
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA nonaqueous potassium-ion hybrid capacitor enabled by two-dimensional diffusion pathways of dipotassium terephthalate†
Yuwen
Luo‡
ab,
Luojia
Liu‡
b,
Kaixiang
Lei
b,
Jifu
Shi
*c,
Gang
Xu
a,
Fujun
Li
 *b and
Jun
Chen
b
*b and
Jun
Chen
b
aGuangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, China
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: fujunli@nankai.edu.cn
cSiyuan Laboratory, Department of Physics, Jinan University, Guangzhou 510632, China. E-mail: shijifu2017@126.com
First published on 10th December 2018
Abstract
Nonaqueous potassium-ion hybrid capacitors (KIHCs) are faced with limited redox reaction kinetics of electrodes for accommodation of large-sized K+. Here, dipotassium terephthalate (K2TP) is applied as an organic negative electrode to provide comparable reaction kinetics with a non-faradaic activated carbon (AC) positive electrode to boost the electrochemical performance of KIHCs. It is revealed that the large exchange current density and fast two-dimensional (2D) diffusion pathways of K+ in K2TP determined by density functional theory (DFT) calculations ensure its fast redox reaction and transport kinetics. The as-constructed KIHC presents both high energy and power densities of 101 W h kg−1 and 2160 W kg−1 based on the mass of the two electrodes (41.5 W h kg−1 and 885.2 W kg−1 based on the mass of the two electrodes and electrolyte), respectively, and a superior capacity retention of 97.7% after 500 cycles. The excellent electrochemical performance is attributed to the fast kinetics, good structural flexibility, and small volume change (9.4%) of K2TP upon K+ insertion/extraction, and its good compatibility with the AC positive electrode in 1,2-dimethoxyethane (DME)-based electrolyte. This will promote application of organic materials in hybrid capacitors and the development of KIHCs.
Introduction
Nonaqueous hybrid capacitors have recently attracted extensive research interest, and are generally composed of battery-type negative electrodes and electric double-layer capacitor (EDLC)-type positive electrodes.1,2 They are endowed with the advantages of both high energy density of batteries and high power density and long lifespan of EDLCs.3–6 The equilibrium potential of K+/K is −2.88 V in propylene carbonate (PC), which is lower than that of Li+/Li (−2.79 V) or Na+/Na (−2.56 V), providing wider operating voltage windows;7 K+ has a smaller Stokes' radius (3.6 Å) than those of Li+ and Na+ in PC (4.8 and 4.6 Å) due to its weaker Lewis acidity, suggesting its high mobility, that is, high ionic conductivity of K+ in electrolytes.8 The abundance of K resources in the Earth's crust is comparable to that of their Na counterparts and about three orders of magnitude higher than that of their Li counterparts, endowing it with potentially low cost and sustainability.9,10 Therefore, developing nonaqueous potassium-ion hybrid capacitors (KIHCs) is highly desirable. However, KIHCs are plagued by poor kinetics of redox reactions and diffusion of large-sized K+ in the bulky electrode materials, which are usually much slower than the non-faradaic capacitive sorption of ions in EDLCs, and construction of KIHCs is faced with a materials challenge.Carbon-based materials are attractive negative electrode candidates for K+ storage, but their low redox potentials and rate capability preclude their usage in KIHCs.5,6,11–14 Alloys and conversion-reaction-type negative electrode materials exhibit high capacity, but their large volume change during charge and discharge induce decaying performance.15–19 Organic materials with structural flexibility enable fast K+ diffusion and redox response, and have been attempted in potassium-ion batteries (KIBs).20–24 Chen et al. demonstrated the feasibility of 3,4,9,10-perylene-tetracarboxylic acid-dianhydride for K+ storage.20 However, its capacity retention is only 66.1% after 200 cycles, which is attributed to its dissolution in electrolytes. Li's group has revealed that organic carboxyls are stable in electrolytes, and their redox potentials are between 0.5 and 1.0 V vs. K+/K, far from the deposition potential of K.25 This can effectively circumvent the formation of K dendrites, and thus will promote safety, especially in high-rate devices. However, although they have been explored in Li-ion and Na-ion batteries,26,27 organic materials for KIHCs and the correlation of their molecular structures with kinetics of ions have never been reported, and are essential for construction of high-performance KIHCs.
Herein, we report a nonaqueous KIHC with an organic negative electrode of K2TP, a positive electrode of AC and DME-based electrolyte. K2TP is found to show fast 2D diffusion pathways for K+, structural flexibility, and a small volume change upon K+ insertion/extraction by DFT calculations. The facile transport of K+ in K2TP and its fast redox reaction kinetics are comparable to those of the non-faradaic AC positive electrode, and boost the electrochemical performance of the KIHC. Maximal power and energy densities of 2160 W kg−1 and 101 W h kg−1 are obtained, respectively, and it shows a high capacity retention of 97.7% after 500 cycles, which corresponds to a capacity loss of 0.0046% per cycle. Such excellent performance will encourage more investigations on organic materials in hybrid capacitors and promote the development of KIHCs.
Results and discussion
The KIHC was composed of a K2TP negative electrode, AC positive electrode, and DME-based electrolyte, as shown in Fig. 1a. The synthesis of K2TP is described in the ESI.† It is micrometers in size as displayed in the inset of Fig. 1b. The X-ray diffraction (XRD) pattern of K2TP in Fig. 1b is consistent with the standard JCPDS no. 52-2142, suggesting high purity. The Rietveld refinement fits well with the experimental data, and it is assigned to monoclinic K2TP with calculated cell parameters of a = 10.552 Å, b = 3.935 Å, c = 11.520 Å, β = 113.08°, and V = 440.05 Å3, as shown in Table S1.† Fourier transform infrared (FTIR) spectroscopy results in Fig. S1† further confirm the formation of K2TP. AC features microporous bulky particles (see the inset), and shows a typical type I isotherm of N2 sorption (Fig. 1c).28 Its specific surface area is calculated to be 1699 m2 g−1 by the Brunauer–Emmett–Teller (BET) method, and its pore size is centered at 1.2 nm in Fig. S2† by Barrett–Joyner–Halenda analysis. The large surface area will favor reversible sorption of ions in KIHCs.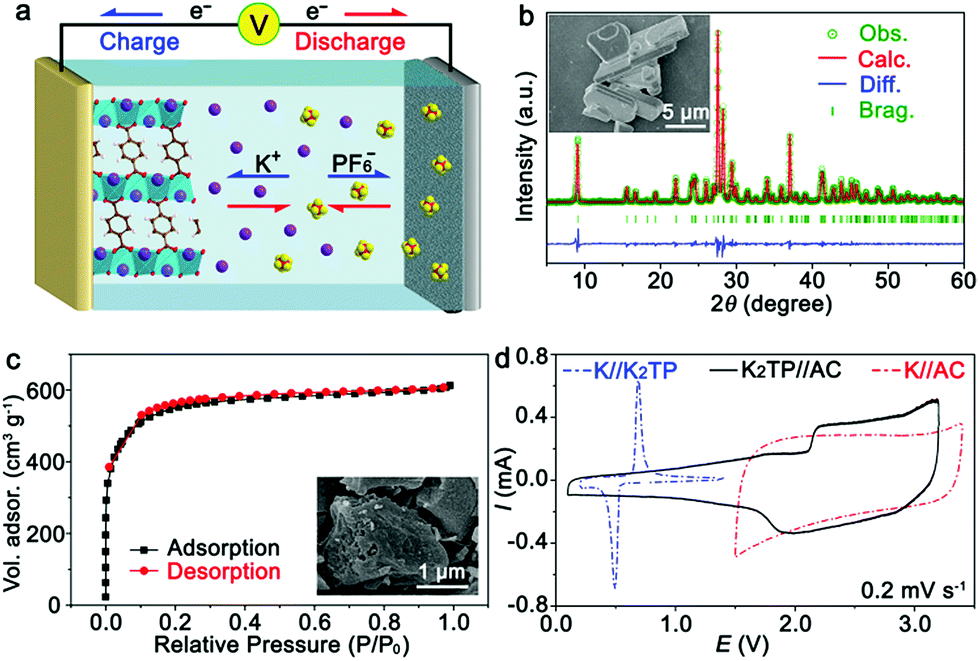 | ||
| Fig. 1 (a) Schematic illustration of the KIHC with the configuration K2TP//DME-based electrolyte//AC; (b) XRD pattern, Rietveld-refined profile, and SEM image (inset) of K2TP; (c) nitrogen sorption isotherm and SEM image (inset) of AC; and (d) CV curves of half batteries and the KIHC. | ||
K2TP was first ball-milled with Super P in a ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to promote electron conduction. It is 50–200 nm in size, as shown in the SEM image in Fig. S3.† The as-prepared K2TP electrode presents a couple of redox peaks in the cyclic voltammetry (CV) curve in a half battery of K//K2TP at 0.2 mV s−1, as shown in Fig. 1d. The discrepancy between the first and succeeding scans in Fig. S4a† is attributed to the formation of a solid-electrolyte interphase (SEI). The redox peaks at ∼0.50 and 0.67 V correspond to K+ insertion/extraction. The discharge/charge profiles of K2TP are displayed in Fig. S4b.† The representative plateaus are in agreement with the redox peaks in Fig. 1d. It shows good rate capability from 0.1 to 5 A g−1, and maintains a stable discharge/charge capacity of 220 mA h g−1 for 100 cycles with coulombic efficiencies of ∼100% beyond the initial cycles in Fig. S4c.† These indicate high reversibility and rate capability of the K2TP electrode for K+ storage.
:3 to promote electron conduction. It is 50–200 nm in size, as shown in the SEM image in Fig. S3.† The as-prepared K2TP electrode presents a couple of redox peaks in the cyclic voltammetry (CV) curve in a half battery of K//K2TP at 0.2 mV s−1, as shown in Fig. 1d. The discrepancy between the first and succeeding scans in Fig. S4a† is attributed to the formation of a solid-electrolyte interphase (SEI). The redox peaks at ∼0.50 and 0.67 V correspond to K+ insertion/extraction. The discharge/charge profiles of K2TP are displayed in Fig. S4b.† The representative plateaus are in agreement with the redox peaks in Fig. 1d. It shows good rate capability from 0.1 to 5 A g−1, and maintains a stable discharge/charge capacity of 220 mA h g−1 for 100 cycles with coulombic efficiencies of ∼100% beyond the initial cycles in Fig. S4c.† These indicate high reversibility and rate capability of the K2TP electrode for K+ storage.
According to the Randles–Sevcik equation (ESI†), the peak currents are plotted against the square roots of scan rates of CV curves in Fig. S5.† The diffusion coefficient of K+ (DK+) in K2TP is estimated to be 1.32 × 10−11 cm2 s−1, which is three orders of magnitude larger than that of Li+ in LiFePO4 (10−14 cm2 s−1).29 Simultaneously, the exchange current density of K2TP can be derived from electrochemical impedance spectroscopy (EIS, Fig. S6†) based on the equation j0 = RT/(nAFRct), where j0 is the exchange current density, R is the gas constant, T is the temperature, n is the charge-transfer number, A is the contact area between the electrode and the electrolyte, F is the Faraday constant, and Rct is the charge-transfer resistance.30 Accordingly, the exchange current density of K2TP is calculated to be 1.25 × 10−3 A cm−2, which is superior to that of LiFePO4 (3.41 × 10−4 A cm−2).29 All of this suggests facile kinetics of K+ transport and redox reaction of K2TP, which are consistent with its superior performance in half batteries.
On the other hand, the capacitive behavior of the AC positive electrode was evaluated in a half battery of K//AC and the results are shown in Fig. 1d. It shows a typical CV curve with a rectangular shape, which is maintained at various scan rates, as shown in Fig. S7a† and indicative of good capacitive performance. The charge/discharge profiles at different current densities in Fig. S7b† exhibit triangular shapes, in good accordance with CV curves. A discharge capacity as high as 55.2 mA h g−1 is retained with a capacity retention of ∼100% after 100 cycles at 0.5 A g−1, as shown in Fig. S7c.† This is attributed to the large surface area and facile capacitive behavior of the microporous AC.
For the KIHC of K2TP//AC, the mass ratio of K2TP to AC is optimized to be 1:4. The operating voltage window of the KIHC is determined to be 0.1–3.2 V. Its CV curve in Fig. 1d exhibits combined behavior of redox reaction of K2TP and capacitive sorption of AC. In situ FTIR was applied to explore the reactions of K2TP, which is based on its conjugated carboxyl groups, as illustrated in Fig. 2a. Each K2TP molecule accepts two K+, and the conjugated carboxyls are transformed into enolates (K4TP). The reversible transformation is monitored by in situ FTIR with a home-made cell in Fig. 2b, where the K2TP negative electrode is in close contact with an attenuated total reflection crystal of the spectrometer. As shown in Fig. 2c, the charge/discharge curve of the KIHC presents sloping plateaus. In the charging process, the intensity of the non-benzene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration at 1660 cm−1 gradually increases, corresponding to potassiation of K2TP, as shown in Fig. 2d. This is ascribed to the formation of C–O bonds and conjugated non-benzene CC double bonds with weaker aromaticity, as shown in Fig. 2a. In the subsequent discharging process, the peak intensity reversibly decreases due to depotassiation. This suggests reversible conversion of non-benzene CC double bonds to C–C bonds and formation of CO bonds. In addition, ex situ Raman spectra in Fig. 2e were also used to investigate the reaction process. In the fully charged state, a broad peak appears at 1516 cm−1, which is assigned to a combination of stretching vibrations of non-benzene CC and C–O bonds of K4TP. This is in good accordance with the theoretically simulated Raman spectrum of K4TP in Fig. S8.† In the discharging process, the broad peak gradually decreases, and the characteristic peaks of K2TP appear as the simulated Raman spectrum of K2TP in Fig. S9.† Both FTIR and Raman spectra clearly verify the electrochemical redox reaction of K2TP via the reversible transformation of conjugated carboxyls and enolates.
C vibration at 1660 cm−1 gradually increases, corresponding to potassiation of K2TP, as shown in Fig. 2d. This is ascribed to the formation of C–O bonds and conjugated non-benzene CC double bonds with weaker aromaticity, as shown in Fig. 2a. In the subsequent discharging process, the peak intensity reversibly decreases due to depotassiation. This suggests reversible conversion of non-benzene CC double bonds to C–C bonds and formation of CO bonds. In addition, ex situ Raman spectra in Fig. 2e were also used to investigate the reaction process. In the fully charged state, a broad peak appears at 1516 cm−1, which is assigned to a combination of stretching vibrations of non-benzene CC and C–O bonds of K4TP. This is in good accordance with the theoretically simulated Raman spectrum of K4TP in Fig. S8.† In the discharging process, the broad peak gradually decreases, and the characteristic peaks of K2TP appear as the simulated Raman spectrum of K2TP in Fig. S9.† Both FTIR and Raman spectra clearly verify the electrochemical redox reaction of K2TP via the reversible transformation of conjugated carboxyls and enolates.
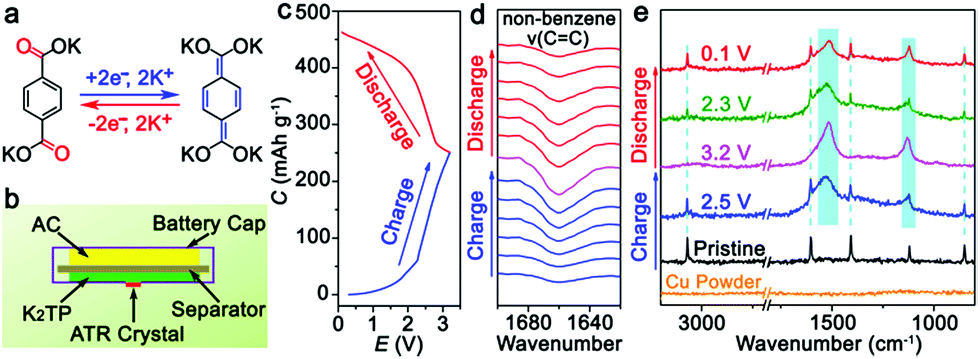 | ||
| Fig. 2 (a) Redox reaction mechanism of K+ with K2TP; (b) diagram of a cell for in situ FTIR test; (c) charge/discharge profile of the KIHC at 20 mA g−1 (based on the mass of K2TP); (d) in situ FTIR spectra of the K2TP negative electrode in a charge/discharge cycle; and (e) Raman spectra of the K2TP negative electrode at selected states with copper powder. | ||
The crystal structures of K2TP and its fully potassiated K4TP are optimized by DFT calculations and depicted in Fig. 3. The XRD pattern of K4TP in the discharge state agrees well with the simulated one in Fig. S10,† confirming the consistency of the crystal structure of K4TP with the experimental result. The structure of K2TP is shown in Fig. 3a and b, where K–O inorganic layers and π-stacking aromatic organic layers are alternatively arranged along the a direction. In each K2TP molecule, the bond lengths of the adjacent C–O bonds are 1.25 and 1.20 Å, respectively. The shorter C–O bond is identified as carbonyl (CO). The structure of K4TP, with optimized cell parameters in Table S2,† is displayed in Fig. 3c–e. The bond lengths of all C–O bonds in each K4TP molecule are 1.35 Å. This is attributed to the disappearance of CO and formation of enolate. The two original K+ of K2TP are marked K1 and K2, and the two inserted K+ are marked K3 and K4, which push K1 and K2 into the K1′ and K2′ sites during potassiation. Meanwhile, the coordination of K1′ and K2′ with O is changed into KO4 coordination from KO6. The thickness of the K–O layer and organic layer increases by 6.2% and 3.0%, respectively, and then the volume expansion is only 9.4%. When comparing the terephthalate motifs between K2TP and K4TP, a distinct structural distortion can be observed in Fig. 3f and g. In K2TP, the dihedral angle between the planes of carboxyl groups and the carbon ring is 6.62°, as shown in Fig. 3f, while it increases to 15.30° in K4TP in Fig. 3g. This indicates good structural flexibility of the terephthalate motif, which facilitates diffusion of K+. Of note, the volume expansion of K2TP after potassiation is significantly smaller than for the negative electrode materials reported so far, for instance, 61% for graphite31 and 409% for Bi.32 The small volume change and high structural flexibility of K2TP favor the diffusion and storage of K+.
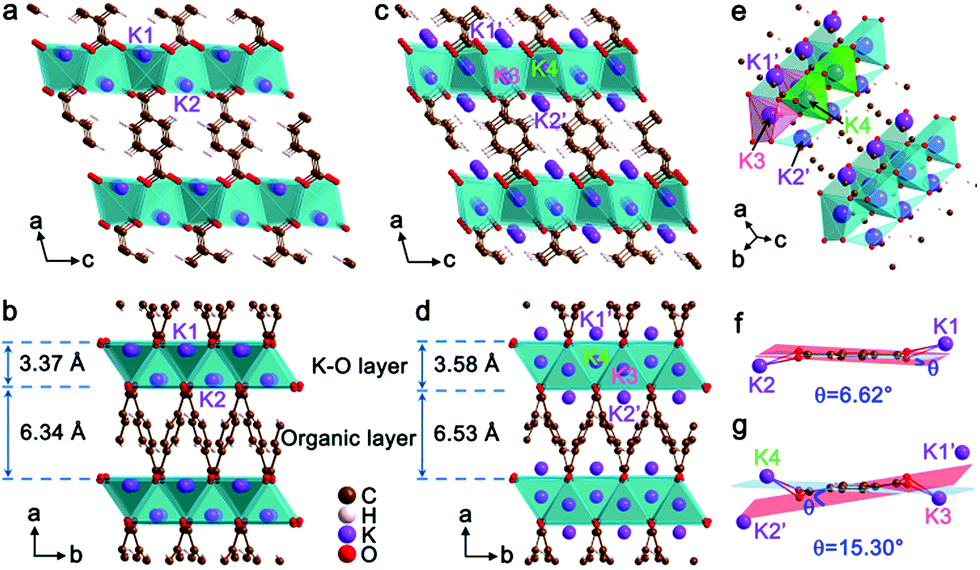 | ||
| Fig. 3 Crystal structures of (a and b) K2TP and (c–e) K4TP; and structural transformation of the terephthalate motif in (f) K2TP and (g) K4TP. The blue and red planes are defined by the carbon ring and carboxyl groups, respectively. | ||
The charge evolution, apart from K+ ions, in the reversible transformation of K2TP to K4TP is revealed by a Bader charge change on their carbon and oxygen atoms, as shown in Fig. S11 and Table S3.† The charge transfer mainly occurs on carbon atoms with an almost constant Bader charge on oxygen atoms after full potassiation. In particular, the carbon atoms in the carboxyl groups of K4TP are localized with a large amount of charge. This suggests that the carbon atoms of carboxyl groups are charge transfer centers rather than oxygen atoms. It is implicated that the π-stacking aromatic organic layer ensures electron conduction and the inorganic layer of K–O polyhedrons linked through edge sharing is responsible for K+ diffusion.
The diffusion pathways of K+ in the optimized crystal structure of K4TP, based on dispersion-corrected density functional theory (DFT-D2),33 are depicted by molecular dynamics simulation at 2000 K in all the possible diffusion directions. The connected trajectories of K+ during the 10 ps (10000 steps) simulation represent the migration pathways of K+, as shown in Fig. 4a and S12.† It is clear that K+ migrates in the two-dimensional ac plane. The diffusion pathways of K+ were studied using the climbing-image nudged elastic band method to find the path with the minimum energy and barrier between certain diffusion starting and ending points. The diffusion was treated as a K+ migrating from a stable position to the created K+ vacancy in K4TP. Three different diffusion pathways of K+ were designed, denoted as path A (4 → 3 → 5 → 1 → 0), path B (4 → 3 → 2 → 1 → 0) and path C (1 → 1′) in Fig. 4b and S13,† respectively. In path A of Fig. 4c, the diffusion barrier was calculated to be 0.46 eV, which is the minimum. Moreover, the diffusion barrier of K+ migrating from position 1 → 0 and 4 → 3 was calculated to be as low as 0.08 eV, which favors facile migration between these positions. Path B in Fig. 4d and path C in Fig. 4e present higher diffusion barriers of 1.08 and 0.98 eV, respectively. The calculated energy profiles along the three pathways are comparatively shown in Fig. 4f. It is obvious that path A has overall low energy barriers and is the optimal diffusion pathway for K+. This guarantees outstanding ionic conductivity and contributes to the excellent electrochemical performance, especially high rate capability. According to an empirical estimation criterion, a migration barrier of 0.525 eV corresponds to a typical ionic diffusivity of 10−12 cm2 s−1 at room temperature, and a 0.06 eV increase/decrease in the migration energy leads to an order of magnitude decrease/increase in the ionic diffusivity.34 Thus, the 0.46 eV diffusion barrier results in a diffusion coefficient of ∼10−11 cm2 s−1, matching well with the experimental DK+ in K2TP of 1.32 × 10−11 cm2 s−1.
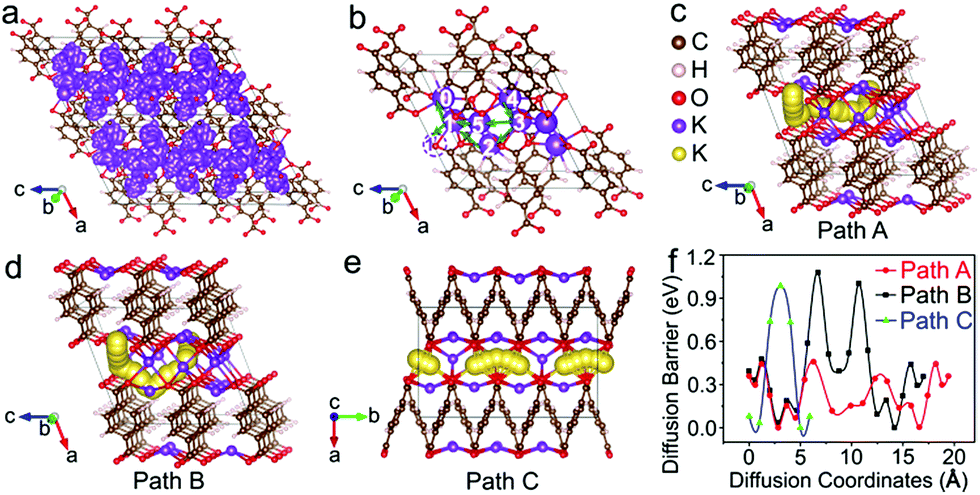 | ||
| Fig. 4 (a) Trajectories of K+ in K4TP obtained by MD simulation at 2000 K for 10 ps; (b) designed K+ diffusion path A (4 → 3 → 5 → 1 →0), B (4 → 3 → 2 → 1 → 0), and C (1 → 1′) in K4TP; (c) path A of K+ diffusion; (d) path B of K+ diffusion; (e) path C of K+ diffusion; and (f) energy profiles of K+ migration in path A, B and C. K+ is displayed in different sizes and colors in (a–e) for clarity. | ||
Capacitive adsorption/release of PF6− onto/from the surface of the AC positive electrode is investigated by elemental mapping. When the KIHC was fully charged, fluorine and phosphorus can be clearly detected as shown in Fig. S14a† with atomic ratios of 1.49% and 0.68% with respect to carbon, respectively, suggesting that PF6− is adsorbed on the AC surface from the electrolyte. After discharge, the atomic ratios of fluorine and phosphorus are dramatically decreased to 0.23% and 0.07%, respectively, in Fig. S14b,† indicating release of PF6− from the AC surface into the electrolyte. The reversible charge storage stems from reversible adsorption and release of PF6− in EDLs of the AC positive electrode.
Fig. 5a presents the rate capability of the KIHC at different current densities. Capacities of 43.5, 41.3, 35.9, 32.1, 28.2, and 23.9 mA h g−1 are obtained at current densities of 20, 40, 100, 200, 400, and 1000 mA g−1, respectively. The selected charge/discharge curves are depicted in Fig. 5b. The sloping plateaus are clearly observed in the charging/discharging process, and are in good agreement with CVs in Fig. 1d. The cycle life of the KIHC is displayed in Fig. 5c. After 500 cycles, the capacity retention is 97.7% and the coulombic efficiencies are close to 100%, implying superior cycling stability of the KIHC. The logo “KIHC” in a pattern of light-emitting diodes is powered by one single KIHC as shown in the inset of Fig. 5c, indicating its potential for application. The KIHC delivered a high energy density of 101 W h kg−1 at a power density of 46 W kg−1 based on the total mass of active materials in both electrodes, as shown in Fig. 5d. Remarkably, a moderate energy density of 52 W h kg−1 can be achieved even at an exceptionally high power density of 2160 W kg−1, demonstrating a good balance between energy and power density. Its overall gravimetric performance is higher than that of many recently reported potassium-ion based capacitors and batteries, such as soft carbon//K0.7Fe0.5Mn0.5O2 (KIB),12 soft carbon//AC (KIHC),6 graphite//AC (KIHC),5 and AC//AC (EDLC).35 Besides, taking the electrolyte into account, the KIHC of K2TP//AC possesses a little lower energy density (41.5 W h kg−1) than 54 W h kg−1 of Ni–Cd batteries but a much higher power density of 885.2 W kg−1vs. 125 W kg−1 of Ni–Cd batteries.36 This is due to the fast kinetics of capacitive behavior of the carbon electrode and redox reactions of K2TP, and shows the advantage of hybrid capacitors.
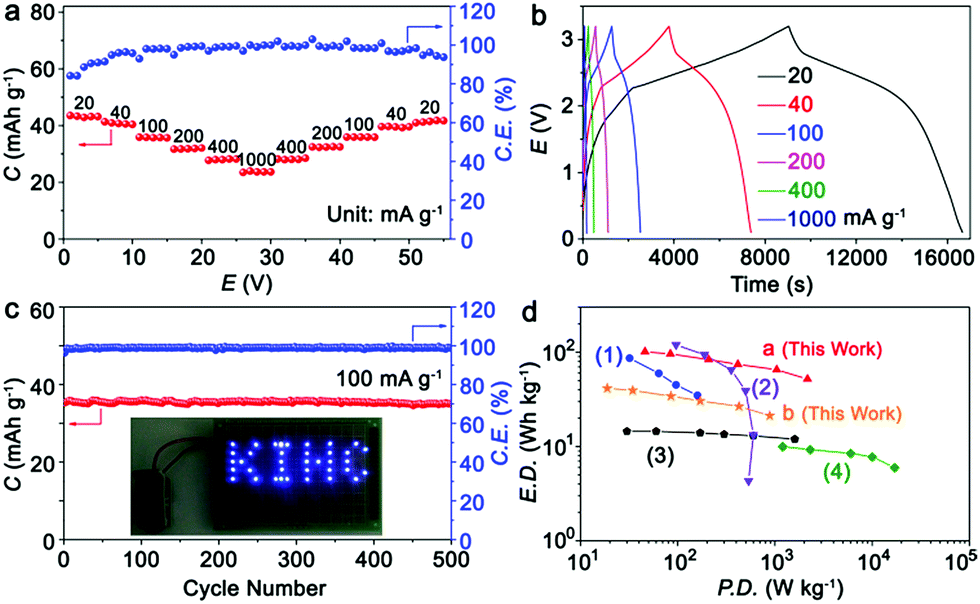 | ||
| Fig. 5 (a) Rate capability; (b) charge/discharge curves at different rates; (c) cycling performance of the KIHC at 100 mA g−1 for 500 cycles; and (d) Ragone plots of the KIHC of K2TP//AC, and other reported KIHCs, a KIB, and an EDLC. (1) Soft carbon//K0.7Fe0.5Mn0.5O2 (KIB), ref. 12; (2) soft carbon//AC (KIHC), ref. 6; (3) graphite//AC (KIHC), ref. 5; (4) AC//AC (EDLC), ref. 35. The current density, capacity, power density and energy density are all based on the total mass of active materials in both electrodes. Exceptionally, plot b is based on the mass of both electrodes and the electrolyte. | ||
Conclusions
In summary, a high-performance KIHC was successfully constructed using a K2TP organic negative electrode and AC positive electrode with DME-based electrolyte. It works with redox reaction of K2TP via reversible transformation of conjugated carboxyls and enolates and capacitive sorption of PF6− in EDLs of AC. DFT calculations indicate a small volume change of 9.4% during discharge/charge of K2TP, thanks to the flexibility of the terephthalate motifs. It is found that the π-stacking aromatic organic layer and the K–O inorganic layer are responsible for electron conduction and K+ diffusion, respectively. The 2D diffusion pathways of K+ have the lowest diffusion barrier of 0.46 eV, and ensure facile K+ diffusion to match with the non-faradaic capacitive kinetics of the AC positive electrode. The KIHC demonstrates a high energy density of 101 W h kg−1, high power density of 2160 W kg−1 based on the mass of the two electrodes, and long lifetime with a capacity retention of 97.7% after 500 cycles. These are attributed to the fast kinetics and high exchange current density of the organic electrode of K2TP and its small volume change during discharge/charge. This investigation reveals promising applications of KIHCs for large scale electric energy storage, and will promote the development of organic functional materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21603108 and 21673243), the Fundamental Funds for the Central Universities, and the NSFC-RGC Joint Program (51761165025) is acknowledged.Notes and references
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1605336 CrossRef PubMed.
- P. Jeżowski, O. Crosnier, E. Deunf, P. Poizot, F. Béguin and T. Brousse, Nat. Mater., 2018, 17, 167 CrossRef PubMed.
- L. Shen, H. Lv, S. Chen, P. Kopold, P. Aken, X. Wu, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1700142 CrossRef PubMed.
- B. Yang, J. Chen, S. Lei, R. Guo, H. Li, S. Shi and X. Yan, Adv. Energy Mater., 2018, 8, 1702409 CrossRef.
- A. Comte, Y. Reynier, C. Vincens, C. Leys and P. Azaïs, J. Power Sources, 2017, 363, 34 CrossRef.
- L. Fan, K. Lin, J. Wang, R. Ma and B. Lu, Adv. Mater., 2018, 30, 1800804 CrossRef PubMed.
- Y. Marcus, Pure Appl. Chem., 1985, 57, 1129 CAS.
- Y. Matsuda, H. Nakashima, M. Morita and Y. Takasu, J. Electrochem. Soc., 1981, 128, 2552 CrossRef CAS.
- Q. Zhao, J. Wang, Y. Lu, Y. Li, G. Liang and J. Chen, Angew. Chem., Int. Ed., 2016, 55, 12528 CrossRef CAS PubMed.
- K. Beltrop, S. Beuker, A. Heckmann, M. Winter and T. Placke, Energy Environ. Sci., 2017, 10, 2090 RSC.
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137, 11566 CrossRef CAS PubMed.
- X. Wang, X. Xu, C. Niu, J. Meng, M. Huang, X. Liu, Z. Liu and L. Mai, Nano Lett., 2017, 17, 544 CrossRef CAS PubMed.
- C. Vaalma, G. Giffin, D. Buchholz and S. Passerini, J. Electrochem. Soc., 2016, 163, A1295 CrossRef CAS.
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef PubMed.
- I. Sultana, T. Ramireddy, M. M. Rahman, Y. Chen and A. M. Glushenkov, Chem. Commun., 2016, 52, 9279 RSC.
- R. Zhang, J. Bao, Y. Wang and C. Sun, Chem. Sci., 2018, 9, 6193 RSC.
- W. Zhang, J. Mao, S. Li, Z. Chen and Z. Guo, J. Am. Chem. Soc., 2017, 139, 3316 CrossRef CAS PubMed.
- Y. Lu and J. Chen, Sci. China: Chem., 2017, 60, 1533 CrossRef CAS.
- X. Ren, Q. Zhao, W. McCulloch and Y. Wu, Nano Res., 2017, 10, 1313 CrossRef CAS.
- Y. Chen, W. Luo, M. Carter, L. Zhou, J. Dai, K. Fu, S. Lacey, T. Li, J. Wan, X. Han, Y. Bao and L. Hu, Nano Energy, 2015, 18, 205 CrossRef CAS.
- Z. Jian, Y. Liang, I. Rodríguez-Pérez, Y. Yao and X. Ji, Electrochem. Commun., 2016, 71, 5 CrossRef CAS.
- J. Pramudita, D. Sehrawat, D. Goonetilleke and N. Sharma, Adv. Energy Mater., 2017, 7, 1602911 CrossRef.
- Y. Liang, Y. Gheytani, K. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841 CrossRef CAS PubMed.
- Z. Song, Y. Qian, X. Liu, T. Zhang, Y. Zhu, H. Yu, M. Otani and H. Zhou, Energy Environ. Sci., 2014, 7, 4077 RSC.
- K. Lei, F. Li, C. Mu, J. Wang, Q. Zhao, C. Chen and J. Chen, Energy Environ. Sci., 2017, 10, 552 RSC.
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribière, P. Poizot and J.-M. Tarascon, Nat. Mater., 2009, 8, 120 CrossRef CAS PubMed.
- A. Jouhara, N. Dupré, A.-C. Gaillot, D. Guyomard, F. Dolhem and P. Poizot, Nat. Commun., 2018, 9, 4401 CrossRef PubMed.
- E. Raymundo-Piñero, K. Kierzek, J. Machnikowski and F. Béguin, Carbon, 2006, 44, 2498 CrossRef.
- D. Li, T. Zhang, X. Liu, P. He, R. Peng, M. Wang, M. Han and H. Zhou, J. Power Sources, 2013, 233, 299 CrossRef CAS.
- H. Ma, S. Zhang, W. Ji, Z. Tao and J. Chen, J. Am. Chem. Soc., 2008, 130, 5361 CrossRef CAS PubMed.
- J. Yang, Z. Ju, Y. Jiang, Z. Xing, B. Xi, J. Feng and S. Xiong, Adv. Mater., 2018, 30, 1700104 CrossRef PubMed.
- K. Lei, C. Wang, L. Liu, Y. Luo, C. Mu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 46877 Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- M. Liu, Z. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder and K. A. Persson, Energy Environ. Sci., 2015, 8, 964 RSC.
- D. Dubal, N. Chodankar, R. Holze, D. Kim and P. Gomez-Romero, ChemSusChem, 2017, 10, 1771 CrossRef CAS PubMed.
- G. P. Hammond and T. Hazeldine, Appl. Energy, 2015, 138, 559 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04489a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |