
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Significantly improved electrocatalytic oxygen reduction by an asymmetrical Pacman dinuclear cobalt(II) porphyrin–porphyrin dyad†
Yanju
Liu
ab,
Guojun
Zhou
a,
Zongyao
Zhang
b,
Haitao
Lei
a,
Zhen
Yao
 c,
Jianfeng
Li
c,
Jun
Lin
b and
Rui
Cao
*a
c,
Jianfeng
Li
c,
Jun
Lin
b and
Rui
Cao
*a
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, China. E-mail: ruicao@ruc.edu.cn
bDepartment of Chemistry, Renmin University of China, Beijing 100872, China
cCollege of Materials Science and Optoelectronic Technology, University of Chinese Academy of Science, Beijing 101408, China
First published on 4th November 2019
Abstract
Pacman dinuclear CoII triphenylporphyrin-tri(pentafluorophenyl)porphyrin 1 and dinuclear CoII bis-tri(pentafluorophenyl)porphyrin 2, anchored at the two meso-positions of a benzene linker, are synthesized and examined as electrocatalysts for the oxygen reduction reaction (ORR). Both dinuclear Co bisporphyrins are more efficient and selective than corresponding mononuclear CoII tetra(pentafluorophenyl)porphyrin 3 and CoII tetraphenylporphyrin 4 for the four-electron electrocatalytic reduction of O2 to water. Significantly, although the ORR selectivities of the two dinuclear Co bisporphyrins are similar to each other, 1 outperforms 2, in terms of larger catalytic ORR currents and lower overpotentials. Electrochemical studies showed different redox behaviors of the two Co sites of 1: the CoIII/CoII reduction of the Co-TPP (TPP = triphenylporphyrin) site is well-behind that of the Co-TPFP (TPFP = tri(pentafluorophenyl)porphyrin) site by 440 mV. This difference indicated their different roles in the ORR: CoII-TPFP is likely the O2 binding and reduction site, while CoIII-TPP, which is generated by the oxidation of CoII-TPP on electrodes, may function as a Lewis acid to assist the O2 binding and activation. The positively charged CoIII-TPP will have through-space charge interactions with the negatively charged O2-adduct unit, which will reduce the activation energy barrier for the ORR. This effect of Co-TPP closely resembles that of the CuB site of metalloenzyme cytochrome c oxidase (CcO), which catalyzes the biological reduction of O2. This work represents a rare example of asymmetrical dinuclear metal catalysts, which can catalyze the 4e reduction of O2 with high selectivity and significantly improved activity.
Introduction
Efficient and selective catalysts for the oxygen reduction reaction (ORR) are required in many kinds of energy conversion and storage devices, such as fuel cells and metal–air batteries, because the kinetically slow ORR is the key cathode reaction in these systems.1–7 Research on molecular catalysts is significant to provide fundamental knowledge on reaction mechanisms and structure–function relationships,8–11 which is essential for the development of new catalysts with improved performance. Among these studies, synthetic modeling of cytochrome c oxidase (CcO) has attracted particular attention.12–16 CcO belongs to a superfamily of heme/Cu oxidases and catalyzes the selective biological reduction of O2 to water at the dinuclear heme a3 and CuB site, which is the destination of electrons from the electron transport chain in respiration.17 The dinuclear heme/Cu site, in cooperation with the surrounding structures, including the tyrosine residue in the vicinity, the distal pocket, and also the trans ligand of the heme a3 Fe ion, is suggested to play crucial roles in minimizing the release of partially reduced oxygen species.4,18 Significantly, the CuI state of the CuB ion can provide an electron to reduce O2, and moreover, its CuII state can function as a Lewis acid to facilitate the O2 binding and activation at the heme site (Scheme 1).16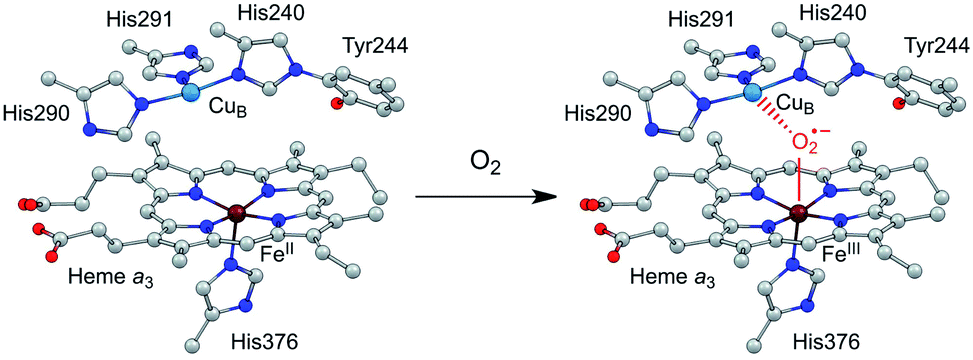 | ||
| Scheme 1 Illustration of the O2 binding and activation at the dinuclear heme a3 and CuB site of CcO. | ||
As inspired by nature, metal porphyrins have been extensively studied as ORR catalysts.4,19–33 In addition to biologically relevant Fe porphyrins, Co porphyrins have also received great attention because of their high activity and stability.19–26,32–34 However, mononuclear Co porphyrins are much less selective than Fe analogues to catalyze the 4e reduction of O2 to water. Unlike Fe complexes, Co complexes in general catalyze the 2e reduction of O2 because the peroxo intermediates of Co are challenging to undergo heterolytic O–O bond cleavage to generate terminal Co-oxo species that have large d electron counts and thus are high in energy.35–37 Several molecular design strategies have been reported to improve the selectivity for the 4e ORR, including (1) formation of intramolecular hydrogen bonding interactions to stabilize O2 adducts,27,30,38,39 (2) appending acid groups to assist proton delivery,19,28,29,40 and (3) introduction of functional groups to ensure rapid electron transfer between catalyst molecules and electrodes.34,41–44 Additionally, for complexes of late-transition metals, such as Co, it is found that dinuclear complexes are typically more efficient than mononuclear analogues to catalyze the 4e reduction of O2.20–26,45–53 This improvement is likely caused by the formation of peroxo-bridged dinuclear intermediates,45,47 whose subsequent reduction can result in the 4e ORR. Despite the improvement in the selectivity, however, from mononuclear to dinuclear complexes, the enhancement of catalytic activity, in terms of both catalytic currents and overpotentials, is unsatisfactory. In general, asymmetrical dinuclear complexes are rarely reported,48,50,51 while symmetrical dinuclear complexes are usually designed and studied with the two metal ions bearing very similar coordination environments. This will lead to parallel redox behaviors of the two metal ions. In other words, the two metal ions will be reduced at close potentials for O2 binding and activation. This will weaken the Lewis acid role of the second metal ion. Therefore, few dinuclear metal complexes can catalyze the ORR with both high activity and selectivity.
As mentioned above, in nature, the Lewis acidic and redox active features of the CuB site are essential for CcO to catalyze the ORR with both high activity and selectivity (Scheme 1). The presence of a positively charged CuII ion can decrease the activation energy barrier for the formation of the negatively charged O2-adduct unit at the heme site.4,16,17 Similar through-space charge interaction effects have been demonstrated for synthetic model complexes to catalyze artificial small molecule activation reactions,54,55 including O2 reduction and CO2 reduction. In order to develop ORR catalysts with satisfactory activity and selectivity, we herein report the synthesis and catalytic features of asymmetrical Pacman dinuclear CoII triphenylporphyrin-tri(pentafluorophenyl)porphyrin 1. Our results show that 1 is significantly more efficient and selective than corresponding mononuclear CoII tetra(pentafluorophenyl)porphyrin 3 and CoII tetraphenylporphyrin 4 to catalyze the 4e reduction of O2 to water, and more notably, it outperforms symmetrical dinuclear CoII bis-tri(pentafluorophenyl)porphyrin 2, in terms of both larger catalytic ORR currents and lower overpotentials. Electrochemical studies indicated that in 1, CoII-TPFP is likely the O2 binding and reduction site, while CoIII-TPP may function as a positively charged Lewis acid to assist the O2 binding and activation. This work represents a rare example of asymmetrical dinuclear metal catalysts with significantly improved performance for electrocatalytic 4e reduction of O2.
Results
Synthesis and characterization
We choose a benzene unit to link two porphyrin moieties at its two meso-positions (Fig. 1a). The resulting Pacman structure will enable the two porphyrin moieties to have a face-to-face configuration, which is considered to be suitable for the binding and activation of an O2 molecule.20,21 The synthetic routes to bisporphyrins and corresponding metal complexes are depicted in Fig. 2, and the details of synthetic conditions and characterizations are described in the ESI.† As shown in Fig. 2, methyl 2-formylbenzoate was used for the synthesis of intermediate a to introduce the first tri(pentafluorophenyl)porphyrin macrocycle (Fig. S2 and S3†). In order to reduce methyl benzoate to benzyl alcohol, we first treated a with lithium aluminium hydride (LiAlH4, route 1). However, in addition to the reduction of methyl benzoate, we found that the fluorine atom at the para-position of the three pentafluorophenyl units is also replaced by a hydrogen atom, leading to the formation of intermediate b. This undesired reduction was confirmed by 1H NMR spectroscopy and high-resolution mass spectrometry (HRMS). In 1H NMR, these three hydrogen atoms can be found with chemical shifts δ = 7.61 (Fig. S4†). The HRMS of b shows an ion with a mass-to-charge ratio of 861.1532 (Fig. S5†). This value matches the calculated value of 861.1518 for the monocation of [b + H+]+ with identical isotopic distribution. Subsequent oxidation of benzyl alcohol to benzaldehyde (Fig. S6 and S7†), and the next conversion to the second tri(pentafluorophenyl)porphyrin macrocycle resulted in bisporphyrin ligand 5′ (Fig. S8 and S9†). Attempts to obtain high quality single crystals of the dinuclear Co complex of 5′ were unsuccessful. However, we were able to get the single crystal X-ray structure of its dinuclear Ni analogue (see below), which clearly shows the replacement of three para-F atoms by H atoms.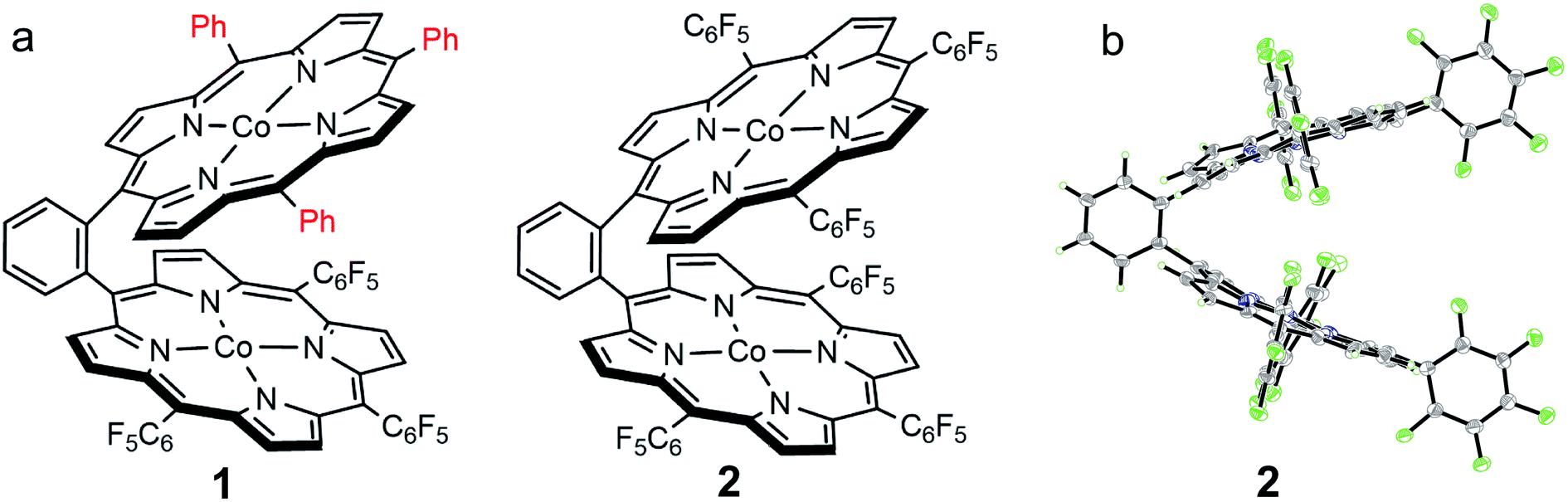 | ||
| Fig. 1 (a) Molecular structures of 1 and 2. (b) Thermal ellipsoid plot of the X-ray structure of 2 (30% probability). | ||
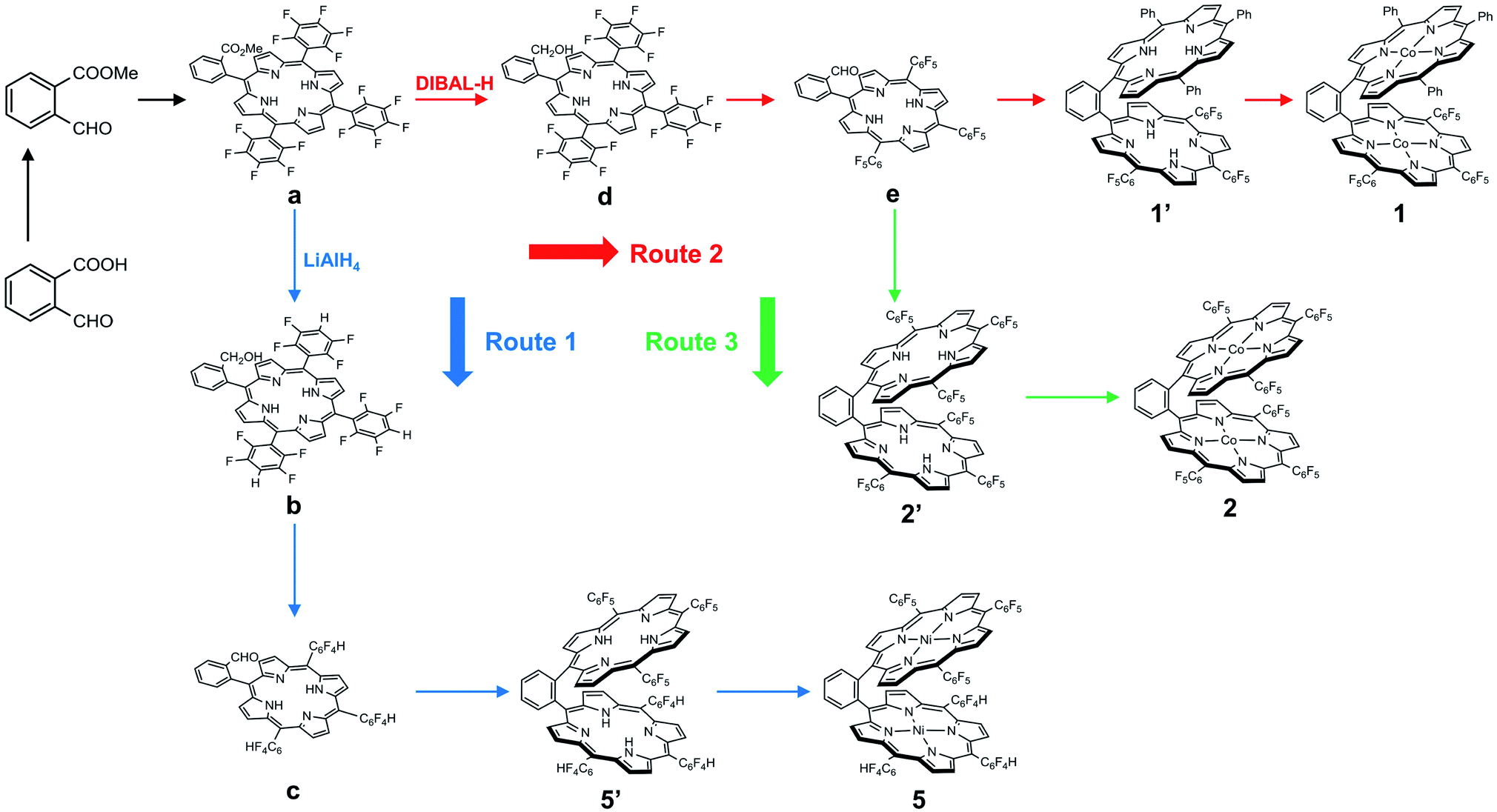 | ||
| Fig. 2 Synthetic routes to dinuclear metal complexes of 1, 2, and 5. | ||
In order to prevent this undesired replacement, we chose diisobutylaluminum hydride (DIBAL-H) to reduce intermediate a. With the use of this relatively weak hydride-donating reagent, we were able to selectively convert methyl benzoate to benzyl alcohol, giving intermediate d. Subsequent oxidation of benzyl alcohol to benzaldehyde leads to the formation of intermediate e. Both d and e are fully characterized by 1H NMR (Fig. S11 and S13,† respectively) and HRMS (Fig. S12 and S14,† respectively), confirming the preservation of the three pentafluorophenyl units during the treatment with DIBAL-H. Starting from e, we can synthesize the asymmetrical bisporphyrin ligand 1′ (Fig. S15,† route 2) and symmetrical analogue 2′ (Fig. S18,† route 3). The identity and purity of both 1′ and 2′ are confirmed by using 1H NMR spectroscopy and HRMS. In HRMS, 1′ displays an ion with a mass-to-charge ratio of 1421.3137 (Fig. S16†), matching the calculated value of 1421.3131 for the monocation of [1′ + H+]+; 2′ displays an ion with a mass-to-charge ratio of 1691.1699 (Fig. S19†), matching the calculated value of 1691.1718 for the monocation of [2′ + H+]+.
Reaction of 1′ with cobalt chloride CoCl2 gave dinuclear Co bisporphyrin 1 with a yield of 87%. The UV-vis spectrum of 1 in dimethylformamide shows a characteristic Soret band at 412 nm and a Q band at 560 nm (Fig. S21†). These bands are in accordance with those from corresponding mononuclear Co porphyrins 3 and 4. In HRMS, 1 shows an ion with a mass-to-charge ratio of 1535.1461 (Fig. S17†). This value matches the calculated value of 1535.1437 for the monocation of [1 + H+]+ with identical isotopic distribution. Reaction of 2′ with CoCl2 resulted in 2 with a yield of 85%. The UV-vis spectrum of 2 is similar to that of 1, showing a Soret band at 401 nm and a Q band at 545 nm (Fig. S21†). The HRMS of 2 shows an ion with a mass-to-charge ratio of 1805.0038 (Fig. S20†), matching the calculated value of 1805.0023 for the monocation of [2 + H+]+. All these results are in agreement with the expected structures for 1 and 2. Notably, we were able to characterize the structure of 2 by using the single crystal X-ray diffraction method (see below), which confirmed the dinuclear Co bisporphyrin structure.
Crystallographic studies
Although we were not able to structurally characterize complex 1, its key structural information could be obtained from crystallographic studies of its analogues 2 and 5. Single crystals of 2 that are suitable for X-ray structure analysis were obtained by slow vapor diffusion of hexane to a toluene solution of 2 at room temperature for two weeks. Complex 2 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with Z = 2. Crystal data and structure refinement details can be found in Table S1.† In the X-ray structure of 2, two symmetrical TPFP moieties are connected together at the two meso-positions of a benzene linker, providing a Pacman configuration (Fig. 1b). In each TPFP moiety, the three pentafluorophenyl substituents are well resolved. Particularly, the three para-F atoms can be well defined. This result further confirms the preservation of the three pentafluorophenyl units during synthesis. Each TPFP ligand coordinates a Co ion at the center of the porphyrin macrocycle through four N atoms. The Co–N bond distances are in the range of 1.908(2) Å to 2.007(2) Å. There are no axial ligands on the Co ions, leading to a distorted square-planar coordination geometry.
with Z = 2. Crystal data and structure refinement details can be found in Table S1.† In the X-ray structure of 2, two symmetrical TPFP moieties are connected together at the two meso-positions of a benzene linker, providing a Pacman configuration (Fig. 1b). In each TPFP moiety, the three pentafluorophenyl substituents are well resolved. Particularly, the three para-F atoms can be well defined. This result further confirms the preservation of the three pentafluorophenyl units during synthesis. Each TPFP ligand coordinates a Co ion at the center of the porphyrin macrocycle through four N atoms. The Co–N bond distances are in the range of 1.908(2) Å to 2.007(2) Å. There are no axial ligands on the Co ions, leading to a distorted square-planar coordination geometry.
The bond valence sum (BVS) calculation suggested the d7 CoII oxidation state. This result is consistent with the charge balance calculation because the porphyrin ligands are doubly negatively charged and no more counter anions are found in the X-ray structure of 2. In addition, we measured the electron paramagnetic resonance (EPR) spectrum of 1 and 2 in toluene (Fig. S22† and 3a, respectively). The axial spectra confirmed the S = 1/2 low spin states of the four-coordinated CoII porphyrin complexes with g = 2.26. The parallel components display a septet superhyperfine splitting pattern, which is due to the interaction of CoII with N atoms. The Co⋯Co distance is 5.762 Å. Interestingly, in the Pacman cleft of 2, there is a toluene solvent molecule, which is located at and is parallel to the angle bisector of the two porphyrin macrocycles.
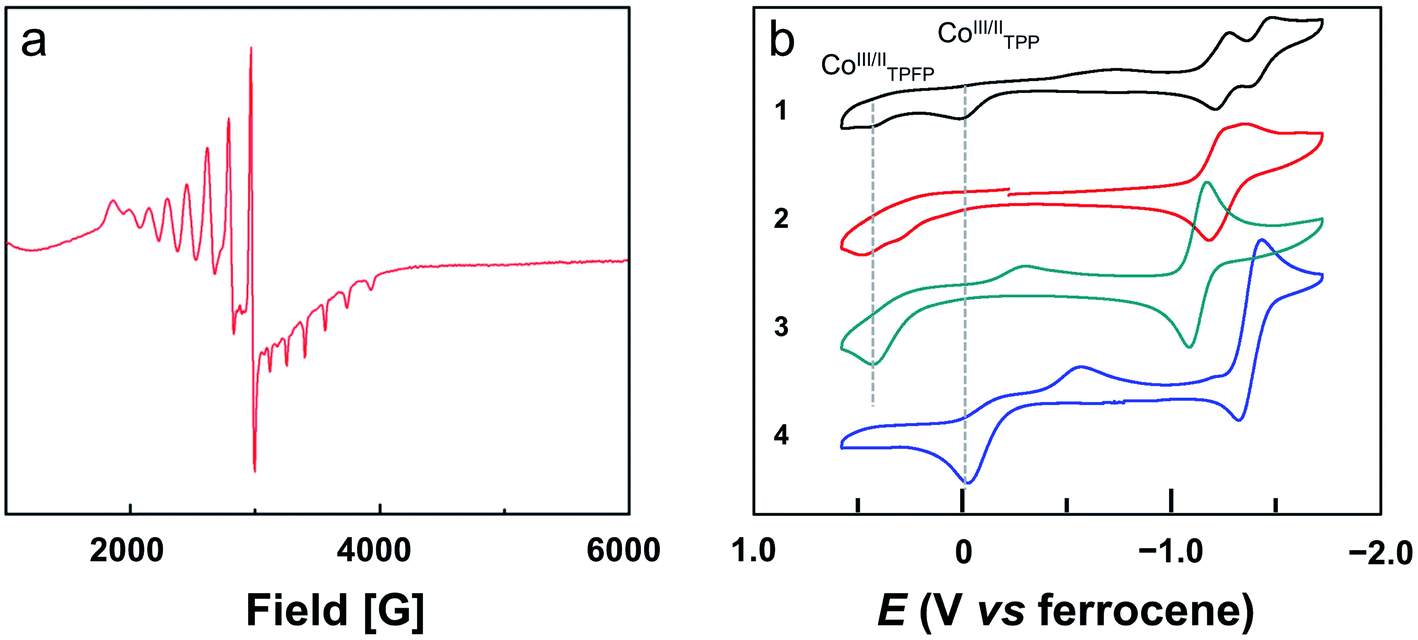 | ||
| Fig. 3 (a) EPR spectrum of 2 in toluene at 90 K. (b) CVs of 0.5 mM benzonitrile solutions of 1 and 2, and 1.0 mM benzonitrile solutions of 3 and 4. Conditions: GC working electrode, 0.1 M (Bu4N)PF6 benzonitrile solution, scan rate 50 mV s−1. | ||
Moreover, we obtained high-quality single crystals of dinuclear Ni bisporphyrin 5 by slow vapor diffusion of pentane to a chloroform solution of 5 at room temperature for two weeks. Complex 5 crystallized in the triclinic space group P with Z = 2 (see Table S1† for crystal data and structure refinement details). The structure of 5 closely resembles that of 2 (Fig. S23†), except that, in one TPFP moiety, the para-F atom of the three pentafluorophenyl substituents is each replaced by a para-H atom. This result is consistent with the findings in synthesis as mentioned above. Each Ni ion is located at the center of a porphyrin ligand with Ni–N bond distances ranging from 1.931(4) Å to 1.947(4) Å. The distorted square-planar coordination geometry of Ni and the BVS calculation all suggest the d8 NiII electronic configuration. The Ni⋯Ni distance is 6.182 Å. In the Pacman cleft of 5, there is a pentane solvent molecule, which is located at the angle bisector of the two porphyrin macrocycles.
Electrochemical and electrocatalytic ORR studies
We next measured the cyclic voltammograms (CVs) of dinuclear and mononuclear Co complexes in benzonitrile using a glassy carbon (GC) working electrode. The CV of mononuclear Co porphyrin 3 shows a CoII-to-CoIII oxidation wave at Ep,a = 0.43 V versus ferrocene (all potentials reported in benzonitrile in this work are referenced to ferrocene) and its corresponding reduction wave at Ep,c = −0.29 V (Fig. 3b). This irreversibility of the CoIII/CoII redox couple is indicative of the binding and dissociation of solvent benzonitrile on the Co ion during the CoIII/CoII conversion.56 Additionally, there is a reversible 1e reduction wave at E1/2 = −1.13 V, which is assigned to the CoII/CoI redox couple. For mononuclear complex 4, the CoII-to-CoIII oxidation wave is at −0.03 V, and its two corresponding reduction waves are at −0.15 and −0.56 V. The reversible CoII/CoI redox couple is found at E1/2 = −1.38 V. As compared to 3, the CoII-to-CoIII oxidation wave of 4 is shifted to the cathodic direction by 460 mV. This large cathodic shift is caused by the replacement of four strong electron-withdrawing pentafluorophenyl units by four phenyl units.56Dinuclear Co complex 1 displays two CoII-to-CoIII oxidation waves at Ep,a = 0.43 and −0.03 V using the GC working electrode in benzonitrile. These values are identical to those represented by mononuclear complexes 3 and 4, respectively. On the basis of this comparison, we can assign the oxidation wave at Ep,a = 0.43 V to the CoII-to-CoIII process of Co-TPFP, and assign the oxidation wave at Ep,a = −0.03 V to the CoII-to-CoIII process of Co-TPP. There are two reversible 1e reduction waves at E1/2 = −1.25 and −1.43 V, which can be assigned to the CoII/CoI redox couple of Co-TPFP and Co-TPP, respectively. For dinuclear Co complex 2, there are two close CoII-to-CoIII oxidation waves at Ep,a = 0.45 and 0.30 V. The two reversible 1e reduction waves of 2 are overlapped with E1/2 = −1.25 V. These results suggest that the electrochemical behaviors of the two Co porphyrin moieties in dinuclear complexes 1 and 2 are mainly determined by the electronic structures of the porphyrin macrocycles and that the interaction between the two porphyrin moieties is weak, which is consistent with the large Co⋯Co distance as revealed by crystallographic studies.
The electrocatalytic ORR features of complexes 1–4 loaded on carbon nanotubes were examined in 0.50 M H2SO4 aqueous solutions at room temperature. Their CVs under N2 and O2 clearly show that all four complexes are active for the ORR (Fig. 4 and S24†). Significantly, the CV of 1 displayed a large catalytic wave with the half-wave potential E1/2 = 0.72 V versus the reversible hydrogen electrode (RHE, all potentials reported in aqueous solutions in this work are referenced to RHE) (Fig. 4a). This value suggests that 1 represents the state-of-the-art electrocatalytic ORR performance among reported molecular catalysts operating under similar acidic conditions (see Table 1 for details). For complex 2, the ORR half-wave potential E1/2 is 0.55 V (Fig. 4b), while for 3 and 4, these values are E1/2 = 0.46 V and 0.41 V, respectively. Two conclusions can be made from these results. First, mononuclear complex 3 is superior to 4 for ORR catalysis, which is a result of the strong electron-withdrawing pentafluorophenyl substituents in 3. Second, dinuclear Co catalysts are more efficient than mononuclear analogues, and 1 outperforms all other Co catalysts examined in this work. Considering that complex 2 contains more pentafluorophenyl substituents than 1, the much higher efficiency of 1 is noteworthy.
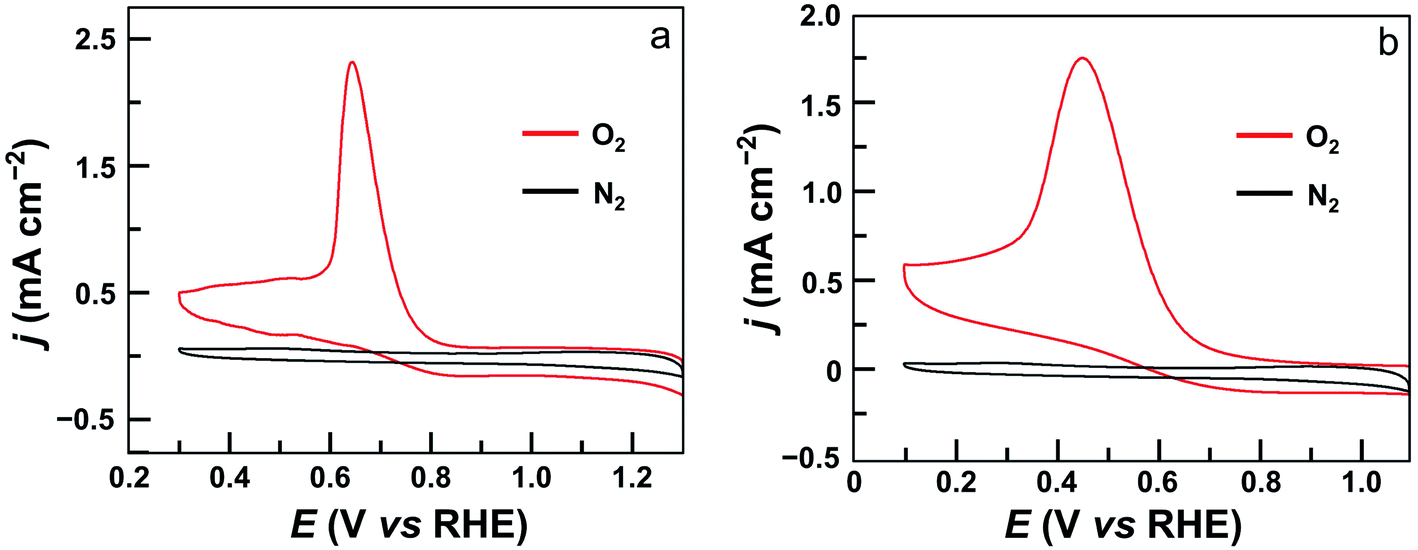 | ||
| Fig. 4 CVs of GC electrodes loaded with 5 μL ink of 1 (a) and 2 (b) under N2 (black) and O2 (red) in 0.5 M H2SO4 solution. Conditions: GC working electrode, 50 mV s−1 scan rate, 20 °C. | ||
| Catalyst category | Catalyst | Solution | η at E1/2a (V) | n | Ref. |
|---|---|---|---|---|---|
| a Half-wave overpotential. Although the overpotentials for the 2e and 4e ORR are different, for simplicity, we treated all entries as the 4e ORR for overpotential calculation. | |||||
| Dinuclear Co bisporphyrin dyads | 1 | 0.5 M H2SO4 | 0.51 | 3.9 | This work |
| 2 | 0.70 | 3.8 | |||
| Co2(DPX) | 0.5 M HClO4 and 1.5 M CF3COOH | 0.65 | 3.7 | 21 and 22 | |
| Co2(DPD) | 0.66 | 3.8 | |||
| Co2(DPXM) | 0.5 M HClO4 and 1.5 M CF3COOH | 0.75 | 3.6 | 20 | |
| Co2(DPDM) | 0.74 | 3.5 | |||
| Ox-Co | 0.5 M H2SO4 | 0.84 | 3.5 | 25 | |
| Oxa-Co | 0.70 | 3.9 | |||
| Benzo-Co | 0.87 | 3.2 | |||
| Co2PP0 | pH 6.8 phosphate buffer | 0.93 | 2.3 | 57 | |
| Co2PP2 | 0.94 | 2.2 | |||
| Co2PP5 | 0.77 | 2.0 | |||
| Co2PP8 | 0.81 | 1.9 | |||
| Dinuclear Co biscorrole dyads | (BCA)Co2 | 1 M HClO4 | 0.60 | 3.4 | 48 |
| (BCB)Co2 | 0.61 | 2.4 | |||
| (BCX)Co2 | 0.61 | 2.9 | |||
| (BCO)Co2 | 0.64 | 3.4 | |||
| (BCS)Co2 | 0.64 | 3.1 | |||
| Dinuclear Co porphyrin-corrole dyads | (PCA)Co2 | 1 M HClO4 | 0.52 | 3.9 | 48 |
| (PCB)Co2 | 0.53 | 3.7 | |||
| (PCX)Co2 | 0.54 | 3.7 | |||
| (PCO)Co2 | 0.58 | 3.5 | |||
| (PCOX)Co2 | 0.59 | 3.1 | 50 | ||
| (PMes2CX)Co2 | 0.67 | 2.5 | |||
| (PMes2CO)Co2 | 0.67 | 2.4 | |||
| (PMes2COX)Co2 | 0.66 | 2.5 | |||
| Heterobimetallic Fe/Mn porphyrin-Co corrole dyads | (PCA)FeClCoCl | 1 M HClO4 | 0.60 | 2.8 | 51 |
| (PCO)FeClCoCl | 0.64 | 2.8 | |||
| (PCX)FeClCoCl | 0.66 | 2.6 | |||
| (PCB)FeClCoCl | 0.69 | 2.6 | |||
| (PCX)MnClCoCl | 0.66 | 2.6 | |||
| (PCO)MnClCoCl | 0.63 | 2.5 | |||
| (PCB)MnClCoCl | 0.67 | 2.8 | |||
| Mononuclear Co porphyrin | 3 | 0.5 M H2SO4 | 0.73 | 2.9 | This work |
| 4 | 0.83 | 2.9 | |||
The ORR selectivity was then evaluated by determining the number of electrons n transferred per O2 molecule using rotating disk electrode (RDE) and rotating ring-disk electrode (RRDE) measurements. In the RRDE, the partially reduced oxygen species (i.e., H2O2) that is possibly produced during the ORR at the GC disk electrode can be detected at the Pt ring electrode (Fig. 5a and S25†). According to the current densities detected at the disk electrode and the ring electrode, the n value can be determined to be 3.90 for 1, 3.80 for 2, 2.90 for 3, and 2.90 for 4 (Fig. 5b). This result shows that in addition to much improved activity, dinuclear Co complexes 1 and 2 are also more selective than mononuclear analogues 3 and 4 to catalyze the 4e ORR. Moreover, the n value can be evaluated using the Koutecky–Levich (K–L) method in RDE measurements (Fig. 5c, S26 and S27†), giving n = 3.70 for 1, 3.60 for 2, 2.60 for 3, and 2.60 for 4. These values are in good agreement with those calculated from RRDE experiments. Under applied potentials of 0.10–0.40 V with catalyst 1, the linear plots of j−1 (j = current density) on ω−1/2 (ω = the rotation rate) and the near-parallelism of these fitting lines suggested a first-order reaction dependence on dissolved O2 concentrations and similar ORR n values under different potentials (Fig. 5d).
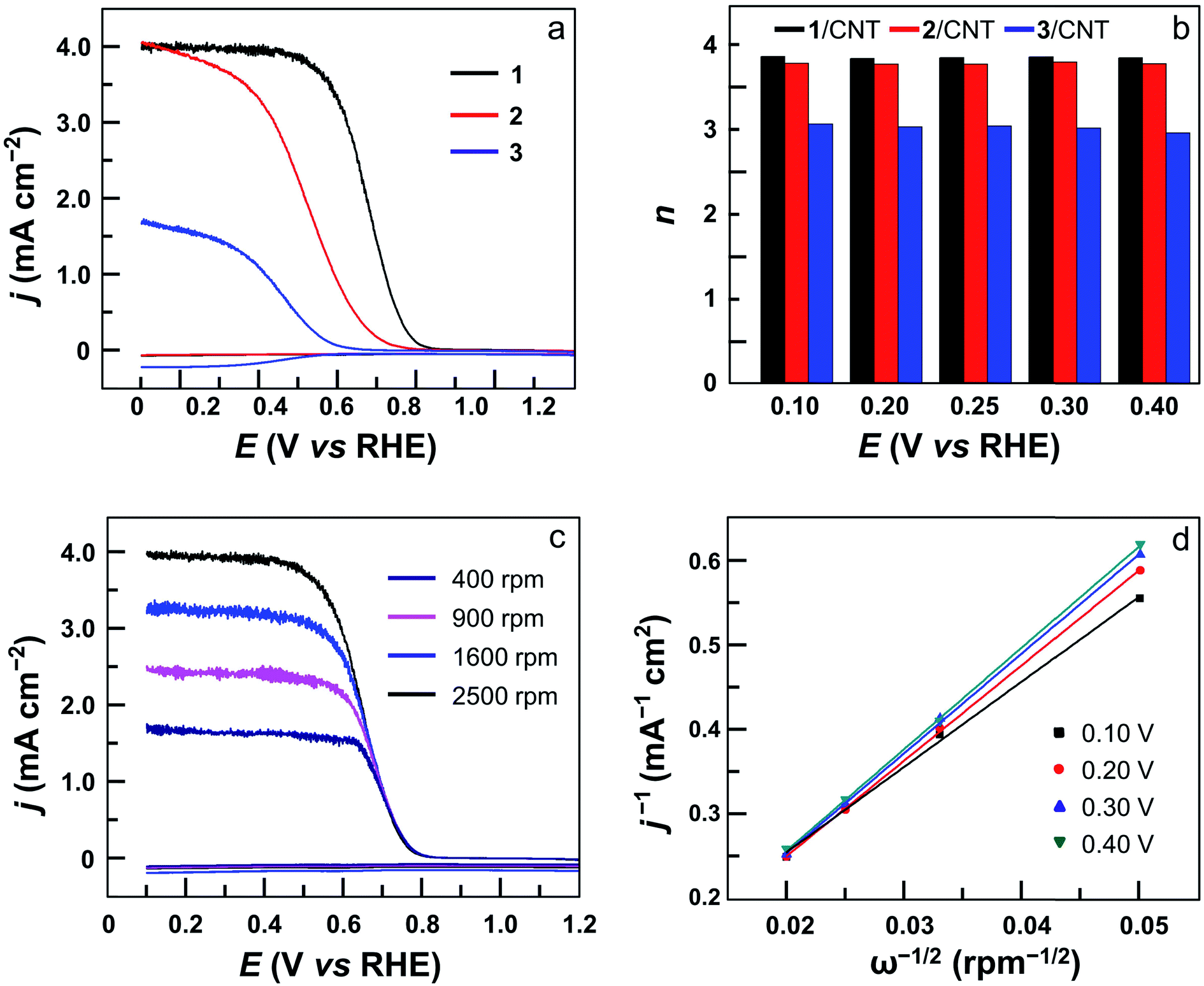 | ||
| Fig. 5 (a) RRDE measurements for the ORR at the GC disk electrode coated with 1 (black), 2 (red), and 3 (blue) in an O2-saturated 0.5 M H2SO4 solution at 2500 rpm. The ring electrode was polarized at 1.0 V. (b) n value of the ORR with 1 (black), 2 (red), and 3 (blue). (c) RRDE measurements for the ORR with 1 in O2-saturated 0.5 M H2SO4 solution at various rotation rates. (d) K–L plots for the ORR with 1. Conditions: GC disk electrode (area 0.125 cm2), Pt ring electrode (area 0.188 cm2), 10 mV s−1 scan rate, 20 °C. | ||
The improved selectivity of dinuclear Co complexes for the 4e ORR shows that the Pacman structure constructed at the two meso-positions of a benzene linker enables efficient cooperation between the two Co ions. For late-transition metals, such as Co, the electrostatic repulsion between their d electrons and the p electrons of the oxo ligand will make terminal late-transition metal-oxo species unfavorable from an energy point of view. As a consequence, mononuclear Co complexes in general catalyze the 2e ORR, because the key peroxo intermediates are challenging to undergo heterolytic O–O bond cleavage to generate high-energy terminal Co-oxo species. Nevertheless, the bimetallic mechanism with the formation of peroxo-bridged dinuclear Co complexes is likely to happen. Subsequent homolytic O–O bond cleavage can lead to the improved 4e ORR.
Discussion
Despite that dinuclear Co complexes 1 and 2 are both highly selective for the 4e reduction of O2, the superior activity of 1 is noteworthy. Strong electron-withdrawing groups, such as the meso-pentafluorophenyl substituents, can cause large shifts of the redox couples to the anodic direction and thus are usually favored for ORR catalysis,58,59 because this effect will lead to the formation of catalytically active species at relatively smaller overpotentials. Complex 2 has two Co-TPFP units, while complex 1 has one Co-TPFP unit and one Co-TPP unit. The CoIII/CoII reduction of the Co-TPP site of 1 is well behind that of the Co-TPFP site by 440 mV, which is in agreement with the replacement of three pentafluorophenyl groups by three phenyl groups. Although the two Co sites in 2 are more easily reduced to the catalytically active CoII state, the half-wave overpotential of 2 is larger than that of 1 by 165 mV. Additionally, to reach the plateau currents, the potential required by 2 is larger than that of 1 by >300 mV. These results are in sharp contrast to the fact that 2 has more electron withdrawing groups.On the basis of these results, we propose that, upon reduction to the CoII state, the Co-TPFP site of 1 becomes catalytically active for binding and activating the O2 molecule,4,48,50 while at this stage, the Co-TPP site of 1 still maintains the CoIII oxidation state (Fig. 6). The reaction of CoII-TPFP and O2 leads to the formation of a formally CoIII–O2˙− species.60,61 This negatively charged O2-adduct unit is likely to interact with the positively charged CoIII-TPP unit. Such through-space charge interaction effects have been demonstrated to assist the formation and stabilization of charged intermediates.54,55 As a consequence, the CoIII-TPP unit is suggested to be able to decrease the activation energy barrier for the binding and activation of O2 in the Pacman cleft of 1. However, for complex 2, its two Co-TPFP sites are reduced to the CoII state at close potentials. Subsequent reaction with an O2 molecule will generate the peroxo-bridged dinuclear CoIII intermediate. The different redox behaviors of 1 and 2 indicated that the CoIII-TPP unit is likely responsible for the stabilization of the O2 adduct and thus for the improved ORR performance of 1.
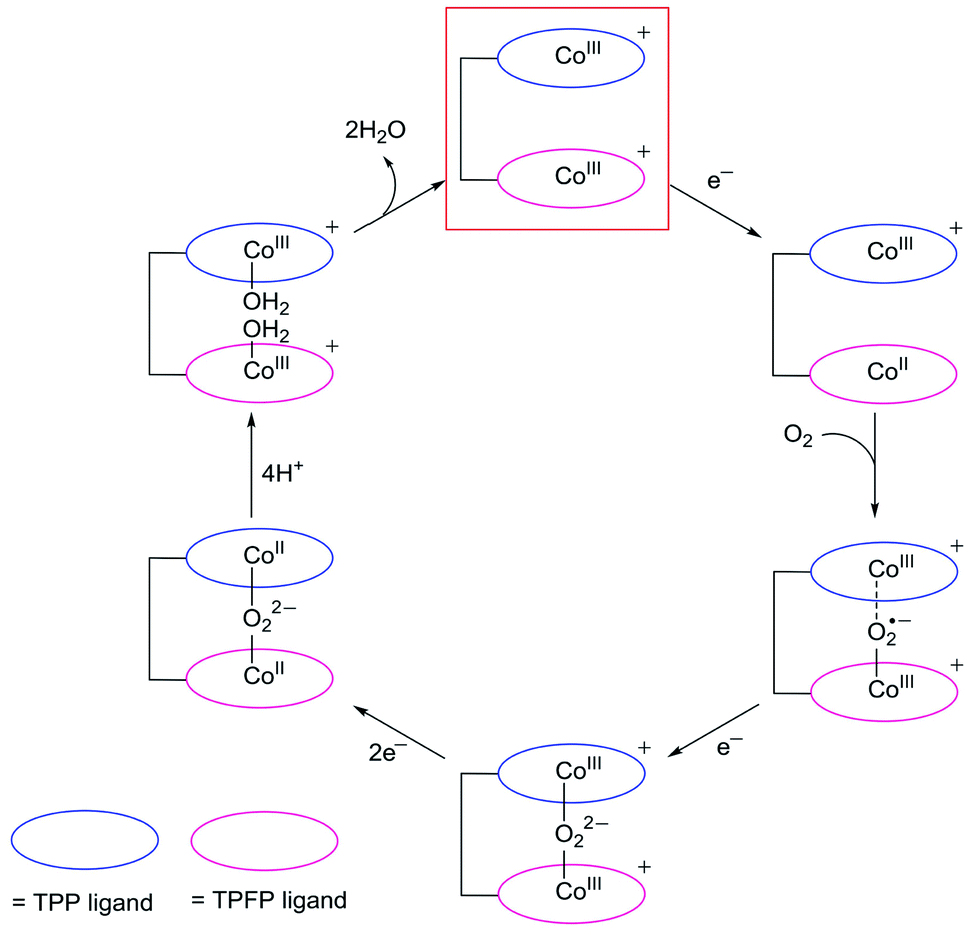 | ||
| Fig. 6 Proposed catalytic cycle for the ORR with 1. Complex 1 will be instantly oxidized to the CoIIICoIII oxidation state on the electrode upon applying large positive potentials, and this 2e-oxidized species (highlighted with a red box) is the real starting catalyst. | ||
In addition to assisting the formation of formally CoIII–O2˙− species, the positively charged CoIII-TPP unit in the vicinity will also benefit the subsequent 1e reduction of the superoxo unit to generate peroxo, leading to the formation of CoIII–O22−–CoIII species. Note that there are two possible pathways for this 1e reduction: the superoxo may be reduced directly, or the CoIII-TPP is first reduced to its CoII oxidation state and then transfers this electron to the superoxo. After the formation of peroxo, for both 1 and 2, the peroxo-bridged dinuclear CoIII intermediates will be further reduced by two electrons to break the O–O bonds.
Comparing complex 1 with reported dinuclear Co bisporphyrins shows the state-of-the-art ORR performance of 1 (Table 1). The n value catalyzed with 1 is 3.90. This value is larger than the n values obtained with previous dinuclear Co bisporphyrins. In addition to the increased selectivity, the ORR half-wave overpotential of 510 mV with 1 is also smaller than the half-wave overpotentials obtained with previous dinuclear Co bisporphyrins. These results clearly demonstrate that 1 is superior to other dinuclear Co bisporphyrins in catalyzing the 4e ORR with higher activity and selectivity.
Comparison of 1 with other dinuclear catalysts, including dinuclear Co biscorrole dyads, dinuclear Co porphyrin-corrole dyads, and heterobimetallic Fe/Mn porphyrin-Co corrole dyads, is also made (Table 1). Three conclusions can be obtained from this table. First, heterobimetallic Fe/Mn porphyrin-Co corrole dyads show quite poor selectivity for the 4e ORR, although their half-wave overpotentials are modest among all catalysts. Second, dinuclear Co biscorroles show moderate selectivity for the 4e ORR, indicating the presence of parallel 4e and 2e ORR pathways. Third, dinuclear Co porphyrin-corrole dyads display both high activity and selectivity for the 4e ORR. For dinuclear Co bisporphyrins, only complex 1 can show comparable performance to the dinuclear Co porphyrin-corrole dyads, while other dinuclear Co bisporphyrins all show relatively larger half-wave overpotentials for the ORR. By using electrochemical and spectroscopic methods, Kadish and co-workers established that the catalytically active species of Co corroles for the ORR are generated at relatively more anodic potentials than Co porphyrins with similar substituents.48–50 As a consequence, dinuclear catalysts, which bear Co corrole moieties, in general will display smaller half-wave overpotentials than dinuclear Co bisporphyrins. Therefore, it is remarkable that the ORR performance of dinuclear Co bisporphyrin 1 (ηE1/2 = 510 mV, n = 3.9) is almost identical to that of the dinuclear Co porphyrin-corrole dyad (PCA)Co2 (ηE1/2 = 520 mV, n = 3.9, Table 1), in terms of both activity and selectivity.
In summary, we report the synthetic procedures and electrocatalytic ORR features of asymmetrical Pacman dinuclear Co porphyrin–porphyrin dyad 1. Complex 1 is highly efficient and selective to catalyze the 4e ORR in aqueous solutions, and its performance is superior to those of corresponding mononuclear Co porphyrins and also all other symmetrical dinuclear Co bisporphyrins reported in this work and in the literature. The significantly improved performance is due to the asymmetrical structure, in which one Co porphyrin moiety functions as a positively charged Lewis acid to assist the binding and activation of the negatively charged O2-adduct unit at its Pacman cleft. This work demonstrates the benefit of asymmetrical dinuclear metal catalysts in electrocatalytic reduction of O2, and this catalyst design strategy may be valuable to be explored in other small molecule activation reactions.
Experimental section
General materials and methods
Mononuclear Co porphyrins 3 and 4 were synthesized according to reported procedures.19,24 Synthetic details and characterization of dinuclear Co porphyrins 1 and 2 and also dinuclear Ni porphyrin 5 are described in the ESI.† All reagents were purchased from commercial suppliers and were used as received. Dry solvents, including dimethylformamide, dichloromethane, acetonitrile, and tetrahydrofuran, were purified by passage through activated alumina. Manipulations of air and moisture-sensitive materials were performed under nitrogen gas using standard Schlenk line techniques. All aqueous solutions were prepared freshly using Milli-Q water. NMR measurements were made on a Brüker spectrometer operating at 400 MHz. High-resolution mass spectra were acquired using a Brüker Fourier transform ion cyclotron resonance mass spectrometer APEX IV. UV-vis spectra were recorded using a Cary 50 spectrophotometer. EPR measurements were carried out on a Bruker EMX plus 10/12 CW X-band EPR spectrometer at a microwave frequency of 9.43 GHz using a liquid nitrogen cooling system. The EPR spectra were measured at 90 K with a modulation amplitude of 0.6 mT, a modulation frequency of 100 kHz, and a microwave power of 6.325 mW. Typically, ∼5 mg of sample of complex 1 or 2 was transferred into an EPR tube in a dry box. A toluene solution with a concentration of 1.63 μM for 1 and 1.39 μM for 2 is used for measurements.Crystallographic studies
Complete datasets for 2 (CCDC 1956352) and 5 (CCDC 1956353) were collected. Single crystals suitable for X-ray analysis were each coated with Paratone-N oil, suspended in a small fiber loop, and placed at 150(2) K on a Bruker D8 Venture X-ray diffractometer. Diffraction intensities were measured using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Data collection, indexing, data reduction, and final unit cell refinements were carried out using APEX2;62 absorption corrections were applied using the program SADABS.63 The structure was solved by direct methods using SHELXS64 and refined against F2 on all data by full-matrix least-squares using SHELXL,65 following established refinement strategies. In the X-ray structure of 2 and 5, all non-hydrogen atoms were refined anisotropically. All hydrogen atoms binding to carbon were included in the model at geometrically calculated positions and refined using a riding model. The isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U value of the atoms they are linked to (1.5 times for methyl groups). Details of the data quality and a summary of the residual values of the refinements are listed in ESI Table S1.†Electrochemical studies
All electrochemical experiments were carried out using a CH Instruments electrochemical analyzer (model CHI660E) at room temperature. The solution was bubbled with N2 gas for at least 30 min before analysis. CVs were recorded in benzonitrile (0.1 M Bu4NPF6) using a three-compartment cell possessing a 0.07 cm2 GC electrode as the working electrode, graphite rod as the auxiliary electrode, and Ag/AgNO3 as the reference electrode (BASi, 10 mM AgNO3, 0.1 M Bu4NPF6 in acetonitrile). Ferrocene was added at the end of the measurement as an internal standard. In aqueous solutions, an Ag/AgCl (KCl saturated) electrode was used as the reference electrode. All potentials measured in aqueous solutions are referenced to the reversible hydrogen electrode (RHE). The GC working electrode was polished with α-Al2O3 of decreasing sizes (1.0 μm to 50 nm) and washed with distilled water and absolute ethanol. For rotating ring-disk electrode measurements, a bipotentiostat (Model CHI 832 Electrochemical Analyzer) and a rotating ring-disk electrode with a rotating GC disk electrode and a platinum ring electrode (ALS RRDE-2) were used. The disk electrode was scanned from 1.20 to 0 V, while the ring potential was held at 1.15 V to detect H2O2 that was possibly generated during the ORR. The collection efficiency of the ring-disk electrode was evaluated with the [Fe(CN)6]3−/4− redox couple and was calculated to be 0.47. Stock dimethylformamide solutions (2 mL) were prepared by mixing multi-walled carbon nanotubes (2 mg), Nafion (20 μL 5%), and Co porphyrin catalysts with the final concentration of 0.1 mM for 1 and 2 and 0.2 mM for 3 and 4. The mixture solution was sonicated for 30 min, and then well-mixed solutions were applied to the surface of the GC (5 μL) and the GC disk (10 μL) electrodes. Evaporation of the solvents at room temperature produced thin catalyst films.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We are grateful for support from the “Thousand Talents Program” of China, Fok Ying-Tong Education Foundation for Outstanding Young Teachers in University, National Natural Science Foundation of China (Grants 21101170, 21573139, and 21773146), Fundamental Research Funds for the Central Universities, and Research Funds of Shaanxi Normal University.References
- M. L. Pegis, C. F. Wise, D. J. Martin and J. M. Mayer, Chem. Rev., 2018, 118, 2340–2391 CrossRef CAS.
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 0098 CrossRef CAS.
- Y. Nie, L. Li and Z. D. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- W. Zhang, W. Z. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- Z. Z. Liang, X. Fan, H. T. Lei, J. Qi, Y. Y. Li, J. P. Gao, M. L. Huo, H. T. Yuan, W. Zhang, H. P. Lin, H. Q. Zheng and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 13187–13191 CrossRef CAS PubMed.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- J. W. Han, J. Jung, Y.-M. Lee, W. Nam and S. Fukuzumi, Chem. Sci., 2017, 8, 7119–7125 RSC.
- H. T. Lei, X. L. Li, J. Meng, H. Q. Zheng, W. Zhang and R. Cao, ACS Catal., 2019, 9, 4320–4344 CrossRef CAS.
- M. Guo, Y.-M. Lee, R. Gupta, M. S. Seo, T. Ohta, H.-H. Wang, H.-Y. Liu, S. N. Dhuri, R. Sarangi, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2017, 139, 15858–15867 CrossRef CAS PubMed.
- S. L. Hooe, A. L. Rheingold and C. W. Machan, J. Am. Chem. Soc., 2018, 140, 3232–3241 CrossRef CAS PubMed.
- Y. J. Liu, Y. Z. Han, Z. Y. Zhang, W. Zhang, W. Z. Lai, Y. Wang and R. Cao, Chem. Sci., 2019, 10, 2613–2622 RSC.
- J. P. Collman, N. K. Devaraj, R. A. Decréau, Y. Yang, Y. L. Yan, W. Ebina, T. A. Eberspacher and C. E. D. Chidsey, Science, 2007, 315, 1565–1568 CrossRef CAS PubMed.
- J. P. Collman, R. A. Decréau, H. W. Lin, A. Hosseini, Y. Yang, A. Dey and T. A. Eberspacher, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7320–7323 CrossRef CAS PubMed.
- J. P. Collman, L. Fu, P. C. Herrmann and X. M. Zhang, Science, 1997, 275, 949–951 CrossRef CAS PubMed.
- S. Chatterjee, K. Sengupta, S. Hematian, K. D. Karlin and A. Dey, J. Am. Chem. Soc., 2015, 137, 12897–12905 CrossRef CAS PubMed.
- T. Chishiro, Y. Shimazaki, F. Tani, Y. Tachi, Y. Naruta, S. Karasawa, S. Hayami and Y. Maeda, Angew. Chem., Int. Ed., 2003, 42, 2788–2791 CrossRef CAS.
- S. Yoshikawa and A. Shimada, Chem. Rev., 2015, 115, 1936–1989 CrossRef CAS.
- S. M. Adam, G. B. Wijeratne, P. J. Rogler, D. E. Diaz, D. A. Quist, J. J. Liu and K. D. Karlin, Chem. Rev., 2018, 118, 10840–11022 CrossRef CAS.
- R. McGuire Jr, D. K. Dogutan, T. S. Teets, J. Suntivich, Y. Shao-Horn and D. G. Nocera, Chem. Sci., 2010, 1, 411–414 RSC.
- C. J. Chang, Z. H. Loh, C. N. Shi, F. C. Anson and D. G. Nocera, J. Am. Chem. Soc., 2004, 126, 10013–10020 CrossRef CAS.
- C. J. Chang, Y. Q. Deng, C. N. Shi, C. K. Chang, F. C. Anson and D. G. Nocera, Chem. Commun., 2000, 1355–1356 RSC.
- S. Fukuzumi, K. Okamoto, C. P. Gros and R. Guilard, J. Am. Chem. Soc., 2004, 126, 10441–10449 CrossRef CAS.
- S. Fukuzumi, K. Okamoto, Y. Tokuda, C. P. Gros and R. Guilard, J. Am. Chem. Soc., 2004, 126, 17059–17066 CrossRef CAS.
- A. N. Oldacre, A. E. Friedman and T. R. Cook, J. Am. Chem. Soc., 2017, 139, 1424–1427 CrossRef CAS.
- A. N. Oldacre, M. R. Crawley, A. E. Friedman and T. R. Cook, Chem.–Eur. J., 2018, 24, 10984–10987 CrossRef CAS.
- P. Peljo, L. Murtomäki, T. Kallio, H. J. Xu, M. Meyer, C. P. Gros, J. M. Barbe, H. H. Girault, K. Laasonen and K. Kontturi, J. Am. Chem. Soc., 2012, 134, 5974–5984 CrossRef CAS.
- B. D. Matson, C. T. Carver, A. Von Ruden, J. Y. Yang, S. Raugei and J. M. Mayer, Chem. Commun., 2012, 48, 11100–11102 RSC.
- C. T. Carver, B. D. Matson and J. M. Mayer, J. Am. Chem. Soc., 2012, 134, 5444–5447 CrossRef CAS.
- M. L. Rigsby, D. J. Wasylenko, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2015, 137, 4296–4299 CrossRef CAS.
- K. Mittra, S. Chatterjee, S. Samanta, K. Sengupta, H. Bhattacharjee and A. Dey, Chem. Commun., 2012, 48, 10535–10537 RSC.
- G. Passard, D. K. Dogutan, M. T. Qiu, C. Costentin and D. G. Nocera, ACS Catal., 2018, 8, 8671–8679 CrossRef CAS.
- I. Hijazi, T. Bourgeteau, R. Cornut, A. Morozan, A. Filoramo, J. Leroy, V. Derycke, B. Jousselme and S. Campidelli, J. Am. Chem. Soc., 2014, 136, 6348–6354 CrossRef CAS PubMed.
- Q. Zuo, G. Z. Cheng and W. Luo, Dalton Trans., 2017, 46, 9344–9348 RSC.
- L. N. Ye, Y. Y. Fang, Z. P. Ou, S. L. Xue and K. M. Kadish, Inorg. Chem., 2017, 56, 13613–13626 CrossRef CAS PubMed.
- B. Wang, Y.-M. Lee, W.-Y. Tcho, S. Tussupbayev, S.-T. Kim, Y. Kim, M. S. Seo, K.-B. Cho, Y. Dede, B. C. Keegan, T. Ogura, S. H. Kim, T. Ohta, M.-H. Baik, K. Ray, J. Shearer and W. Nam, Nat. Commun., 2017, 8, 14839 CrossRef CAS.
- S. Liu, K. Mase, C. Bougher, S. D. Hicks, M. M. Abu-Omar and S. Fukuzumi, Inorg. Chem., 2014, 53, 7780–7788 CrossRef CAS.
- S. Fukuzumi, Y.-M. Lee and W. Nam, ChemCatChem, 2018, 10, 9–28 CrossRef CAS.
- S. Bhunia, A. Rana, P. Roy, D. J. Martin, M. L. Pegis, B. Roy and A. Dey, J. Am. Chem. Soc., 2018, 140, 9444–9457 CrossRef CAS.
- K. Sengupta, S. Chatterjee, S. Samanta and A. Dey, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8431–8436 CrossRef CAS.
- D. K. Dogutan, S. A. Stoian, R. McGuire Jr, M. Schwalbe, T. S. Teets and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 131–140 CrossRef CAS.
- H. T. Lei, C. Y. Liu, Z. J. Wang, Z. Y. Zhang, M. N. Zhang, X. M. Chang, W. Zhang and R. Cao, ACS Catal., 2016, 6, 6429–6437 CrossRef CAS.
- J. Meng, H. T. Lei, X. L. Li, J. Qi, W. Zhang and R. Cao, ACS Catal., 2019, 9, 4551–4560 CrossRef CAS.
- J. J. Tang, Z. P. Ou, R. Guo, Y. Y. Fang, D. Huang, J. Zhang, J. X. Zhang, S. Guo, F. M. McFarland and K. M. Kadish, Inorg. Chem., 2017, 56, 8954–8963 CrossRef CAS.
- A. Friedman, L. Landau, S. Gonen, Z. Gross and L. Elbaz, ACS Catal., 2018, 8, 5024–5031 CrossRef CAS.
- S. Fukuzumi, S. Mandal, K. Mase, K. Ohkubo, H. Park, J. Benet-Buchholz, W. Nam and A. Llobet, J. Am. Chem. Soc., 2012, 134, 9906–9909 CrossRef CAS.
- G. Passard, A. M. Ullman, C. N. Brodsky and D. G. Nocera, J. Am. Chem. Soc., 2016, 138, 2925–2928 CrossRef CAS.
- C. Di Giovanni, C. Gimbert-Suriñach, M. Nippe, J. Benet-Buchholz, J. R. Long, X. Sala and A. Llobet, Chem.–Eur. J., 2016, 22, 361–369 CrossRef CAS.
- K. M. Kadish, L. Frémond, Z. P. Ou, J. G. Shao, C. N. Shi, F. C. Anson, F. Burdet, C. P. Gros, J. M. Barbe and R. Guilard, J. Am. Chem. Soc., 2005, 127, 5625–5631 CrossRef CAS.
- K. M. Kadish, J. G. Shao, Z. P. Ou, L. Frémond, R. Q. Zhan, F. Burdet, J. M. Barbe, C. P. Gros and R. Guilard, Inorg. Chem., 2005, 44, 6744–6754 CrossRef CAS.
- K. M. Kadish, L. Frémond, J. Shen, P. Chen, K. Ohkubo, S. Fukuzumi, M. El Ojaimi, C. P. Gros, J. M. Barbe and R. Guilard, Inorg. Chem., 2009, 48, 2571–2582 CrossRef CAS.
- K. M. Kadish, L. Frémond, F. Burdet, J. M. Barbe, C. P. Gros and R. Guilard, J. Inorg. Biochem., 2006, 100, 858–868 CrossRef CAS.
- M. Quernheim, H. W. Liang, Q. Su, M. Baumgarten, N. Koshino, H. Higashimura and K. Müllen, Chem.–Eur. J., 2014, 20, 14178–14183 CrossRef CAS.
- C. Y. Liu, H. T. Lei, Z. Y. Zhang, F. F. Chen and R. Cao, Chem. Commun., 2017, 53, 3189–3192 RSC.
- S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Nat. Chem., 2014, 6, 934–940 CrossRef CAS.
- I. Azcarate, C. Costentin, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS.
- Y. Y. Fang, Z. P. Ou and K. M. Kadish, Chem. Rev., 2017, 117, 3377–3419 CrossRef CAS.
- W. B. Lu, C. X. Wang, Q. Y. Lv and X. H. Zhou, J. Electroanal. Chem., 2003, 558, 59–63 CrossRef CAS.
- A. Schechter, M. Stanevsky, A. Mahammed and Z. Gross, Inorg. Chem., 2012, 51, 22–24 CrossRef CAS.
- N. Levy, A. Mahammed, M. Kosa, D. T. Major, Z. Gross and L. Elbaz, Angew. Chem., Int. Ed., 2015, 54, 14080–14084 CrossRef CAS.
- Y.-H. Wang, P. E. Schneider, Z. K. Goldsmith, B. Mondal, S. Hammes-Schiffer and S. S. Stahl, ACS Cent. Sci., 2019, 5, 1024–1034 CrossRef CAS.
- J. F. Li, B. C. Noll, A. G. Oliver and W. R. Scheidt, J. Am. Chem. Soc., 2012, 134, 10595–10606 CrossRef CAS PubMed.
- APEX2 v2009, Bruker AXS., Madison, WI, 2009 Search PubMed.
- G. M. Sheldrick, SADABS, 2008/1, University of Göttingen, Göttingen, Germany, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S27, Table S1, and crystallographic data in CIF format for complexes 2 and 5. The X-ray crystallographic coordinates for reported structures in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 1956352 and 1956353. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05041h |
| This journal is © The Royal Society of Chemistry 2020 |