
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium-catalysed tetradehydro-Diels–Alder reactions of enediynes via a rhodium-stabilized cyclic allene†
Srinivas
Thadkapally
a,
Kaveh
Farshadfar
 bc,
Melanie A.
Drew
a,
Christopher
Richardson
a,
Alireza
Ariafard
*b,
Stephen G.
Pyne
*a and
Christopher J. T.
Hyland
*a
bc,
Melanie A.
Drew
a,
Christopher
Richardson
a,
Alireza
Ariafard
*b,
Stephen G.
Pyne
*a and
Christopher J. T.
Hyland
*a
aSchool of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: chris_hyland@uow.edu.au
bSchool of Physical Sciences, University of Tasmania, Hobart, TAS 7018, Australia
cDepartment of Chemistry, Islamic Azad University, Central Tehran Branch, Poonak, Tehran 1469669191, Iran
First published on 4th September 2020
Abstract
Efficient methods for the synthesis of fused-aromatic rings is a critical endeavour in the creation of new pharmaceuticals and materials. A direct method for preparing these systems is the tetradehydro-Diels–Alder reaction, however this is limited by the need for harsh reaction conditions. A potential, but underdeveloped, route to these systems is via transition metal-catalysed cycloaromatisation of ene-diynes. Herein, tethered unconjugated enediynes have been shown to undergo a facile room-temperature RhI-catalysed intramolecular tetradehydro-Diels–Alder reaction to produce highly substituted isobenzofurans, isoindolines and an indane. Furthermore, experimental and computational studies suggest a novel mechanism involving an unprecedented and complex RhI/RhIII/RhI/RhIII redox cycle involving the formation of an unusual strained 7-membered rhodacyclic allene intermediate and a RhIII-stabilized 6-membered ring allene complex.
Introduction
New methods for the construction of functionalized aromatic rings is an important area of research due to their wide occurrence in bioactive molecules and materials. An advanced strategy for preparing functionalized aromatic rings is cycloaromatisation of polyunsaturated acyclic building blocks, including, enediynes, enyne-allenes and dienynes.1 The tetradehydro-Diels–Alder (TDDA) reaction of polyunsaturated enediyne systems is of particular note as it allows for the direct formation of a benzenoid ring in a single step, via an intriguing high energy cyclic allene intermediate, which typically aromatises via a hydride shift (Scheme 1a).2,3 The thermal mode of this reaction has been investigated for many years, but its synthetic applications are severely limited by the requirement of the process for very forcing reaction conditions and particular substitution patterns on the starting materials.3 The transition metal-catalysed variant of the reaction has demonstrated potential for unveiling tetradehydro-Diels–Alder reactions of enediynes that proceed under milder conditions for a greater range of substrates. For example, several recent reports have harnessed dual gold-catalysts in cycloaromatisations via TDDA reactions of enediynes.3–7 In these cases the enediyne is not fully conjugated but has the alkyne and enyne units linked via a heteroatom tether leading to a diverse range of fused heterocyclic benzenoid structures of biological significance (Scheme 1b).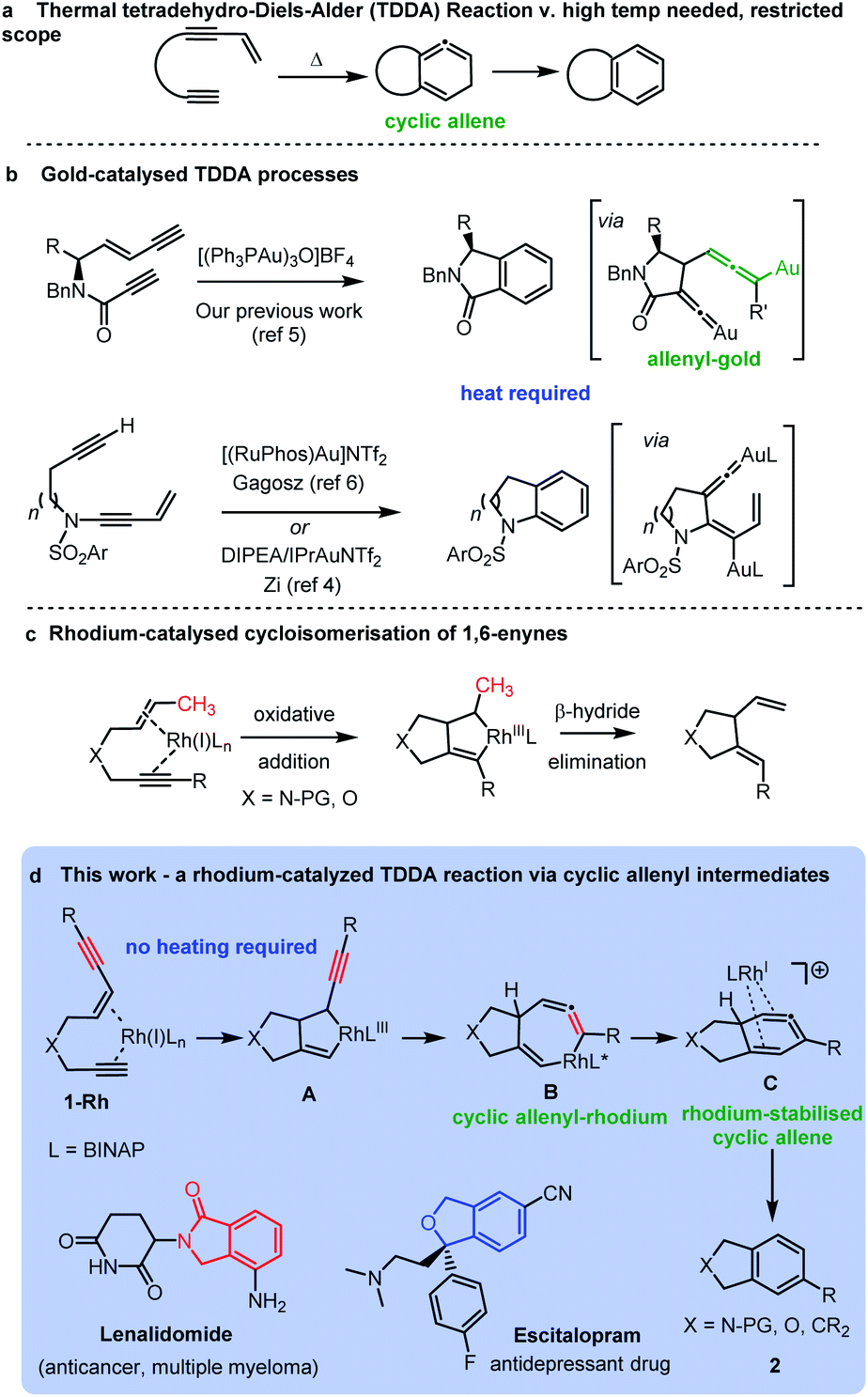 | ||
| Scheme 1 Tetradehydro-Diels–Alder reactions of enediynes: (a) thermal; (b) gold-catalysed; and (c) rhodium catalysed processes. | ||
In the case of our previous work,5 the key reaction intermediate incorporated a vinylidene gold8 intermediate and a rare allenyl gold species (Scheme 1c).
Curiously, there have been no investigations of rhodium-catalysed cycloaromatisation reactions of tethered non-conjugated enediynes – and just a single report of a cycloaromatisation reaction of a conjugated enediyne.9 We hypothesized that a rhodium-catalysed cycloaromatisation reaction could be realized by harnessing the well-known oxidative cyclization process observed in Rh-catalysed 1,6-enyne reactions (Scheme 1c).10–16 The triple bond of an enediyne opens up the possibility of forming a cyclic vinyl-rhodium species Avia oxidative cyclization of ene-diyne 1-Rh. Isomerization of the oxidative cyclization product A should form B – providing a direct route to the intriguing allenyl-rhodium intermediate B and an alternative route to a cyclic allene cf. the thermal TDDA reaction (Scheme 1d). A reductive elimination to the same cyclic allene proposed in the thermal TDDA reaction could then be envisaged en-route to the final fused-aromatic products 2via a potential final hydride shift. Herein, we report the successful implementation of this proposed pathway and provide experimental and computational evidence for a rhodium-catalysed TDDA reaction that proceeds via an unprecedented cyclic allenyl-rhodium species B and a rhodium-stabilised 6-membered cyclic allene C involving a complex RhI/RhIII/RhI/RhIII redox cycle. The resulting isobenzofuran,17 isoindoline,18,19 isoindolinone20 and indane21 products are important heterocycles that are found in an array of important bioactive and medicinally important compounds.
Results and discussion
To examine our hypothesis the NTs-tethered (Z)-enediyne 1a was synthesized (see ESI†) as a model substrate and treated with [RhCl(COD)]2/AgSbF6 and (rac)-BINAP at rt in DCE – a general system that has previously proved successful in catalyzing the cycloisomerization of 1,6-enynes.16 Gratifyingly, this proved a highly efficient catalyst for the cycloaromatisation of 1a to give isoindoline 2a in high yield (Scheme 2a – structure determined unambiguously by X-ray crystallography).22 Replacing the ligand BINAP with dppb, which has a similar bite angle, had a deleterious effect on the yield (Scheme 2b). Starting material was recovered when 1a was heated at reflux in DCE in the absence of catalyst, indicating that there is no competing non-catalysed TDDA reaction occurring (Scheme 2c).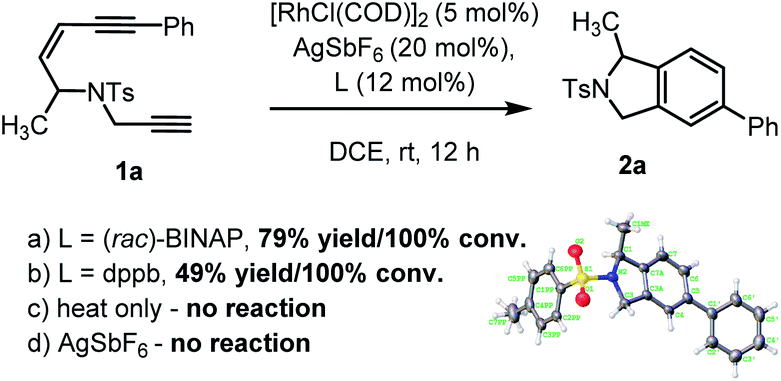 | ||
| Scheme 2 Initial cycloaromatisation reaction discovery using: (a) L = (rac)-BINAP; (b) L = dppb; (c) heat only; or (d) AgSbF6 alone. | ||
The presence of AgSbF6 was essential as the reaction with [RhCl(COD)]2 alone resulting in no conversion. Replacing AgSbF6 with AgBF4 also proved detrimental giving only a 60% yield of isoindoline 2a (80% conversion). Furthermore, lowering the Rh-catalyst loading to 2.5 mol% resulted in a significantly reduced conversion (75%) and yield (44%) over 12 h. This is even more striking for substrate 1b to give 2b (see Chart 1), where 2.5 mol% of the Rh-catalyst gave the product in 10% yield (17% conversion) or 19% yield (38% conversion) at 80 °C. Finally, changing the solvent to DCM reduced the yield to 62%.
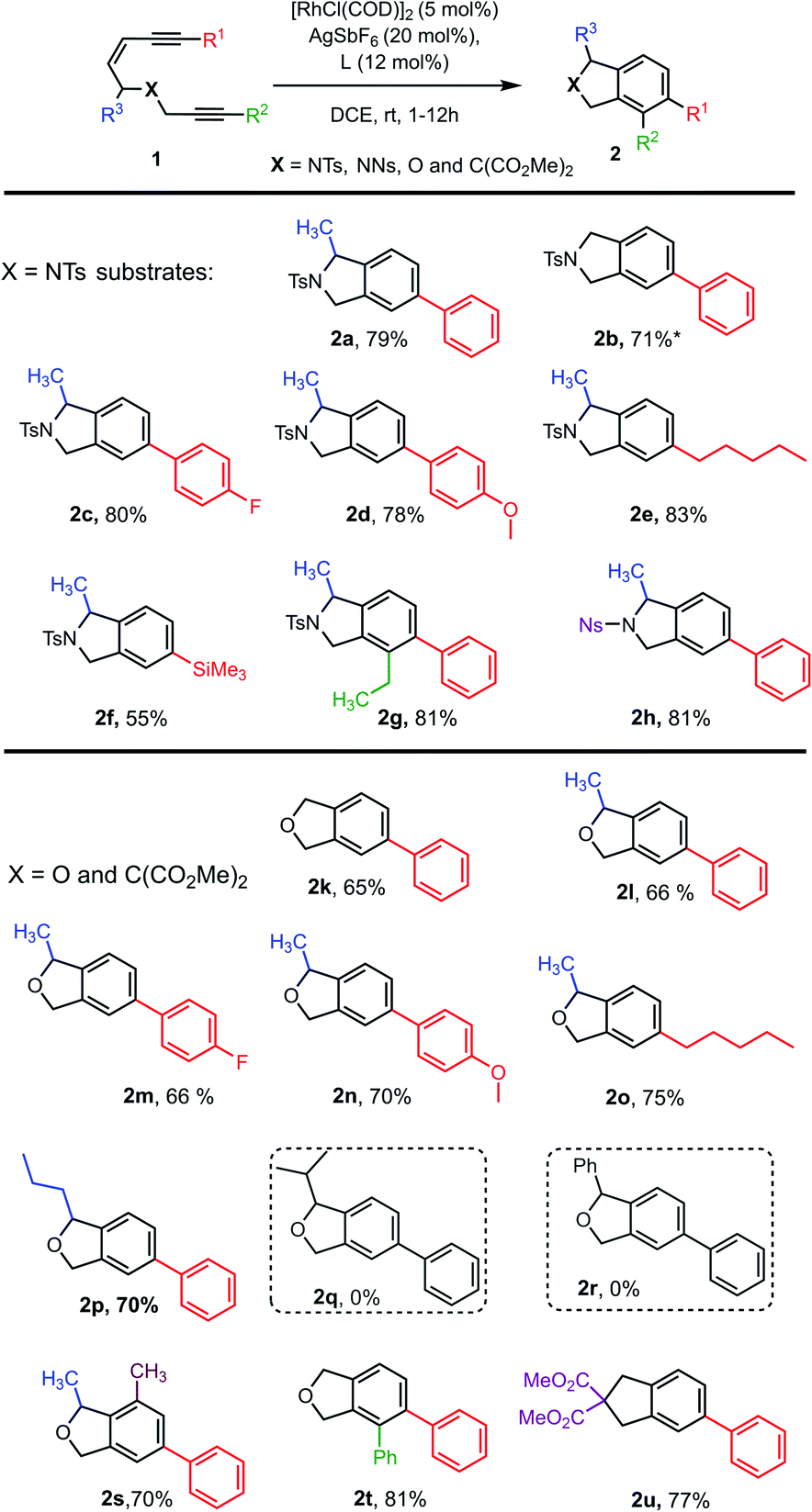 | ||
| Chart 1 Substrate scope for Rh-catalysed tetradehydro-Diels–Alder reactions of enediynes 1. aThe starting material for 2d and 2f were a mixture of E/Z isomers. *2.5 mol% of the Rh-catalyst gave the product in 10% yield (17% conversion) or 19% yield (38% conversion) at 80 °C. | ||
With an effective catalyst system identified for the Rh-catalysed TDDA reaction, the substrate scope was established with a range of N- and O-tethered enediynes 1, as shown in Chart 1. For both the N- and O-tethered enediynes, aryl (products 2a–d, 2g–h, 2k–n, 2p and 2s–2u), alkyl (products 2e and 2o) and silyl (product 2f) groups at R1 could be accommodated. Both R1 and R2 could be simultaneously substituted to provide more densely functionalised isoindolines and isobenzofurans 2g and 2t, respectively. The R3 substituent could be a hydrogen (products 2b, 2k and 2t) or accommodate an n-propyl group in place of the methyl group (product 2p). However, branched substituents or a phenyl ring (to give predicted products 2q and 2r) were not tolerated, presumably for steric reasons – in these cases only decomposition of starting material was observed. The dimethyl substituted O-tethered enediyne 1s was also well tolerated under the cycloaromatisation reaction conditions to give trisubstituted isobenzofuran 2s in good yield. Isoindoline 2h formed smoothly from nosyl-protected enediyne 1h. Other protecting groups on N were not investigated due to the established instability of isoindolines without an electron-withdrawing N group.23–27 Finally, a carbon-tethered enediyne 1u was prepared and found to undergo smooth cycloaromatisation to trisubstituted indane 2u.
Our attention then turned to experimentally and computationally investigating the reaction mechanism (Scheme 3 and Fig. 1). The C3-position in the ene-diyne starting material 1 (X = O or NTs) is key to understanding the reaction mechanism as it becomes one of the carbons (C7a) fusing the two rings of the product 2via migration of the attached hydrogen. For the thermal TDDA reaction this typically occurs via one or more hydride shifts. To understand this process for the current system, a C3-deuterated analogue 1i and a C3-methylated analogue 1j were prepared (Scheme 3). To determine the fate of the C3-hydrogen, deuterated substrate 1i was subjected to the reaction conditions, leading to predominant deuterium incorporation at the C6-position of the isoindoline benzenoid ring of 2i. The partial loss of deuterium at the C6-position of the product pointed away from a hydride shift in intermediate B or C to yield the aromatic product and hinted at the potential formation of a rhodium-hydride species. A substrate (1j) with a methyl group on the enyne double bond stopped the reaction completely – again suggesting a key role for this position in the reaction. The sensitivity of the cycloaromatisation reaction towards the double bond geometry was also probed by utilizing (E)-enediyne 1l′, which led to the formation of the isobenzofuran 2l in comparable yield to its corresponding (Z)-enediyne starting material 1l.
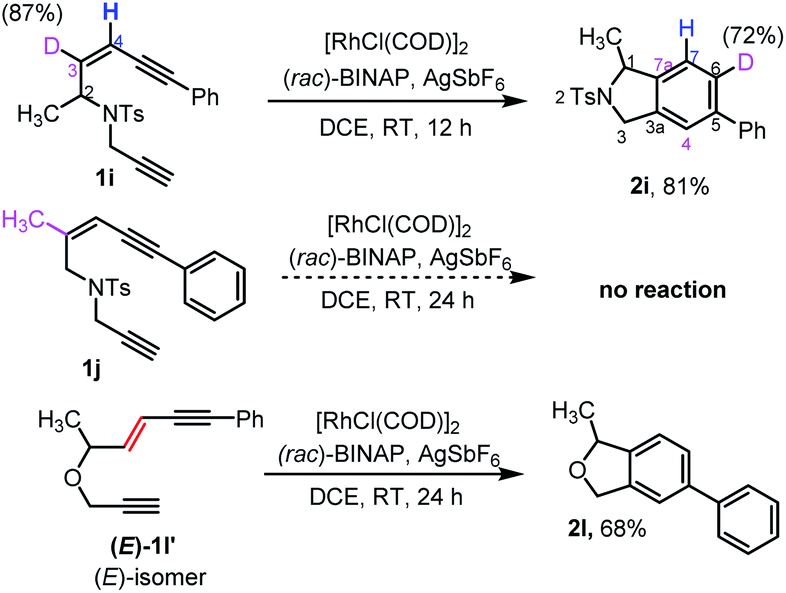 | ||
| Scheme 3 Experiments carried out to probe the reaction mechanism. | ||
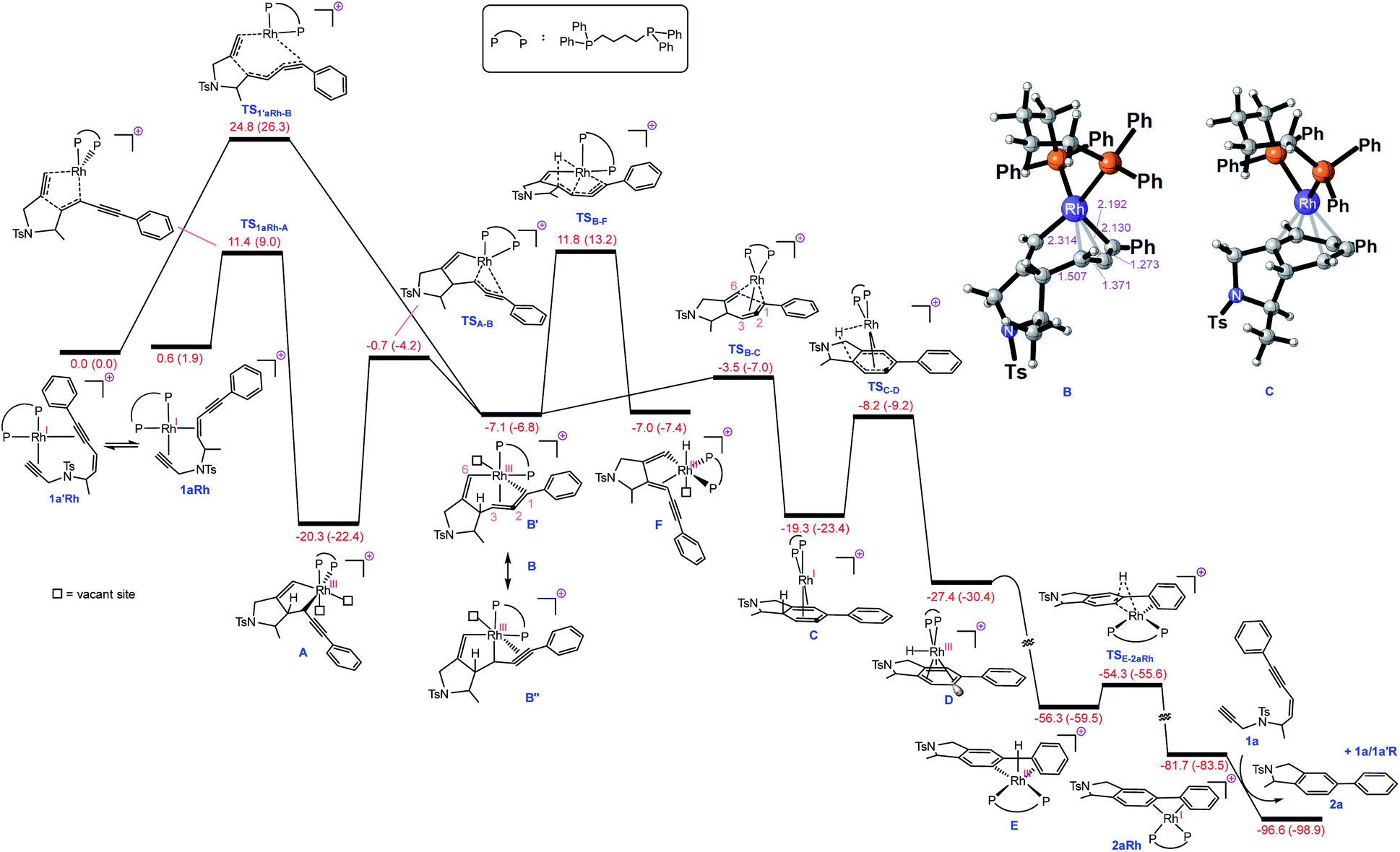 | ||
| Fig. 1 Free energy profile for Rh-catalysed tetradehydro-Diels–Alder reaction of a (Z)-enediyne. Free energies (potential energies) are given in kcal mol−1. | ||
With this set of key experimental results in place, we turned attention to DFT calculations for further mechanistic insight, considering our proposed cyclic allenyl-Rh intermediate B (Fig. 1d).28 In contrast to simple 1,6-enynes, the initial rhodium-catalysed oxidative cyclization has two coordination possibilities – coordination between the enyne double bond and the triple bond of the propargyl sulfonamide (1aRh), or between the two triple bonds of the enediyne (1a′Rh). While oxidative cyclization via1a′Rh would directly afford the proposed allenyl-RhIII intermediate B, the barrier for this process was significantly higher than that for the oxidative cyclization via1aRh to provide RhIII metallacyclopentene A. Following this exergonic process, an endergonic ring-expansion transformation into the key RhIII intermediate B occurs. This unusual intermediate can be represented by two resonance contributors: an allenyl-RhIII intermediate B′ and a propargyl RhIIIB′′ and its hybrid structure (top corner of Fig. 1) is indicative of a bent allenyl rhodium species. The ensuing allenyl-Rh intermediate is a branching point for two different reaction channels: (a) C–H elimination via transition structure TSB-F affording F and (b) C–C reductive elimination via transition structure TSB-C yielding strained RhI-stabilised cyclic allene C.
Surprisingly, the C–C reductive elimination process is computed to be significantly more favourable than the C–H elimination. This is because the allenyl Rh resonance contributor B′ makes a significant contribution to the overall structure B and this means that there is significant σ-bond character between the Rh and C1, thus promoting reductive elimination. The intermediate resulting from the reductive elimination can be considered as a stabilized version of the cyclic allene invoked in the thermal TDDA reactions of enediynes – thereby playing a key role in making the rhodium catalysed variant so facile (Scheme 1a). The catalytic cycle continues with the allene-bound RhI intermediate C undergoing oxidative addition to the C–H bond to form RhIII-hydride D, with the rhodium being bound to one face of the aromatic ring, having the formal anion at C6. This result explains the failure of substrate 1j in the reaction due to the low propensity of C–C bonds to undergo oxidative addition compared with C–H bonds.29 This is also an interesting mechanistic point of difference to the thermal TDDA reaction, wherein formation of the final aromatic ring typically occurs via hydride-migration. In the current case, coordination of Rh to the π-system of intermediate C directs reactivity away from such a hydride migration and towards a C–H insertion.
The hydride complex D undergoes rearrangement to E, which is primed for a highly favourable H–C6 reductive elimination. Once this has taken place, the organic product 2a is formed and the RhI catalyst re-enters the catalytic cycle. This mechanism explains the incorporation of deuterium at C6, with some scrambling likely occurring between rhodium hydrides D/E and solvent/adventitious water. These calculations and the experimental studies enable the catalytic cycle in Fig. 2 to be proposed. Attempts to trap cyclic allene (derived from ene-diyne 1a or 1j) intermediate Cvia Diels–Alder reactions with furan were not successful, which is due to the favorable oxidative addition towards intermediate D – which is a fast intramolecular process – as well as the coordination of Rh to the resulting cyclic allene.
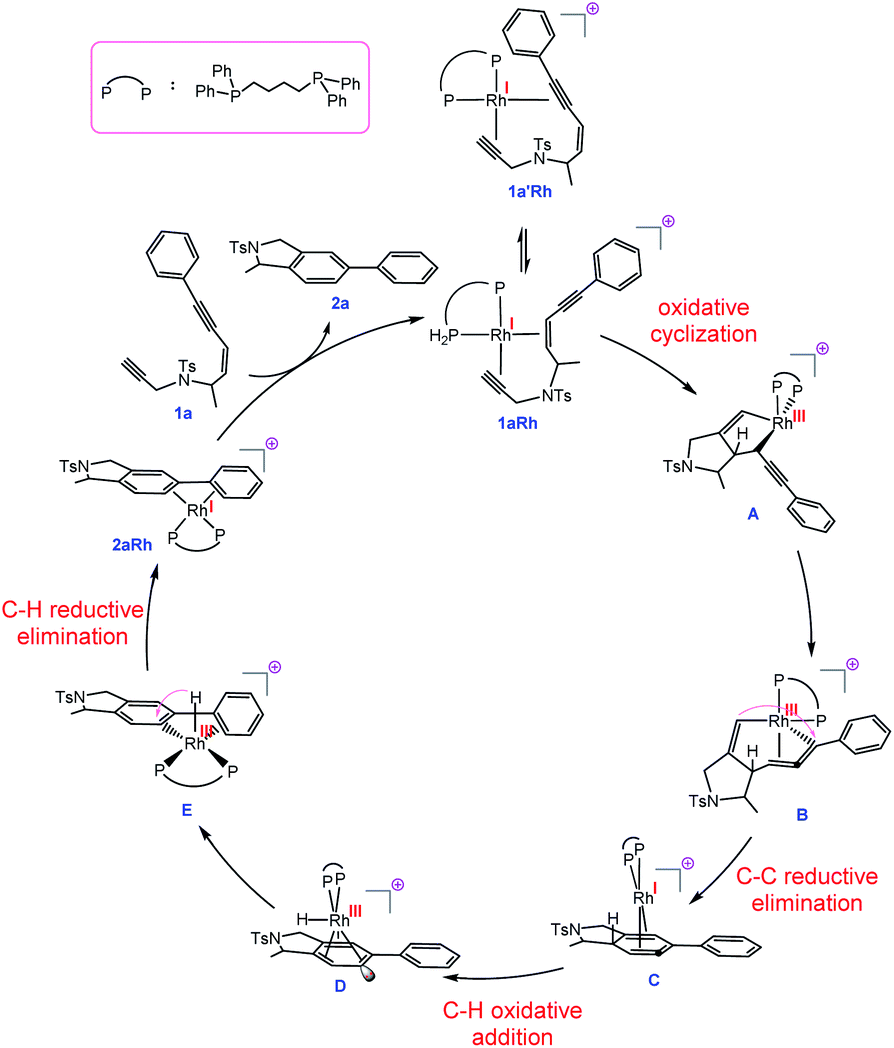 | ||
| Fig. 2 Proposed mechanism for the transformation 1a → 2a catalysed by a Rh complex involving RhI/RhIII/RhI/RhIII cycle and allenic-intermediate C. | ||
Synthetic transformations were performed on the isoindoline and isobenzofuran products to demonstrate their utility (Scheme 4). Initially, 2a and 2k were converted to biologically important isoindolinone scaffolds 3 and isobenzofuranone 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4′ respectively, using sodium chlorite as a mild and green oxidizing reagent.30 Gratifyingly, the N-Ts group could be readily removed from 2a upon sonication in the presence of Mg/MeOH to produce 1-methyl-5-phenylisoindoline 5 in excellent yield – it should be noted, however, that this product is unstable and cannot be purified by chromatography.31
:4′ respectively, using sodium chlorite as a mild and green oxidizing reagent.30 Gratifyingly, the N-Ts group could be readily removed from 2a upon sonication in the presence of Mg/MeOH to produce 1-methyl-5-phenylisoindoline 5 in excellent yield – it should be noted, however, that this product is unstable and cannot be purified by chromatography.31
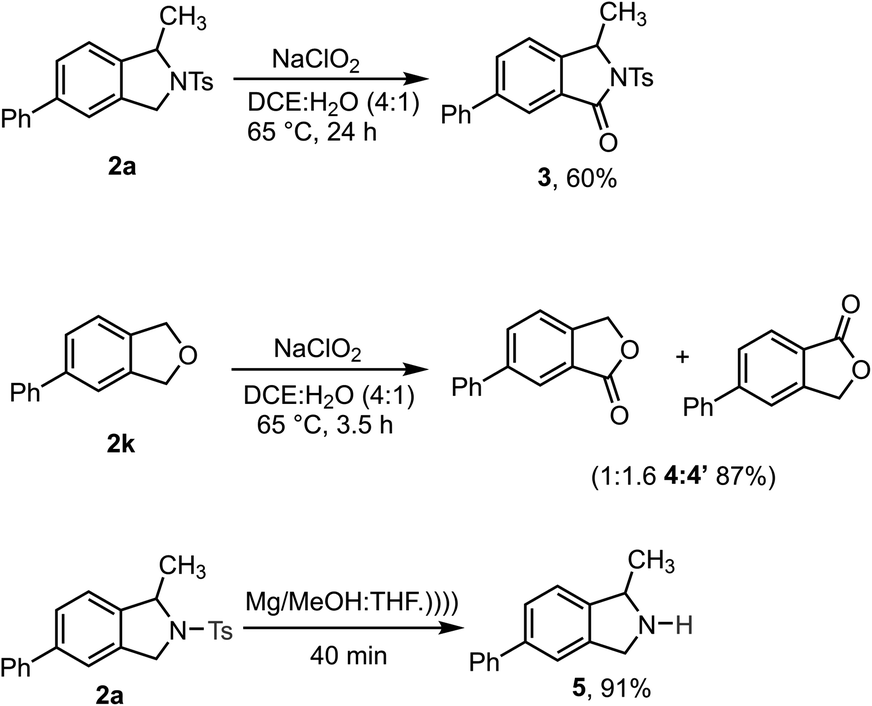 | ||
| Scheme 4 Synthetic modifications of isoindoline and isobenzofuran scaffolds. | ||
Conclusions
In conclusion, a mild and versatile rhodium-catalysed tetradehydro-Diels–Alder reaction of enediynes has been developed allowing the synthesis of substituted, biologically important isoindolines, isobenzofurans and indanes. DFT calculations show that the reaction proceeds through an unprecedented RhI/RhIII/RhI/RhIII catalytic cascade (Fig. 2) that includes two unusual organometallic allene-intermediates. As such, the reaction represents a conceptually new way to harness in situ generated metal-allenyl species through catalytic cycloisomerization reactions at room temperature. It also allows for formation of a low energy rhodium-stabilized cyclic allene intermediate and contributes to this catalytic process being more favourable and wider in scope than the thermal TDDA reaction. This intermediate is of particular interest given that cyclic allenes are emerging as powerful building blocks in modern synthesis and the new conceptual approach described here should lead to further developments in this field.32–34 Further work on generating and harnessing allenic-rhodium and rhodium-stabilized allene species in catalytic processes is currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Australian Research Council (grant number DP180101332) for the generous funding for this research. ST thanks the University of Wollongong for a PhD scholarship. The authors also acknowledge the Australian National Computational Infrastructure and the University of Tasmania for the generous allocation of computing time.Notes and references
- C. Raviola, S. Protti, D. Ravelli and M. Fagnoni, Chem. Soc. Rev., 2016, 45, 4364–4390 RSC.
- W. Li, L. Zhou and J. Zhang, Chem.–Eur. J., 2016, 22, 1558–1571 CrossRef CAS.
- P. Wessig and G. Müller, Chem. Rev., 2008, 108, 2051–2063 CrossRef CAS.
- H.-F. Wang, S.-Y. Wang, T.-Z. Qin and W. Zi, Chem.–Eur. J., 2018, 24, 17911–17914 CrossRef CAS.
- F. Zamani, R. Babaahmadi, B. F. Yates, M. G. Gardiner, A. Ariafard, S. G. Pyne and C. J. T. Hyland, Angew. Chem., Int. Ed., 2019, 58, 2114–2119 CrossRef CAS.
- Q. Zhao, D. F. León Rayo, D. Campeau, M. Daenen and F. Gagosz, Angew. Chem., Int. Ed., 2018, 57, 13603–13607 CrossRef CAS.
- W. Xu, G. Wang, X. Xie and Y. Liu, Org. Lett., 2018, 20, 3273–3277 CrossRef CAS.
- B. M. Trost and A. McClory, Chem.–Asian J., 2008, 3, 164–194 CrossRef CAS.
- K. Ohe, M.-a. Kojima, K. Yonehara and S. Uemura, Angew. Chem., Int. Ed., 1996, 35, 1823–1825 CrossRef CAS.
- A. S. K. Hashmi, P. Haufe and A. Rivas Nass, Adv. Synth. Catal., 2003, 345, 1237–1241 CrossRef CAS.
- K. Mikami, S. Kataoka, K. Wakabayashi and K. Aikawa, Tetrahedron Lett., 2006, 47, 6361–6364 CrossRef CAS.
- P. Cao, B. Wang and X. Zhang, J. Am. Chem. Soc., 2000, 122, 6490–6491 CrossRef CAS.
- P. Cao and X. Zhang, Angew. Chem., Int. Ed., 2000, 39, 4104–4106 CrossRef CAS.
- X. Deng, S.-F. Ni, Z.-Y. Han, Y.-Q. Guan, H. Lv, L. Dang and X.-M. Zhang, Angew. Chem., Int. Ed., 2016, 55, 6295–6299 CrossRef CAS.
- M. Fujiwara and I. Ojima, Rhodium(I)-Catalyzed Cycloisomerization and Cyclotrimerization Reactions, in Modern Rhodium-Catalyzed Organic Reactions, ed. P. A. Evans, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005 Search PubMed.
- V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS.
- R. Karmakar, P. Pahari and D. Mal, Chem. Rev., 2014, 114, 6213–6284 CrossRef CAS.
- P. J. Kukkola, N. A. Bilci, T. Ikler, P. Savage, S. S. Shetty, D. DelGrande and A. Y. Jeng, Bioorg. Med. Chem. Lett., 2001, 11, 1737–1740 CrossRef CAS.
- C. A. Luckhurst, L. A. Stein, M. Furber, N. Webb, M. J. Ratcliffe, G. Allenby, S. Botterell, W. Tomlinson, B. Martin and A. Walding, Bioorg. Med. Chem. Lett., 2011, 21, 492–496 CrossRef CAS.
- K. Speck and T. Magauer, Beilstein J. Org. Chem., 2013, 9, 2048–2078 CrossRef.
- H.-F. Luo, L.-P. Zhang and C.-Q. Hu, Tetrahedron, 2001, 57, 4849–4854 CrossRef CAS.
- CCDC Number: 1999577.†.
- D. Solé and O. Serrano, J. Org. Chem., 2010, 75, 6267–6270 CrossRef.
- J. L. Jeffrey, E. S. Bartlett and R. Sarpong, Angew. Chem., Int. Ed., 2013, 52, 2194–2197 CrossRef CAS.
- J. Kopecký, J. E. Shields and J. Bornstein, Tetrahedron Lett., 1967, 8, 3669–3674 CrossRef.
- I. Ahmed, G. W. H. Chesseman and B. Jaques, Tetrahedron, 1979, 35, 1145–1149 CrossRef CAS.
- L. J. Kricka and J. M. Vernon, J. Chem. Soc. C, 1971, 2667–2670 RSC.
- DPPB was used in place of BINAP to simplify calculations, noting that it is also able to catalyze the reaction in combination with the Rh-catalyst.
- R. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, 2005 Search PubMed.
- C. Zhang, A.-R. Song and J. Yu, Synthesis, 2012, 44, 2903–2909 CrossRef.
- M. A. Drew, S. Arndt, C. Richardson, M. Rudolph, A. S. K. Hashmi and C. J. T. Hyland, Chem. Commun., 2019, 55, 13971–13974 RSC.
- M. M. Yamano, R. R. Knapp, A. Ngamnithiporn, M. Ramirez, K. N. Houk, B. M. Stoltz and N. K. Garg, Angew. Chem., Int. Ed., 2019, 58, 5653–5657 CrossRef CAS.
- V. A. Lofstrand, K. C. McIntosh, Y. A. Almehmadi and F. G. West, Org. Lett., 2019, 21, 6231–6234 CrossRef CAS.
- J. S. Barber, M. M. Yamano, M. Ramirez, E. R. Darzi, R. R. Knapp, F. Liu, K. N. Houk and N. K. Garg, Nat. Chem., 2018, 10, 953–960 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1999577. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04390g |
| This journal is © The Royal Society of Chemistry 2020 |