 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanism of ammonia synthesis on Fe3Mo3N†
Michael D.
Higham
 *ab,
Constantinos D.
Zeinalipour-Yazdi
d,
Justin S. J.
Hargreaves
e and
C. Richard A.
Catlow
abc
*ab,
Constantinos D.
Zeinalipour-Yazdi
d,
Justin S. J.
Hargreaves
e and
C. Richard A.
Catlow
abc
aUniversity College London, Department of Chemistry, Kathleen Lonsdale Building, Gower Place, London, WC1E 6BT, UK. E-mail: m.higham@ucl.ac.uk
bResearch Complex at Harwell, Rutherford Appleton Laboratory, Harwell, Oxon, OX11 0FA, UK
cSchool of Chemistry, Cardiff University, Park Place, Cardiff, CF10 1AT, UK
dUniversity of East London, University Way, London, E16 2RD, UK
eUniversity of Glasgow, School of Chemistry, Glasgow, G12 8QQ, UK
First published on 25th November 2022
Abstract
Ammonia (NH3) synthesis is an essential yet energy-demanding industrial process. Hence, there is a need to develop NH3 synthesis catalysts that are highly active under milder conditions. Metal nitrides are promising candidates, with the η-carbide Co3Mo3N having been found to be more active than the industrial Fe-based catalyst. The isostructural Fe3Mo3N catalyst has also been identified as highly active for NH3 synthesis. In the present work, we investigate the catalytic ammonia synthesis mechanisms in Fe3Mo3N, which we compare and contrast with the previously studied Co3Mo3N. We apply plane-wave density functional theory (DFT) to investigate surface N vacancy formation in Fe3Mo3N, and two distinct ammonia synthesis mechanisms. The calculations reveal that whilst N vacancy formation on Fe3Mo3N is more thermodynamically demanding than for Co3Mo3N, the formation energies are comparable, suggesting that surface lattice N vacancies in Fe3Mo3N could facilitate NH3 synthesis. N2 activation was found to be enhanced on Fe3Mo3N compared to Co3Mo3N, for adsorption both at and adjacent to the vacancy. The calculated activation barriers suggest that, as for Co3Mo3N, the associative Mars van Krevelen mechanism affords a much less energy-demanding pathway for ammonia synthesis, especially for initial hydrogenation processes.
Introduction
Ammonia (NH3) synthesis is one of the most important industrial chemical processes, most notably for the production of fertilisers, which are crucial to supporting agriculture, and therefore ensuring global food security. With the world population estimated at 7.6 billion people in 2021, and projected to reach over 11 billion people by the end of the century, it is all but certain that ammonia synthesis will remain a high priority for the chemical industry.1,2 Indeed, industry reports conclude that 80% of all ammonia produced is consumed by the agricultural sector, with an estimated 178![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 330 kilotons being produced in 2021 alone,3 and it is estimated that over the past century, 27% (ref. 4) of the world's population has been supported by food produced using ammonia-based fertilisers.4,5 Consequently, ammonia synthesis is also one of the most environmentally challenging industrial processes, with ammonia production being responsible for an estimated 1.8% of global energy consumption, and the same proportion of global CO2 emissions;6 producing one tonne of NH3 consumes an estimated 30 GJ of energy, and releases 2.3 tonnes of CO2.7,8 Furthermore, between 3–5% of global consumption of natural gas can be attributed to ammonia production, making reducing the environmental impact of ammonia synthesis even more urgent in light of instability of global gas supplies.9
330 kilotons being produced in 2021 alone,3 and it is estimated that over the past century, 27% (ref. 4) of the world's population has been supported by food produced using ammonia-based fertilisers.4,5 Consequently, ammonia synthesis is also one of the most environmentally challenging industrial processes, with ammonia production being responsible for an estimated 1.8% of global energy consumption, and the same proportion of global CO2 emissions;6 producing one tonne of NH3 consumes an estimated 30 GJ of energy, and releases 2.3 tonnes of CO2.7,8 Furthermore, between 3–5% of global consumption of natural gas can be attributed to ammonia production, making reducing the environmental impact of ammonia synthesis even more urgent in light of instability of global gas supplies.9
Since the early 20th century, the Haber–Bosch process10 has been employed to produce ammonia on an industrial scale, relying on promoted iron-based catalysts and high temperature and pressure conditions (400–500 °C, 100–200 atm), which account for the high energy consumption of this process. As such, the quest to develop highly active and stable catalysts for ammonia synthesis that can operate under milder conditions is a key scientific challenge, with even relatively minor enhancements in performance translating into significant reductions in environmental and economic cost. It has been demonstrated that the ammonia synthesis reaction on Fe-based catalysts proceeds via a dissociative Langmuir–Hinshelwood (LH) mechanism, which involves the first process, the dissociation of adsorbed N2, being the most activated, and therefore rate determining, process,11,12 with a similar mechanism being identified for the more active, but more costly, Ru-based system.13,14 The high activation barrier associated with the rate determining N2 dissociation process can be attributed to the high bond dissociation energy (946 kJ mol−1), and accounts for the high temperature and pressure conditions required by the Haber–Bosch process. Hence, novel catalysts that provide an alternative, more energetically accessible, reaction mechanism, afford a viable avenue for developing highly active ammonia synthesis catalysts that can operate under milder conditions.15
Transition metal nitrides have emerged as promising candidate catalysts for ammonia synthesis,16 spanning a wide range of materials of different structure and composition.17–25 The ternary metal nitride, Co3Mo3N, is an especially promising candidate material,26–31 exhibiting even greater activity than the industrial Fe-based catalyst when suitably promoted.27 It has been suggested that the remarkable activity of this system can be wholly attributed to the inherent activities of metallic Co and Mo towards N2 binding; both of these elements are expressed in the Co3Mo3N(111) surface, and linear scaling relations from previous studies show that the average of the N2 binding energies for metallic Co and Mo is close to the supposed optimum for ammonia synthesis.29 However, further experimental studies show that lattice N in Co3Mo3N is highly active,32 and could therefore facilitate ammonia synthesis by enabling a Eley–Rideal–Mars–van-Krevelen (ER–MvK) type mechanism, whereby lattice nitrogen is continually consumed (and evolved as the product, ammonia) and replenished (from the reactant gas phase nitrogen). A series of detailed computational studies explored the possibility of nitrogen vacancy formation on Co3Mo3N surfaces, and investigated both the traditional LH mechanism, and the novel ER–MvK mechanism, for ammonia synthesis; the calculations revealed that surface N vacancy formation is indeed highly plausible, and that the novel ER–MvK mechanism affords a highly kinetically accessible mechanism for ammonia synthesis, with significantly lower activation barriers for key elementary processes.15,33–36
The Fe3Mo3N system, which is isostructural to Co3Mo3N, has also been found to be highly active for ammonia synthesis.37,38 However, experimental studies show that, in contrast to the isostructural Co system, Fe3Mo3N appears to be highly resistant to reduction, with no appreciable loss of lattice N being observed even under harsh conditions.39 Furthermore, experimental studies also show that the quaternary metal nitride system (Co3−xFexMo3N) is highly active towards ammonia synthesis,38 opening the possibility to finely tune ammonia synthesis activity by determining the optimal Co:Fe ratio. Clearly, a detailed understanding of both the Co and Fe ternary systems is required before embarking on the rational design of an optimal quaternary metal nitride catalyst.
It is therefore of considerable interest to explore the mechanisms for ammonia synthesis on Fe3Mo3N, analogous to the previous studies for Co3Mo3N, both to rationalise the high activity of the Fe3Mo3N ternary metal nitride, and to inform future investigations into the quaternary Co3−xFexMo3N system. In the present work, plane-wave density functional theory techniques are applied to explore surface N vacancy formation, and both the dissociative LH mechanism, and associative ER–MvK mechanism, for Fe3Mo3N. The calculations reveal that surface N vacancy formation is feasible for the Fe3Mo3N system, and that N2 activation takes place both at and adjacent to surface N vacancy sites. The calculations also show that, as for Co3Mo3N, the associative ER–MvK mechanism provides a more energetically accessible mechanistic pathway for ammonia synthesis. The present work, in addition to providing key insights into the behaviour of the Fe3Mo3N system, will serve as a benchmark study for future work exploring promotion of the Fe3Mo3N catalyst, and (in tandem with the previous work for the Co3Mo3N system) for investigations concerning the quaternary Co3−xFexMo3N system, supporting the rational design of optimal metal nitride catalysts for ammonia synthesis.
Methodology
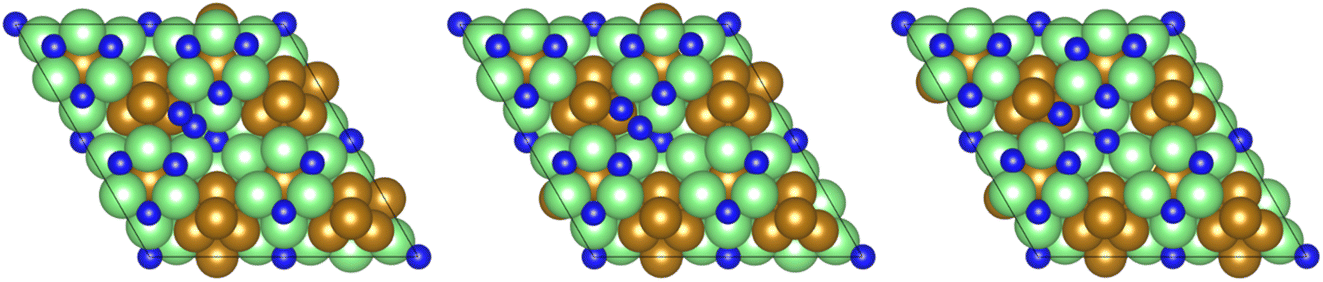
All calculations were performed using plane-wave density functional theory (DFT) as implemented in the VASP code (version 5.4.4).40–43 A thin-film Fe3Mo3N (111) model was constructed exposing a nitrogen-rich termination, based on the model successfully developed for the isostructural Co3Mo3N system reported in previous computational studies.15,33,35 The optimised lattice parameter for bulk Fe3Mo3N was determined in previous studies33 and was used to define the dimensions of the surface slab periodic cell. The thin-film slab consists of a 2 × 2 surface supercell, to minimise lateral interactions between adsorbed species in adjacent periodic images, and three N-containing layers forming a non-stoichiometric symmetric slab model, as illustrated in Fig. 1. The thin film slab was centred in the periodic cell, with a vacuum separation of 30 Å between slabs in adjacent periodic images, ensuring sufficient slab separation to avoid any spurious interactions. During geometry optimisation, all atomic positions were allowed to relax. In line with the previous computational studies for Co3Mo3N, a Γ-centred (4 × 4 × 1) Monkhorst–Pack k-point sampling scheme was used.15,44 Inner electrons were replaced by projector-augmented waves (PAW),45,46 and the valence states were expanded in plane-waves with a cut-off energy of 650 eV. The revised PBE (RPBE) exchange correlation functional was used throughout, having previously been demonstrated to accurately reproduce N2 bond dissociation energy,15 with a dispersion correction applied using the D3 scheme devised by Grimme, and Becke–Johnson damping, in order to account for the weak van der Waals interactions responsible for the physisorption behaviour of species such as N2.47,48 | ||
| Fig. 1 Graphic illustrating the symmetric, non-stoichiometric thin-film slab model used to model the Fe3Mo3N surface, from the top (left) and side (right), as viewed across the cell diagonal. The pristine surface is illustrated, i.e. no N vacancies are present. Key: green, Mo; brown, Fe; blue, N. | ||
In order to investigate the elementary reaction processes involved in the LH and ER–MvK mechanisms for ammonia synthesis, optimised geometries were obtained for all adsorbed reactants, products, and intermediates, on the Fe3Mo3N (111) surface model featuring a single surface N vacancy, with atomic forces converged to within 0.01 eV Å−1. In each case, electronic wavefunctions were converged such that the total energy was converged to within 10−5 eV. Transition states were identified using the climbing image nudged elastic bands (CI-NEB) and dimer methods, with vibrational analysis confirming that a saddle point had indeed been located.
For the ab initio thermodynamics study, calculated DFT electronic energies were combined with gas-phase thermochemistry data (obtained from the NIST Webbook49) using the general approach described by Reuter et al.50 to obtain the Gibbs free energy of vacancy formation as a function of H2 partial pressure and temperature, to estimate typical surface N vacancy concentrations under a range of hydrogenating conditions. Further details of the ab initio thermodynamics model can be found in the ESI.†
Results
Formation of surface N vacancies
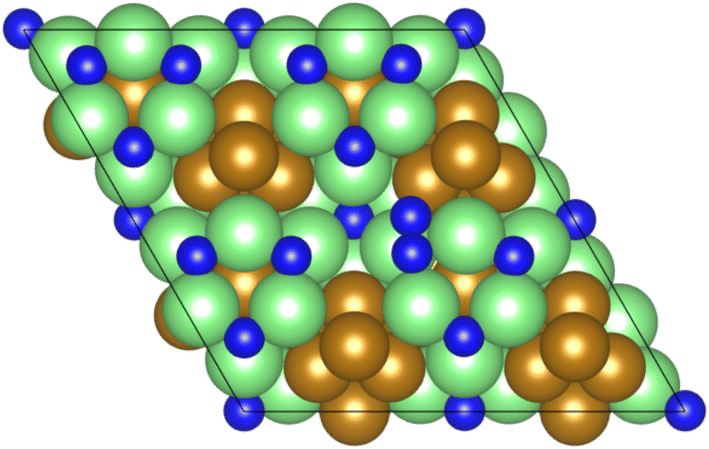
As illustrated in the previous studies investigating the Co3Mo3N system, the possibility of an associative MvK-type mechanism depends on the presence of surface N vacancies to activate adsorbed N2. As such, it is of interest to calculate the N vacancy formation energy, both with respect to N2 and NH3 arising from hydrogenation of surface lattice N under appropriate conditions, to determine the overall thermodynamic feasibility of the vacancy formation. The calculated nitrogen vacancy formation energies are presented in Table 1, for both a single N vacancy, and two adjacent N vacancies to determine the extent of lateral instability; Fig. 2 illustrates both the surface with a single N vacancy and two adjacent N vacancies.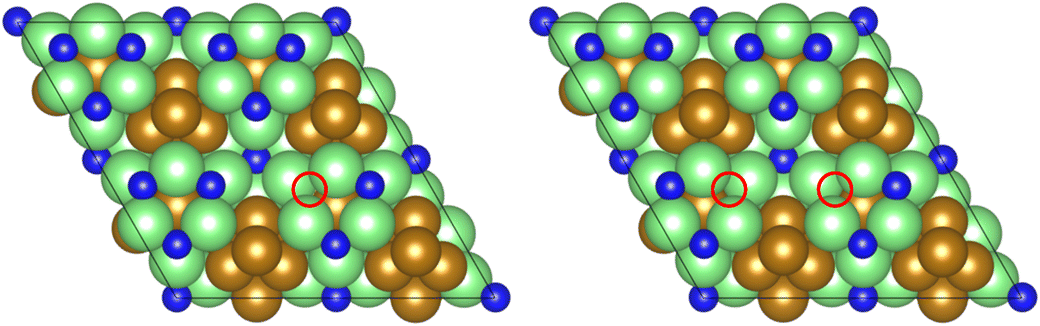 | ||
| Fig. 2 Graphic illustrating the model surface with a single N vacancy present (left) and two adjacent N vacancies (right). The locations of the N vacancies are highlighted by the red circles. | ||
| N vacancy | ΔEvac. ref. N2/eV | ΔEvac. ref. NH3/eV | ΔEvac. ref. N2 per N/eV | ΔEvac. ref. NH3 per N/eV |
|---|---|---|---|---|
| 1 | 1.893 | 0.972 | 1.893 | 0.972 |
| 2 | 4.247 | 2.406 | 2.124 | 1.203 |
The calculated vacancy formation energies show that, with respect to the formation of N2, the formation of a single N vacancy on the model surface is endothermic by 1.893 eV. For the formation of two adjacent vacancies, a value of 4.247 eV was obtained, giving a formation energy per N vacancy of 2.124 eV, indicating that there is only a moderate extent of repulsive interaction between adjacent N vacancies, with the vacancy formation energy per vacancy differing by only 0.231 eV; hence, it may be possible to achieve appreciable surface N vacancy concentrations under appropriate conditions.
For the vacancy formation energies with respect to H2 and NH3, we find that the vacancy formation energy is considerably less endothermic, with a vacancy formation energy of only 0.972 eV for a single N vacancy, and 1.203 eV per vacancy for two adjacent N vacancies, suggesting that, under ammonia synthesis conditions where H2 is present, formation of N vacancies via evolution of NH3 should be more thermodynamically feasible compared to the formation of N vacancies via evolution of N2 under comparable conditions where H2 is not present.
Ab initio thermodynamics of vacancy formation under hydrogenation conditions
Whilst the calculated vacancy formation energies for both Co3Mo3N and Fe3Mo3N are endothermic, the DFT calculated energies do not account for temperature and pressure dependent thermodynamic contributions under real conditions; in the case of lattice N being evolved into the gas phase to form surface vacancies, it is clear that entropy contributions arising both from translational and rotational degrees of freedom of gas phase molecules, and configurational entropy arising from many possible permutations of larger numbers of surface vacancies, will have a considerable impact on the thermodynamic feasibility of vacancy formation. To this end, we apply ab initio thermodynamics techniques to extrapolate the DFT results to provide approximate insights into the extent of surface N vacancy formation under a range of possible experimental conditions, based on the established approach detailed in previous works investigating a variety of different systems;51–54 details of the ab initio thermodynamics model are described in detail in the ESI.†The thin-film Fe3Mo3N (111) model used in the present work consists of two surfaces of 248.46 Å2, which corresponds to 2.49 × 10−22 cm2, giving a total surface area of 4.97 × 10−22 cm2. The (2 × 2) surface supercell presents a total of 24 surface lattice N atoms, 12 for each side of the slab. Hence, the range of vacancy concentrations represented by the model spans from 2.01 × 1021 cm−2 (for a single vacancy) to 4.83 × 1022 cm−2 (for the maximum surface vacancy concentration, i.e. all 24 surface N atoms in the thin film model having been removed). Fig. 3 shows the approximate vacancy concentration for varying H2 partial pressure and temperatures,55 based on the calculated Gibbs free energy of vacancy formation for each vacancy concentration representable within the thin-film model.
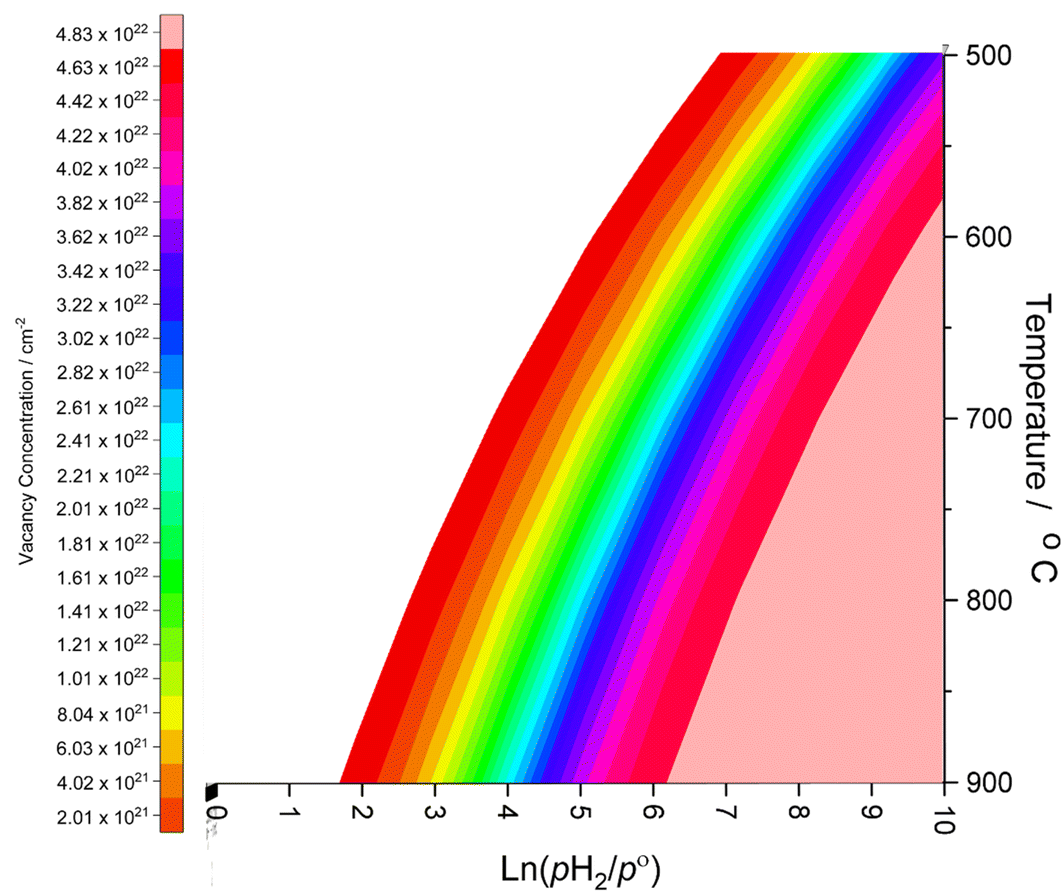 | ||
| Fig. 3 Surface N vacancy concentration as a function of H2 partial pressure (where p° is ambient pressure) and temperature, as estimated from ab initio thermodynamics. | ||
The ab initio thermodynamic studies summarised in Fig. 3 show that N vacancy concentration is higher at higher H2 partial pressures and higher temperatures, as expected. Whilst the model is necessarily limited to a minimum vacancy concentration of 2.01 × 1021 cm−2 (this being the vacancy concentration corresponding to a single surface N vacancy within the 2 × 2 thin film model), it can still be seen that one would not necessarily expect an appreciable number of vacancies to be formed under ambient pressure  for any range of temperatures. However, even at relatively modest H2 partial pressures, such as 10 mPa,56,57 which is at the lower range of pressures applied for ammonia synthesis using the industrially well-establish Fe-based catalysts
for any range of temperatures. However, even at relatively modest H2 partial pressures, such as 10 mPa,56,57 which is at the lower range of pressures applied for ammonia synthesis using the industrially well-establish Fe-based catalysts  , the ab initio thermodynamics suggest that the 2.01× 1021 cm−2 vacancy concentration threshold would be reached at 650 °C. It is therefore highly plausible that lower concentrations of surface N vacancies would be present at somewhat lower temperatures and pressures and could therefore facilitate ammonia synthesis via the ER–MvK mechanism.
, the ab initio thermodynamics suggest that the 2.01× 1021 cm−2 vacancy concentration threshold would be reached at 650 °C. It is therefore highly plausible that lower concentrations of surface N vacancies would be present at somewhat lower temperatures and pressures and could therefore facilitate ammonia synthesis via the ER–MvK mechanism.
Associative Eley–Rideal–Mars–van-Krevelen mechanism
As illustrated from the DFT calculations for vacancy formation, and resulting ab initio thermodynamics studies, it is likely that there is an appreciable vacancy concentration on the Fe3Mo3N (111) surface under relatively mild experimental conditions. Hence, it is of interest to explore the energetics of the ER–MvK mechanism for this system that was found to afford a more kinetically accessible pathway for ammonia synthesis for the isostructural Co3Mo3N system.35The calculated reaction energies and activation barriers with corresponding frequencies for the unstable mode are presented in Table 2.
| Process number | Process | ΔE/eV | E A/eV | ν/cm−1 |
|---|---|---|---|---|
| 1 | □ + N2(g) → N□2 | −1.746 | — | — |
| 2 | N□2 + H2(g) → N2H□2 | −0.753 | — | — |
| 3 |
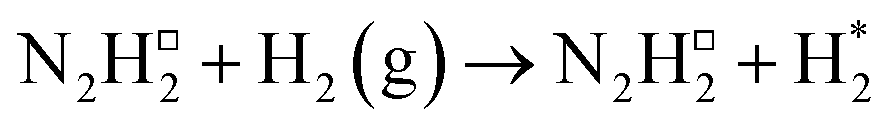
|
−0.286 | — | — |
| 4 |

|
0.290 | 1.269 | 1361.892 |
| 5 | N2H□3 + H* → N□ + H* + NH3(g) | −0.274 | — | — |
| 6 | N□ + H* → NH□ | −0.812 | 0.373 | 1310.662 |
| 7 |
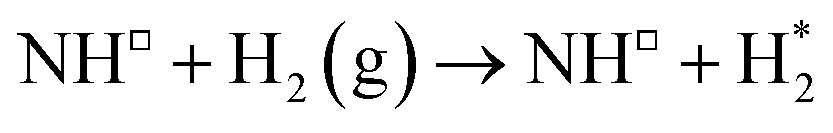
|
−0.315 | — | — |
| 8 |
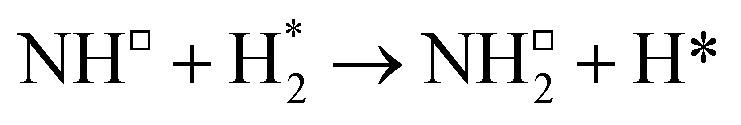
|
1.013 | 1.590 | 1195.889 |
| 9 | NH□2 + H* → NH□3 | −0.229 | 0.430 | 1191.220 |
| 10 | NH□3 → □ + NH3(g) | 1.272 | — | — |
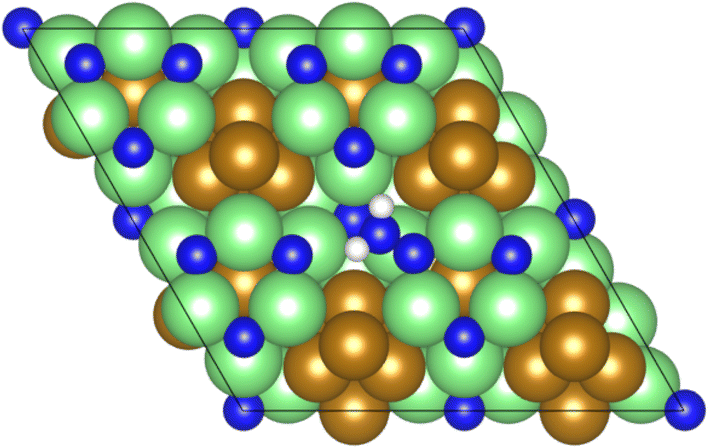
The mechanism proceeds with the initial associative adsorption of the N2 molecule at the vacancy site (process 1), as illustrated in Fig. 4. This adsorption process is highly exothermic, at −1.746 eV, and induces a considerable elongation of the N2 bond, to 1.320 Å, compared to the 1.098 Å determined experimentally,58 indicating a substantial degree of activation of the N2 molecule.
 | ||
| Fig. 4 Graphic illustrating the associative adsorption of N2 at the surface N vacancy site. | ||
The initial hydrogenation process takes place via an Eley–Rideal mechanism with direct hydrogenation of the adsorbed N2 species by H2 from the gas phase (Table 2, process 2), yielding the N2H□2 intermediate (Fig. 5). The process is exothermic by −0.753 eV and lacks an intrinsic barrier due to the high energy of the incident H2 molecule.
 | ||
| Fig. 5 Graphic illustrating the adsorption configuration of the N2H□2 intermediate. | ||
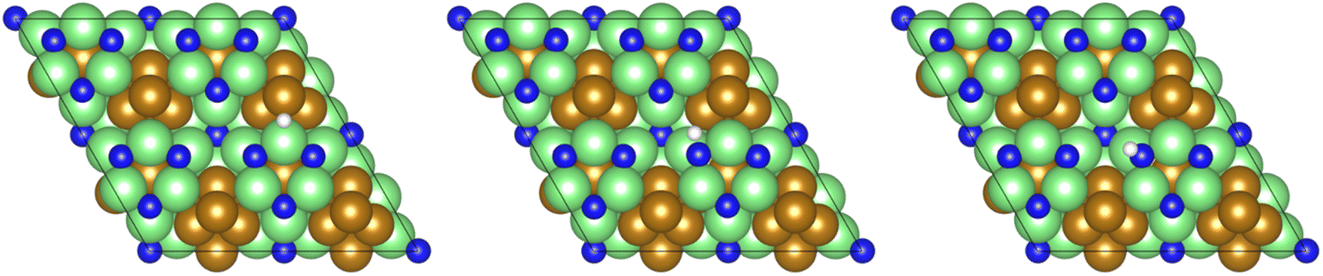
Subsequently, a second equivalent of H2 is exothermically adsorbed (−0.286 eV) adjacent to the N2H□2 (Table 2, process 3), undergoing dissociation synchronously with the formation of a new N–H bond to yield the N2H□3 intermediate (Table 2, process 4), with this process having a much higher activation barrier (1.269 eV), reflecting the saturation of the N–N bond with successive hydrogenation, although the process is only mildly endothermic (0.290 eV), as illustrated in Fig. 6.
 | ||
| Fig. 6 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with hydrogenation of the N2H□2 intermediate to yield the N2H□3 intermediate (Table 2, process 4). | ||
Dissociation of the N–N bond of the N2H□3 intermediate evolves the first equivalent of ammonia, the process is exothermic (Table 2, process 5, −0.274 eV) and leaves the remaining N atom originating from the adsorbed N2 molecule in process 1 located in the vacancy site, essentially replacing the lattice N lost during vacancy formation, and a co-adsorbed H atom. These co-adsorbed species subsequently form the NH□ intermediate (Table 2, process 6, Fig. 7), the process being moderately exothermic (−0.812 eV), and again, only weakly activated (0.373 eV).
 | ||
| Fig. 7 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with hydrogenation of surface lattice N (regenerated after the evolution of a first equivalent of NH3) to yield the NH□ intermediate (Table 2, process 6). | ||
The third and final equivalent of H2 required to complete the catalytic cycle is then adsorbed exothermically (−0.315 eV) in a similar manner to process 3 (Table 2, process 7), before again dissociating and forming a second N–H bond to yield the NH□2 (Table 2, process 8); the process is moderately endothermic (1.013 eV) and has a considerable activation barrier of 1.590 eV, making this the most energy-demanding elementary process in the ER–MvK mechanism by a considerable margin, and thus we would expect this process to also be the rate determining elementary step in this catalytic cycle (Fig. 8). It must also be noted that whilst the activation barrier for this hydrogenation process is high, and therefore represents a significant kinetic obstacle to formation of the second equivalent of ammonia to regenerate the surface vacancy, at this stage in the reaction mechanism a first equivalent of ammonia has already been evolved. Hence, it is entirely possible that ammonia synthesis could proceed identically to that described by processes 1 to 5 in Table 2 at a different N vacancy site. Such behaviour would correspond to N looping, rather than true catalysis, since in this scenario the N vacancies are not regenerated within the same cycle, but could be under a different set of conditions.
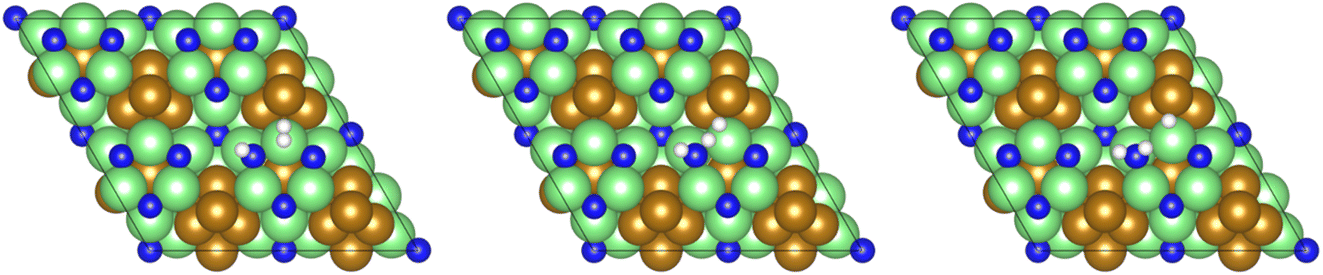 | ||
| Fig. 8 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with hydrogenation of the NH□ intermediate to yield the NH□2 intermediate (Table 2, process 8). | ||
The remaining hydrogenation process then takes place, yielding the NH□3 intermediate (Table 2, process 9), which is conversely mildly exothermic (−0.229 eV) and has only a moderate activation barrier (0.430 eV), illustrated in Fig. 9.
 | ||
| Fig. 9 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with hydrogenation of the NH□2 intermediate to yield the NH□3 (Table 2, process 9). | ||
Evolution of the second equivalent of ammonia completes the catalytic cycle, regenerating the surface lattice N vacancy, this process being considerably endothermic by 1.272 eV.
The calculated reaction profile for the ER–MvK mechanism reveals that the presence of a surface lattice N vacancy is indeed able to significantly activate N2, and the initial hydrogenation processes are facile, with generally low activation barrier. Conversely, higher activation barriers and more energy-demanding processes are observed for the final hydrogenation processes and evolution of the second equivalent of ammonia. Hence, whilst the surface lattice N vacancies are clearly highly active, their formation (with the latter processes of the mechanism essentially corresponding to the hydrogenation of lattice N to yield the surface vacancy) presents a greater challenge for overall feasibility of the reaction mechanism.
Dissociative Langmuir–Hinshelwood mechanism
Having explored the associative ER–MvK mechanism, it is also of interest to consider the possibility of the dissociative LH mechanism providing an alternative pathway for ammonia synthesis, to afford comparison both with the ER–MvK on Fe3Mo3N, and the LH mechanism on the Co3Mo3N system which has been extensively investigated previously.35The calculated reaction energies and activation barriers with corresponding frequencies for the unstable mode are presented in Table 3. The initial process, as for the associative ER–MvK mechanism, consists of adsorption of the N2 molecule, although in the associative LH mechanism, N2 is adsorbed adjacent to, rather than at, the surface N vacancy site.
| Process number | Process | ΔE/eV | E A/eV | ν/cm−1 |
|---|---|---|---|---|
| 1 |
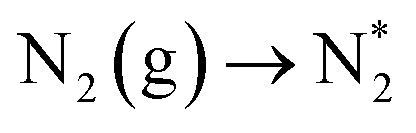
|
−0.172 | — | — |
| 2 |
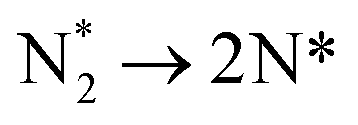
|
−0.436 | 0.708 | 453.952 |
| 3 | 2N* + H2(g) → 2N* + 2H* | −1.372 | — | — |
| 4 | 2N* + 2H* → NH* + N* + H* | 0.027 | 1.480 | 1263.294 |
| 5 |
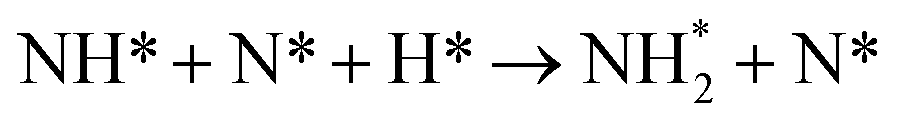
|
0.104 | 1.143 | 1095.821 |
| 6 |
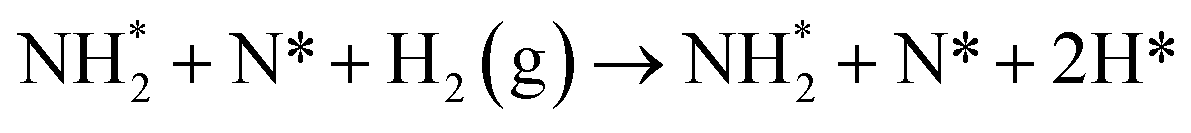
|
−0.885 | — | — |
| 7 |

|
0.282 | 1.329 | 886.360 |
| 8 |

|
1.405 | — | — |
| 9 | N* + H* → NH* | −0.600 | 0.532 | 1192.142 |
| 10 | NH* + H2(g) → NH* + 2H* | −1.349 | — | — |
| 11 |

|
−0.012 | 1.206 | 999.554 |
| 12 |
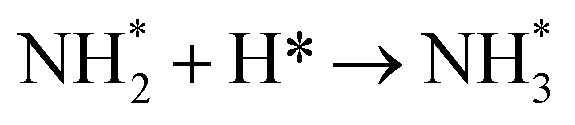
|
0.910 | 1.636 | 1169.424 |
| 13 |
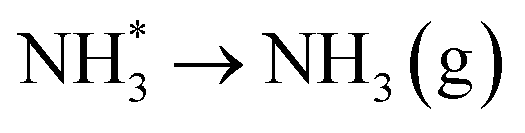
|
0.258 | — | — |
The adsorption of N2 adjacent to the vacancy site is weakly exothermic (−0.172 eV, Table 3, process 1), considerably less so than for adsorption at the vacancy, although notably there is still evidence of N2 activation even when the N2 molecule is merely adsorbed near, rather than at, the surface N vacancy site, with an elongation of the N2 bond length being observed, 1.359 Å compared to 1.098 Å determined experimentally,58 even greater than that observed for N2 adsorbed at the N vacancy site as investigated in the ER–MvK mechanism. This result suggests that the presence of surface N vacancies is able to facilitate N2 activation even when adsorption takes place outside the immediate vicinity of surface N vacancies.
The dissociation of the adsorbed N2 molecule (Table 3, process 2), which has been identified as the rate-limiting elementary process for ammonia synthesis on Fe11,12 and Ru13,14 based catalysts, is only moderately activated on the Fe3Mo3N (111) surface, with a calculated activation barrier of 0.708 eV, and the process is exothermic by −0.436 eV, reflecting the considerable N2 activation even when N2 is not adsorbed directly at the surface N vacancy site (Fig. 10). The resulting N* species are adsorbed at two distinct surface sites: the first being a 3-coordinate Mo–Fe–Mo site, subsequently referred to as the Fe–Mo interface; whilst the second is located at a 3-coordinate Mo-only site, adjacent to the lattice N vacancy. We will first consider hydrogenation of N* at the Fe–Mo interfacial site.
 | ||
| Fig. 10 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with N2 dissociation (Table 3, process 2). | ||
Exothermic dissociative adsorption of H2 (−1.372 eV, Table 3, process 3) is followed by hydrogenation of one of the N* resulting from N2 dissociation (Table 3, process 4). Whilst the process is almost thermoneutral (0.027 eV), it also has a high activation barrier (1.480 eV), presenting a significant energetic barrier to initial N hydrogenation, as illustrated in Fig. 11.
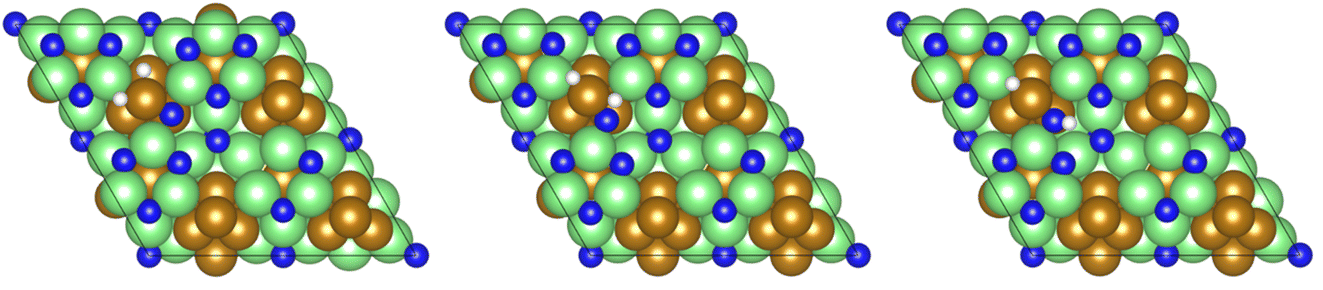 | ||
| Fig. 11 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with the first N* hydrogenation process (Table 3, process 4). | ||
This pattern continues for the second hydrogenation process to yield  (Fig. 12), which is also mildly endothermic (0.104 eV) and has a high activation barrier of 1.143 eV (Table 3, process 5), albeit somewhat lower than for the preceding hydrogenation process. It was also seen that during the hydrogenation process, NH* shifts from the 3-coordinate Fe–Mo interfacial site with
(Fig. 12), which is also mildly endothermic (0.104 eV) and has a high activation barrier of 1.143 eV (Table 3, process 5), albeit somewhat lower than for the preceding hydrogenation process. It was also seen that during the hydrogenation process, NH* shifts from the 3-coordinate Fe–Mo interfacial site with  being formed at a 2-coordinate Fe–Mo site.
being formed at a 2-coordinate Fe–Mo site.
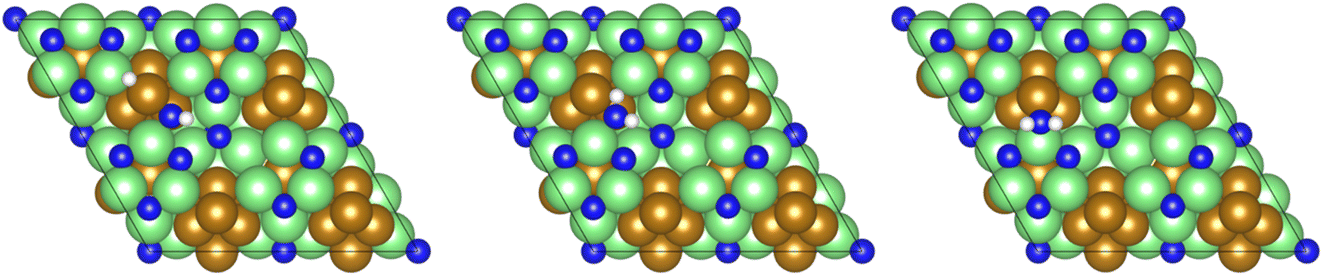 | ||
| Fig. 12 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with the first NH* hydrogenation process (Table 3, process 5). | ||
A second equivalent of H2 is then dissociatively adsorbed on the surface exothermically (−0.885 eV, Table 3, process 6), and subsequently hydrogenation of the  intermediate takes place to yield
intermediate takes place to yield  (Table 3, process 7). As for the preceding hydrogenation processes,
(Table 3, process 7). As for the preceding hydrogenation processes,  formation is only slightly endothermic (0.282 eV) but has a high activation barrier of 1.329 eV. During hydrogenation,
formation is only slightly endothermic (0.282 eV) but has a high activation barrier of 1.329 eV. During hydrogenation,  shifts once again such that the resulting
shifts once again such that the resulting  species adopts a 1-coordinate adsorption environment located slightly offset from the top of the Fe atom, as illustrated in Fig. 13.
species adopts a 1-coordinate adsorption environment located slightly offset from the top of the Fe atom, as illustrated in Fig. 13.
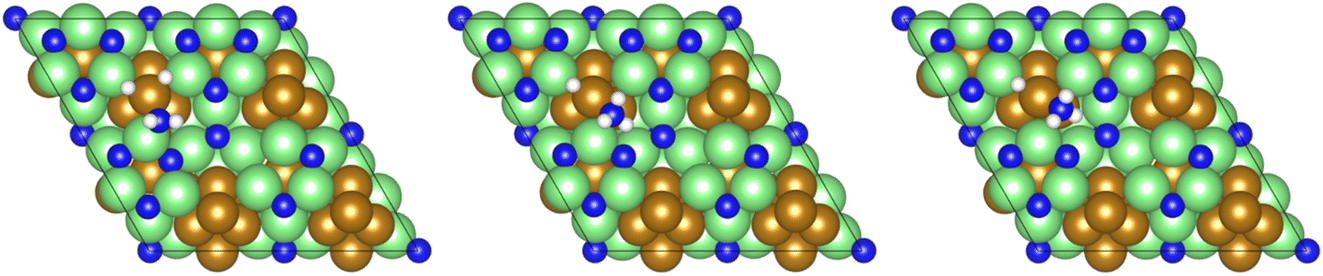 | ||
Fig. 13 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with the first  hydrogenation process (Table 3, process 7). hydrogenation process (Table 3, process 7). | ||
Evolution of the first equivalent of ammonia is strongly endothermic (1.405 eV, Table 3, process 8), and is followed by the first hydrogenation of the second N* (Table 3, process 9); in contrast to the corresponding process for the first N* (Table 3, process 4), this process is exothermic by −0.600 eV, and has only a moderate activation barrier of 0.532 eV. It is notable that the first series of hydrogenation processes (Table 3, processes 4, 5, 7), which were all determined to be endothermic and highly activated, took place at the N* species located at the Fe–Mo interface, whilst hydrogenation of the second N* species (Table 3, process 9; Fig. 14), involves N* migrating from a 3-coordinate Mo site to the Fe–Mo interface. This implies that the N*, NH*, and  species are more stabilised at the Fe–Mo interface site, and therefore these species are more resistant to hydrogenation at this site, compared to at the 3-coordinate Mo site.
species are more stabilised at the Fe–Mo interface site, and therefore these species are more resistant to hydrogenation at this site, compared to at the 3-coordinate Mo site.
 | ||
| Fig. 14 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with the second N* hydrogenation process (Table 3, process 9). | ||
Dissociative adsorption of the third and final equivalent of H2 takes place exothermically (−1.349 eV, Table 3, process 10). Subsequently, NH* undergoes hydrogenation to  (Table 3, process 11), this process being found to be almost thermoneutral (−0.012 eV), and a return to the high activation barriers calculated for the first series of hydrogenation processes is observed, with an activation barrier of 1.206 eV being determined; once again, during hydrogenation the NH* species moves from the 3-coordinate Fe–Mo interfacial site to form
(Table 3, process 11), this process being found to be almost thermoneutral (−0.012 eV), and a return to the high activation barriers calculated for the first series of hydrogenation processes is observed, with an activation barrier of 1.206 eV being determined; once again, during hydrogenation the NH* species moves from the 3-coordinate Fe–Mo interfacial site to form  at the adjacent 2-coordinate Fe–Mo bridging site (Fig. 15), in a manner virtually identical to that determined for process 4.
at the adjacent 2-coordinate Fe–Mo bridging site (Fig. 15), in a manner virtually identical to that determined for process 4.
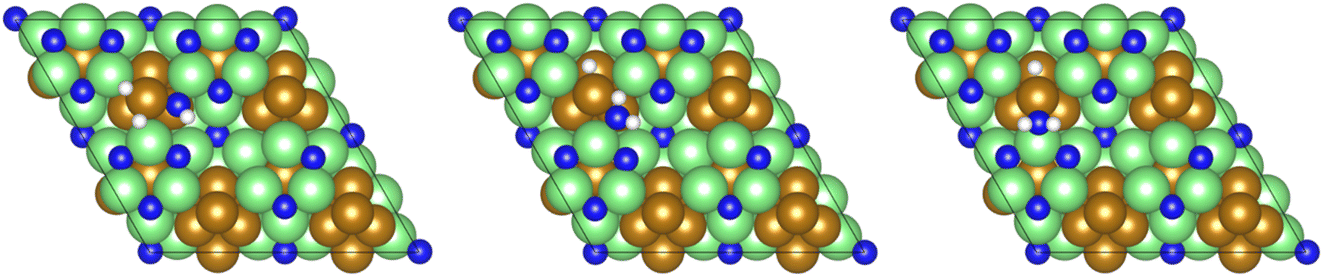 | ||
| Fig. 15 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with the second NH* hydrogenation process (Table 3, process 11). | ||
The final hydrogenation process (Table 3, process 12; Fig. 16) to form  occurs analogously to process 7, being even more endothermic (0.910 eV) and having a high activation barrier of 1.636 eV, making this the most energy-demanding elementary process in the dissociative LH mechanism.
occurs analogously to process 7, being even more endothermic (0.910 eV) and having a high activation barrier of 1.636 eV, making this the most energy-demanding elementary process in the dissociative LH mechanism.
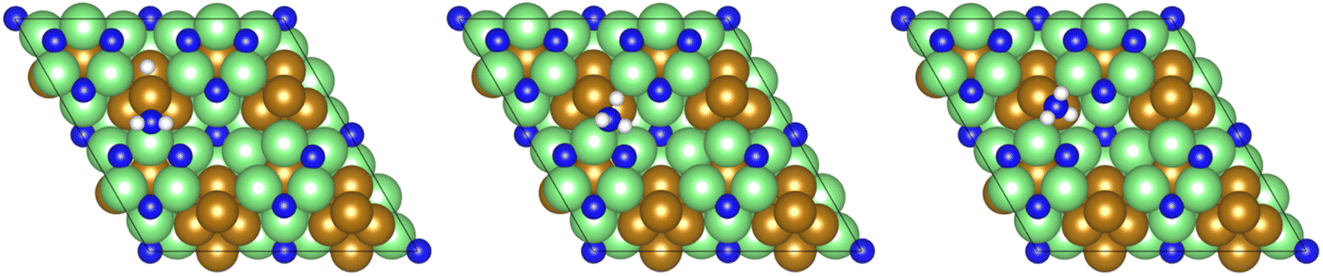 | ||
Fig. 16 Graphics illustrating the initial (left), transition (centre), and final (right) state geometries associated with the second  hydrogenation process (Table 3, process 12). hydrogenation process (Table 3, process 12). | ||
Finally, evolution of the second equivalent of ammonia (Table 3, process 13) completes the catalytic cycle, and is mildly endothermic at 0.258 eV.
The calculations for the dissociative LH mechanism for ammonia synthesis reveal that whilst the initial adsorption and dissociation of N2 is enhanced on the Fe3Mo3N (111) surface, subsequent hydrogenation processes are largely highly activated and endothermic, especially at the Mo–Fe interfacial surface site; indeed the only hydrogenation process that deviates from this is process 9, which involves hydrogenation of the N* species from the Mo-only site, with the remaining endothermic hydrogenation processes largely being compensated by highly exothermic dissociative adsorption of H2.
Discussion
The computed reaction profiles for the two mechanisms are presented in Fig. 17. It is clear that whilst N2 adsorption and activation (and dissociation, for the LH type mechanism) is enhanced both at and adjacent to the surface N vacancy, the subsequent hydrogenation processes are far less activated for the associative ER–MvK mechanism, at least for the initial hydrogenation processes; indeed, the only elementary process in the ER–MvK mechanism with an activation barrier greater than 1 eV is the second NH* hydrogenation process (Table 2, process 8), whilst for the LH mechanism, the only hydrogenation process with an activation barrier less 1 eV is the second N* hydrogenation process (Table 3, process 9). Hence, it appears likely that the high activity of Fe3Mo3N can be attributed to this mechanism, and therefore N vacancy formation in Fe3Mo3N is confined to the surface (as suggested by the bulk N/vacancy diffusion calculations), explaining the negligible loss of lattice N in Fe3Mo3N reported experimentally, in stark contrast with Co3Mo3N. Here, we will compare the two mechanisms and the two ternary metal nitride systems, comparing and contrasting the results, and offering some insight to rationalise the experimentally reported phenomena in light of the calculations presented in the present work and previous literature.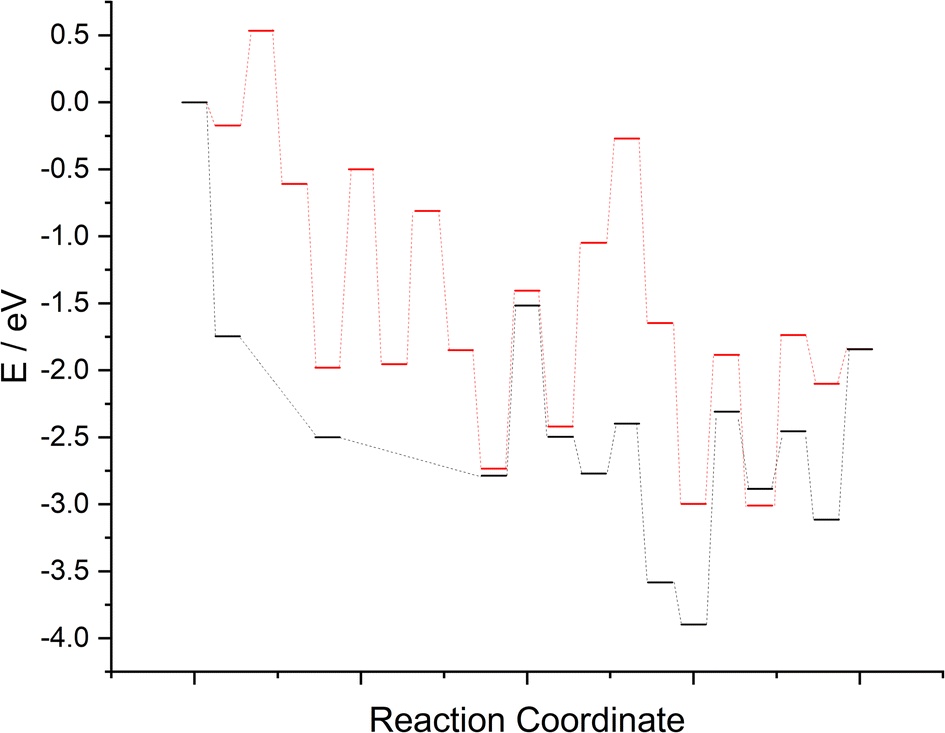 | ||
| Fig. 17 Computed reaction profile for the two mechanisms for ammonia synthesis on Fe3Mo3N, for the associative Eley–Rideal–Mars–van-Krevelen mechanism (black) and the dissociative Langmuir–Hinshelwood mechanism (red). | ||
Surface formation and bulk diffusion of N vacancies
In comparison with the vacancy formation energies for Co3Mo3N in the literature,15 the corresponding Co3Mo3N(111) surface N vacancy formation energy was calculated to be 1.68 eV, only 0.21 eV less endothermic than for Fe3Mo3N. Whilst this is in agreement with the experimental findings that lattice N in Fe3Mo3N is generally less active compared to Co3Mo3N,38,39,59 it is notable that the modest difference in the calculated vacancy formation energies for the two compounds suggests that formation of surface N vacancies on Fe3Mo3N is at the very least highly plausible, which is consistent with the experimental observation that bulk lattice N in Fe3Mo3N is inactive, if loss of lattice N is confined only to the surface layers, and therefore accounts for only a negligible loss of total lattice nitrogen from the experimental sample. Hence, this finding suggests that the experimentally observed resistance of Fe3Mo3N to loss of lattice N under high temperature hydrogenation conditions can be attributed to the diffusion of bulk and subsurface lattice nitrogen to the surface being kinetically, rather than thermodynamically, unfeasible.N2 activation
Comparison of the N2 adsorption process between the Fe3Mo3N and Co3Mo3N shows that N2 activation is considerably enhanced on Fe3Mo3N, with N2 adsorption both at the surface N vacancy site, and adjacent to the N vacancy site, being more exothermic than for Co3Mo3N. Adsorption of N2 at the surface N vacancy is exothermic by −1.746 eV (Table 2, process 1), compared to a mere ∼−0.73 eV for Co3Mo3N, whilst at the site adjacent to the vacancy, the process is exothermic by −0.172 eV for Fe3Mo3N (Table 3, process 1), and slightly endothermic for Co3Mo3N (+0.42 eV).35 The enhanced N2 activation for Fe3Mo3N can be interpreted in terms of its more endothermic vacancy formation energy (Table 1) compared to Co3Mo3N, implying that the defective system is inherently less stable, and therefore more active, for the Fe system compared to the Co system. Despite the considerably more exothermic adsorption energies on the Fe3Mo3N surface, the elongation of the N2 bond is comparable between the Fe and Co systems, both when N2 is adsorbed at the N vacancy site (1.320 Å for Fe3Mo3N, compared to 1.297 Å for Co3Mo3N) and when N2 is adsorbed adjacent to the vacancy site (1.359 Å for Fe3Mo3N, compared to 1.363 Å for Co3Mo3N). For both the Fe and Co systems, activation of N2 adjacent to the N vacancy also can be seen to play a key role in facilitating N2 dissociation (Table 3, process 2), a key process in the LH mechanism for ammonia synthesis; for the Fe system the process has an activation barrier of 0.708 eV, and is exothermic by −0.436, whereas a lower barrier and even greater exothermicity was reported for the Co system (EA = 0.24 eV, ΔE = −1.86 eV). Whilst N2 dissociation appears to be both more kinetically and thermodynamically favourable over the Co system compared to the Fe system, one must bear in mind that the preceding N2 adsorption is more exothermic for the Fe system, hence it is likely that the overall pathway for N2 adsorption from the gas phase, and subsequently dissociation, is more facile for the Fe system, given that both processes are moderately exothermic and the dissociation process has only a moderate activation barrier.Comparison of hydrogenation processes and insights into activity of quaternary metal nitrides
Whilst an examination of the reaction profiles for the two mechanisms for both the Fe system investigated in the present work, and the Co system in previous computational studies, shows broadly similar behaviour, there are a few key differences that could help to identify limitations to the activity of both catalysts, and thus inform the future development of, for example, highly active ammonia synthesis catalysts based on the quaternary Co3−xFexMo3N system, with an optimal Co:Fe ratio.
As has already been discussed, the N2 activation behaviour of the Co and Fe systems is broadly similar, although in terms of hydrogenation, it appears that these processes are activated more for the Fe system. Indeed, examining the LH pathways for the two systems, it can be seen that activation barriers are consistently higher for the Fe system than the corresponding activation barriers for the Co system. The key structural difference appears to lie in the coordination environment of the NH*,  and
and  intermediates, with the calculations implying a greater stabilisation of these intermediates at the Fe–Mo interfacial sites compared to the corresponding Co–Mo sites in the Co system. Similarly, whilst the ER–MvK mechanism provides a less activated mechanistic pathway for both systems, it can be seen that whilst the initial hydrogenation processes are less activated for the Fe system, the latter hydrogenation processes are more so, once again reflecting the greater stability of these intermediates at the N vacancy site.
intermediates, with the calculations implying a greater stabilisation of these intermediates at the Fe–Mo interfacial sites compared to the corresponding Co–Mo sites in the Co system. Similarly, whilst the ER–MvK mechanism provides a less activated mechanistic pathway for both systems, it can be seen that whilst the initial hydrogenation processes are less activated for the Fe system, the latter hydrogenation processes are more so, once again reflecting the greater stability of these intermediates at the N vacancy site.
To compare the more accessible ER–MvK mechanism for both the Fe3Mo3N and Co3Mo3N systems, a reaction profile illustrating this mechanism for the two systems is presented in Fig. 18, based on the results presented in this work and the previous literature on Co3Mo3N.35 Whilst the reaction profiles are broadly similar, the general trend of the initial hydrogenation processes being less activated, and latter ones more activated, for the Fe3Mo3N system is clearly evident. In particular, it is notable that the highest activation barriers, which are likely to correspond to the overall rate-limited elementary processes, are comparable, even if they do not correspond to the same process. Namely, the activation barrier for the NH* hydrogenation process is much higher for Fe3Mo3N than for Co3Mo3N, and is the most energy-demanding process for Fe3Mo3N (Table 1, process 8); yet as illustrated in Fig. 18, the most energy-demanding processes for Co3Mo3N correspond to  hydrogenation, and evolution of the second equivalent of NH3 are comparable to that of the most energy-demanding process for Fe3Mo3N. This explains the high activity observed for Fe3Mo3N experimentally; indeed, it seems likely, based on the calculations performed in the present work, that the key difference between the two systems lies primarily in the greater resistance of Fe3Mo3N to loss of lattice N, which in turn reduces the concentration of active N vacancy sites compared to Co3Mo3N, resulting in overall lower ammonia production.
hydrogenation, and evolution of the second equivalent of NH3 are comparable to that of the most energy-demanding process for Fe3Mo3N. This explains the high activity observed for Fe3Mo3N experimentally; indeed, it seems likely, based on the calculations performed in the present work, that the key difference between the two systems lies primarily in the greater resistance of Fe3Mo3N to loss of lattice N, which in turn reduces the concentration of active N vacancy sites compared to Co3Mo3N, resulting in overall lower ammonia production.
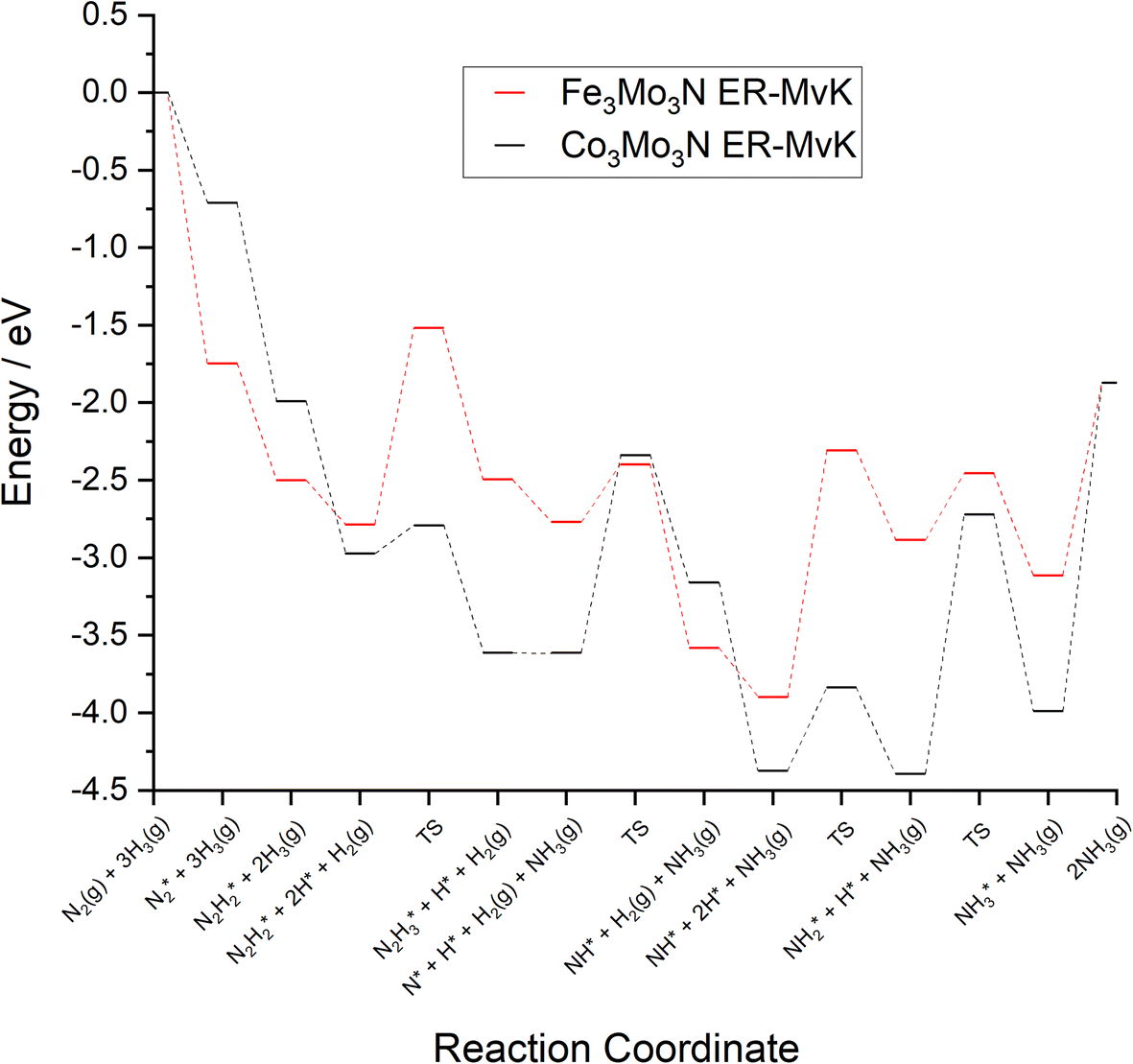 | ||
| Fig. 18 Computed reaction profiles comparing the ER–MvK mechanism for ammonia synthesis on Fe3Mo3N (red) and for Co3Mo3N (black).35 | ||
Based on these observations, we can suggest that a possible means by which the mixed quaternary Co/Fe system can enhance ammonia synthesis is by combining the greater ease of N vacancy formation (thereby increasing the concentration of active sites for the ER–MvK mechanism, which is the less activated for both the Fe and Co systems) associated with the Co system, along with the weaker binding of key intermediates at the Co–Mo interface and N vacancy site, with the greater extent of N2 activation and lower barriers for initial hydrogenation processes associated with the Fe system. We therefore speculate that optimal catalytic activity could be tuned by careful control of the Co:Fe ratio in the quaternary system, which combined with the promotion and doping strategies applied to the ternary systems, could enable highly active and stable ammonia synthesis catalysts that can operate under mild conditions.
Conclusions
The calculations reported in the present work reveal that, like the isostructural Co3Mo3N system, the Fe3Mo3N system affords low activation barriers for ammonia synthesis via the ER–MvK mechanism, and in particular the Fe system has lower barriers for initial hydrogenation processes via the hydrazine-like intermediate. However, the vacancy formation energy for Fe3Mo3N is more endothermic than for Co3Mo3N, and as illustrated by the ab initio thermodynamics study, it is anticipated that Fe3Mo3N would feature lower concentrations of surface N vacancies compared to Co3Mo3N, in line with the experimentally well-observed tendency of the Fe system to be resistant to loss of lattice nitrogen. It is also notable that whilst the ER–MvK mechanism was determined to be the less activated pathway for both systems, the dissociative LH mechanism is more accessible on the Fe system since the initial adsorption of N2 is considerably more exothermic compared to the Co system, thus enabling the initial N2 activation process, although higher activation barriers were reported for latter hydrogenation processes in the LH mechanism. Considering this, and the broadly similar features of the ammonia synthesis mechanisms for the two ternary metal nitride systems, it seems likely that the high activity of the Fe3Mo3N system reported experimentally can be attributed to the lower vacancy concentration, and thus lower concentration of active sites for the favoured ER–MvK mechanism, being partially compensated by the existing possibility for N2 hydrogenation to take place via the LH mechanism, reflecting the well-established high activity of promoted Fe catalysts used in industry.The present work provides useful insights into the activity of metal nitride catalysts and it is intended that future work will explore not only promotion of the Fe3Mo3N system, but also the quaternary Co3−xFexMo3N system, which will be informed by the present work and the existing previous literature for the Co3Mo3N system.
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The authors thank the EPSRC/UKRI (EP/T028629/1) for financial support of this project, and the UK Catalysis Hub Consortium (funded by EPSRC (Grants EP/R026815/1)) for the provision of additional resources. The authors acknowledge the STFC (SCARF) and UK National Supercomputing Service (ARCHER2) for the provision of computational resources, the latter enabled via membership of the MCC (EP/L000202).References
- Scaling Ammonia Production For The World's Food Supply, https://www.forbes.com/sites/mitsubishiheavyindustries/2021/10/29/scaling-ammonia-production-for-the-worlds-food-supply/?sh=63a47ef968d1, accessed 28 September 2022 Search PubMed.
- World population projected to reach 9.8 billion in 2050, and 11.2 billion in 2100, https://www.un.org/development/desa/en/news/population/world-population-prospects-2017.html, accessed 28 September 2022 Search PubMed.
- Ammonia Market – Growth, Trends, COVID-19 Impact, and Forecasts (2022–2027), https://www.mordorintelligence.com/industry-reports/ammonia-market, accessed 28 September 2022 Search PubMed.
- J. Humphreys, R. Lan and S. Tao, Advanced Energy and Sustainability Research, 2021, 2, 2000043 CrossRef CAS.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- Ammonia: Zero-Carbon Fertiliser, Fuel and Energy Store, https://royalsociety.org/-/media/policy/projects/green-ammonia/green-ammonia-policy-briefing.pdf, accessed 28 September 2022 Search PubMed.
- S. Ghavam, M. Vahdati, I. A. G. Wilson and P. Styring, Front. Energy Res., 2021, 9, 1–19 Search PubMed.
- Electrochemical Ammonia Synthesis from Water and Nitrogen using Solid State Ion Conductors, https://www.ammoniaenergy.org/paper/electrochemical-ammonia-synthesis-from-water-and-nitrogen-using-solid-state-ion-conductors/, accessed 28 September 2022 Search PubMed.
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1055–1074 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S2542435118301788.
- F. Haber, in Nobel Lecture, 1920, pp. 328–340 Search PubMed.
- G. Ertl, Catal. Rev. Sci. Eng., 1980, 21, 201–223 CrossRef CAS.
- M. Bowker, Catal. Today, 1992, 12, 153–163 CrossRef CAS.
- Á. Logadóttir and J. K. Nørskov, J. Catal., 2003, 220, 273–279 CrossRef.
- C. J. Zhang, M. Lynch and P. Hu, Surf. Sci., 2002, 496, 221–230 CrossRef CAS.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2015, 119, 28368–28376 CrossRef CAS.
- J. S. J. Hargreaves, Appl. Petrochem. Res., 2014, 4, 3–10 CrossRef CAS.
- L. Volpe and M. Boudart, J. Phys. Chem., 1986, 90, 4874–4877 CrossRef CAS.
- W. Zheng, T. P. Cotter, P. Kaghazchi, T. Jacob, B. Frank, K. Schlichte, W. Zhang, D. S. Su, F. Schüth and R. Schlögl, J. Am. Chem. Soc., 2013, 135, 3458–3464 CrossRef CAS PubMed.
- N. Segal, J. Catal., 1967, 8, 105–112 CrossRef CAS.
- N. Segal and F. Sebba, J. Catal., 1967, 8, 113–119 CrossRef CAS.
- D. A. King, J. Catal., 1965, 4, 253–259 CrossRef CAS.
- D. A. King and F. Sebba, J. Catal., 1965, 4, 430–439 CrossRef CAS.
- R. Kojima and K. I. Aika, Appl. Catal., A, 2001, 209, 317–325 CrossRef CAS.
- R. Kojima, H. Enomoto, M. Muhler and K. I. Aika, Appl. Catal., A, 2003, 246, 311–322 CrossRef CAS.
- G. I. Panov and A. S. Kharitonov, React. Kinet. Catal. Lett., 1985, 29, 267–274 CrossRef CAS.
- R. Kojima and K. Aika, Appl. Catal., A, 2001, 215, 149–160 CrossRef CAS.
- R. Kojima and K. Aika, Appl. Catal., A, 2001, 218, 121–128 CrossRef CAS.
- R. Kojima and K.-I. Aika, Appl. Catal., A, 2001, 219, 157–170 CrossRef CAS.
- C. J. H. Jacobsen, S. Dahl, B. G. S. Clausen, S. Bahn, A. Logadottir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404–8405 CrossRef CAS PubMed.
- C. J. H. Jacobsen, Chem. Commun., 2000, 1057–1058 RSC.
- A. Boisen, S. Dahl and C. J. H. Jacobsen, J. Catal., 2002, 208, 180–186 CrossRef CAS.
- S. M. Hunter, D. H. Gregory, J. S. J. Hargreaves, M. Richard, D. Duprez and N. Bion, ACS Catal., 2013, 3, 1719–1725 CrossRef CAS PubMed.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2016, 120, 21390–21398 CrossRef CAS.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves, S. Laassiri and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2018, 20, 21803–21808 RSC.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2018, 122, 6078–6082 CrossRef CAS.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves, S. Laassiri and C. R. A. Catlow, R. Soc. Open Sci., 2021, 8 DOI:10.1098/rsos.210952.
- D. Mckay, PhD thesis, University of Glasgow, 2008.
- S. M. Hunter, PhD thesis, University of Glasgow, 2012.
- A. Daisley, L. Costley-Wood and J. S. J. Hargreaves, Top. Catal., 2021, 64, 1021–1029 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- H. Monkhorst and J. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- NIST Webbook, https://webbook.nist.gov/chemistry/, accessed 28 September 2022.
- K. Reuter, Catal. Lett., 2016, 146, 541–563 CrossRef CAS.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 205424 CrossRef.
- J. Rogal and K. Reuter, Ab Initio Atomistic Thermodynamics for Surfaces: A Primer, Experiment, Modeling and Simulation of Gas-Surface Interactions for Reactive Flows in Hypersonic Flights, 2007, pp. 2-1–2-18 Search PubMed.
- M. Bollinger, K. Jacobsen and J. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 85410–85427 CrossRef.
- M. Moser, V. Paunović, Z. Guo, L. Szentmiklósi, M. G. Hevia, M. Higham, N. López, D. Teschner and J. Pérez-Ramírez, Chem. Sci., 2016, 7, 2996–3005 RSC.
- A. Daisley and J. S. J. Hargreaves, J. Energy Chem., 2019, 39, 170–175 CrossRef.
- T. Wang and F. Abild-Pedersen, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 1–5 Search PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. Morris Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, 873–880 CAS.
- K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure, Springer US, Boston, MA, 1979 Search PubMed.
- D. Mckay, J. S. J. Hargreaves, J. L. Rico, J. L. Rivera and X. L. Sun, J. Solid State Chem., 2008, 181, 325–333 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2fd00148a |
| This journal is © The Royal Society of Chemistry 2023 |