delay chemical and physical slurry gelation by hindering PVdF binder adsorption through neutralization and complexation of the NMC622 surface basic groups. Through the use of these additives, gelation was actively tuned so that processing of super high 75.5% wt. SC NMC622 slurries became possible, going beyond the standard processability limits previously reported in the literature.69 LiTFA proved itself to be the most effective additive in controlling gelation and its usage guaranteed much lower viscosity and Ecoh increases upon 3 days of slurry storage compared to the other additives at the same concentration. Manufacturing of the additive-free electrode was possible only using a much lower 70.0 wt% SC slurry due to extremely high slurry viscosity at 75.5 wt% SC. Moreover, continuous casting of the additive-free slurry was not possible due to fast (right after mixing) slurry gelation hindering the ongoing roll-to-roll process (Fig. S1, ESI†). The long-term cycling in a full cell with a graphite anode (Fig. 6(B)) showed faster degradation for all the additive-containing cells compared to the reference and, in particular, the LiTFSI cathode cell performed close to the additive-free sample in terms of capacity retention after 1000 cycles. Post-mortem EIS measurements were performed on fatigued cathodes after 1000 cycles and they demonstrated how contact resistance at the Al current collector is a main source of impedance growth upon long cycling for Li-salt containing cathodes compared to the additive-free reference. Indeed, Li-salts localized at the Al surface can accelerate electrode delamination from the current collector (as shown in SEM images of ion-milled cross-sections) and favour a more pronounced Al passivation by replacement of adsorbed Li-salt anions with electrolyte species. In this respect, LiODFB and LiTFA scored higher R0 values due to their stronger adsorption tendency towards the Al current collector when compared to LiTFSI. One main cause behind faster capacity fading, higher overpotentials and active material losses for all the additive-containing cells compared to the additive-free cell was identified as a positive electrode effect; in particular, owing to increased contact-resistance when employing slurry additives and to the concomitant growth, upon long cycling, of insulating layers at the Al/composite interface. Secondly, as suggested by the dQ/dV curves analysis and by the post-mortem EDX investigation of cycled anodes, an augmented SEI formation at the graphite anode is envisaged as a second main aging mechanism when employing Li-salts, in particular LiTFA. Negative electrode exacerbated degradation when employing slurry additives could stem from Li plating at the anodic side or from co-intercalation of Li-salts’ anions into the layered graphitic structure. Therefore, contrary to the previously reported results in the literature,16 this work highlights a common problem70 in battery research as experienced these days: laboratory scale results are poorly transferable to the pilot scale. Despite the decreased capacity retention upon long-cycling, both process and material-related benefits are found from including electrolyte salts in the slurry processing step, such as drastically reduced slurry viscosity as well as diminished particle cracking over an extended number of cycles. This work seeks to support the research community to further develop and advance ideas with respect to multi-purpose additives, by providing insightful details into failure mechanisms and by pointing out potentials, limitations and problems of this new challenging approach.Author contributions
F. C. carried out the experimental work and wrote the manuscript. M. M., A. W. and N. K. contributed to the experimental work. J. F., W. B. and H. E. supervised the project and contributed with findings interpretation, reviewing, and revision of the final manuscript.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
We thank all the KIT-IAM-ESS colleagues who helped the development of this work through critical reviews and discussions. This work contributes to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe).References
- H. A. Barnes, J. F. (John) Hutton and K. Walters, An introduction to rheology, Elsevier, 1989 Search PubMed.
- B. Ameduri and B. Améduri, Macromol. Chem. Phys., 2020, 221 Search PubMed.
- W. Bauer and D. Nötzel, Ceram. Int., 2014, 40, 4591–4598 CrossRef CAS.
- J. A. Lewis, J. Am. Ceram. Soc., 2000, 83, 2341–2359 CrossRef CAS.
- W. Bauer and D. Nötzel, Ceram. Int., 2014, 40, 4591–4598 CrossRef CAS.
- L. Ouyang, Z. Wu, J. Wang, X. Qi, Q. Li, J. Wang and S. Lu, RSC Adv., 2020, 10, 19360–19370 RSC.
- F. Jeschull, D. Brandell, M. Wohlfahrt-Mehrens and M. Memm, Energy Technol., 2017, 5, 2108–2118 CrossRef CAS.
- A. A. Potanin, R. De Rooij, D. Van Den Ende and J. Mellema, J. Chem. Phys., 1995, 102, 5845–5853 CrossRef CAS.
- S. H. Sung, S. Kim, J. H. Park, J. D. Park and K. H. Ahn, Materials, 2020, 13, 4544 CrossRef CAS PubMed.
- J. J. Lee and G. G. Fuller, J. Colloid Interface Sci., 1985, 103, 569–577 CrossRef CAS.
- G. J. Fleer, M. A. Cohen Stuart, J. M. H. M. Scheutjens, T. Cosgrove, B. V. Chapman and H. London, Polym. Int., 1995, 36, 102 Search PubMed.
- Y. Adachi, Encycl. Biocolloid Biointerface Sci., 2013, 931–944 Search PubMed.
- E. Dickinson and S. R. Euston, J. Chem. Soc., Faraday Trans., 1991, 87, 2193–2199 RSC.
- M. G. Rasteiro, F. A. P. Garcia, P. Ferreira, A. Blanco, C. Negro and E. Antunes, Chem. Eng. Process., 2008, 47, 1323–1332 CrossRef.
- J. Lee, S. J. Lee, K. H. Ahn and S. J. Lee, Langmuir, 2015, 31, 13639–13646 CrossRef CAS PubMed.
- S. S. Zhang, X. Fan and C. Wang, ChemElectroChem, 2019, 6, 1536–1541 CrossRef CAS.
- S. S. Zhang, Energy Storage Mater., 2020, 24, 247–254 CrossRef.
- H. Arai, S. Okada, H. Ohtsuka, M. Ichimura and J. Yamaki, Solid State Ionics, 1995, 80, 261–269 CrossRef CAS.
- K. Matsumoto, R. Kuzuo, K. Takeya and A. Yamanaka, J. Power Sources, 1999, 81–82, 558–561 CrossRef CAS.
- Y. Kim, J. Solid State Electrochem., 2013, 17, 1961–1965 CrossRef CAS.
- H. A. Sodano, Ferroelectric polymers from dehydrofluorinated PVDF, Google Patents, US10584189B2. Published online 2020. Accessed June 3, 2022.
- G. Kutringer and G. Weill, Polymer, 1991, 32, 877–883 CrossRef.
- C. Van Goethem, M. Mertens and I. F. J. Vankelecom, J. Membr. Sci., 2019, 572, 489–495 CrossRef CAS.
- L. Pagliaro and D. A. Lowy, Granja, 2019, 29, 18–32 Search PubMed.
- K. Park, J. H. Park, S. G. Hong, B. Choi, S. Heo, S. W. Seo, K. Min and J. H. Park, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- X. Xiong, Z. Wang, P. Yue, H. Guo, F. Wu, J. Wang and X. Li, J. Power Sources, 2013, 222, 318–325 CrossRef CAS.
- H. G. Song, J. Y. Kim, K. T. Kim and Y. J. Park, J. Power Sources, 2011, 196, 6847–6855 CrossRef CAS.
- C. H. Jo, D. H. Cho, H. J. Noh, H. Yashiro, Y. K. Sun and S. T. Myung, Nano Res., 2014, 8, 1464–1479 CrossRef.
- V. Aravindan, J. Gnanaraj, S. Madhavi and H. K. Liu, Chem. – Eur. J., 2011, 17, 14326–14346 CrossRef CAS PubMed.
- D. Aurbach, B. Markovsky, M. D. Levi, E. Levi, A. Schechter, M. Moshkovich and Y. Cohen, J. Power Sources, 1999, 81–82, 95–111 CrossRef CAS.
- A. Webber, J. Electrochem. Soc., 1991, 138, 2586–2590 CrossRef CAS.
- J. Foropoulos and D. D. Desmarteau, Inorg. Chem., 1984, 23, 3720–3723 CrossRef CAS.
- H. Zhou, Z. Fang and J. Li, J. Power Sources, 2013, 230, 148–154 CrossRef CAS.
- S. S. Zhang, ECS Trans., 2007, 3, 59–68 CrossRef CAS.
- J. Huang, L. Z. Fan, B. Yu, T. Xing and W. Qiu, Ionics, 2010, 16, 509–513 CrossRef CAS.
- H. Zhou, F. Liu and J. Li, J. Mater. Sci. Technol., 2012, 28, 723–727 CrossRef CAS.
- N. Ehteshami, L. Ibing, L. Stolz, M. Winter and E. Paillard, J. Power Sources, 2020, 451, 227804 CrossRef CAS.
- H. Zhou, D. Xiao, C. Yin, Z. Yang, K. Xiao and J. Li, J. Electroanal. Chem., 2018, 808, 293–302 CrossRef CAS.
- S. Li, Y. Liang, J. Xie, L. Ai, Y. Xie, C. Li, C. Wang and X. Cui, J. Electroanal. Chem., 2018, 823, 688–696 CrossRef CAS.
- A. Hofmann, A. Höweling, N. Bohn, M. Müller, J. R. Binder and T. Hanemann, ChemElectroChem, 2019, 6, 5255–5263 CrossRef CAS.
- Z. Wang, F. Qi, L. Yin, Y. Shi, C. Sun, B. An, H. M. Cheng and F. Li, Adv. Energy Mater., 2020, 10, 1903843 CrossRef CAS.
- A. Abend, V. Illich and J. Rétey, Eur. J. Biochem., 1997, 249, 180–186 CrossRef CAS PubMed.
- K. Oka, C. Strietzel, R. Emanuelsson, H. Nishide, K. Oyaizu, M. Strømme and M. Sjödin, Electrochem. Commun., 2019, 105, 106489 CrossRef CAS.
- D. Megías-Alguacil, Appl. Rheol., 2004, 14, 57–126 Search PubMed.
- F. Matsumoto and T. Gunji, Water in Lithium-Ion Batteries, Springer Nature, Singapore, 2022 Search PubMed.
- A. Purwanto, S. Nisa, I. P. Lestari, M. N. Ikhsanudin, C. S. Yudha and H. Widiyandari, KONA Powder Part. J., 2022, 39, 130–149 CrossRef CAS.
- Y. Li, M. Abdel-Monem, R. Gopalakrishnan, M. Berecibar, E. Nanini-Maury, N. Omar, P. van den Bossche and J. Van Mierlo, J. Power Sources, 2018, 373, 40–53 CrossRef CAS.
- J.-M. Atebamba, J. Moskon, S. Pejovnik and M. Gaberscek, J. Electrochem. Soc., 2010, 157, A1218 CrossRef CAS.
- Y. Ruan, X. Song, Y. Fu, C. Song and V. Battaglia, J. Power Sources, 2018, 400, 539–548 CrossRef CAS.
- L. Zhang, J. Jiang and W. Zhang, Energies, 2017, 10, 1147 CrossRef.
- T. Ohzuku, Y. Iwakoshi and K. Sawai, J. Electrochem. Soc., 1993, 140, 2490–2498 CrossRef CAS.
- S. S. Zhang, J. Electrochem. Soc., 2020, 167, 100510 CrossRef CAS.
- M. Müller, L. Schneider, N. Bohn, J. R. Binder and W. Bauer, ACS Appl. Energy Mater., 2021, 4, 1993–2003 CrossRef.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef CAS.
- J. Zhu, M. S. Dewi Darma, M. Knapp, D. R. Sørensen, M. Heere, Q. Fang, X. Wang, H. Dai, L. Mereacre, A. Senyshyn, X. Wei and H. Ehrenberg, J. Power Sources, 2020, 448, 227575 CrossRef CAS.
- J. P. Meyers, M. Doyle, R. M. Darling and J. Newman, J. Electrochem. Soc., 2000, 147, 2930 CrossRef CAS.
- M. Doyle, J. P. Meyers and J. Newman, J. Electrochem. Soc., 2000, 147, 99 CrossRef CAS.
- B. C. Han, A. Van Der Ven, D. Morgan and G. Ceder, Electrochim. Acta, 2004, 49, 4691–4699 CrossRef CAS.
- S. Lopez, J. Petit, G. Tourillon, H. M. Dunlop and J. Butruille, J. Electrochem. Soc., 1998, 145, 829–834 CrossRef CAS.
- A. Schulze, M. F. Maitz, R. Zimmermann, B. Marquardt, M. Fischer, C. Werner, M. Went and I. Thomas, RSC Adv., 2013, 3, 22518–22526 RSC.
- E. McCafferty, J. Electrochem. Soc., 2003, 150, B342 CrossRef CAS.
- W. Bauer, F. A. Çetinel, M. Müller and U. Kaufmann, Electrochim. Acta, 2019, 317, 112–119 CrossRef CAS.
- R. Jung, F. Linsenmann, R. Thomas, J. Wandt, S. Solchenbach, F. Maglia, C. Stinner, M. Tromp and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A378–A389 CrossRef CAS.
- H. Tsunekawa, S. Tanimoto, R. Marubayashi, M. Fujita, K. Kifune and M. Sano, J. Electrochem. Soc., 2002, 149, A1326 CrossRef CAS.
- X. Xiao, Z. Liu, L. Baggetto, G. M. Veith, K. L. More and R. R. Unocic, Phys. Chem. Chem. Phys., 2014, 16, 10398–10402 RSC.
- C. Delacourt, A. Kwong, X. Liu, R. Qiao, W. L. Yang, P. Lu, S. J. Harris and V. Srinivasan, J. Electrochem. Soc., 2013, 160, A1099–A1107 CrossRef CAS.
- J. A. Gilbert, I. A. Shkrob and D. P. Abraham, J. Electrochem. Soc., 2017, 164, A389–A399 CrossRef CAS.
- T. Placke, S. Rothermel, O. Fromm, P. Meister, S. F. Lux, J. Huesker, H.-W. Meyer and M. Winter, J. Electrochem. Soc., 2013, 160, A1979–A1991 CrossRef CAS.
- L. Ouyang, Z. Wu, J. Wang, X. Qi, Q. Li, J. Wang and S. Lu, RSC Adv., 2020, 10, 19360–19370 RSC.
- A. Smith, P. Stüble, L. Leuthner, A. Hofmann and F. Jeschull, Chem. Eng. J., 2023, 6, e202300080, DOI:10.1002/batt.202300080.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00246b |
| This journal is © The Royal Society of Chemistry 2023 |