
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanomedicine strategies to improve therapeutic agents for the prevention and treatment of preterm birth and future directions
Jessica
Taylor
a,
Andrew
Sharp
 b,
Steve P.
Rannard
ad,
Sarah
Arrowsmith
*c and
Tom O.
McDonald
*ae
b,
Steve P.
Rannard
ad,
Sarah
Arrowsmith
*c and
Tom O.
McDonald
*ae
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: Thomas.Mcdonald@liverpool.ac.uk
bHarris-Wellbeing Preterm Birth Research Centre, Department of Women's and Children's Health, Liverpool Women's Hospital, University of Liverpool, Crown Street, Liverpool, L8 7SS, UK
cDepartment of Life Sciences, Manchester Metropolitan University, Chester Street, Manchester, M1 5GD, UK. E-mail: S.Arrowsmith@mmu.ac.uk
dCentre of Excellence in Long-acting Therapeutics (CELT), University of Liverpool, Liverpool, L7 3NY, UK
eDepartment of Materials, Henry Royce Institute, The University of Manchester, Manchester, M13 9PL, UK
First published on 4th March 2023
Abstract
The World Health Organisation (WHO) estimates 15 million babies worldwide are born preterm each year, with 1 million infant mortalities and long-term morbidity in survivors. Whilst the past 40 years have provided some understanding in the causes of preterm birth, along with development of a range of therapeutic options, notably prophylactic use of progesterone or uterine contraction suppressants (tocolytics), the number of preterm births continues to rise. Existing therapeutics used to control uterine contractions are restricted in their clinical use due to pharmacological drawbacks such as poor potency, transfer of drugs to the fetus across the placenta and maternal side effects from activity in other maternal systems. This review focuses on addressing the urgent need for the development of alternative therapeutic systems with improved efficacy and safety for the treatment of preterm birth. We discuss the application of nanomedicine as a viable opportunity to engineer pre-existing tocolytic agents and progestogens into nanoformulations, to improve their efficacy and address current drawbacks to their use. We review different nanomedicines including liposomes, lipid-based carriers, polymers and nanosuspensions highlighting where possible, where these technologies have already been exploited e.g. liposomes, and their significance in improving the properties of pre-existing therapeutic agents within the field of obstetrics. We also highlight where active pharmaceutical agents (APIs) with tocolytic properties have been used for other clinical indications and how these could inform the design of future therapeutics or be repurposed to diversify their application such as for use in preterm birth. Finally we outline and discuss the future challenges.
Introduction
Preterm birth
Preterm birth is defined as the birth of a child before 37 weeks' gestation; a full-term pregnancy lasts 39–41 week. According to the 2018 report from the World Health Organisation (WHO), preterm birth globally affects up to 15 million babies each year, with a total of 1 million infant mortalities.1,2 Globally the preterm birth rate is 11%,3,4 ranging from 8.7% in Europe to 13.4% in North Africa.4 Preterm birth is categorised according to gestation at delivery as; extremely preterm (<28 weeks), very preterm (28–32 weeks) and moderate to late preterm (32 to 37 weeks); with late preterm accounting for 70% of cases.5 Infants born at 22 weeks or with a birth weight of ≤500 g are considered to be at the lower limit of viability having very limited chances of survival.6,7 However, as gestational age at delivery increases, so does survival rate: Larroque et al. found neonates born at 24 weeks had an increased survival rate at 31%, followed by 78% at 28 weeks and 97% at 32 weeks.8 Survival rate however, is also dependent on the social and medical conditions in which the infant is born. Additionally, although the largest concern linked to preterm birth is immediate mortality, preterm infants often experience long-term morbidity including neurodevelopmental delay, cerebral palsy as well as respiratory disorders, cardiovascular disorders, infections, visual and hearing problems and learning disabilities owing to the underdeveloped fetal organs and systems.9 Indeed, preterm birth is one of the largest contributors to the disease burden and concomitant economic burden globally due to the lifelong disability often associated with being born preterm.10Clinical risk factors for preterm birth
Clinically, preterm birth is classified into different subtypes: In ∼30–35% of cases, preterm labour is pre-planned (medically indicated (iatrogenic)) and instigated by obstetricians; these planned cases occur primarily because the mother or the fetus is suffering from a potentially life-threatening condition and hence earlier delivery is safer.11 This includes poor fetal growth (fetal growth restriction, FGR) often as a result of a poor functioning placenta, fetal anatomical concerns, or fetal distress. Alternatively, planned preterm birth can occur if the mother has a health condition such as preeclampsia or other obstetrical complications including placental abruption. In these situations, obstetricians induce birth early given that there is a clear clinical advantage of early labour based on maternal and fetal considerations.11,12 Given that here, labour onset is pre-planned and somewhat controlled, pharmacological intervention is not necessary and hence iatrogenic preterm births are not the focus of this review.The majority (∼65–70%) of preterm births are classed as sudden or unexpected and are referred to as spontaneous in onset.11,13 Spontaneous preterm birth results from either preterm premature rupture of membranes (PPROM) or idiopathic (no known cause) spontaneous labour onset involving uterine contractions and cervical dilation. Around 40–45% of all preterm births are idiopathic and are the main focus of the treatments described here.
Risk factors for spontaneous preterm birth include preterm birth in a previous pregnancy, having a short (≤25 mm) cervix (cervical insufficiency), either naturally or because of cervical surgery, and multiple pregnancy (e.g. twins). Other possible etiologies include intrauterine infection and inflammation. That previous spontaneous preterm delivery increases the risk of subsequent preterm birth, suggests that there may be a genetic factor involved. Many studies to date have identified multiple genetic variants linked to spontaneous preterm birth, however, in most cases, outcomes cannot be replicated across different population cohorts.14 Furthermore, the function of the genes identified are often not validated and therefore a biological role is not provided. In addition, many genetic studies to do not consider the gene–environment interactions including external and in utero environmental factors.
Multiple gestations, i.e. twins and triplets are considerably over-represented within preterm birth statistics accounting for 20% of all preterm births and 11.2% of all neonatal intensive care unit admissions despite representing only 3% of all live births. Up to 60% of multiple pregnancies will deliver <37 weeks' gestation and 10% <32 weeks. Nearly two thirds of multiple pregnancy preterm births are due to spontaneous onset of labour with the remaining third being iatrogenic due to maternal or fetal complications. Additionally, multiple gestations, account for more than 50% of preterm birth complications and 10–12% of all fetal deaths.15–17 Twins are at increased risk of low birth weight and as a result of preterm birth, have a five times higher risk of early neonatal and infant death than when only one baby is carried (singletons).18,19 The increased incidence of preterm birth in twins is multifactorial and includes maternal complications (discussed above) as well as uterine overdistention (stretch) which can trigger uterine contractions.
Interestingly maternal age, both ≤19 years and ≥35 years, is often described as high risk for preterm birth. For those of a young maternal age (≤19 years), spontaneous preterm birth may be due to biological immaturity increasing their risk of PPROM.20 Conversely recent studies suggest that advanced maternal age (≥35 years) and concomitant increase in risk of gestational diabetes and preeclampsia, may contribute to the increased risk of preterm birth.21 Pregnancies conceived by assisted reproductive technologies (e.g. In vitro fertilization/intracytoplasmic sperm injection, IVF/ICSI) are also at higher risk of preterm birth.22
Prevention and treatment of preterm birth
The treatment of preterm birth can be divided into two categories; either prophylactic treatment for preterm birth prevention for women at high risk of preterm birth, such as those with a history of previous preterm birth and/or current evidence of a short cervix, or treatment at the onset of spontaneous preterm labour to suppress uterine contractions, known as tocolysis. The types of active pharmaceutical ingredients (APIs) used, and the clinical findings will be briefly discussed in this section.The APIs used for prophylactic treatment to reduce the risk of preterm birth occurring are progestogens (based on the hormone progesterone, Fig. 1) and include a synthetic derivative of progesterone, 17-hydroxyprogesterone caproate (OHPC) and vaginal progesterone. These two APIs have different physiochemical properties which influences their route of administration. For example, OHPC has a higher octanol–water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) and lower aqueous solubility than progesterone and is therefore administered as an intramuscular injection (Table 1). For women with a singleton gestation and a history of spontaneous preterm birth, OHPC (250 mg) can be administered weekly, starting at 16–20 gestational weeks until 36 weeks or delivery. For women with a short cervix, vaginal progesterone, either as a vaginal gel (90 mg) or micronised vaginal soft capsules (200 mg) is offered, however, the benefits of treatment are not clear: the largest study on vaginal administration of progesterone (200 mg capsules) (a multicentre, randomised and double-blind trial) reported that “Vaginal progesterone was not associated with reduced risk of preterm birth or composite neonatal adverse outcomes, and had no long-term benefit or harm on outcomes in children at 2 years of age.”23 However, a very recent network meta-analysis of clinical trials has shown that vaginal progesterone to be the treatment of choice for women with a singleton pregnancy and with a short cervical length.24 Indeed, in the United Kingdom, the National Institute for Health and Care Excellence (NICE) have recommended vaginal progesterone for high risk women. For OHPC, in 2003, a multi-centre, double-blind, placebo-controlled trial evaluating progesterone in women with prior history of spontaneous preterm birth found clear benefit with significant reduction in the rate of recurrent preterm birth and significantly prolonged the duration of pregnancy25 and was granted approval by the U.S. Food and Drug Administration (FDA). However, a subsequent follow-up confirmatory trial could not replicate the initial findings and raised other safety concerns.26 In October 2022, the FDA held a hearing to discuss the proposal to withdraw the approval of OHPC for the treatment of preterm birth, the decision has not yet been announced.
P) and lower aqueous solubility than progesterone and is therefore administered as an intramuscular injection (Table 1). For women with a singleton gestation and a history of spontaneous preterm birth, OHPC (250 mg) can be administered weekly, starting at 16–20 gestational weeks until 36 weeks or delivery. For women with a short cervix, vaginal progesterone, either as a vaginal gel (90 mg) or micronised vaginal soft capsules (200 mg) is offered, however, the benefits of treatment are not clear: the largest study on vaginal administration of progesterone (200 mg capsules) (a multicentre, randomised and double-blind trial) reported that “Vaginal progesterone was not associated with reduced risk of preterm birth or composite neonatal adverse outcomes, and had no long-term benefit or harm on outcomes in children at 2 years of age.”23 However, a very recent network meta-analysis of clinical trials has shown that vaginal progesterone to be the treatment of choice for women with a singleton pregnancy and with a short cervical length.24 Indeed, in the United Kingdom, the National Institute for Health and Care Excellence (NICE) have recommended vaginal progesterone for high risk women. For OHPC, in 2003, a multi-centre, double-blind, placebo-controlled trial evaluating progesterone in women with prior history of spontaneous preterm birth found clear benefit with significant reduction in the rate of recurrent preterm birth and significantly prolonged the duration of pregnancy25 and was granted approval by the U.S. Food and Drug Administration (FDA). However, a subsequent follow-up confirmatory trial could not replicate the initial findings and raised other safety concerns.26 In October 2022, the FDA held a hearing to discuss the proposal to withdraw the approval of OHPC for the treatment of preterm birth, the decision has not yet been announced.
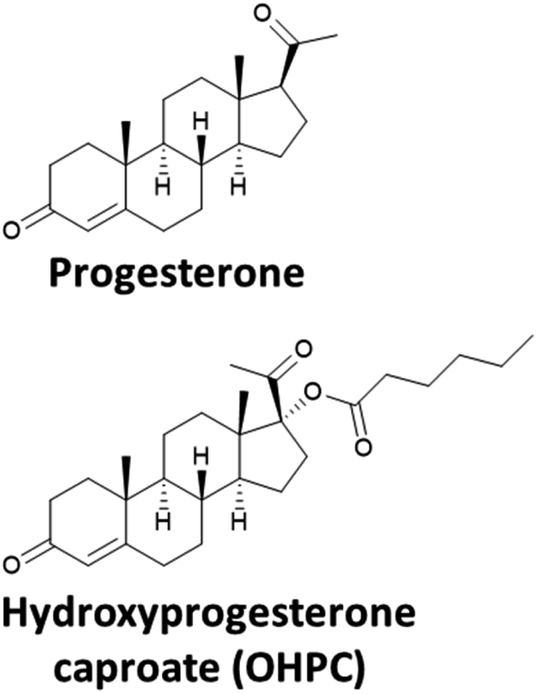 | ||
| Fig. 1 The structures, of progesterone and hydroprogesterone caproate (OHPC). | ||
| API | logP |
Solubility (mg ml−1) | Dose | Route of administration | Side effects |
|---|---|---|---|---|---|
| Progesterone | 3.58 | 0.00546 | 200–400 mg | Oral, vaginal | - Maternal headache, breast tenderness, nausea, cough |
| Hydroxyprogesterone caproate (OHPC) | 4.81 | 0.00086 | 250 mg | Intramuscular | - Maternal headache, breast tenderness, nausea, cough |
| - Local irritation at injection site |
For multiple gestations the aetiology of preterm birth is likely to be multifactorial and different to singleton pregnancies (for a review on this topic see Murray et al.27). For these pregnancies, there is currently a lack of evidence-based interventions for the prevention of preterm birth. The most studied is prophylactic progesterone administration (intramuscular OHPC and vaginal suppository or gel) in twin or triplet pregnancy. Neither vaginal progesterone in twin nor OHPC in twins or triplets has been shown to reduce preterm birth before 34 weeks.28,29 However, a higher dosage of daily vaginal progesterone (600 mg) for women with twins may improve preterm birth outcomes.30 Additionally, in a subgroup of women with twins and a short cervix, vaginal progesterone reduced the risk of delivering before 34 weeks.31 The same, however, has not been found for use of OHC in twin pregnancies and short cervix.32 Guidelines, such as those by the International Society of Ultrasound in Obstetrics and Gynecology (ISUOG), therefore recommend screening for preterm birth in twin pregnancy with cervical assessment on vaginal ultrasound but without clear guidance on management regime to follow. Due to insufficient evidence, routine use of intramuscular or vaginal progesterone for prevention of preterm birth in multiple pregnancies is not recommended.33 Unlike in singleton pregnancies, the use of tocolytics in multiple pregnancies has been less studied.34 Questions also arise over whether tocolytic treatments demonstrate the same efficacy in preventing or delaying preterm birth or improving infant outcomes for multiple gestation pregnancies. There is insufficient evidence for the prophylactic use of β2 adrenergic receptor agonists in twins,35 whilst nifedipine appears safe and effective in delaying labour in both singleton and twin pregnancies with spontaneous preterm labour onset.36 Indomethacin has not been investigated specifically in multiple pregnancy, but twin gestations were included in a network meta-analysis comparing tocolytic therapies for preterm delivery. Indomethacin showed the highest delay in delivery by 48 hours and least maternal side effects,37 albeit its use was restricted to before 32 weeks due to known fetal complications. Given the weaker evidence basis for the preterm birth treatments for multiple gestations compared to singleton pregnancies more research is needed to identify the best treatment options.
In cases of spontaneous labour onset with uterine contraction and/or cervical dilation, women are primarily treated with APIs named tocolytics, uterine contraction suppressants. The primary role of tocolytics is to reduce the frequency and intensity of uterine contractions.39–41 The resulting delay in labour is usually sufficient to enable the transfer of the woman to a care setting with appropriate neonatal facilities, or for administration of corticosteroids (betamethasone or dexamethasone) to enhance fetal lung maturation prior to delivery; to reduce neonatal respiratory complications. Furthermore, the woman may also be administered magnesium sulphate to provide neuroprotection and reduce brain injury in the infant. These APIs can be divided into subcategories of tocolytics based on their mechanism of action (Fig. 2). Tocolytics include calcium channel blockers (e.g. nifedipine), β2 adrenoceptor agonists (betamimetics) (e.g. salbutamol, ritodrine, terbutaline), oxytocin receptor antagonists (e.g. atosiban, nolasiban and barusiban) and non-steroidal anti-inflammatory drugs (NSAIDs) (e.g. indomethacin).37,42,43 For further discussion of mechanism of action of these drugs, see Arrowsmith et al. 2010 and Wray et al. 2022.44,45 These tocolytic APIs, with the exception of salbutamol, generally possess limited aqueous solubility (with predicted values ≤0.05 mg ml−1), while all except atosiban can be administered orally (Table 2). Each API presents side effects to the mother and/or the fetus which can be seen in Table 2. It is important to note however, that very few tocolytics are approved for the treatment of preterm birth. Hence, given the very limited range of clinical options, and the limited evidence of the benefit of the approved treatments, most are used off-label for the treatment of preterm birth. The only approved treatment is atosiban, which was approved by the European Medicines Agency (EMA) in 2000, but not the FDA. Atosiban is an oxytocin-receptor antagonist (ORAs) developed especially for the treatment of preterm birth. Because the primary targets for oxytocin are the uterine muscle (myometrium) and decidua, it was anticipated that atosiban would be highly organ specific with limited adverse side effects. Atosiban displayed mixed results in clinical trials; it has been shown to reduce the likelihood of birth within the first 48 hours,46 however without improvement in neonatal outcomes. An upcoming multi-centre trial of atosiban and steroid vs. placebo has been approved which may address the neonatal benefit question.47 Several other ORAs are also in development including barusiban and nolasiban which confer different pharmacokinetic profiles and potencies.
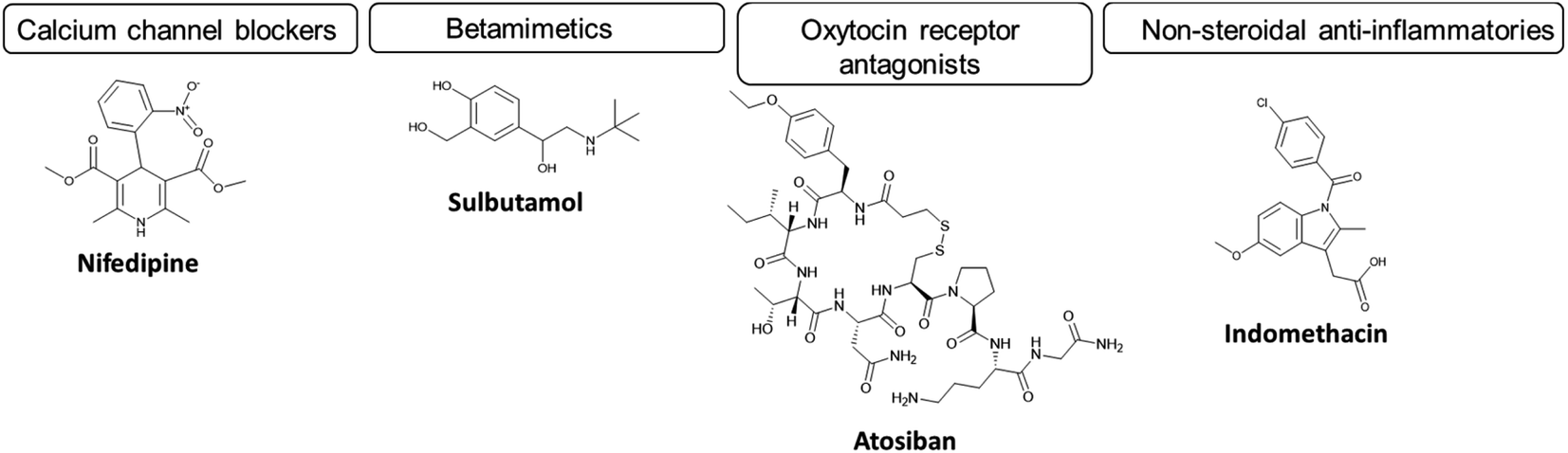 | ||
| Fig. 2 The main subcategories of commonly used tocolytics and their structures. | ||
P values and calculated solubilities were ALOGPS values51
| API | logP |
Solubility (mg ml−1) | Dose | Route of administration | Side effects |
|---|---|---|---|---|---|
| Nifedipine | 2.49 | 0.0177 | 10–30 mg, then 10–20 mg every 4–6 h | Oral | - Maternal headache and hypotension |
| - Tachycardia (maternal and fetal) | |||||
| Sulbutamol | 0.44 | 2.15 | 10 mcgs min−1, titrated to response until contractions cease (max rate 45 mcgs/min), then gradually reduce, max duration 48 h | Oral or IV | - Maternal tachycardia, hyperglycaemia and pulmonary tachycardia |
| - Fetal tachycardia and respiratory distress syndrome | |||||
| Atosiban | −0.17 | 0.0517 | 6.75 mg bolus, then 300 mg min−1 infusion for 3 h, followed by 100 mg min−1 infusion for up to 45 h | IV | - Maternal tachycardia and chest pain |
| Indomethacin | 4.25 | 0.0024 | 100 mg, then 25 mg orally 4–6 hourly for 48 h | Oral | - Fetal reduced amniotic fluid, premature closure of ductus arteriosus |
Despite there being several different tocolytics available, they have a number of clinical drawbacks. Firstly, tocolytics have repetitive dosing and time restrictions; with many requiring the termination of administration after 48 hours.52 Secondly, their efficacy is variable amongst different patient cohorts with some displaying uterine quiescence for up to 7 days, whereas others only maintain quiescence for 24 hours after the maximum dosing of 48 hours.53 Thirdly, further speculation around the safety and efficacy of tocolytics is raised due to a number of associated side effects in the mother and/or the fetus, the nature of which depend on the subclass of tocolytic used (see Fig. 2).
Currently in clinic, calcium channel blockers are the most common first line treatment due to their enhanced tocolytic activity over other tocolytic categories and reduced fetal effects.54,55 However, owing to the fact that the same calcium channels are present in the heart and cardiovascular system, calcium channel blockers can cause substantial maternal side effects including hypotension and tachycardia.50 Hence, calcium channel blockers suffer from time restrictions of use up to 48 hours and dosage restrictions to a maximum of 160 mg day−1.56 Indomethacin is another commonly used tocolytic. It reduces the activity of the type 2 isoform of cyclo-oxygenase (COX-2), which is important for the production of prostaglandins within intrauterine tissues, and has been shown to reduce uterine contractility; thus, extending gestation. However, it also poses concerning side effects including premature closure of a major blood vessel, the ductus arteriosus, in the fetus as well as intraventricular bleeding.57,58
With significant disadvantages still associated with the most potent tocolytics, there is a clear requirement for the development of new tocolytics or new formulations of pre-existing APIs that can be engineered to improve their pharmacokinetic and safety properties. The key desirable outcomes are to improve the in vivo pharmacological performance of tocolytic agents, whilst addressing dosage restrictions and maternal and fetal safety concerns. The development of new APIs for the treatment of preterm birth is outside the scope of this review, instead the reader is directed to the review by Zierden et al.59
Potential advantages of using nanomedicine for preterm birth
Nanomedicine, the application of nanotechnology for therapeutics and diagnostics, has provided significant advantages for medicine in recent decades.60–62 Most notably, nanomedicine technologies offer the potential to alter the pharmacokinetics and biodistribution of APIs. An example of this is in the clinical success of targeted liposomes for the delivery of anticancer therapies: paclitaxel loaded liposomes for lung cancer, using d-α-tocopheryl polyethylene glycol 1000 succinate triphenylphosphine conjugate to actively target the mitochondria in cancerous cells, have shown a strong inhibitory effect of 73% and increased apoptosis of cancerous cells.63 In the setting of preterm birth, better drug pharmacokinetics and biodistribution to reduce the dosage profile is clearly attractive for improving the clinical performance of tocolytic agents where side-effects in both the mother and the fetus are often limiting factors for their use. As noted for the example in cancer therapy, the targeting of nanomedicines can be used to further enhance the pharmacokinetics of an API, typically using intravenous (IV) as the route of administration. Nanomedicines can be designed to incorporate passive and/or active targeting. Passive targeting uses the properties of the nanoparticles themselves (diameter, surface charge, surface coating) in combination with the physiology of the disease site to increase the percentage of the API that reaches the target site. An example of this such of targeting is the enhanced permeation and retention effect which exploits the pathophysiological characteristics observed in some solid tumours.64,65 Active targeting involves the conjugation of a high-affinity ligand onto the surface of the nanoparticle. This ligand is then able to bind to a specific receptor that is found on the targeted cells.66 Targeting of nanomedicines is a considerable opportunity for improving the efficacy of treatments.Another opportunity offered by nanomedicines is the potential to open new routes of administration. For ‘conventional’ (non-nano) formulations of an API the route of administration is limited by the physicochemical parameters of the API. For example, for oral dosing the API needs to typically possess physiochemical parameters within Lipinski's rule of 5 (molecular weight ≤500, logP ≤ 5, H-bond donors ≤5, H- bond acceptors ≤10).67 This is because the oral bioavailability is controlled by the aqueous solubility and permeability of the drug through the gastrointestinal membrane; APIs with low solubility and low permeability are often associated with poor therapeutic efficacy after oral administration.68 However, the use of nanomedicines can be used to circumvent the limits caused by an API's physicochemical properties. For example, nanosuspensions can be used to enhance the dissolution kinetics of poorly solubility APIs,69 while lipid nanoparticles can show mucoadhesive behaviour in the gut or drive lymphatic uptake of their payload.70 Such approaches are of interest for a wide range of conditions including cancer.71 Conversely, moving from an oral dose to an IV nanomedicine can be beneficial, as the properties of the nanomedicine can influence the elimination mechanisms. Thus the properties of the nanomedicine will also influence the pharmacokinetics of the treatment rather than these being purely controlled by the physiochemical properties of the dissolved API. In the treatment of preterm birth, where therapies are given in a clinical setting (compatible with IV administration), it may be attractive to move from the oral dosing of an API to IV dosing of a nanomedicine with the aim of improving targeting to the uterus. This could potentially allow for a dose reduction and reduced side-effects. Furthermore, because nanomedicines can offer different behaviour in terms of penetration across biological barriers there is also the potential to delivery APIs topically or vaginally. Vaginal administration is an attractive route for the treatment of preterm birth as it avoids hepatic first-pass metabolism that is seen for oral dosing and is also successful in delivering a local effect at the uterus.72,73 The vagina is lined with mucosal tissue and as such the efficacy of vaginally administered APIs is strongly influenced by the interaction of the formulation with the mucus lining in the vagina. In terms of nanomedicine, both mucoadhesive or mucoinert nanoparticles can be designed, each with its own strengths and weaknesses. Mucoadhesion can be used to prolong the residence time of an API in the vagina, however, this can also limit the penetration of the API through the mucus to the underlying cells.74 Conversely mucoinert/mucus penetrating nanoparticles can improve the delivery of an API to the vaginal epithelium.75 The ability to introduce specific moieties to target specific organs and the ability to address the aqueous solubility issues surrounding the lipophilic character of many drugs, including tocolytics, makes this technology exciting and viable for novel therapies for preterm birth, as discussed next.76
Nanomedicine opportunities for tocolytics
The opportunities for tocolytic nanomedicine formulations are vast, particularly due to the hydrophobic nature of the majority of the tocolytic APIs. Some of the nanomedicine types that are best suited to the delivery of poorly water-soluble APIs are liposomes, solid lipid nanoparticles, nanostructured lipid carriers, nanoemulsions, polymer nanoparticles and nanosuspensions (Fig. 3).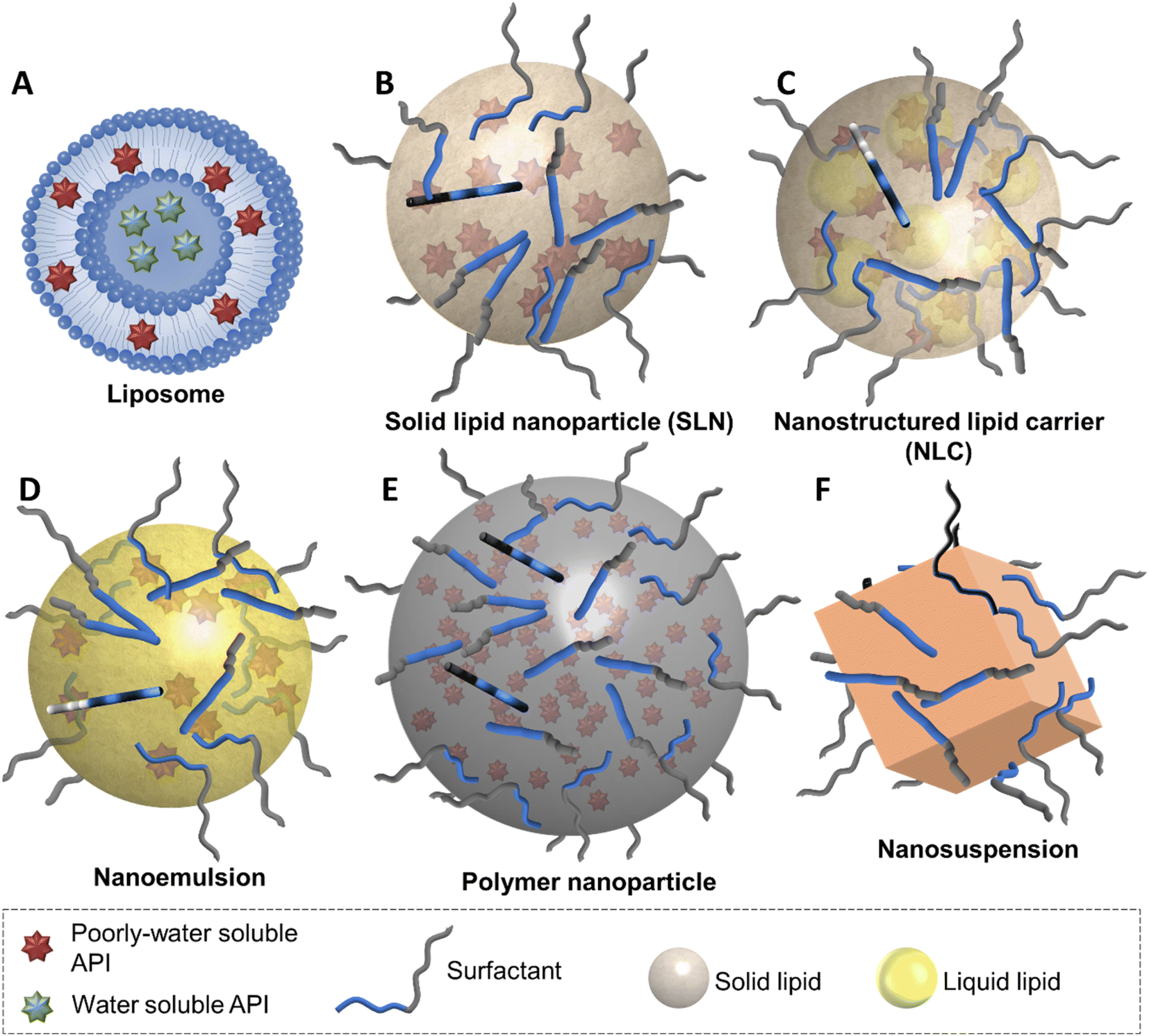 | ||
| Fig. 3 Illustration of liposomes, solid lipid nanoparticles, nanostructured lipid carriers, nanoemulsions, polymer nanoparticles and nanosuspension nanomedicines showing the differences in the structure and composition of the different nanoparticles. | ||
Whilst beneficial, the ability of nanomedicines to alter the pharmacokinetics and biodistribution of an API also introduces additional complexity to the formulation. A nanomedicine has many additional physical properties when compared to a dissolved API including the size, shape, surface coating and particle composition, which may all influence the biological behaviour of the nanomedicine.62,77,78 Additionally, these particle properties can typically interact with one another and so it can be challenging to tailor the nanomedicine properties to a given application. Furthermore, the efficacy of a nanomedicine is strongly influenced by its behaviour in vivo, including drug release from the nanoparticles, nanomedicine accumulation in the target cells (e.g. the uterus in preterm birth) and its biodistribution. Typically, initial development of nanomedicine formulations focusses on producing stable nanoparticles with the required surface coating, the highest possible drug loadings (measured as a percentage of the drug in the total formulation) and the appropriate drug release kinetics. The ability to obtain the highest drug loadings possible for a formulation is important for the potential translation of a nanomedicine formulation to clinical use. While nanomedicines offer the potential to increase the therapeutic index of an API,79 allowing for dose reduction. Nanomedicine formulations also can consist of a considerable mass of excipients which reduce the drug loading in the formulation; ultimately low drug loadings can make it impractical to dose the therapeutically relevant amount of the API. Nanoparticle diameter is another key property and is possibly the most studied parameter with numerous studies investigating how this influences the biological behaviour of nanoparticles. The diameters of the nanoparticles produced are often driven by what is possible due to the processing method and nanomedicine type used; typically, it is possible to produce samples with diameters that vary up to five-fold. While the size of a nanomedicine is not the only factor that controls its biological properties, some generalisations have been provided. For IV dosing, a the distribution of particle sizes can not exceed 5 μm in diameter, this is because particles larger than this may lead to embolization.80 Nanoparticles in the diameter range of 20–200 nm do not generally leave the capillaries, while nanoparticles with diameters ≤10 nm can be filtered out of the circulation by the kidneys.81 For oral administration, there is no upper particle size limit and generally smaller sizes of nanoparticles can increase bioavailability of the API.82 The understanding of the effect of nanoparticle size on less common administration routes (such as vaginal pessaries) is more limited; we will discuss the findings of specific studies in the later sections of the review.
In the treatment of preterm birth with tocolytics, the ultimate objective is to target the distribution of the API to the uterus to stop contractions and avoid effects in other organs and systems in the mother, as well as critically, to prevent the API from crossing the placenta and entering the fetal circulation. In terms of applications in the treatment of preterm birth, there are very few studies that have systematically varied particle size and investigated biodistribution. One study has shown that smaller gold nanoparticles (20 or 50 nm) displayed higher accumulation in the uterus than nanoparticles with diameters of either 100 or 200 nm.83 However, this behaviour might not be generalisable to other nanoparticles with different surface properties. In the following sections we review the different nanomedicine systems and discuss how they have been developed and investigated to date for the treatment of preterm birth, including the in vitro and in vivo models used to evaluate their efficacy. We also discuss nanomedicine formulations that have been developed for indications other than preterm birth but with APIs that have known tocolytic properties and could therefore be repurposed for use in treating preterm birth. To aid comparison of the different nanomedicine formulations each section contains a table that summarises the key feature of the formulations.
Liposomes
Liposomes are artificial spherical vesicles made of one or more lipid bilayers.84 Due to this structure, a liposome possesses an aqueous core that is surrounded by a hydrophobic lipid bilayer(s). As such, liposomes can encapsulate both hydrophilic drugs (in the aqueous core) and hydrophobic drugs (within the lipid bilayers(s)) (Fig. 3A). Liposomes are the nanomedicine technology platform that has been investigated most extensively for the delivery of tocolytics. To the best of our knowledge, Refuerzo et al. were the first to develop liposomal formulations containing indomethacin, for the prevention of preterm birth.85 Indomethacin is an attractive tocolytic to formulate into the lipid bilayers of liposomes due its lipophilic nature (logP of 4.5). Refuerzo et al. developed fluorescently labelled multilamellar liposomes (150–170 nm) (Fig. 4A) synthesised from phosphatidylcholine and cholesterol via the lipid hydration-extrusion technique and the liposomes contained 3.7 wt% indomethacin.85 Fluorescently-labelled particles were imaged to identify particle accumulation, which showed successful, predominantly uterine accumulation (Fig. 4B) and minimal evidence of placental transfer (Fig. 4C).85 The liposomes were further developed to specifically target oxytocin receptors (OTRs) in the uterus, where they are abundantly expressed and are responsible for mediating the contraction stimulating effect of oxytocin during labour.76 Refuerzo et al. targeted the liposomes to the OTRs, by conjugating atosiban, a competitive OTR antagonist, to the surface of the liposome (Fig. 4D).76 Analysis of these OTR-targeted liposomes in pregnant mice via tail vein injection showed predominant distribution in the uterus compared to other sites such as the liver, placenta and fetus (Fig. 4E). When comparing the indomethacin-loaded, targeted liposomes to free indomethacin, the targeted liposomes demonstrated a 4-fold reduction in placental indomethacin concentration and hence reduced placental transfer (Fig. 4F). Finally, in an infection-induced preterm birth mouse model using lipopolysaccharide (LPS), the targeted liposomes reduced preterm birth rates compared to control (Fig. 4G).76 Further to these studies, Paul et al. developed uterine-targeted liposomes of ∼200 nm diameter containing other tocolytic agents: nifedipine, salbutamol, rolipram and indomethacin.86 The liposomes contained excipients cholesterol and the phospholipid 1,2-distearoyl-sn-glycero-2-phosphocholine (DSPC), as adapted from a method developed by Hua et al. for the development of antibody-conjugated liposomes.87 To add a uterine targeting functionality, a lipid 1,2-diastearoyl-sn-glycero-3-phospho-ethanolamine coupled to polyethylene glycol (PEG) with a maleimide chain end functionality was conjugated to an anti-OTR antibody. The liposomes were then synthesised by high pressure extrusion.86 Their key findings showed that OTR-targeted liposomes containing nifedipine, salbutamol or rolipram were able inhibit ex vivo human uterine contractions, whilst non-targeted liposomes did not reduce contractions at the equivalent concentrations tested (Fig. 4H).86 A reduction in contractility was only observed when the targeting approach was used (Fig. 4I). In a similar inflammation-induced mouse model of preterm birth, indomethacin-loaded OTR-targeted liposomes reduced preterm birth rates to 18%, whereas non-targeted liposomes had no significant effect and remained at a preterm birth rate of 58%.86 Further work published by Hua et al. exploring the in vitro mechanisms of the cellular uptake, internalisation and toxicity profiles of liposomes with a mean diameter of 200 nm.88 While the encapsulation efficiency was reported in the work, the drug loadings in the liposomes were not given. They showed that the incorporation of OTR targeting enhanced cellular interactions with uterine tissues, with the potential to increase tocolytic efficacy and decrease dosage profiles.76,86,89 This was true regardless of the targeting ligand (either an OTR monoclonal antibody or the OTR antagonist, atosiban).88 Both targeting approaches enhanced cellular internalisation compared to conventional liposomes; +78% with OTR antibody targeting and +82% with atosiban targeting.88
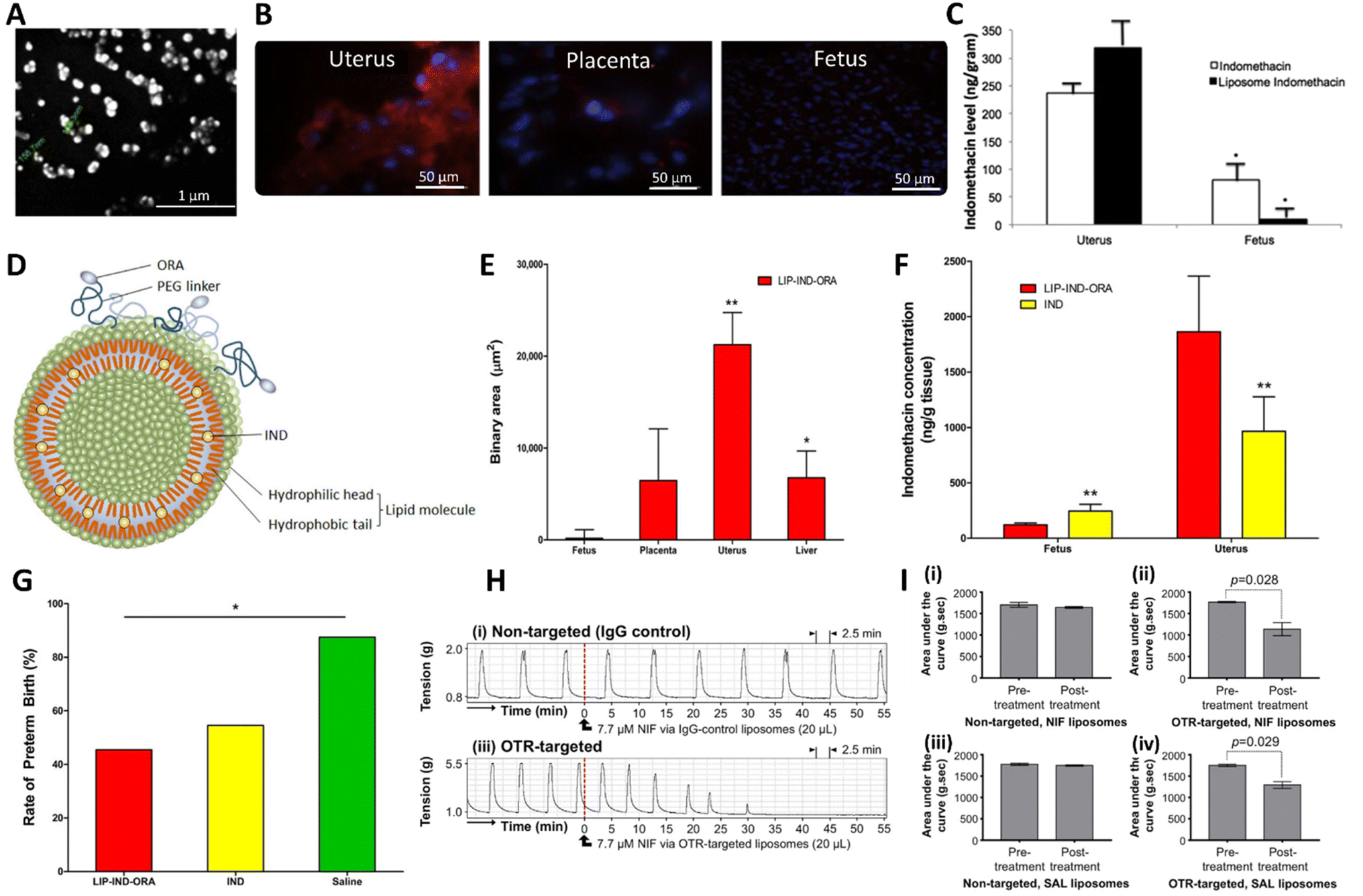 | ||
| Fig. 4 Liposomes developed for the treatment of preterm birth. (A) Scanning electron microscopy of indomethacin loaded liposome. (B) Analysis of the distribution of fluorescently labelled (red) indomethacin loaded liposomes in uterus, placenta, and fetus. (C) Quantification of the indomethacin concentration within uterus and fetus 4 hours following administration of indomethacin or indomethacin loaded liposomes. (D) Illustration of indomethacin (IND) loaded liposomes with oxytocin receptor antagonist (ORA) targetting ligands. (E) Biodistribution of liposomes containing indomethacin and targeted with oxytocin receptor antagonist components (LIP-IND-ORA) in vivo in pregnant mice. (F) A comparison of the targeted indomethacin liposomes (LIP-IND-ORA) and free indomethacin (IND) in terms of the concentrations of indomethacin in the maternal uterus and fetus, both were dosed at the same concentration (1 mg kg−1). (G) Comparison of the in vivo therapeutic efficacy in prevention of LPS induced preterm birth, free indomethacin and indomethacin liposomes were both dosed at 1 mg kg−1. (H) Effect of non-targeted and OTR-targeted nifedipine loaded liposomes on myometrial contractions in vitro. (I) Analysis of the contractility testing to obtain area under the curve (AUC) values for 30 minutes immediately prior to and 30 minutes after treatment with nifedipine (NIF) loaded liposomes (pre-treatment and post-treatment, respectively), (i) non-targeted liposomes containing NIF, (ii) targeted liposomes containing NIF, (iii) non-targeted liposomes containing salbutamol (SAL) and (iv) targeted liposomes containing SAL. (A), (B) and (C) are adapted with permission from Elsevier reference,85 (D), (E), (F) and (G) are taken from reference under Creative Commons CC BY license,76 (H) and (I) are taken with permission from Elsevier ref. 86. | ||
From the targeted liposomal studies, it is evident that the conjugating of a targeting moiety such as one directed to the OTR and exploiting the upregulation of OTRs in pregnant uterine tissue is a significant angle to explore for the progression of tocolytic agent development. Nevertheless, although the OTR has shown significant targeting benefit, other receptors that are over expressed and/or are uterine- or gestationally-specific should also be considered as future therapeutic targets.90 However, to date, other than the family of prostaglandin receptors which also change expression with gestation91 there have been very few alternatives put forward.
While liposomes have shown exciting data in terms of enhancing the efficacy of tocolytic APIs, further work is required to progress these systems through the preclinical translation pathway. Additionally, liposomes typically show limited drug loading, e.g. for indomethacin the highest drug loading reported is only 3.7% (a summary of all the liposomal systems is given in Table 3).76 Furthermore, long term storage instability in the dispersed form can present another potential barrier to the clinical use of liposomes.92 Therefore, alternative nanomedicine platforms may help to address these challenging areas by providing increased drug loading and greater storage stability.
| API | Lipid(s) | Targeting | Mean diameter (nm) | Drug loading (wt%) | Administration route | Ref. |
|---|---|---|---|---|---|---|
| a * values were not reported in the paper and could not be calculated based on the information given in the experimental. | ||||||
| Indomethacin | Soy bean phosphatidyl- choline & cholesterol | N/A | 150–170 | 3.7 | IV | 85 |
| Indomethacin | Soy bean phosphatidyl- choline, cholesterol & 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) | Atosiban-PEG2000-DSPE | 124 | 3.7 | IV | 76 |
| Nifedipine, salbutamol, rolipram or indomethacin | 1,2-Distearoyl-sn-glycero- 2-phosphocholine (DSPC), cholesterol & DSPE | Polyclonal anti-OTR antibody-PEG2000-DSPE | ∼200 | * | IV | 86 |
| Nifedipine or salbutamol hemisulfate | DSPC, cholesterol & DSPE | Polyclonal anti-OTR antibody-PEG2000-DSPE | 200 | * | Not tested in vivo (however route of administration would be IV) | 58 |
| Atosiban-PEG2000-DSPE | ||||||
Lipid-based nanomedicines
Lipid-based nanomedicines are nanoscale particles that are made of a lipid core. An attractive feature of these nanomedicines is that the lipid core of the particle can be made from physiological or biodegradable lipids.93 As with liposomes, the lipids within the particles can then processed within existing biological pathways within the body. Lipid-based nanomedicines can be categorised based on their core composition; solid lipid nanoparticles (SLNs) are made of lipid(s) that are solid at body temperature (Fig. 3B), nanostructured lipid nanocarriers (NLCs) have a core that contains both solid and liquid lipids (Fig. 3C) and nanoemulsions have a lipid core that is liquid (Fig. 3D). In all cases, the lipid-based nanomedicines have a coating of a stabiliser that provides colloidal stability.To our knowledge, there are no reported examples of any of these three lipid-based nanomedicines having been used to trial the delivery of tocolytics for the treatment of preterm birth in any in vitro, ex vivo or in vivo model. However, lipid-based nanomedicines containing APIs with tocolytic properties have been produced but for different indications. Formulations of progesterone have also been developed with potential prophylactic uses.
SLNs were introduced as a new potential nanocarrier system in 1991.94 They sparked further interest within the nanomedicine field following a publication by Muller et al. in 2000, that successfully highlighted the opportunities amongst SLN systems for pharmaceutical translation.94 SLNs were developed to combine the advantages of liposomes such as high chemical and physical stability, biocompatibility, specific targeting capabilities and sustained release.95–97 Moreover, their composition of physiologically-compatible excipients affords other pharmacological benefits such as improvement of bioavailability and decreased toxicity, with a simultaneous increase in efficacy for comparable dosages to non-formulated APIs.98,99 The ability for lipid-based nanoparticles to encapsulate an API is strongly influenced by the logP of the API and its compatibility with the lipids used in the formulation. Typically, high logP APIs are more successfully incorporated into lipid nanoparticles.100,101 Additionally, the surfactants selected in formulation, play a significant role in the formation of colloidally-stable nanoparticles. Recently, it has also been found that subtle differences in the relative block lengths of polymer surfactants can influence the polarity inside a lipid nanoparticle.102 Therefore, the insights offered by successful formulations reported in the literature could potentially be used to inform the design of future formulations.
SLNs have been successful at encapsulating a range of therapeutic agents for several acute and chronic conditions including ocular diseases and anticancer therapeutics.103,104 Of particular interest to the treatment of preterm birth, is lipid-based nanoformulations of indomethacin which were developed originally for ocular inflammation and rheumatoid arthritis due to its anti-inflammatory properties.103,105 For the treatment of chronic ocular inflammation, Hippalgaonkar et al. synthesised indomethacin loaded SLNs via a hot homogenisation technique with Compritol 888 ATO as the lipid to produce 1.9 wt% indomethacin nanoparticles with a mean diameter of 140 nm. They showed that the mean particle size of the SLNs could be tuned by varying the composition of the Tween 80 and poloxamer 188 surfactant mixture used. The SLN formulation was compared directly against Indocollyre®- a commercially available indomethacin eye drop containing 0.1 wt% of drug; showing an increased corneal permeation without effecting corneal integrity, and thus suggested improved safety and efficacy for the treatment of ocular inflammation.103 Balguri et al. then published using indomethacin SLNs to the deliver the API to posterior segment ocular tissues for the treatment of conditions such as macular oedema and ocular inflammation. They used the same excipients as Hippalgaonkar et al. but also functionalised the nanoparticles with chitosan chloride to enhance ocular tissue penetration. Chitosan is a linear polysaccharide derived from the chitin shells of crustaceans with adhesive-like properties and has been shown to increase mucoadhesion and potentially increase nanoparticle transport across the epithelium.106 The chitosan functionalised SLNs had a mean diameter of 265 nm and were found to increase the ocular penetration of indomethacin across the cornea compared to non-functionalised SLNs.107 Another API with tocolytic properties that has been formulated as an SLN is nifedipine. Barman et al. made particles of hydrogenated soybean phosphatidylcholine and dipalmitoylphosphatidyl glycerol with a mean diameter of 69 nm and a drug loading of nifedipine of 2.7%. By using trehalose as a cryoprotectant, the SLNs could be freeze dried to give a re-dispersible formulation which was stable under storage in the dry form for at least 6 months.108 Cassano and Trombino also processed progesterone to produce SLNs with the aim to develop a formulation with sustained release.109 They functionalised the SLNs with L-cysteine amino acid units designed to increase the time enabled for SLNs to anchor into the mucus layer of the uterus. The particles were produced by a microemulsion method, and the unloaded nanoparticles had a mean diameter of 740 nm (Fig. 5A) (the progesterone loaded particles were slightly smaller with a mean diameter of 640 nm). The drug loading was not specifically reported but we calculated it to be 3%. This study concluded that prolonged progesterone release was achieved for up to 72 hours in acidic in vitro media (representative of the vaginal cavity pH), therefore highlighting that progesterone-SLNs may be a viable route to provide sustained intravaginal progesterone administration, such as that which is required during assisted reproduction treatment e.g. for luteal support during embryo transfer and implantation.109 Importantly however, these studies also demonstrate the potential for SLNs to act as viable drug delivery vehicles for APIs for the treatment of preterm birth.
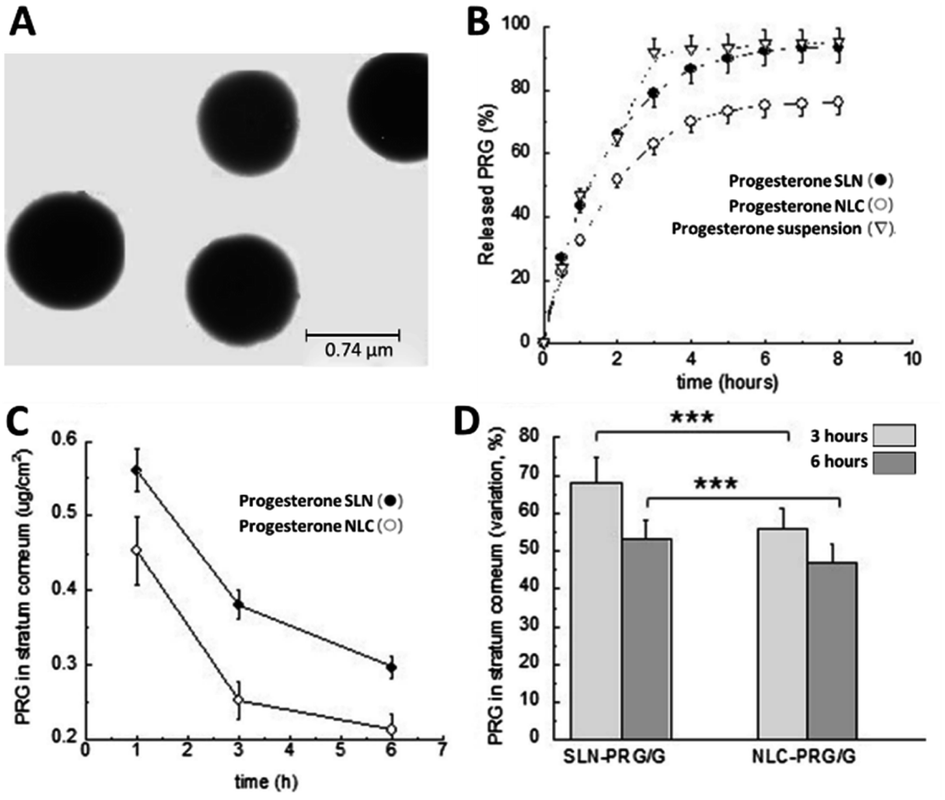 | ||
| Fig. 5 Lipid-based nanomedicines containing APIs relevant to the treatment of preterm birth. (A) TEM analysis of solid lipid nanoparticles based on L-cysteine. (B) In vitro release kinetics of progesterone from SLNs, NLCs compared to the suspension of progesterone in poloxamer 188 (2.5% w/w). (C) Analysis of the amount of progesterone in the stratum corneum after the application of either the SLNs or NLCs. (D) Comparison of the progesterone concentration in the skin after treatment with either the SLNs or NLCs after 3 hours or 6 hours in stratum corneum, as determined by tape-stripping experiments. (A) was adapted from reference under Creative Commons license,109 (B), (C) and (D) were adapted with permission from Elsevier ref. 111. | ||
Nanostructured Lipid Carriers (NLCs) were developed as ‘second generation lipid nanoparticles’ to overcome drawbacks associated with some SLN formulations118,119 including potential for low drug loading and drug expulsion resultant of the highly crystalline nature of the solid lipid cores.119,120 As a result, NLC formulations are very closely linked to SLNs in terms of their composition, however with an additional liquid lipid in the core of the carrier (Fig. 3), NLCs tend to display higher drug loading capabilities. NLCs of indomethacin have been produced by modifying an SLN formation with the addition of a liquid lipid, Miglyol 812, to the solid lipid of Compritol 888 ATO. The resulting nanoparticles had a mean diameter of 227 nm and a drug loading of 9.9%.107 This drug loading was much higher than that of the SLNs with a similar composition (excluding the liquid lipid) which had a 1.9% drug loading. In vivo application of the NLC formulation produced a 5-fold higher concentration of indomethacin to ocular tissues than the SLN formulation, which was attributed to the higher drug loading.107 NLCs of indomethacin and a non-tocolytic API (celastrol) have also been produced for potential applications in the treatment of rheumatoid arthritis as a transdermal drug delivery system. The optimised formulation contained as Precirol ATO 5 (solid lipid) and Labrasol ALF (lipid) and had a mean diameter of 27 nm with a total drug loading of 3.6%. The particles showed no irritation on rat skin but successfully penetrated through the top ∼300 μm of skin, demonstrating the potential of these lipid carriers to enhance API penetration into epithelial tissue.105
There are a few examples of NLCs of progesterone, although none specifically focussed on preterm birth treatment. Yuan et al. in 2007 produced NLCs by melt-emulsification using monostearin and stearic acid as the solid lipids, with oleic acid as the liquid lipid. Mean particle sizes of 340–385 nm were obtained (reported as a volume average rather than intensity average as is typical for dynamic light scattering (DLS)) and drug loadings were in the range of 3–11%.110 In 2017, Esposito et al. published work comparing both SLN and NLC formulations of progesterone using both ultrasound homogenisation and high-pressure homogenisation. The high-pressure homogenisation approach avoided the production of agglomerates and produced progesterone loaded SLNs of 181 nm or NLCs of 137 nm in diameter. The drug loading of the two formulations was ∼2%. This work showed that the composition of the lipids could be used to control the rate of progesterone release from the nanoparticles; the API was released more slowly when formulated into NLCs (Fig. 5B). The type of lipid nanocarrier used for the progesterone also influenced the penetration into skin (Fig. 5C); the amount of progesterone in the top layer of the skin (the stratum corneum) was higher for SLNs than NLCs (Fig. 5D).111 In 2018, Elmowafy demonstrated a NLCs formulation of progesterone with fatty alcohols as the lipids. The NLCs were prepared by high shear homogenisation and the mean diameter of the nanoparticles varied between ∼135–225 nm based on the different fatty alcohols used in the formulation. The drug loading however was not reported. They investigated the permeation across ex vivo rabbit duodenum and found enhanced permeation versus a suspension of progesterone, demonstrating the potential of this approach for enhancing oral delivery of progesterone.112
An NLC formulation of nifedipine, combined with simvastatin (medication to treat elevated cholesterol), has been produced for potential treatment of cardiovascular disease. Hassan et al. used a quality by design and a design of experiments approach to optimise the formulation.113 The nanomedicine was produced by a combined solvent evaporation and hot homogenisation method. The optimised nanoparticles had a mean diameter of 200 nm, however, it was not clear what drug loading was achieved in this formulation.113 This article demonstrated the benefit of using a design of experiments approach; they varied a large number of factors in their formulations to effectively identify the optimal formulation. Something that would have likely been much more time consuming to achieve with a traditional univariant testing method.
Nanoemulsions are the third type of lipid-based nanomedicine, they are nanoscale particles of liquid lipids. Similar to the other lipid-based nanomedicines previously discussed, the dispersed lipid phase is stabilised by stabiliser molecules. There are a few examples of nanoemulsions which have been investigated as carriers for APIs associated with the treatment of preterm birth. Patki et al. developed a lipid-based nanoemulsion with OHPC as the API with dimethylacetamide and medium chain triglycerides as the liquid core and Kolliphor HS 15 as the surfactant.114 Their system was a self-nanoemulsifying drug delivery system, an isotropic mixture of oil, surfactant and solvent that forms a stable nanoemulsion when dispersed in aqueous media.121 Their optimised system possessed a drug loading of 9% wt., had a mean diameter of 50 nm and was absorbed onto a powder of poly(vinyl alcohol) to allow processing into tablets along with other tabletting excipients. The vaginal administration of their nanoemulsion to a LPS infection-induced preterm birth model in mice showed a reduction in the number of mice delivering preterm: 60% compared to 100% for the mice treated with a blank formulation (no API). The mean time to delivery was increased from 14.5 hours for the blank formulation to 20.2 hours for the OHPC containing nanoemulsion.114 Much of the same team of researchers led by Guisto have demonstrated another nanoemulsion that used a liquid mixture of triglyceride and the co-solvent dimethylacetamide as the lipid carrier. The API used was a novel compound, a sphingosine kinase (SphK), inhibitor, 4-[[4-(4-chlorophenyl)-2-thiazolyl]amino]phenol, SKI II. SKI II had been shown to inhibit SphK, a key enzyme in the pathways that triggers contraction of myometrial tissue in response to lipopolysaccharide exposure.122 SKI II is a lipophilic compound with a solubility in aqueous media of less than 1 μg ml−1.115 The nanoemulsion was also a self-nanoemulsifying drug-delivery system. The authors studied different compositions of lipid, co-solvent and surfactant in order to determine an optimised nanoemulsion which had a mean diameter of 37 nm.115 Upon administration of the SKI II nanoemulsion via the vaginal route to LPS-infected mice, there was a significant reduction in rate of preterm birth. This work shows that SKI II could be a useful future option for treating preterm birth triggered by bacterial inflammation. However, SKI II is a compound still in early development and further work is needed to progress the API towards clinical trials.
There are also published examples of the development of nanoemulsions of other APIs with tocolytic properties, although with a focus on use for other clinical indications. For example, indomethacin nanoemulsions have been shown for ocular applications.116,117 Klang et al. showed that positively charged nanoemulsions of Lipoid E-80 (mainly comprising of phosphatidylcholine) with stearyl amine providing the positive surface charge, produced prolonged duration of the API on the cornea.116 Yamaguchi et al. produced nanoemulsions of castor oil by high-pressure homogenisation and used chitosan to provide mucoadhesive properties. The resulting nanoparticles had a mean diameter of 117 nm and a drug loading of 0.048% with regards to total solids mass in the formulation. The use of chitosan as a coating enhanced the attachment of the formulation onto mucin and increased the residence time of the indomethacin in the tear fluid.117
From the successful encapsulation of APIs with tocolytic properties such as indomethacin and nifedipine as well as progestogens in SLNs, NLCs and nanoemulsions, it is clear that there is also potential for these systems to be engineered specifically for applications in preterm birth (a summary of all the lipid-based nanomedicines can be seen in Table 4). The examples of how stabilisers such as chitosan can be used to alter the biological behaviour demonstrate that lipid-based nanomedicines also have the potential for uterine targeting, thus providing enhanced tocolytic activity. The existing publications on the APIs relevant to preterm birth have shown a number of potential benefits including: (1) the ability to freeze dry and redisperse some lipid-based nanomedicines provides the opportunity to improve formulation storage stability. (2) The composition of the lipids in the nanoparticles can be modified to tune drug release behaviour. (3) The drug loadings of lipid-based nanoparticles are typically ∼2–3% with upper values of 11% reported for NLCs, which is much higher than those reported for liposomes and hence provide greater drug delivery. (4) The ability of lipid-based nanoparticles, particularly NLCs, to enhance permeability across biological barriers may allow the nanomedicines to open new administration routes for these tocolytic APIs.
| API | Carrier type | Lipid(s) | Stabiliser(s) | Mean diameter (nm) | Drug loading (wt%) | Administration route | Ref. |
|---|---|---|---|---|---|---|---|
| a NE-nanoemulsion. b * values were not reported in the paper and could not be calculated based on the information given in the experimental. | |||||||
| Indomethacin | SLNs | Compritol 888 ATO | Tween 80 and poloxamer 188 | 140 | 1.9 | Topical (ocular) | 103 |
| Indomethacin | SLNs | Compritol 888 ATO | Tween 80, poloxamer 188 and chitosan | 265 | 1.8 | Topical (ocular) | 107 |
| Nifedipine | SLNs | Hydrogenated soybean phosphatidylcholine and dipalmitoylphosphatidyl glycerol | Stabilisation provided by the charge by of the phospholipid | 69 | 2.7 | None specified | 108 |
| Progesterone | SLNs | 2(R)-2,3-dihydroxypropanoate of octadecyl 2,3-dihydroxypropano-ate | Tween 20 | 640 | 3 | Vaginal | 109 |
| Indomethacin | NLCs | Miglyol 812 and Compritol 888 ATO | Tween 80 | 227 | 9.9 | Topical (ocular) | 107 |
| Indomethacin and a non-tocolytic API (celastrol) | NLCs | Precirol ATO 5 (solid lipid) and Labrasol ALF | Cremophor RH40 | 27 | 3.6 | Transdermal | 105 |
| Progesterone | NLCs | Monostearin, stearic acid and oleic acid | Polyethylene glycol | 340–385 | 3–11 | Oral | 110 |
| Monostearate and Tween 20 | |||||||
| Progesterone | SLNs | Tristearin | Poloxamer 188 | 180 | 2 | Topical (skin) | 111 |
| Progesterone | NLCs | Tristearin and Miglyol 812 N | Poloxamer 188 | 140 | 2 | Topical (skin) | 111 |
| Progesterone | NLCs | Stearic acid, sesame oil and cetyl alcohol, cetostearyl alcohol | Tween 80 and PEG mixture (400, 1500, 4000) | 135–225 | * | Oral | 112 |
| Nifedipine and a non-tocolytic API (simvastatin) | NLCs | Precirol ATO 5 and Capryol 90 | Poloxamer 407, Gelucire 44/14, Tween 20, and lecithin | 200 | * | Oral | 113 |
| OHPC | NE | Dimethylacetamide and medium chain triglyceride | Kolliphor HS 15 | 50 | 10 | Vaginal | 114 |
| SKI II | NE | Dimethylacetamide and medium chain triglyceride | Kolliphor HS 15 | 37 | * | Vaginal | 115 |
| Indomethacin | NE | Lipoid E-80 | Poloxamer 188 and stearylamine | 110 | 0.1 | Topical (ocular) | 116 |
| Indomethacin | NE | Castor oil | Polysorbate 80 | 117 | 0.05 | Topical (ocular) | 117 |
Polymer nanoparticles
Polymer nanoparticles are submicron particles with a core consisting of a hydrophobic (non-water soluble) polymer. These nanoparticles are coated with stabilisers that have a polar character and provide colloidal stability.123 The polymer core is made of a degradable polymer to ensure that the nanoparticle does not persist in the body after the drug has been delivered. While a wide range of polymers have been investigated this application, polyesters, specifically polylactic acid (PLA), polyglycolic acid (PGA), their copolymer poly(lactic-co-glycolic acid) (PLGA) and polycaprolactone (PCL) are the most widely used polymers due to their well-documented use in biomedical applications.124 Whilst we were not able to find any examples of polymer nanoparticles that had been designed for application in preterm birth per se, APIs used to treat preterm birth have been formulated as polymer nanoparticles for use in other indications. For example, Alkholief et al. produced a PLGA nanoformulation of indomethacin; they used an emulsion solvent evaporation method to investigate the production parameters using two different surfactants (polyvinylpyrrolidone and polyvinyl alcohol), at a range of concentrations and different organic solvents. Depending on the conditions used, mean particle sizes of ∼280–610 nm were obtained, with higher surfactant concentrations yielding larger particle size. The indomethacin drug loadings were 5–6.8% of the formulations. Cytotoxicity tests showed that encapsulating the indomethacin in the polymer nanoparticles reduced the toxicity in HepG2 liver cells compared to the free indomethacin.125 The reduced toxicity of their formulation might have been caused by a reduction in the concentration of free indomethacin, or differences in the accumulation of the nanoparticles into the cells, although accumulation was not investigated. Muljajew et al. produced polymer nanoparticles containing indomethacin by a nanoprecipitation approach. They assessed six poly(ester amides) (Fig. 6A) as the carrier and combined a modelling and experimental high throughput screening method to determine the optimal conditions for obtaining indomethacin polymer nanoparticles. Particles with mean diameters of 100–400 nm (Fig. 6B) and modest drug loadings of 0.9–1.5% (Fig. 6C) were obtained.126 The combined modelling and high throughput experimental approach may provide an opportunity to develop future nanoformulations in a more informed manner than the traditional screening methods typically used elsewhere; this combination of modelling may also accelerate the development of new formulations.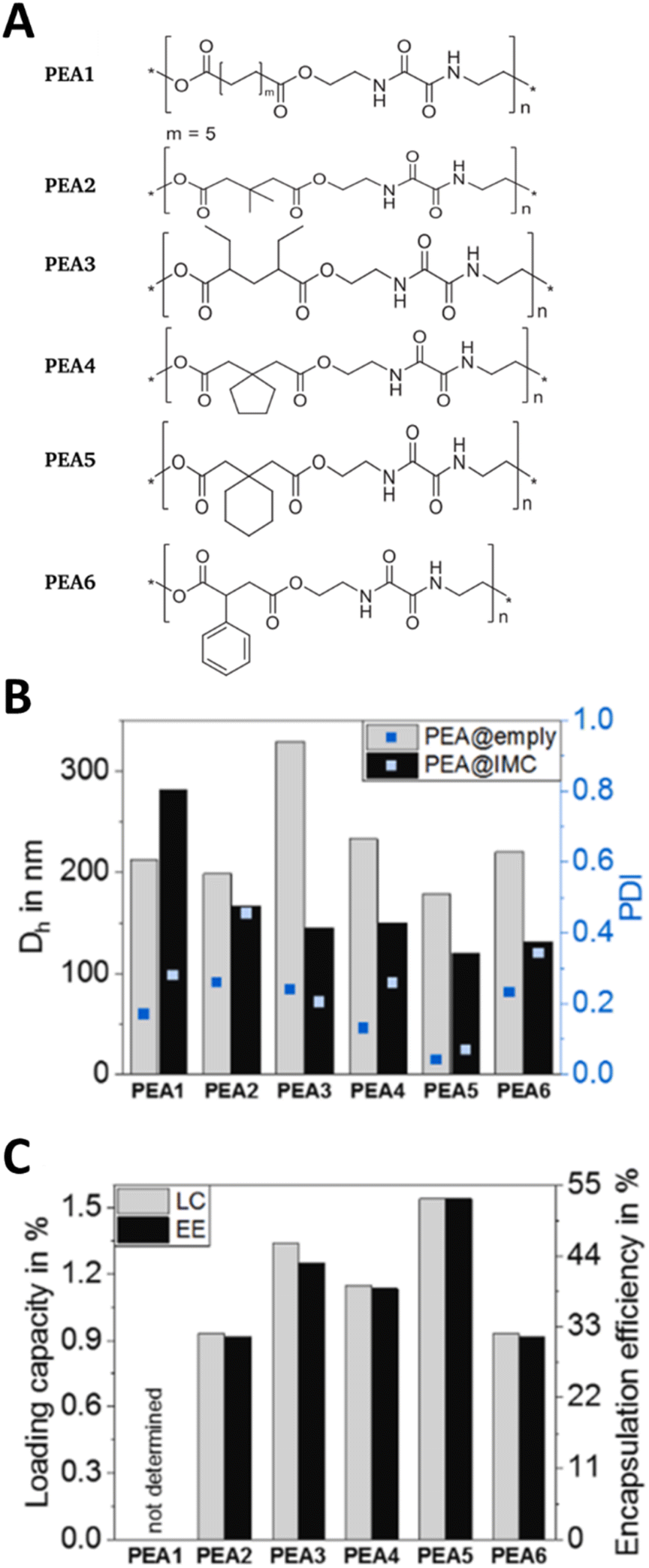 | ||
| Fig. 6 Polymer nanoparticles using poly(ester amide)s nanoparticles and indomethacin at the API. (A) The repeat units of the poly(ester amide)s used in the research. (B) Characterisation of the unloaded poly(ester amide)s nanoparticles (PEA@empty) and indomethacin loaded poly(ester amide)s nanoparticles (PEA@IMC NPs) by DLS to give the mean diameter (Dh) and polydispersity index (PDI). (C) The drug loading capacity (LC) and encapsulation efficiency (EE) of indomethacin loaded poly(ester amide)s nanoparticles. All parts of this figure are taken with permission from Elsevier ref. 126. | ||
The potential to produce polymer nanoparticles containing indomethacin is attractive for the treatment of preterm birth, particularly as the examples show that it is possible to obtain higher drug loadings than in liposomes (see Table 5 for a summary of the polymer nanoparticle formulations). Polymer nanoparticles have also been produced with targeting ligands.127 Therefore, it is highly feasible to utilise the OTR targeting (or others as they are identified) approaches as shown in liposomes and apply them to polymer nanoparticles for use in the treatment of preterm birth. To further investigate this however, it is imperative that the biodistribution and efficacy of the nanomedicines in vivo is examined.
| API | Polymer | Stabiliser(s) | Mean diameter (nm) | Drug loading (wt%) | Administration route | Ref |
|---|---|---|---|---|---|---|
| Indomethacin | PLGA | Polyvinylpyrrolidone and polyvinyl alcohol | 280–610 | 5–6.8 | Not specified | 125 |
| Indomethacin | Poly(ester amides) (repeat units can be seen in Fig. 6A) | No surfactant or polyvinyl alcohol | 100–400 | 0.9–1.5 | Not specified | 126 |
Nanosuspensions
Nanosuspensions, also known as nanocrystals or solid drug nanoparticles, are sub-micron particles of water insoluble (high logP) compounds. The hydrophobic surfaces of the particles are coated by the adsorption of stabilisers which provide colloidal stability.128 Nanosuspensions differ from the other nanomedicines in that they do not contain a carrier material, rather the nanoparticles are themselves composed entirely of the poorly water-soluble API. This means that nanosuspensions offer the highest drug loadings out of all nanomedicines. Nanosuspensions of a wide range of poorly water-soluble APIs can be produced via a number of routes, with milling,129–131 nanoprecipitation,132–134 high pressure homogenisation135 and emulsion-templated freeze drying136–139 being some of the most common methods.
Hoang et al. reported the successful formulation of a nanosuspension of progesterone. Nanosuspensions were prepared by three different processing methods, nanoprecipitation, high pressure homogenisation and wet milling, all using poloxamer 407 (Pluronic® F127) as the stabiliser. Wet milling produced the smallest particle size with a mean diameter of ∼260 nm (Fig. 7A). The drug loading in the formulation was not given but we calculated it to be 80%. The authors carried out an in vivo comparison of their nanosuspension formulation to a gel formulation of progesterone (Crinone® 8%). They showed that when administered vaginally the gel formulation displayed significantly less progesterone uptake in the cervix compared to a nanosuspension formulation of progesterone. The maximum concentration (Cmax) of progesterone in the cervix using the nanosuspension was increased 5-fold compared to the branded gel (Fig. 7B). The authors speculated that the improvement in drug delivery was due to particle size and the PEG coating which made the nanoparticles mucoinert and therefore able to better penetrate through the mucus lining of the cervix. In an in vivo trial of progesterone rescue in mice where preterm birth was initiated using a progesterone antagonist, RU486, the nanosuspension increased the number of pups delivering full term compared to the commercial progesterone gel (Fig. 7C).140 Further work by ensign exploring nanosuspensions for the treatment of preterm birth, has focussed on the use of histone deacetylase (HDAC) inhibitors.141 Histone deacetylation has been found to increase at term in both the myometrium (smooth muscle of the uterus) and the cervix. Trichostatin A is a potent HDAC inhibitor. Recently, Zierden et al. produced a nanosuspension formulation by milling trichostatin A with Pluronic® F127 to give a mean diameter of ∼200 nm as measured by DLS. They investigated the vaginal delivery of a combination of the trichostatin A and progesterone nanosuspensions (in 1.5:100 mass ratio of the two APIs) in an LPS-induced mouse model. The combination treatment was more effective at preventing preterm birth than either of the nanosuspensions independently. They also found combined application of the two APIs inhibited cell contractility in human myometrial cells in vitro.141
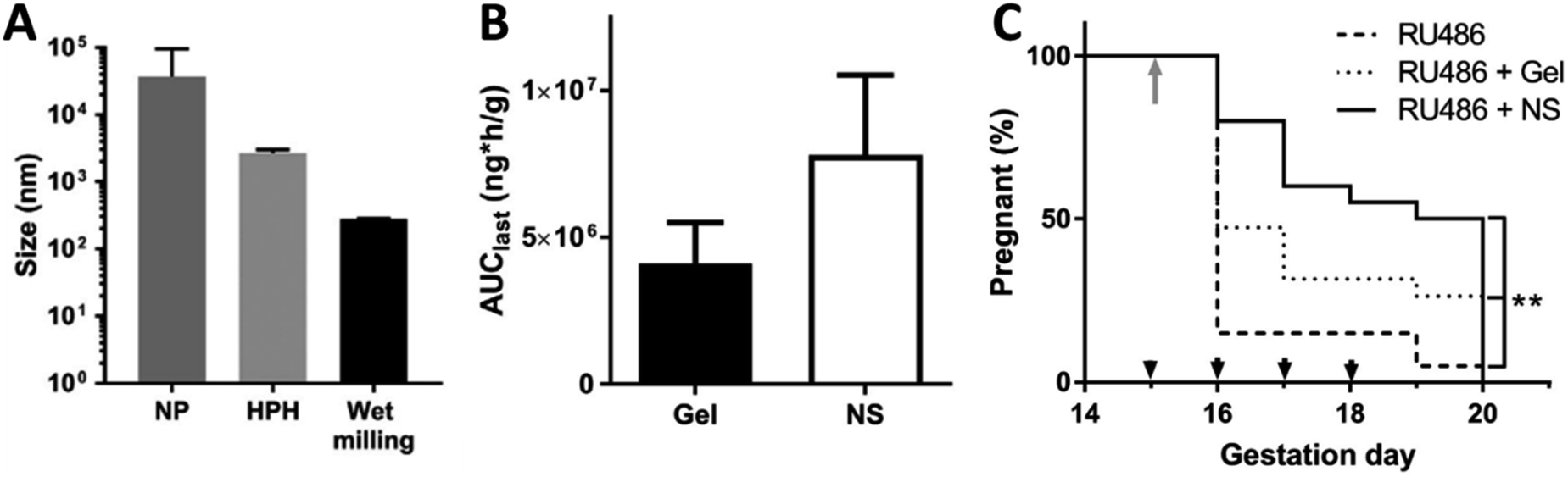 | ||
| Fig. 7 Nanosuspensions containing progesterone investigated for the treatment of preterm birth. (A) Mean diameter of progesterone nanosuspension produced by different size reduction techniques, nanoprecipitation (NP), high pressure homogenisation (HPH) and wet milling (the wet milling duration used was 9 h). (B) Area under the curve (AUC) in the cervix after 24 h (AUClast) after a single vaginal dose in healthy pregnant mice on gestation day 15. (C) Comparison of the percentage of animals remaining pregnant after RU486 injection on gestation day 15 (grey arrow) then treated with either the progesterone nanosuspension (NS) or the progesterone gel. Animals in the treatment groups received daily vaginal doses from (black arrowheads). ** denotes log-rank p = 0.001 for all curve comparison. All parts of the figure are adapted with permission from Elsevier ref. 140. | ||
A nanosuspension of indomethacin has been produced by Styliari et al. by using block copolymer of PEG and PCL (PEG–PCL) as the polymer stabiliser.142 They combined molecular simulations with experimental testing to investigate how different PEG chain lengths of block copolymers of PEG-PCL controlled the formation of polymer coated nanosuspensions using a nanoprecipitation type method. In this work, the indomethacin was processed as a coarse aqueous nanosuspension by sonication (mean diameter by DLS of ∼700 nm), then this suspension was mixed with an acetone solution of PEG–PCL in a T-piece. The authors hypothesised that the acetone solution of the polymer would partially dissolve the larger nanoparticles of indomethacin and also provide colloidal stabilisation. They observed a reduction in particle diameter, obtaining particles of ∼250 nm diameter, and drug loadings of up to 78% were also reported.
Nanosuspensions offer the highest drug loadings out of all the nanomedicine options discussed in this review, with values of 80% reported (see Table 6 for a summary of the nanosuspension formulations). This attribute of high drug loadings for nanosuspensions is very useful for APIs with lower potency values, where considerable amounts of a nanomedicine may need to be administered to achieve a therapeutic dose. Furthermore, there are many examples of nanosuspensions that can be processed into a dry form that can be redispersed upon use, and hence overcome some storage stability issues.137,143,144 While examples of targeted nanosuspensions are less common than other nanomedicines, progress has been made in this area with the design of targeting ligands that can adsorb onto the surface of the nanoparticles.145
| API | Stabiliser | Mean diameter (nm) | Drug loading (wt%) | Administration route | Ref. |
|---|---|---|---|---|---|
| a * values were not reported in the paper and could not be calculated based on the information given in the experimental. | |||||
| Progesterone | Pluronic F127 | 260 | 80 | Vaginal | 140 |
| Trichostatin A | Pluronic F127 | 200 | * | Vaginal | 141 |
| Indomethacin | PEG-PCL | 250 | 78 | Not specified | 142 |
Conclusions and future work
Preterm birth represents a major, pressing, global healthcare problem with an urgent need for more effective treatments. New formulations of APIs are required to prolong gestation, to improve dosage profiles, enhance targeting to the uterine tissue, reduce feto-maternal side effects and ultimately improve safety and efficacy. The use of nanomedicine for the delivery of tocolytics is still at an early stage; the first example of a nanomedicine delivering a tocolytic in vivo was published in 2015.85 Over the past 8 years there has been considerable improvements in nanomedicine technology for tocolytic therapy, this has been led by Refuerzo, Paul and Hua showing promising manipulation of several tocolytics in liposomal formulations.76,88,90 Collectively, their key findings have shown increased uterine targeting, increased uterine uptake, reduced placental transfer and a noteworthy improvement on uterine quiescence with concomitant reduction in preterm birth delivery rates in in vivo mouse models. Liposomes are likely the nanomedicine type that is most progressed in the field of preterm birth because liposomes were amongst the first approved nanomedicines; liposomal doxorubicin (a cancer drug) was approved by the FDA in 1995.146 This success de-risked the development of liposomal formulations; the majority of the earliest clinically approved nanomedicines were liposomes,147 and liposomes initially dominated early clinical nanomedicine success.148 However, more recently other nanomedicine types have now moved through the development pathway with 14% and 8% of clinical trials in investigating lipid nanoparticles and polymer nanoparticles respectively. This is a considerable growth in the development of such systems as lipid nanoparticles and polymer nanoparticles only represent ∼3% and 0% of currently approved nanomedicines.148 While liposomes are attractive nanocarriers they tend to be less stable than the other nanomedicine types, and are not typically suitable for oral dosing due to degradation under acidic conditions.149 As such, the opportunities offered by other nanomedicine types other than liposomes should be more effectively explored for preterm birth. As identified in this review, there are many examples of other lipid-based nanomedicines where tocolytic APIs could be formulated to potentially provide increased drug loading compared to liposomes. Such lipid-based nanomedicines can use the same approaches to incorporating targetting as liposomes, such as the use of lipid-PEG-ligand conjugates. Therefore, the successful targeting identified with liposomes can likely be adapted and used for lipid-based nanoparticles. Polymer nanoparticles are the nanomedicine type that has the fewest examples of the delivery of tocolytic APIs. However, this should not exclude polymer nanoparticles as a potential opportunity, they have good stability during preparation and storage in biological fluids. Additionally, polymer nanoparticles have been shown to offer many of the benefits of nanomedicine types (targeting, the ability to tune size and surface chemistry, etc.)150 which should allow application in the treatment of preterm birth. Finally, nanosuspensions offer a route to deliver high concentrations of the API due to their very high drug loadings of up to ∼80% wt relative to excipients. Nanosuspensions are the second most progressed nanomedicine type (after liposomes) for the treatment of preterm birth with in vivo trials showing significantly more efficient in drug delivery for a nanosuspension of progesterone than unformulated progesterone. There is therefore a considerable potential for non-lipid systems to also be used as potential tocolytic therapeutics. Additionally, repurposing nanomedicine formulations from other indications may provide a considerable opportunity to diversify the range of formulations available for application in preterm birth.Nanomedicines can be produced with many different particle properties including: size, surface charge, surface chemistry and carrier composition, all of which can considerably affect biological behaviour. The studies that show the most positive results in terms of reduction in preterm birth in mice have been using liposomes ∼200 nm in diameter for IV dosing or 200 nm nanosuspensions for vaginal dosing of progesterone. For IV dosing, targeting is critical to achieve a therapeutic effect, however with regards to the other particle properties there have not been studies to ascertain the optimal parameters. In terms of nanomedicine size, studies on the biodistribution of nanoparticles in pregnant animals have shown that there is a considerable difference in the maternal-to-foetal transfer based on the type and size of nanoparticles. Such analysis indicates that smaller nanoparticles (less than ∼10 nm) may be more likely to enter the foetus.90 However, there is not the data to generalise if this is true across all nanomedicine types. Furthermore, the density of the polymer coating on the nanoparticle may considerably alter the ability of nanoparticles to penetrate through tissue. For example, the use of a dense coating of PEG has been found to facilitate an increase in the mobility of polymer nanoparticles in tissue.151 Additionally, studies on targeted nanomedicines for other applications have shown that factors such as the density of the targetting ligand and the length of the polymer used between the nanoparticle and the targetting ligand can play a key role in influencing the targeting efficacy.152,153 Therefore, the careful selection of the polymer stabiliser is a key consideration for targeted nanomedicines as it has the potential to alter targeting efficacy or potential lead to increased transfer to the fetus. Currently, these knowledge gaps make it challenging to efficiently design nanomedicines for the treatment of preterm birth based on IV dosing. For vaginal dosing of nanomedicines with the aim to avoid mucoadhesion, there is data to support some elements of the nanomedicine design. Trends for enhancing muco-penetration have been shown across at least two nanoparticle types, with both polymer nanoparticles75,154 and nanosuspensions140 showing that particles with mean diameters in the range 110–260 nm and coated with polymers that provided a PEG coating have enhanced muco-penetration. In the future, the production of nanomedicines with systematic structural variation would allow more detailed studies in the relationships between particle properties and biological behaviour. Identifying the key design rules for nanomedicines for the treatment of preterm birth would considerably enhance progress in this field.
The drug loading of an API within a nanomedicine is another key factor that needs to be considered in order to allow clinical translation of novel formulations. With the exception of the nanosuspensions, many of the nanomedicines report API loadings of ∼1–4%. If we focus on indomethacin, the most widely investigated nanomedicine formulations of a tocolytic API, it is typically given orally in doses of 25–100 mg as a tocolytic and has a bioavailability of virtually 100%.155 Assuming that a similar IV dose of indomethacin would be used in a new nanomedicine formulation with a 1% drug loading, then potentially 2.5–10.0 g of a nanomedicine formulation would need to be infused. This amount of excipients in the formulation is much larger compared to other clinically used liposome systems such as liposomal doxorubicin (which has a API loading of 16.7%).156 Further optimisation of the current liposome formulations or use of higher drug loading lipid-based nanomedicines such as NLCs may address this issue.
From a clinical perspective, future developments in nanomedicines for the prevention of preterm birth are likely to be beneficial in the acute setting of clinical management of women in threatened spontaneous labour. Preterm birth is an attractive target due to the very high risks of morbidity and mortality to the neonate. As such, preterm birth is associated with a very significant financial cost both in the acute hospital setting of neonatal intensive care unit and to individuals and society in the longer term from caring for infants harmed by preterm birth (neurodevelopmental delay etc.). Women in preterm labour currently receive IV medications such as magnesium sulfate, antibiotics and tocolytics. As such, the administration of nanomedicine by IV should be suitable for a clinical environment. The clinical application of nanomedicines is likely to be beneficial in the acute setting to allow reduction in dosing regimen to prevent side effects. Additionally, improvements in clinical management of preterm birth from the cessation of uterine activity would allow transfer to specialist neonatal facilities, administration of antenatal corticosteroids or potentially the prolongation of pregnancy all of which have strong associations with improved neonatal outcomes.
Ultimately, the treatment of preterm birth is a considerable opportunity for nanomedicine to address, improved therapies would offer significant health benefits globally. Addressing the challenges identified in this review should allow for the accelerated development of nanomedicine technologies that have a greater potential for clinical translation.
Conflicts of interest
SR is a Director of Tandem Nano Ltd and co-inventor of patents relating to drug delivery using nanotechnologies. SR has received research funding from GSK/ViiV, AZ and Gilead and a consultancy from Gilead. TM is a co-inventor of patents relating to drug delivery using nanotechnologies and has carried out consultancy work for Vifor Pharma.References
- World Health Organization (WHO), New Global Estimates on Preterm Birth, 2018 Search PubMed.
- L. Liu, S. Oza, D. Hogan, Y. Chu, J. Perin, J. Zhu, J. E. Lawn, S. Cousens, C. Mathers and R. E. Black, Lancet, 2016, 388, 3027–3035 CrossRef.
- S. R. Walani, Internet J. Gynecol. Obstet., 2020, 150, 31–33 CrossRef PubMed.
- S. Chawanpaiboon, J. P. Vogel, A. B. Moller, P. Lumbiganon, M. Petzold, D. Hogan, S. Landoulsi, N. Jampathong, K. Kongwattanakul, M. Laopaiboon, C. Lewis, S. Rattanakanokchai, D. N. Teng, J. Thinkhamrop, K. Watananirun, J. Zhang, W. Zhou and A. M. Gülmezoglu, Lancet Global Health, 2019, 7, e37–e46 CrossRef PubMed.
- W. A. Engle, K. M. Tomashek and C. Wallman, Pediatrics, 2007, 120, 1390–1401 CrossRef PubMed.
- S. Al-Alaiyan, Ann. Saudi Med., 2008, 28, 1–3 CrossRef PubMed.
- P. I. Macfarlane, S. Wood and J. Bennett, Arch. Dis. Child. Fetal Neonatal Ed., 2003, 88, 199–202 CrossRef PubMed.
- P. T. B. Larroque, G. Breart, M. Kaminski, M. Dehan, M. Andre, A. Burguet, H. Grandjean, B. Ledesert, C. Leveque, F. Maillard, J. Matis and J. Roze, Arch. Dis. Child. Fetal Neonatal Ed., 2004, 89, 139–144 CrossRef PubMed.
- J. Chatterjee, J. Gullam, M. Vatish and S. Thornton, Arch. Dis. Child. Fetal Neonatal Ed., 2007, 92, 88–93 CrossRef PubMed.
- R. Lozano, M. Naghavi, K. Foreman, S. Lim, K. Shibuya, V. Aboyans, J. Abraham, T. Adair, R. Aggarwal, S. Y. Ahn, M. A. AlMazroa, M. Alvarado, H. R. Anderson, L. M. Anderson, K. G. Andrews, C. Atkinson, L. M. Baddour, S. Barker-Collo, D. H. Bartels, M. L. Bell, E. J. Benjamin, D. Bennett, K. Bhalla, B. Bikbov, A. Bin Abdulhak, G. Birbeck, F. Blyth, I. Bolliger, S. Boufous, C. Bucello, M. Burch, P. Burney, J. Carapetis, H. Chen, D. Chou, S. S. Chugh, L. E. Coffeng, S. D. Colan, S. Colquhoun, K. E. Colson, J. Condon, M. D. Connor, L. T. Cooper, M. Corriere, M. Cortinovis, K. Courville De Vaccaro, W. Couser, B. C. Cowie, M. H. Criqui, M. Cross, K. C. Dabhadkar, N. Dahodwala, D. De Leo, L. Degenhardt, A. Delossantos, J. Denenberg, D. C. Des Jarlais, S. D. Dharmaratne, E. R. Dorsey, T. Driscoll, H. Duber, B. Ebel, P. J. Erwin, P. Espindola, M. Ezzati, V. Feigin, A. D. Flaxman, M. H. Forouzanfar, F. G. R. Fowkes, R. Franklin, M. Fransen, M. K. Freeman, S. E. Gabriel, E. Gakidou, F. Gaspari, R. F. Gillum, D. Gonzalez-Medina, Y. A. Halasa, D. Haring, J. E. Harrison, R. Havmoeller, R. J. Hay, B. Hoen, P. J. Hotez, D. Hoy, K. H. Jacobsen, S. L. James, R. Jasrasaria, S. Jayaraman, N. Johns, G. Karthikeyan, N. Kassebaum, A. Keren, J. P. Khoo, L. M. Knowlton, O. Kobusingye, A. Koranteng, R. Krishnamurthi, M. Lipnick, S. E. Lipshultz, S. Lockett Ohno, J. Mabweijano, M. F. MacIntyre, L. Mallinger, L. March, G. B. Marks, R. Marks, A. Matsumori, R. Matzopoulos, B. M. Mayosi, J. H. McAnulty, M. M. McDermott, J. McGrath, Z. A. Memish, G. A. Mensah, T. R. Merriman, C. Michaud, M. Miller, T. R. Miller, C. Mock, A. O. Mocumbi, A. A. Mokdad, A. Moran, K. Mulholland, M. N. Nair, L. Naldi, K. M. V. Narayan, K. Nasseri, P. Norman, M. O'Donnell, S. B. Omer, K. Ortblad, R. Osborne, D. Ozgediz, B. Pahari, J. D. Pandian, A. Panozo Rivero, R. Perez Padilla, F. Perez-Ruiz, N. Perico, D. Phillips, K. Pierce, C. A. Pope, E. Porrini, F. Pourmalek, M. Raju, D. Ranganathan, J. T. Rehm, D. B. Rein, G. Remuzzi, F. P. Rivara, T. Roberts, F. Rodriguez De León, L. C. Rosenfeld, L. Rushton, R. L. Sacco, J. A. Salomon, U. Sampson, E. Sanman, D. C. Schwebel, M. Segui-Gomez, D. S. Shepard, D. Singh, J. Singleton, K. Sliwa, E. Smith, A. Steer, J. A. Taylor, B. Thomas, I. M. Tleyjeh, J. A. Towbin, T. Truelsen, E. A. Undurraga, N. Venketasubramanian, L. Vijayakumar, T. Vos, G. R. Wagner, M. Wang, W. Wang, K. Watt, M. A. Weinstock, R. Weintraub, J. D. Wilkinson, A. D. Woolf, S. Wulf, P. H. Yeh, P. Yip, A. Zabetian, Z. J. Zheng, A. D. Lopez and C. J. L. Murray, Lancet, 2012, 380, 2095–2128 CrossRef PubMed.
- National Institute for Health and Care Excellence (NICE), Preterm Labour and Birth, 2016 Search PubMed.
- L. Lightstone, Med., 2015, 43, 550–555 CrossRef.
- L. nan Zeng, L. li Zhang, J. Shi, L. ling Gu, W. Grogan, M. M. Gargano and C. Chen, Taiwan. J. Obstet. Gynecol., 2014, 53, 443–451 CrossRef PubMed.
- E. Bhattacharjee and A. Maitra, npj Genomic Med., 2021, 6(43) DOI:10.1038/s41525-021-00209-5.
- F. M. Ubaldi, D. Cimadomo, A. Vaiarelli, G. Fabozzi, R. Venturella, R. Maggiulli, R. Mazzilli, S. Ferrero, A. Palagiano and L. Rienzi, Front. Endocrinol., 2019, 10, 1–18 CrossRef PubMed.
- K. S. Montgomery, S. Cubera, C. Belcher, D. Patrick, H. Funderburk, C. Melton and M. Fastenau, J. Perinat. Educ., 2005, 14, 26–35 CrossRef PubMed.
- F. S. M. Fuchs, Semin. Fetal Neonat. Med., 2016, 21, 113–120 CrossRef CAS PubMed.
- G. Puccio, M. Giuffré, M. Piccione, E. Piro, V. Malerba and G. Corsello, Ital. J. Pediatr., 2014, 40, 1–7 CrossRef PubMed.
- B. S. Muhlhausler, S. N. Hancock, F. H. Bloomfield and R. Harding, Pediatr. Res., 2011, 70, 117–122 CrossRef PubMed.
- M. L. Hediger, T. O. Scholl, J. I. Schall and P. M. Krueger, Ann. Epidemiol., 1997, 7, 400–406 CrossRef CAS PubMed.
- I. Glick, E. Kadish and M. Rottenstreich, Int. J. Women's Health, 2021, 13, 751–759 CrossRef PubMed.
- T. Sha, X. Yin, W. Cheng and I. Y. Massey, Fertil. Steril., 2018, 109, 330–342 CrossRef PubMed.
- J. E. Norman, N. Marlow, C. M. Messow, A. Shennan, P. R. Bennett, S. Thornton, S. C. Robson, A. McConnachie, S. Petrou, N. J. Sebire, T. Lavender, S. Whyte and J. Norrie, Lancet, 2016, 387, 2106–2116 CrossRef CAS PubMed.
- A. Care, S. J. Nevitt, N. Medley, S. Donegan, L. Good, L. Hampson, C. Tudur Smith and Z. Alfirevic, BMJ, 2022, 376, 1–12 Search PubMed.
- R. Gaiser, P. J. Meis, M. Klebanoff, E. Thom, M. P. Dombrowski, B. Sibai, A. H. Moawad, C. Y. Spong, J. C. Hauth, M. Miodovnik, M. W. Varner, K. J. Leveno, S. N. Caritis, J. D. Iams, R. J. Wapner, D. Conway, M. J. O'Sullivan, M. Carpenter, B. Mercer, S. M. Ramin, J. M. Thorp and A. M. Peaceman, N. Engl. J. Med., 2003, 348, 2379–2385 CrossRef PubMed.
- S. C. Blackwell, S. P. Chauhan, C. Gyamfi-Bannerman, J. R. Biggio, B. L. Hughes, J. M. Louis, T. A. Manuck, H. S. Miller, A. F. Das, G. R. Saade, P. Nielsen, J. Baker, O. M. Yuzko, G. I. Reznichenko, N. Y. Reznichenko, O. Pekarev, N. Tatarova, J. Gudeman, M. Duncan, L. Williams, J. Krop, R. Birch and M. J. Jozwiakowski, Am. J. Perinatol., 2020, 37, 127–136 CrossRef PubMed.
- S. R. Murray, S. J. Stock, S. Cowan, E. S. Cooper and J. E. Norman, Obstet. Gynecol., 2018, 20, 57–63 Search PubMed.
- J. M. Dodd, R. M. Grivell, C. M. OBrien, T. Dowswell and A. R. Deussen, Cochrane Database Syst. Rev., 2019, 2019(11) DOI:10.1002/14651858.CD012024.pub3.
- L. A. Stewart, M. Simmonds, L. Duley, A. Llewellyn, S. Sharif, R. A. Walker, L. Beresford, K. Wright, M. M. Aboulghar, Z. Alfirevic, A. Azargoon, R. Bagga, E. Bahrami, S. C. Blackwell, S. N. Caritis, C. A. Combs, J. M. Croswell, C. A. Crowther, A. F. Das, K. Dickersin, K. C. Dietz, A. Elimian, W. A. Grobman, A. Hodkinson, K. A. Maurel, D. S. McKenna, B. W. Mol, K. Moley, J. Mueller, A. Nassar, J. E. Norman, J. Norrie, J. M. O'Brien, R. Porcher, S. Rajaram, L. Rode, D. J. Rouse, C. Sakala, E. Schuit, M. V. Senat, J. L. Simpson, K. Smith, A. Tabor, E. A. Thom, M. A. van Os, E. P. Whitlock, S. Wood and T. Walley, Lancet, 2021, 397, 1183–1194 CrossRef PubMed.
- S. Walter, A. Rehal, Z. Benkő, C. De Paco Matallana, A. Syngelaki, D. Janga, S. Cicero, R. Akolekar, M. Singh, P. Chaveeva, J. Burgos, F. S. Molina, M. Savvidou, M. De La Calle, N. Persico, M. S. Quezada Rojas, A. Sau, E. Greco, N. O'Gorman, W. Plasencia, S. Pereira, J. C. Jani, N. Valino, M. del Mar Gil, K. Maclagan, A. Wright, D. Wright and K. H. Nicolaides, Am. J. Obstet. Gynecol., 2021, 224, 86.e1–86.e19 CrossRef PubMed.
- R. Romero, A. Conde-Agudelo, A. Rehal, E. Da Fonseca, M. L. Brizot, L. Rode, V. Serra, E. Cetingoz, A. Syngelaki, A. Tabor, A. Perales, S. S. Hassan and K. H. Nicolaides, Ultrasound Obstet. Gynecol., 2022, 59, 263–266 CrossRef CAS PubMed.
- M. V. Senat, R. Porcher, N. Winer, C. Vayssière, P. Deruelle, M. Capelle, F. Bretelle, F. Perrotin, Y. Laurent, L. Connan, B. Langer, A. Mantel, S. Azimi and P. Rozenberg, Am. J. Obstet. Gynecol., 2013, 208(3), 194.e1–194.e8 CrossRef CAS PubMed.
- National Institute for Health and Care Excellence (NICE), Twin and triplet pregnancy, https://www.nice.org.uk/guidance/ng137/chapter/recommendations, (accessed January 2023) Search PubMed.
- A. Wilson, V. A. Hodgetts-Morton, E. J. Marson, A. D. Markland, E. Larkai, A. Papadopoulou, A. Coomarasamy, A. Tobias, D. Chou, O. T. Oladapo, M. J. Price, K. Morris and I. D. Gallos, Cochrane Database Syst. Rev., 2022 DOI:10.1002/14651858.CD014978.pub2.
- W. Yamasmit, S. Chaithongwongwatthana, J. E. Tolosa, S. Limpongsanurak, L. Pereira and P. Lumbiganon, Cochrane Database Syst. Rev., 2015 DOI:10.1002/14651858.CD004733.pub4 , https://www.cochranelibrary.com.
- A. Derbent, S. Simavli, I. I. Gümüş, M. M. Tatli and N. Ö. Turhan, Arch. Gynecol. Obstet., 2011, 284, 821–826 CrossRef CAS PubMed.
- D. Haas, D. M. Caldwell, P. Kirkpatrick, J. J. McIntosh and N. J. Welton, BMJ, 2012, 345, 16–17 CrossRef PubMed.
- J. M. Dodd, L. Jones, V. Flenady and C. A. Crowther, Cochrane Database Syst. Rev., 2013, 31(7) DOI:10.1002/14651858.CD004947.pub3.
- C. D. Cuppett and S. N. Caritis, Uterine Contraction Agents and Tocolytics, Elsevier Inc., 2013 Search PubMed.
- S. Vause and T. Johnston, Arch. Dis. Child. Fetal Neonatal Ed., 2000, 83, 79–85 CrossRef PubMed.
- A. Carlin, J. Norman, S. Cole and R. Smith, Br. Med. J., 2009, 338 DOI:10.1136/bmj.b195.
- D. M. Haas, T. Benjamin, R. Sawyer and S. K. Quinney, Int. J. Women's Health, 2014, 6, 343–349 CrossRef PubMed.
- M. Hanley, L. Sayres, E. Reiff, A. Wood, C. Grotegut and J. Kuller, Obstet. Gynecol. Surv., 2019, 74, 50–55 CrossRef PubMed.
- S. Arrowsmith, A. Kendrick and S. Wray, Obstet., Gynaecol. Reprod. Med., 2010, 20, 241–247 CrossRef PubMed.
- S. Wray, S. Arrowsmith and A. Sharp, Annu. Rev. Pharmacol. Toxicol., 2023, 63, 1–19 CrossRef PubMed.
- T. C. Tan, K. Devendra, L. K. Tan and H. K. Tan, Singapore Med. J., 2006, 47, 361–366 CAS.
- J. Klumper, W. Breebaart, C. Roos, C. A. Naaktgeboren, J. Van Der Post, J. Bosmans, A. Van Kaam, E. Schuit, B. W. Mol, J. Baalman, F. McAuliffe, J. Thornton, M. Kok and M. A. Oudijk, BMJ Open, 2019, 9, 1–6 CrossRef PubMed.
- L. W. Chan, D. S. Sahota, S. Y. Yeung, T. Y. Leung, T. Y. Fung, T. K. Lau and T. N. Leung, Hong Kong Med. J., 2008, 14, 267–272 CAS.
- P. G. Pryde, R. E. Besinger, J. G. Gianopoulos and R. Mittendorf, Semin. Perinatol., 2001, 25, 316–340 CrossRef CAS PubMed.
- R. Gáspár and J. Hajagos-tóth, Pharmaceuticals, 2013, 6, 689–699 CrossRef PubMed.
- I. V Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V Radchenko, N. S. Zefirov, A. S. Makarenko, V. Y. Tanchuk and V. V. Prokopenko, J. Comput.-Aided Mol. Des., 2005, 19, 453–463 CrossRef PubMed.
- Y. Jacquemyn, BJOG: Int. J. Obstet. Gynaecol., 2006, 113, 78–80 CrossRef PubMed.
- N. M. Fisk and J. Chan, BJOG: Int. J. Obstet. Gynaecol., 2003, 110, 98–102 CrossRef.
- J. F. King, Curr. Opin. Obstet. Gynecol., 2004, 16, 459–463 CrossRef PubMed.
- V. Flenady, W. Am, P. Dnm, S. Om, L. Murray, J. La, B. Carbonne, V. Flenady, W. Am, P. Dnm, S. Om, L. Murray, J. La and B. Carbonne, Cochrane Database Syst. Rev., 2014, 6 DOI:10.1002/14651858.CD002255.pub2 , https://www.cochranelibrary.com.
- K. E. Economy and A. Z. Abuhamad, Semin. Perinatol., 2001, 25, 264–271 CrossRef CAS PubMed.
- C. Hammerman, J. Glaser, M. Kaplan, M. S. Schimmel, B. Ferber and A. I. Eidelman, Pediatrics, 1998, 102, 6–9 CrossRef PubMed.
- A. L. Hammers, L. Sanchez-Ramos and A. M. Kaunitz, Am. J. Obstet. Gynecol., 2015, 212, 505.e1–505.e13 CrossRef CAS PubMed.
- H. C. Zierden, R. L. Shapiro, K. DeLong, D. M. Carter and L. M. Ensign, Adv. Drug Delivery Rev., 2021, 174, 190–209 CrossRef CAS PubMed.
- R. Kammari, N. G. Das and S. K. Das, Nanoparticulate Systems for Therapeutic and Diagnostic Applications, Elsevier, 2017 Search PubMed.
- A. Z. Mirza and F. A. Siddiqui, Int. Nano Lett., 2014, 4 DOI:10.1007/s40089-014-0094-7.
- S. Soares, J. Sousa, A. Pais and C. Vitorino, Front. Chem., 2018, 6, 1–15 CrossRef PubMed.
- T. O. B. Olusanya, R. R. H. Ahmad, D. M. Ibegbu, J. R. Smith and A. A. Elkordy, Molecules, 2018, 23, 1–17 CrossRef PubMed.
- H. Nehoff, N. N. Parayath, L. Domanovitch, S. Taurin and K. Greish, Int. J. Nanomed., 2014, 9, 2539–2555 Search PubMed.
- R. Bazak, M. Houri, S. El Achy, W. Hussein and T. Refaat, Mol. Clin. Oncol., 2014, 2, 904–908 CrossRef PubMed.
- A. Wicki, D. Witzigmann, V. Balasubramanian and J. Huwyler, J. Controlled Release, 2015, 200, 138–157 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- R. Ghadi and N. Dand, J. Controlled Release, 2017, 248, 71–95 CrossRef CAS PubMed.
- S. Jacob, A. B. Nair and J. Shah, Biomater. Res., 2020, 24(3), 1–16 Search PubMed.
- S. Talegaonkar and A. Bhattacharyya, AAPS PharmSciTech, 2019, 20 DOI:10.1208/s12249-019-1337-8.
- A. Parodi, P. Buzaeva, D. Nigovora, A. Baldin, D. Kostyushev, V. Chulanov, L. V. Savvateeva and A. A. Zamyatnin, J. Nanobiotechnol., 2021, 19, 1–19 CrossRef PubMed.
- N. J. Alexander, E. Baker, M. Kaptein, U. Karck, L. Miller and E. Zampaglione, Fertil. Steril., 2004, 82, 1–12 CrossRef PubMed.
- N. Einer-Jensen, E. Cicinelli, P. Galantino, V. Pinto and B. Barba, Hum. Reprod., 2002, 17, 3060–3064 CrossRef CAS PubMed.
- A. Mir, R. V. Vartak, K. Patel, S. M. Yellon and S. E. Reznik, Pharmaceutics, 2022, 14 DOI:10.3390/pharmaceutics14102019.
- L. M. Ensign, B. C. Tang, Y. Y. Wang, T. A. Tse, T. Hoen, R. Cone and J. Hanes, Sci. Transl. Med., 2012, 4(138) DOI:10.1126/scitranslmed.3003453.
- J. S. Refuerzo, F. Leonard, N. Bulayeva, D. Gorenstein, G. Chiossi, A. Ontiveros, M. Longo and B. Godin, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- M. S. Taha, S. Padmakumar, A. Singh and M. M. Amiji, Drug Deliv. Transl. Res., 2020, 10, 766–790 CrossRef PubMed.
- J. Wong, A. Brugger, A. Khare, M. Chaubal, P. Papadopoulos, B. Rabinow, J. Kipp and J. Ning, Adv. Drug Delivery Rev., 2008, 60, 939–954 CrossRef CAS PubMed.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Pharmaceutics, 2018, 10, 1–17 CrossRef PubMed.
- Y. Wang, C. Pi, X. Feng, Y. Hou, L. Zhao and Y. Wei, Int. J. Nanomed., 2020, 15, 6295–6310 CrossRef CAS PubMed.
- M. Poley, P. Mora-Raimundo, Y. Shammai, M. Kaduri, L. Koren, O. Adir, J. Shklover, J. Shainsky-Roitman, S. Ramishetti, F. Man, R. T. M. De Rosales, A. Zinger, D. Peer, I. Ben-Aharon and A. Schroeder, ACS Nano, 2022, 16, 5246–5257 CrossRef CAS PubMed.
- A. Wagner and K. Vorauer-Uhl, J. Drug Delivery, 2011, 2011, 1–9 CrossRef PubMed.
- J. S. Refuerzo, J. F. Alexander, F. Leonard, M. Leon, M. Longo and B. Godin, Am. J. Obstet. Gynecol., 2015, 212, 508.e1–508.e7 CrossRef CAS PubMed.
- J. W. Paul, S. Hua, M. Ilicic, J. M. Tolosa, T. Butler, S. Robertson and R. Smith, Am. J. Obstet. Gynecol., 2017, 216, 283.e1–283.e14 CrossRef CAS PubMed.
- S. Hua, H.-I. I. Chang, N. M. Davies and P. J. Cabot, J. Liposome Res., 2011, 21, 95–105 CrossRef CAS PubMed.
- S. Hua and B. Vaughan, Int. J. Nanomed., 2019, 14, 2191–2206 CrossRef CAS PubMed.
- S. Hua, J. Liposome Res., 2019, 29, 357–367 CrossRef CAS PubMed.
- J. W. Paul and R. Smith, Best Pract. Res., Clin. Obstet. Gynaecol., 2018, 52, 48–59 CrossRef PubMed.
- D. P. Fischer, A. L. Griffiths, S. Lui, U. J. Sabar, D. Farrar, P. J. O'Donovan, D. F. Woodward and K. M. Marshall, J. Pharmacol. Exp. Ther., 2020, 373, 381–390 CrossRef CAS PubMed.
- L. Sercombe, T. Veerati, F. Moheimani, S. Y. Wu, A. K. Sood and S. Hua, Front. Pharmacol., 2015, 6, 1–13 Search PubMed.
- S. Samimi, N. Maghsoudnia, R. B. Eftekhari and F. Dorkoosh, Lipid-Based Nanoparticles for Drug Delivery Systems, Elsevier Inc., 2018 Search PubMed.
- R. H. Muller, K. Mader and S. Gohla, Eur. J. Pharm. Biopharm., 2000, 50, 161–177 CrossRef CAS PubMed.
- C. Pardeshi, P. Rajput, V. Belgamwar, A. Tekade, G. Patil, K. Chaudhary and A. Sonje, Acta Pharm, 2012, 62, 433–472 CAS.
- S. Mukherjee, S. Ray and R. S. Thakur, Indian J. Pharm. Sci., 2009, 71, 349–358 CrossRef CAS PubMed.
- P. Ekambaram, A. A. H. Sathali and K. Priyanka, Sci. Rev. Chem. Commun., 2018, 2, 80–102 Search PubMed.
- J. R. Madan, P. A. Khude and K. Dua, Int. J. Pharm. Invest., 2014, 4, 60–64 CrossRef CAS PubMed.
- R. Shtay, C. Ping and K. Schwarz, J. Food Eng., 2018, 231, 30–41 CrossRef CAS.
- Z. Zhu, Mol. Pharm., 2014, 11, 776–786 CrossRef CAS PubMed.
- C. Hogarth, K. Arnold, A. McLauchlin, S. P. Rannard, M. Siccardi and T. O. McDonald, J. Mater. Chem. B, 2021, 9, 9874–9884 RSC.
- J. M. Taylor, K. Scale, S. Arrowsmith, A. Sharp, S. Flynn, S. Rannard and T. O. Mcdonald, Nanoscale, 2020, 2 10.1039/D0NA00582G.
- K. K. Hippalgaonkar, G. R. Adelli, K. K. Hippalgaonkar, M. A. Repka and S. Majumdar, J. Ocul. Pharmacol. Ther., 2013, 29, 216–228 CrossRef CAS PubMed.
- L. Bayón-Cordero, I. Alkorta and L. Arana, Nanomaterials, 2019, 9(3) DOI:10.3390/nano9030474.
- Q. Kang, J. Liu, Y. Zhao, X. Y. Liu, X. Y. Liu, Y. J. Wang, N. L. Mo and Q. Wu, Artif. Cells, Nanomed., Biotechnol., 2018, 46, S585–S597 CrossRef CAS PubMed.
- M. A. Mohammed, J. T. M. Syeda, K. M. Wasan and E. K. Wasan, Pharmaceutics, 2017, 9(4) DOI:10.3390/pharmaceutics9040053.
- S. P. Balguri, G. R. Adelli and S. Majumdar, Eur. J. Pharm. Biopharm., 2016, 109, 224–235 CrossRef CAS PubMed.
- R. K. Barman, Y. Iwao, Y. Funakoshi, A. H. Ranneh, S. Noguchi, M. I. Ibne Wahed and S. Itai, Chem. Pharm. Bull., 2014, 62, 399–406 CrossRef CAS PubMed.
- R. Cassano and S. Trombino, Int. J. Polym. Sci., 2019, 2019 DOI:10.1155/2019/8690145.
- H. Yuan, L. L. Wang, Y. Z. Du, J. You, F. Q. Hu and S. Zeng, Colloids Surf., B, 2007, 60, 174–179 CrossRef CAS PubMed.
- E. Esposito, M. Sguizzato, M. Drechsler, P. Mariani, F. Carducci, C. Nastruzzi and R. Cortesi, Eur. J. Pharm. Biopharm., 2017, 119, 437–446 CrossRef CAS PubMed.
- M. Elmowafy, K. Shalaby, M. M. Badran, H. M. Ali, M. S. Abdel-Bakky and I. El-Bagory, J. Drug Delivery Sci. Technol., 2018, 45, 230–239 CrossRef CAS.
- T. H. Hassan, S. S. Salman, M. M. Elkhoudary and S. Gad, J. Drug Delivery Sci. Technol., 2021, 64, 102570 CrossRef CAS.
- M. Patki, K. Giusto, S. Gorasiya, S. E. Reznik and K. Patel, Pharmaceutics, 2019, 11 DOI:10.3390/pharmaceutics11070335.
- K. Giusto, M. Patki, J. Koya, C. R. Ashby, S. Munnangi, K. Patel and S. E. Reznik, Nanomedicine, 2019, 14, 2835–2851 CrossRef CAS PubMed.
- S. Klang, M. Abdulrazik and S. Benita, Pharm. Dev. Technol., 2000, 5, 521–532 CrossRef CAS PubMed.
- M. Yamaguchi, K. Ueda, A. Isowaki, A. Ohtori, H. Takeuchi, N. Ohguro and K. Tojo, Biol. Pharm. Bull., 2009, 32, 1266–1271 CrossRef CAS PubMed.
- P. Ghasemiyeh and S. Mohammadi-Samani, Res. Pharm. Sci., 2018, 13 Search PubMed.
- P. A. Makoni, K. W. Kasongo and R. B. Walker, Pharmaceutics, 2019, 11(8) DOI:10.3390/pharmaceutics11080397.
- E. B. Souto, W. Mehnert and R. H. Müller, J. Microencapsulation, 2006, 23, 417–433 CrossRef CAS PubMed.
- S. Rani, R. Rana, G. K. Saraogi, V. Kumar and U. Gupta, AAPS PharmSciTech, 2019, 20(3), 129 CrossRef CAS PubMed.
- V. Vyas, C. R. Ashby, N. S. Olgun, S. Sundaram, O. Salami, S. Munnangi, R. Pekson, P. Mahajan and S. E. Reznik, Am. J. Pathol., 2015, 185, 862–869 CrossRef CAS PubMed.
- B. Begines, T. Ortiz, M. Pérez-Aranda, G. Martínez, M. Merinero, F. Argüelles-Arias and A. Alcudia, Nanomaterials, 2020, 10, 1–41 CrossRef PubMed.
- S. K. Prajapati, A. Jain, A. Jain and S. Jain, Eur. Polym. J., 2019, 120, 109191 CrossRef CAS.
- M. Alkholief, M. A. Kalam, M. K. Anwer and A. Alshamsan, Pharmaceutics, 2022, 14 DOI:10.3390/pharmaceutics14040870.
- I. Muljajew, M. Chi, A. Vollrath, C. Weber, B. Beringer-Siemers, S. Stumpf, S. Hoeppener, M. Sierka and U. S. Schubert, Eur. Polym. J., 2021, 156, 110606 CrossRef CAS.
- C. J. Cheng, G. T. Tietjen, J. K. Saucier-Sawyer and W. M. Saltzman, Nat. Rev. Drug Discovery, 2015, 14, 365 CrossRef CAS.
- B. E. Rabinow, Nat. Rev. Drug Discovery, 2004, 3, 785–796 CrossRef CAS PubMed.
- D. Medarević, J. Djuriš, S. Ibrić, M. Mitrić and K. Kachrimanis, Int. J. Pharm., 2018, 540, 150–161 CrossRef PubMed.
- Y. Tian, S. Wang, Y. Yu, W. Sun, R. Fan, J. Shi, W. Gu, Z. Wang, H. Zhang and A. Zheng, Int. J. Pharm., 2022, 623, 121862 CrossRef CAS PubMed.
- M. Malamatari, K. M. G. Taylor, S. Malamataris, D. Douroumis and K. Kachrimanis, Drug Discovery Today, 2018, 23, 534–547 CrossRef CAS PubMed.
- T. O. McDonald, L. M. Tatham, F. Y. Southworth, M. Giardiello, P. Martin, N. J. Liptrott, A. Owen and S. P. Rannard, J. Mater. Chem. B, 2013, 1, 4455 RSC.
- J. J. Hobson, A. C. Savage, A. B. Dwyer, C. Unsworth, J. Massam, U. Arshad, H. Pertinez, H. Box, L. Tatham, R. K. R. Rajoli, M. Neary, J. Sharp, A. Valentijn, C. David, P. Curley, N. J. Liptrott, T. O. McDonald, A. Owen and S. P. Rannard, Nanoscale, 2021, 13, 6410–6416 RSC.
- J. Tao, S. F. Chow and Y. Zheng, Acta Pharm. Sin. B, 2019, 9, 4–18 CrossRef PubMed.
- C. M. Keck and R. H. Müller, Eur. J. Pharm. Biopharm., 2006, 62, 3–16 CrossRef CAS PubMed.
- H. Zhang, D. Wang, R. Butler, N. L. Campbell, J. Long, B. Tan, D. J. Duncalf, A. J. Foster, A. Hopkinson, D. Taylor, D. Angus, A. I. Cooper and S. P. Rannard, Nat. Nanotechnol., 2008, 3, 506–511 CrossRef CAS PubMed.
- T. O. McDonald, M. Giardiello, P. Martin, M. Siccardi, N. J. Liptrott, D. Smith, P. Roberts, P. Curley, A. Schipani, S. H. Khoo, J. Long, A. J. Foster, S. P. Rannard and A. Owen, Adv. Healthcare Mater., 2014, 3, 400–411 CrossRef CAS PubMed.
- M. Giardiello, N. J. Liptrott, T. O. McDonald, D. Moss, M. Siccardi, P. Martin, D. Smith, R. Gurjar, S. P. Rannard and A. Owen, Nat. Commun., 2016, 7, 13184 CrossRef CAS PubMed.
- N. M. Elbaz, L. M. Tatham, A. Owen, S. Rannard and T. O. McDonald, Food Hydrocolloids, 2021, 121, 107005 CrossRef CAS.
- T. Hoang, H. Zierden, A. Date, J. Ortiz, S. Gumber, N. Anders, P. He, J. Segars, J. Hanes, M. Mahendroo and L. M. Ensign, J. Controlled Release, 2019, 295, 74–86 CrossRef CAS PubMed.
- H. C. Zierden, J. I. Ortiz, K. DeLong, J. Yu, G. Li, P. Dimitrion, S. Bensouda, V. Laney, A. Bailey, N. M. Anders, M. Scardina, M. Mahendroo, S. Mesiano, I. Burd, G. Wagner, J. Hanes and L. M. Ensign, Sci. Transl. Med., 2021, 13 DOI:10.1126/scitranslmed.abg7212.
- I. D. Styliari, V. Taresco, A. Theophilus, C. Alexander, M. Garnett and C. Laughton, RSC Adv., 2020, 10, 19521–19533 RSC.
- S. P. R. & A. O. Lee, M. Tatham, A. C. Savage, A. Dwyer, M. Siccardi, T. Scott, Manoli Vourvahis, in Conference on Retroviruses and Opportunistic Infections (CROI) - Poster 482, Boston, USA, 2018 Search PubMed.
- N. M. Elbaz, L. M. Tatham, A. Owen, S. Rannard and T. O. McDonald, Food Hydrocolloids, 2021, 121, 107005 CrossRef CAS.
- Y. Fan, Y. Cui, W. Hao, M. Chen, Q. Liu, Y. Wang, M. Yang, Z. Li, W. Gong, S. Song, Y. Yang and C. Gao, Bioact. Mater., 2021, 6, 4402–4414 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, Angew. Chem., Int. Ed., 2016, 1, 10–29 Search PubMed.
- A. C. Anselmo and S. Mitragotri, Angew. Chem., Int. Ed., 2019, 4, 1–16 Search PubMed.
- S. F. Pantze, J. Parmentier, G. Hofhaus and G. Fricker, Eur. J. Lipid Sci. Technol., 2014, 116, 1145–1154 CrossRef CAS.
- T. Feczkó, J. Drug Delivery Sci. Technol., 2021, 64 DOI:10.1016/j.jddst.2021.102597.
- E. A. Nance, G. F. Woodworth, K. A. Sailor, T. Y. Shih, Q. Xu, G. Swaminathan, D. Xiang, C. Eberhart and J. Hanes, Sci. Transl. Med., 2012, 4 DOI:10.1126/scitranslmed.3003594.
- D. R. Elias, A. Poloukhtine, V. Popik and A. Tsourkas, Nanomed.: Nanotechnol. Biol. Med., 2013, 9, 194–201 CrossRef CAS PubMed.
- C. Pereira Gomes, V. Leiro, C. D. Ferreira Lopes, A. P. Spencer and A. P. Pêgo, Acta Biomater., 2018, 78, 247–259 CrossRef CAS PubMed.
- Y. Y. Wang, S. K. Lai, J. S. Suk, A. Pace, R. Cone and J. Hanes, Angew. Chem., Int. Ed., 2008, 47, 9726–9729 CrossRef CAS PubMed.
- O. Summ and S. Evers, Curr. Pain Headache Rep., 2013, 17 DOI:10.1007/s11916-013-0327-x.
- A. Soundararajan, A. Bao, W. T. Phillips, R. Perez and B. A. Goins, Nucl. Med. Biol., 2009, 36, 515–524 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |