
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 2D and 3D MOFs with tuneable Lewis acidity from preformed 1D hybrid sub-domains†
José María
Moreno
,
Alexandra
Velty
,
Urbano
Díaz
 * and
Avelino
Corma
*
* and
Avelino
Corma
*
Instituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avenida de los Naranjos s/n, E-46022 Valencia, Spain. E-mail: udiaz@itq.upv.es; acorma@itq.upv.es; Tel: +34963877800
First published on 17th December 2018
Abstract
Novel MOF-type materials with different morphologies based on assembled 1D organic–inorganic sub-domains were prepared using specific monodentate benzylcarboxylate spacers with functional substituents in the para-position as structure modulating agents. The combination of electron-withdrawing or electron-donating functions in the organic spacers with suitable solvothermal synthesis conditions allowed modulating the structuration level (2D or 3D), vacancies, physico-chemical properties and Lewis acidity strength of the metal–organic structures. Furthermore, bimetallic (Al/Fe) MOF-type materials were synthesized by a one-pot direct process without modification of the structural framework. The activity of these hybrid materials as Lewis acid catalysts was evaluated to prepare cyanohydrins as precursors for the synthesis of biologically active compounds, and for aerobic oxidation of thiols to disulfides. The catalytic results showed that the derived MOFs exhibited modulatable Lewis acid capacities which are a function of the morphology, functionality of monodentate substituents present in the networks and a cooperative effect between metallic nodes of different nature.
Introduction
Hybrid materials are based not only on the combination of inorganic building units and organic linkers, but also on preformed organic–inorganic monomers. The resultant materials can be of interest for a wide range of applications such as catalysis, gas storage, gas separation, photochemistry, sensing and drug delivery.1–7 Advanced porous materials are built through different pathways such as sol–gel, solvothermal and hydrothermal synthesis processes which promote the synthesis of hybrid materials with a high surface area and free pore volume.8,9 Depending on the organic and inorganic precursors and the synthesis conditions, it is possible to synthesize periodic mesoporous organosilicas (PMOs), covalent organic frameworks (COFs) and metal–organic frameworks (MOFs) with different morphologies and levels of structuration.10–12 In the case of MOFs, the majority of materials grow and crystallize as 3D ordered structures with high homogeneity and regularity, forming low density porous architectures. Many examples of this kind of 3D metal–organic framework with well-known catalytic applications, such as MIL-53, MIL-101, UiO66 or UiO67, have been prepared and characterized, and their properties and reactivity have been explored for different reaction processes.13–17In contrast, there are not many cases where ordered 1D or 2D hybrid metal–organic materials were obtained. However, these types of frameworks would be useful because of the possibility to modify the space between individual chains or layers, facilitating the incorporation of new functions in the materials, without losing their accessibility. The first studies to generate 1D metal–organic materials were based on coordination polymers through the use of specific organic linkers to obtain materials formed from linear chains structured by successive assembly of metallic nanoclusters and organic units.18,19 In the last few years, new 2D metal–organic materials were obtained from different approaches based on the suitable combination between rare earth metallic nodes and specific organic linkers. MOFs formed from Zn layers, through the use of liquid–liquid biphasic systems, and lamellar Cu-MOFs obtained by reduction of reactants through the use of solvent mixtures as a synthesis medium are other examples of 2D metal–organic structures. Very recently, specific organic spacers that act as growth inhibitors of 3D metal–organic structures, being directly coordinated to the inorganic nodes to build the final 2D hybrid material, have also been described.20–24 Furthermore, the importance of using specific organic linkers in metal–organic architectures has been explored for modulating the physico-chemical behaviour of MOFs, improving their reactivity and uptake capacity.31,32 Even the effect of introducing organic linkers with different substituents into their composition has been studied to build metal–organic materials with still more specific properties. For instance, the possibility of increasing their effectiveness in adsorption–desorption processes and catalysis, modulating not only their reactivity, but also their activity and efficiency, together with their hydrophobic–hydrophilic nature has been explored. This effect is based on the modification of the electronic properties of the active sites present on the derived MOFs, improving the affinity of the hybrid catalysts towards some substrates, achieving better yields and selectivities.33,34
Additional synthetic strategies to build bi- or trimetallic metal–organic materials through the incorporation of additional metallic nodes into the framework to improve the characteristics of the pristine materials have been explored. One-pot or post-synthetic metal exchange reactions were carried out for obtaining the final multi-metal MOFs.35,36 Furthermore, the presence of structural defects in the framework together with the cooperative effects between several metallic nodes could improve the intrinsic reactivity of the metal–organic architectures, increasing their potentiality as effective and recoverable solid catalysts.37–40
Finally, even if MOFs have been widely studied as heterogeneous catalysts because of the high concentration of metal ions dispersed in a micro- or mesoporous network, their catalytic performance remains limited because of fully coordinated framework metal ions. Then research work has been directed towards generating defects, breaking metal–linker bonds and generating catalytically active sites or coordinatively unsaturated Lewis acid sites.41 Several tools were proposed such as post-synthetic treatment with inorganic acids that increases the number of Lewis and Brönsted acid sites,42 fast precipitation of materials,43 or the use of modulators that allow controlling the level and nature of defects.44 A modulator presents the same or a similar chemical functionality to the organic spacers typically used, but it has to be monodentate rather than bi- or polydentate to favour the generation of structural defects.
In this work, specific mono-benzylcarboxylate linkers with different substituents in the para-position, containing electron-withdrawing (nitro and bromine groups) and electron-donating groups (amine and ethyl groups), have been used as structural and modulator agents. The suitable combination with a metallic source and optimal solvothermal synthesis conditions, by the use of a solvent mixture with different polarity, promoted the generation of 1D organic–inorganic sub-domains which were auto-assembled to build novel metal–organic materials with several structuration levels, from 3D to 2D hybrid materials. The generation of vacancies, specific physico-chemical properties and differentiated morphology has been possible through the synthesis pathway described that has been directed towards modulating the reactivity of the solids. Moreover, bimetallic metal–organic materials have been synthesized here by a one-pot direct synthesis, and additional metallic nodes have been incorporated without modification of the structural framework.
These metal–organic materials prepared have been characterized by XRD, chemical analysis, TGA, NMR and IR spectroscopies, and FESEM/TEM microscopy in order to study their physico-chemical properties to establish structure–activity correlation. In this sense, the textural properties have been analyzed through CO2 adsorption together with their uptake capacity. Finally, the reactivity of these hybrid materials as Lewis acid catalysts has been demonstrated during the synthesis of several biologically active compounds, and for the aerobic oxidation of thiols to disulfides. The catalytic results showed that the derived MOFs exhibit modulatable acid capacities as a function of the morphology and monodentate functional substituents present in their networks, which modify the final activity of Lewis acid active sites.
Experimental
Chemicals
3D Al-metal–organic materials were obtained using 1,4-benzenedicarboxylic acid (BDC, Aldrich), 4-bromobenzoic acid (BrB, Aldrich) and 4-nitrobenzoic acid (NB, Aldrich) as organic sources. Lamellar hybrid materials were obtained using 4-ethylbenzoic acid (EB, Aldrich) and 4-aminobenzoic acid (AB, Aldrich) as organic sources. Aluminium nitrate nonahydrate (Al(NO3)3·9H2O, Aldrich) and iron nitrate nonahydrate (Fe(NO3)3·9H2O, Aldrich) were used as inorganic sources. A mixture of Milli-Q water/DMF (dimethylformamide, Across) was employed as solvent during the solvothermal process. Finally, the elimination of unreacted linkers and excess of water/DMF molecules occluded in the solids was carried out by washing with methanol.Synthesis of Al-metal–organic materials
Catalytic tests
A typical procedure for cyanosilylation of carbonyl compounds is as follows: the catalyst (0.5 mol% Al) was added into a reactor vessel (2 mL), containing benzaldehyde (5 mmol) and TMSCN (trimethylsilylcyanide, 6 mmol). After that, the reaction mixture was vigorously stirred at 30 °C under a N2 atmosphere.Oxidation of thiophenol was performed in a reactor vessel (2 mL) which was charged with an adequate amount of catalyst (10 mol% Fe) in 1 mL of acetonitrile, adding 0.48 mmol of thiophenol. The reaction mixture was stirred at 70 °C under an O2 atmosphere.
The reactions were followed by gas chromatography analysis using a Varian 3900 equipped with a Sapiens 5MS column (30 m × 0.25 mm × 0.25 μm), being operated at an injector temperature of 280 °C and a detector temperature (FID) of 300 °C.
Characterization techniques
XRD analysis was carried out with a Philips X'PERT diffractometer equipped with a proportional detector and a secondary graphite monochromator. Data were collected stepwise over the 2° ≤ 2θ ≤ 20° angular region, with steps of 0.02° 2θ, 20 s per step accumulation time and CuKα (λ = 1.54178 Å) radiation. Transmission electron microscopy (TEM) micrographs were obtained with a JEOL JEM2100F electron microscope operating at 200 keV. The samples were prepared directly by dispersing the powders onto carbon copper grids. Field-emission scanning electron microscopy (FESEM) micrographs were recorded on a ZEISS Ultra 55 microscope operating at 2 kV, with a 2 × 10−9 A beam current and 2.5 mm as the working distance. C, N and H contents were determined with a Carlo Erba 1106 elemental analyser, while the Al content was obtained by means of atomic absorption spectroscopy (Spectra AA 10 Plus, Varian). Thermogravimetric and differential thermal analyses (TGA-DTA) were conducted in an air stream with a Mettler Toledo TGA/SDTA 851E analyser. IR spectra were obtained on a Nicolet 710 spectrometer (4 cm−1 resolution), using a conventional greaseless cell. Wafers of 10 mg cm−2 were outgassed at 100 °C overnight. Solid state MAS-NMR spectra were recorded at room temperature on a Bruker AV-400 III HD spectrometer. 27Al spectra were obtained using a 4 mm Bruker BL-4 probe. Pulses of 0.5 μs to flip the magnetization π/20 rad and a recycle delay of 1 s were used. The 1H to 13C cross-polarization (CP) spectra were acquired by using a 90° pulse for 1H of 5 μs, a contact time of 2 ms, and a recycle delay of 3 s. The 13C spectra were recorded with a 4 mm Bruker BL-4 probe at a sample spinning rate of 10 kHz. The 1H to 27Al cross-polarization (CP) spectra were acquired with a Bruker 3.2 mm probe using a rf-field for 27Al of 8 kHz, spinning the sample at 16 kHz, a contact time of 1 ms, and a recycle delay of 3 s.13C and 27Al were referenced to adamantane and an aqueous solution of Al(NO3)3, respectively.Results and discussion
Synthesis and characterization
The methodology used to obtain derived metal–organic materials with different structuration levels, formed by coordinated auto-assembly of monodentate organic and polyhedral inorganic builder units, is based not only on the use of specific arylic spacers with only one terminal carboxylate reactive group containing additional functional groups, but also on selecting the more suitable solvothermal synthesis conditions (temperature, pressure, time or solvent nature). Specifically, benzene monocarboxylate compounds with different functional substituents in the para position were used as organic spacers, which are connected to straight chains of metal-centred octahedra, AlO4(OH)2, forming 1D organic–inorganic nanoribbons (Scheme 1). In these sub-structures, octahedral units would be linked together via two hydroxyl groups located in trans positions, the other positions being coordinatively connected with up to four monodentate spacers, shared with adjacent octahedra, following a well-known Kagomé net structural formation for trivalent metallic nodes.45,46 This type of 1D sub-domain allowed obtaining derived MOF architectures with (organic spacer number/aluminium nodes) ratios between 0.5 and 2, depending on whether one to four molecules of monodentate ligands are coordinated to each inorganic node (Table 1). | ||
| Scheme 1 Synthesis routes to obtain Al-MOF-type materials with different morphologies, based on associated 1D organic–inorganic nanoribbons, depending on functional organic spacers used during the solvothermal process. At the bottom of the illustration, details of 1D sub-domains are shown with a (organic spacer number/Al) ratio of 1 for clarity (in blue, chain based on associated aluminium octahedra following a Kagomé conformation; in grey, ethylbenzoate monodentate spacers; in red, oxygen atoms from carboxylate groups). | ||
| Samples | Ca | Ha | Na | Org. cont.a | Ala | Fea | C/NExp | C/NTheor | Organic spacer no./Al | |
|---|---|---|---|---|---|---|---|---|---|---|
| CHNb | TGAc | |||||||||
| a Percentage in weight (% wt). b Organic content from CHNS elemental analysis. c Organic content from thermogravimetric analysis without taking into account hydration water. | ||||||||||
| MIL-53(Al) | 43.8 | 2.6 | 0 | 46.4 | 72.6 | 12.0 | — | — | — | 1.0 |
| Al-ITQ-Br | 37.0 | 2.3 | 0 | 39.3 | 88.0 | 7.8 | — | — | — | 1.5 |
| Al-ITQ-NO2 | 45.7 | 2.9 | 7.0 | 55.6 | 83.1 | 6.8 | — | 7.6 | 7 | 2.1 |
| Al/Fe-ITQ-NO2 | 37.2 | 2.7 | 6.1 | 46.0 | 6.9 | 3.6 | 7.1 | 7 | 1.7 | |
| L-MOF-EB | 61.5 | 5.6 | 0 | 67.1 | 84.4 | 9.0 | — | — | — | 1.7 |
| L-MOF-AB | 31.8 | 3.6 | 4.7 | 40.1 | 64.0 | 12.6 | — | 7.8 | 7 | 0.8 |
Additionally, we report here the importance of the selection of organic spacers together with suitable solvothermal synthesis conditions. Indeed, we show the effect of different organic spacers over not only the structuration level of metal–organic materials, but also over the reactivity of the synthesised materials. We have prepared new metal–organic materials selecting four organic spacers derived from benzoic acid with different functional groups located in the para position: amino, ethyl, bromine and nitro groups.
Firstly, two electron-donating groups (EDG), amine and ethyl, which donate electron density to the conjugated π system via resonance or inductive effects, making the aromatic linkers more nucleophilic, were chosen. With the incorporation of these two groups, we expected to increase the negative charge density of the carboxylic groups. Consequently, the coordinatively unsaturated metal sites (CUS) present on the polyhedral builder units of the metal–organic materials were expected to exhibit lower Lewis acid strength. Moreover, from a structural point of view, one has to take into account the fact that although amino and ethyl groups present different chemical properties, the final morphology achieved should be similar. Because of the presence of nitrogen atoms, amino groups are able to establish hydrogen bonds between themselves. Then, we can expect that a great order and organization level between organic spacers during the synthesis process will occur. Ethyl groups have a tendency to stay straight and pack neatly by van der Waals forces. So, in both cases, we can predict an elevated organization level and order by intercalation of monodentate organic spacers containing amino and ethyl groups.
Secondly, we chose linkers containing electron withdrawing groups (EWGs), nitro and bromine, which remove electron density from a conjugated π system, making it more electrophilic. With the incorporation of these two groups, we can expect an opposite effect to that in the case of EDG, such as a decrease of the negative charge density in the carboxylic groups of the organic spacers. Consequently, the coordinatively unsaturated metal sites (CUS) included in the metallic nodes of MOF-type materials should exhibit higher Lewis acid strength. Moreover, we have to take into account that nitro and bromine groups, because of their electron withdrawing character and electronegativity, present high charge density. Because of this, we can predict the existence of electric fields and random forces between the monodentate organic spacers themselves, inducing disorder in the formed sub-domains, being more compacted and forming low-ordering metal–organic 3D networks.
Experimental results evidenced that the obtained solids, after the solvothermal process, exhibited different structuration levels and morphologies due to the specific properties and polarity of the functional substituent groups present within the organic spacers, and their affinity with the solvent mixture used for the synthesis (Milli-Q water/DMF). Specifically, 3D Al-metal–organic-type materials (Al-ITQ-Br and Al-ITQ-NO2) were obtained when organic spacers with electron-withdrawing groups were incorporated in the materials, i.e. when bromine or nitro substituents were present in the para position (–Br or –NO2). This fact would be associated with the facility to auto-assemble organic spacers with octahedral aluminium nodes due to the higher polar nature of organic spacers which are easily stabilized in the water/DMF synthesis medium. Furthermore, as we predicted, the presence of electric fields and derived random forces generated by the use of organic spacers with functional groups with high charge density produced compacted metal–organic frameworks with 3D structuration and low ordering between organic and inorganic builders. The structuration of the derived 3D organic–inorganic MOF-type materials present XRD patterns similar to those of 3D standard MIL-53(Al) (Fig. 1). It is important to point out that Al-metal–organic materials containing nitro groups (Al-ITQ-NO2) in the organic spacers were obtained with an enhanced crystallinity, probably due to higher regularity achieved in the distribution of organic–inorganic sub-domains compared with materials with bromine groups.
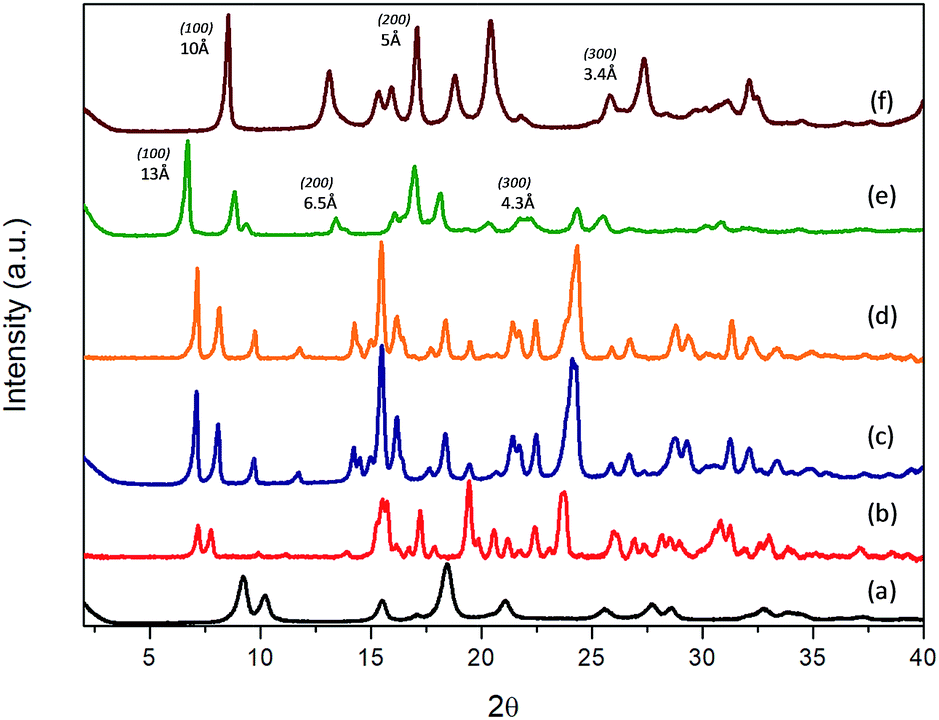 | ||
| Fig. 1 XRD patterns of the derived Al-metal–organic materials: (a) 3D standard MIL-53(Al), (b) Al-ITQ-Br, (c) Al-ITQ-NO2, (d) Al/Fe-ITQ-NO2, (e) L-MOF-EB and (f) L-MOF-AB. | ||
From a point of view of potential applications, the preparation of mixed-metal–organic materials could perform catalytic transformations where each of the catalytically active metals plays a different role or they simply act cooperatively. Then, we explored here the synthesis of Al/Fe bimetallic metal–organic materials, using monocarboxylic acid with nitro functional groups in the para position as an organic spacer and metal sources of Al and Fe. In this case, the preparation of Al/Fe-mixed-metal–organic materials in one step was successfully accomplished following the same synthesis process. So, an Al/Fe metal–organic material with similar 3D structuration was obtained with the effective incorporation of secondary metallic nodes without a significant modification in the organic–inorganic architecture. Indeed, XRD patterns of Al-ITQ-Br, Al-ITQ-NO2 and Al/Fe-ITQ-NO2 were similar (Fig. 1).
On the other hand, when electron-donating functional groups (–NH2) or alkyl groups (–C2H5) were present in the monodentate organic spacers in the para position, 2D materials with a lamellar morphology based on associated octahedral aluminium 1D chains separated by ethylbenzyl (EB) or aminobenzyl (AB) units, normally perpendicularly located to inorganic nodes, were obtained (L-MOF-EB and L-MOF-AB). XRD diffraction patterns clearly exhibited a (100) low angle diffraction band, characteristic of solids with a layered order, formed from individual sheets perpendicularly disposed to the axis a. The lamellar materials showed different basal space due to the variation in the separation between individual layers, depending on the pending functional groups present in the organic spacers. Basically, individual layers were formed from associated 1D organic–inorganic Al-nanoribbons associated between them, favouring bi-dimensional structuration, separated by organic groups (EB and AB) located in the interlayer space. In this case, probably the aluminium octahedral chains are organized along the plane bc, following a Kagomé system and forming sheets composed of hexagonal rings delimited by six triangular rings (Fig. S1†).47 Furthermore, from XRD patterns, it was possible to detect not only the (100) diffraction band, but also further bands assigned to (200) and (300) reflections, confirming the regular and homogeneous separation existing between successive organic–inorganic layers.
Specifically, (100) diffraction bands were observed at ∼13 Å and ∼10 Å for lamellar materials L-MOF-EB and L-MOF-AB, respectively (Fig. 1). Considering that the molecular lengths of the organic spacers are 6.3 Å and 5.4 Å for ethylbenzoate (EB) and aminobenzoate (AB) groups, respectively, and they were directly bonded to 1D Al-chains, it would be possible to estimate the thickness of each individual chain, being close to 4–5 Å, which is coincident with the molecular dimensions of one octahedral aluminium cluster. Moreover, the concentration of organic spacers around the Al-chains can be different depending on the lamellar hybrid material, this fact being corroborated by elemental CHNS analysis, and it will be presented later. As discussed above, the intermolecular interactions of ligands are of great importance, and the presence of hydrogen bonds in the case of amine groups or van der Waals forces in the case of ethyl groups is determinant for the structuration and order achieved in the metal–organic materials. In fact, ethyl chains have a tendency to stay linear due to specific inter-linker interactions that generated hybrid lamellar structures with a marked 2D spatial order.
Therefore, XRD results show that the electronic and polar properties of functional pendant groups present in the organic spacers in para positions are a key factor to modulate the morphology in the hybrid metal–organic materials obtained, even when using the same solvothermal synthesis conditions. In the case of electron-withdrawing groups incorporated in the derived Al-MOFs, the synthesis process was completed in shorter times (24 hours), probably because bromine and nitro benzyl carboxylate spacers are easily and rapidly dispersed in the water/DMF solvothermal medium, together with the inorganic counterparts, obtaining finally a more compact 3D hybrid architecture where organic and inorganic counterparts are close to each other and distributed along the formed framework. This fact is also favoured by the high charge density of EWG that facilitates electronic fields and random forces between organic linkers, not producing ordered and compacted 3D hybrid architectures. Moreover, it was possible to incorporate a secondary metallic node, based on iron polyhedrons, in this 3D Al-MOF-type structure containing nitro groups, without altering the pristine structure.
However, when ethylbenzoate or aminobenzoate moieties were introduced in the solvothermal process, longer synthesis times were necessary to obtain solid materials. In this case, an interphase water/DMF is generated in the synthesis medium that promotes a slower interaction between the building units, obtaining metal–organic materials with a lamellar organization in which individual sheets, based on associated organic–inorganic nanoribbons, are separated in an ordered way by the organic spacers located in the interlayer region, also favoured by hydrogen bonds and van der Waals interactions between them. In fact, XRD patterns of 3D Al-ITQ-NO2, Al-ITQ-Br and Al/Fe-ITQ-NO2 materials were more similar to those of 3D standard MIL-53(Al) than to those of 2D L-MOF-EB and L-MOF-AB materials. Therefore, depending on the organic spacers used during the solvothermal synthesis process, two completely different structuration levels were obtained, 3D or 2D, although all of them are based on the assembled 1D organic–inorganic nanoribbons (Scheme 1).
The morphology proposed above for the two different families of hybrid materials (2D and 3D) was confirmed by EM images (FESEM and TEM). Fig. 2 shows FESEM micrographs of the 3D materials prepared using electron-withdrawing substituents (–NO2 and –Br) which were incorporated in the framework of metal–organic hybrid materials. In these cases, the existence of 3D crystallites was observed with a long square morphology, indicating a more robust and compact topology. Furthermore, FESEM images confirmed that Al-ITQ-NO2, Al-ITQ-Br and Al/Fe-ITQ-NO2 materials exhibited a similar morphology, this being in agreement with the XRD patterns obtained characteristic of more compacted metal–organic frameworks, similar to conventional MIL-53(Al) MOF-type materials. Additionally, a mapping analysis (EDS-TEM) was performed in order to confirm the homogeneous distribution of the two types of metallic nodes, Al and Fe, present in the as-synthesized bimetallic metal–organic materials along the hybrid network (Fig. 3).
 | ||
| Fig. 2 FESEM images of 3D Al-MOFs: (a) and (b) Al-ITQ-Br, (c) and (d) Al-ITQ-NO2 and (e) and (f) Al/Fe-ITQ-NO2. Scale bars correspond to 1 μm for (a), (c), (e) and (f) micrographs, and 200 nm for (b) and (d) micrographs. | ||
 | ||
| Fig. 3 EDS mapping of metallic elements in 3D Al/Fe-ITQ-NO2. | ||
On the other hand, Fig. 4 shows TEM images of the samples obtained when electron-donating amino or alkyl groups were incorporated in the final Al-metal–organic materials. In these cases, it was possible to confirm the lamellar morphology of L-MOF-EB and L-MOF-AB solids. Specifically, layered materials containing ethylbenzoate groups are based on plate-like crystals overlapped between them, forming agglomerated blocks. For L-MOF-AB materials, similar lamellar structuration was observed, it being even possible to detect the presence of ordered piled individual layers, indicating the lamellar character of these derived Al-MOF-type materials.
 | ||
| Fig. 4 TEM images of 2D Al-MOFs: (a) and (b) L-MOF-EB and (c) and (d) L-MOF-AB. Scale bars correspond to 5 μm, 200 nm, 1 μm and 100 nm for (a), (b), (c) and (d) micrographs, respectively. | ||
From elemental CHNS analysis (Table 1) of Al- and Al/Fe metal–organic materials with different structuration levels, organic content was estimated due to the presence of the functionalized organic spacers in the network of hybrid materials. The results indicated that organic linkers are effectively included into the structure of the MOF-type materials. For Al-metal–organics materials with electron-withdrawing groups (–Br and –NO2) in their frameworks (Al-ITQ-Br and Al-ITQ-NO2), the organic content was comparable to that included in 3D standard MIL-53(Al) MOFs (∼40% wt), and similar values of organic content were observed in the bimetallic Al/Fe metal–organic material. These results would corroborate the similar morphology achieved between Al-ITQ-NO2 and Al-ITQ-Br materials, where compacted auto-assembly of 1D associated nanoribbons, formed from octahedral aluminium chains and monodentate organic spacers, occurred to build this type of 3D topology of hybrid materials.
Unlike these 3D structures, lamellar hybrid materials presented different organic contents. Specifically, L-MOF-EB exhibited the highest organic content (∼60% wt) due to the elevated incorporation of organic spacers with a greater number of carbon atoms in the hydrocarbon chain linked to benzoate groups, which were coordinated to aluminum octahedral chains, forming ordered 1D sub-domains. This result would confirm the higher organic content present in this lamellar hybrid material containing ethyl-benzyl groups. In contrast, L-MOF-AB presented the lowest organic content (∼30% wt) due to more reduced incorporation of amino-benzyl spacer groups located between individual layers of the lamellar material. However, in both cases, the organic spacer number/Al ratio was between 1 and 2, indicating that the monodentate spacers were located on both sides of the associated aluminum octahedra, facilitating lamellar structuration (Table 1).
Regarding the nitrogen content, it was almost zero for materials where organic spacers did not contain nitrogen in their composition. For Al-ITQ-NO2, Al/Fe-ITQ-NO2 and L-MOF-AB materials, which contained nitro and amino benzylic groups, the C/N experimental molar ratios were close to the theoretical values, confirming the effective incorporation of these organic spacers into the framework of the hybrid materials, preserving their initial composition.
TG and DTA curves of the MOF-type solids are shown in Fig. S2,† evidencing the weight loss for the corresponding organic spacers incorporated into the Al and Al/Fe metal–organic hybrid materials and their hydrothermal stability. Without considering the hydration water and the residual DMF occluded in the materials (weight loss detected approximately at 80–150 °C), two main weight losses were clearly identified in all cases. The first weight loss (300–400 °C) was assigned not only to the functional substituents present in the organic spacers, but also to the dehydration phenomenon due to the decomposition of AlO4(OH)2 octahedra present in the 1D inorganic Al-chains. This fact would contribute to explaining the higher organic content estimated from TGA compared with CHNS analysis associated with the oxygen elimination from [AlO6] octahedral units. In the temperature range between 450 °C and 600 °C, the last weight loss was detected, being assigned to aromatic fragments present in the monodentate organic spacers, incorporated in the framework, which exhibited the highest thermal stability.
Spectroscopic characterization
Fig. 5 shows the 13C CP/MAS NMR spectra of Al and Al/Fe metal–organic hybrid materials, containing different functionalized organic spacers. The total integrity of the organic spacers in the hybrid materials was corroborated through the assignment of all carbon atoms to different chemical shifts, even the carbon atoms bonded directly to functional substituents. The results showed that chemical shifts located between 110 and 160 ppm were due to carbon atoms from aromatic rings, the signal band being detected at ∼170 ppm associated with aryl-carboxylate groups. Moreover, the spectra showed different bands at low chemical shifts, from 10 to 40 ppm, which can be assigned to carbon atoms –CH2– from the alkyl chain of the ethyl benzyl organic spacers, in the particular case of L-MOF-EB materials. In this last range, chemical shifts with reduced intensity were also detected due to the presence of residual DMF used during the solvothermal synthesis process and some MeOH molecules occluded into the hybrid materials, due to the successive washing steps. Taking this into account, NMR spectra confirmed the total integrity of the monocarboxylate para substituted benzyl groups, incorporated in the material after the solvothermal synthesis process and their effective coordination to aluminum inorganic chains.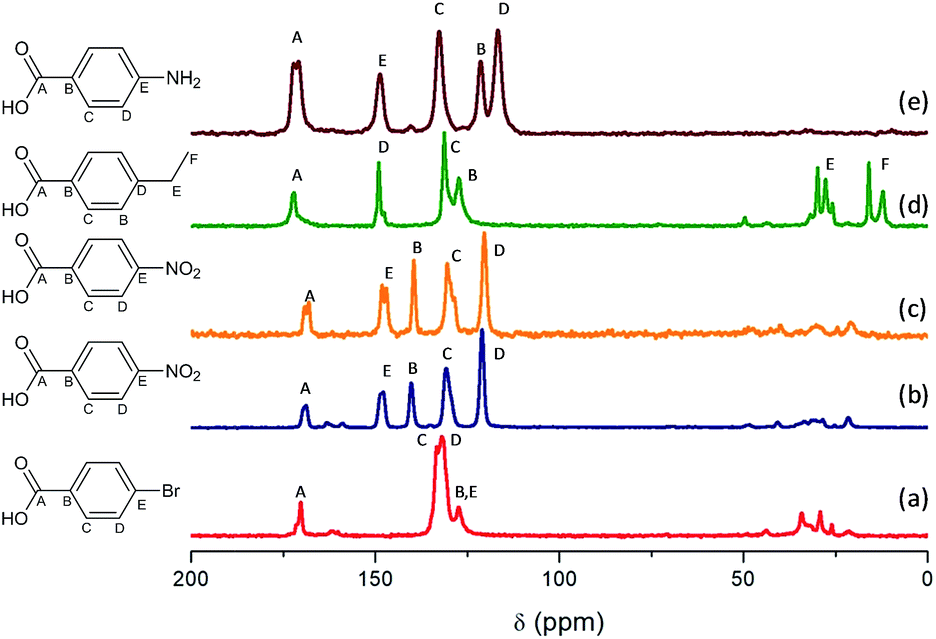 | ||
| Fig. 5 13C MAS NMR spectra: (a) Al-ITQ-Br, (b) Al-ITQ-NO2, (c) Al/Fe-ITQ-NO2, (d) L-MOF-EB and (e) L-MOF-AB. | ||
On the other hand, 27Al NMR spectra show the chemical environment of aluminum in the structure of hybrid metal–organic materials (Fig. 6). From all spectra, a single resonance signal was detected with an isotropic shift around 0 ppm which is in agreement with only one type of [AlO6] octahedron present in the crystal structure for all materials, due to aluminium(oxo)hydroxide species contained in the 1D nanoribbons. Furthermore, a shoulder in the band was detected which is dominated by a second order quadrupole interaction due to the aqueous synthesis medium used during the solvothermal process. This effect is characteristic of solvated octahedral aluminium interacting with water molecules, present in the synthesis medium that favours broad chemical shift signals because there is an electric field gradient at the aluminium structural nodes.48
 | ||
| Fig. 6 27Al MAS NMR spectra: (a) Al-ITQ-Br, (b) Al-ITQ-NO2, (c) Al/Fe-ITQ-NO2, (d) L-MOF-EB and (e) L-MOF-AB. | ||
The IR spectra of the organic–inorganic hybrid metal–organic materials are shown in Fig. 7. In all cases, several signals between 1400 and 1700 cm−1 were detected due to the carboxylate groups from organic spacers attached to 1D Al inorganic chains. Specifically, some bands in this infrared range were assigned to carboxylate asymmetric stretching vibrations (1600–1550 cm−1) and at lower wavenumbers (1450–1400 cm−1) to carboxylate symmetric stretching vibrations. In the same range, vibration bands due to (C–C) units from aromatic rings of monodentate organic ligands were also detected. A wide vibration band was observed between 3600 and 3200 cm−1 due to hydroxyl groups (–OH) from hydration water and hydrated aluminum octahedra present in the 1D nanoribbons (AlO4(OH)2). Specifically, the L-MOF-AB material showed two bands at 3450 and 3370 cm−1 assigned to amino groups incorporated in the interlamellar space from the organic spacers. The aliphatic chain (–CH2) from the ethyl monocarboxylate benzylic group (EB) introduced into L-MOF-EB materials was identified by two signals located at 2960 and 2930 cm−1, confirming the incorporation of this organic spacer. Furthermore, symmetric stretching bands assigned to nitro groups present in Al-ITQ-NO2 and Al/Fe-ITQ-NO2 solids were also detected at ∼1300 cm−1. Therefore, IR spectroscopy corroborated the integrity of the organic compounds to form 3D and 2D metal–organic derived materials, together with NMR results described above.
 | ||
| Fig. 7 IR spectra: (a) Al-ITQ-Br, (b) Al-ITQ-NO2, (c) Al/Fe-ITQ-NO2, (d) L-MOF-EB and (e) L-MOF-AB. On the left, infrared spectra in the hydroxyl stretching region. On the right, infrared spectra in the framework region. | ||
FTIR spectroscopy with CO adsorption was used to verify the Lewis acidity of the hybrid metal–organic structures.49,50 Specifically, in situ IR CO adsorption at low temperature (100 K) was used to obtain information about Lewis acid sites present in the framework of the hybrid materials (Fig. S3†). In all cases, an absorption band between 2130 and 2150 cm−1 was detected, independently of the solid studied, which confirms the presence of weak Lewis acid sites in the frameworks that can be useful for catalysis, as will be shown later. CO adsorption was a reversible process, and the CO adsorption band gradually disappeared after subsequent treatment of the samples at 100 K in a vacuum.
Textural properties
The textural properties of the different Al and Al/Fe metal–organic hybrid materials were studied from CO2 adsorption isotherms (Fig. 8 and Table S1†). It is important to consider that the introduction of different functionalities through the monodentate organic spacers included in the framework and the obtained morphologies (3D and 2D) should influence the adsorption properties of the hybrid materials. The CO2 adsorption properties were investigated at 0 °C up to 1 bar and, in all cases, type-II isotherms, characteristic of solids with reduced porosity, were obtained. In general, in these hybrid metal–organic materials, the high concentration of organic spacers located in the framework of the solids did not favour the generation of free porous cavities with high dimensions. Furthermore, the marked hydrophobic character of the solids made difficult the optimal gas adsorption. All these factors hinder the correct estimation of the textural properties and gas uptake capacities of the derived MOF material based on 1D sub-domains. However, higher values of CO2 uptake were obtained for the 3D materials compared with the lamellar frameworks, probably due to the high contribution of free pores associated with lower steric hindrance. In particular, the highest CO2 uptake capacity was achieved for Al-ITQ-NO2 materials (V = 24.3 cm3 g−1), being lower for Al-ITQ-Br (V = 14.7 cm3 g−1) due to the bulkier bromine groups compared to –NO2 groups. For lamellar materials, a lower CO2 uptake was always obtained because the interlayer space is completely occupied by organic spacers, and CO2 molecules should present diffusional problems to be adsorbed inside the materials. More specifically, the lowest value was found for L-MOF-EB (V = 4.8 cm3 g−1) due to the higher concentration of organic content detected in this type of hybrid material which would avoid the CO2 diffusion into the free pores. | ||
| Fig. 8 CO2 isotherms at 0 °C up to 1 bar of Al and Al/Fe-metal–organic materials. | ||
Catalytic activity
Metal–organic materials have been widely studied as heterogeneous catalysts since they offer numerous advantages such as a large number of different active centres in the hybrid frameworks as metallic clusters, functionalized organic linkers, and a large surface area and pore volume, along with their impressive topological variety, maintained under reaction conditions, which do not compromise their crystal stability. More specifically, metal–organic frameworks have been successfully developed as effective heterogeneous catalysts with coordinatively unsaturated metal sites (CUS) as Lewis acid active sites.50,51In this work, five hybrid materials with different topologies were synthesized through the use of four different functional monocarboxylate organic spacers, which should promote the generation of metal free centres as structural defects. As we discussed above, we expect that the different chemical properties of the chosen organic spacers influence the strength of Lewis acid sites. With the incorporation of electron-donating groups (EDG), amine and ethyl groups that donate electron density to a conjugated π system via resonance or inductive effects, thus making the π system more nucleophilic, we hope to increase the negative charge density of the carboxylic group and so decrease the electrophilicity of the coordinated Al-atom. In other words, the CUS or the Lewis acid sites are expected to exhibit lower Lewis acid strength. On the other hand, with the incorporation of electron-withdrawing groups (EWG), nitro and bromine groups, that remove electron density from the π system making it more electrophilic, we would expect a decrease of the negative charge density of the carboxylic group and so an increase of the electrophilicity of the coordinated Al-atom. Then, CUS or the Lewis acid sites will exhibit higher Lewis acid strength. Together with the difference in the strength of Lewis acid sites generated in the different materials, we also have to consider the different structuration or framework morphology of the materials and thus their textural properties and nature, such as the hydrophobicity/hydrophilicity of the catalytic surface, area, porosity and accessibility towards active sites.
The use of monocarboxylate linkers can afford architectures with a high number of coordinatively unsaturated sites or CUS and thus materials with higher catalytic performance. In order to discuss the importance of the choice of an appropriate monodentate organic spacer, we compared the catalytic behaviour of the different metal–organic materials for the production of cyanohydrins by cyanosilylation of carbonyl compounds that constitute a potential synthetic route to produce derived cyano-compounds through C–C bond reactions. Cyanohydrins are useful as intermediates in the synthesis of biological compounds such as α-hydroxy acids, α-hydroxyl ketones and α-amino-acids.52–54 Several studies showed that the reaction is catalysed by homogeneous catalysts such as metallic Lewis acids/bases, inorganic solid acids/bases and non-metallic organic molecules,54 that presented typical drawbacks such as difficulty in separation and recyclability of the catalyst. Different MOFs such as HKUST-1, V-MIL-47, Zr-UiO-66, Cr-MIL-101, Al-MIL-53 and In-MOFs showed suitable catalytic properties to carry out the cyanosilylation, and high yields and selectivity, comparable to those of other solid catalysts like acid zeolites or highly siliceous mesoporous materials, could be achieved.25–30,55
With the aim of developing clean and environmentally friendly reaction profile in agreement with green chemistry processes,56 cyanosilylation was tested under solvent-free conditions. Specifically, a solvent-free reaction presents some advantages such as less by-product generation, energy consumption reduction and purification step elimination. Thus, the catalytic properties of the different MOF-type materials were evaluated to perform the cyanosilylation with TMSCN and with benzaldehyde as the standard molecule, under solvent-free conditions (Scheme 2).
 | ||
| Scheme 2 Cyanosilylation of benzaldehyde. | ||
As shown in Fig. 9, the rate of reaction is considerably enhanced when the reaction was carried out in the presence of more compacted 3D materials with electron-withdrawing groups on the linker, Al-ITQ-NO2 and Al-ITQ-Br, reaching full conversion after 5 and 7 hours, respectively (Table 2). Therefore, different aspects have to be considered since at the same time two important factors are modified such as the morphology framework, 2D versus 3D, and the electron-donating versus electron-withdrawing character of the spacers. When we compare the catalytic performance of both 3D-structured materials, we can conclude that the more electron-withdrawing organic spacer contributes to the formation of more active Lewis acid sites, Al-ITQ-NO2 being the most active catalyst in comparison to Al-ITQ-Br. This fact was confirmed by plotting the logarithm of the relative initial reaction rates versus the σpara Hammett constant of each substituent on the benzoate spacer (Fig. S4†).34
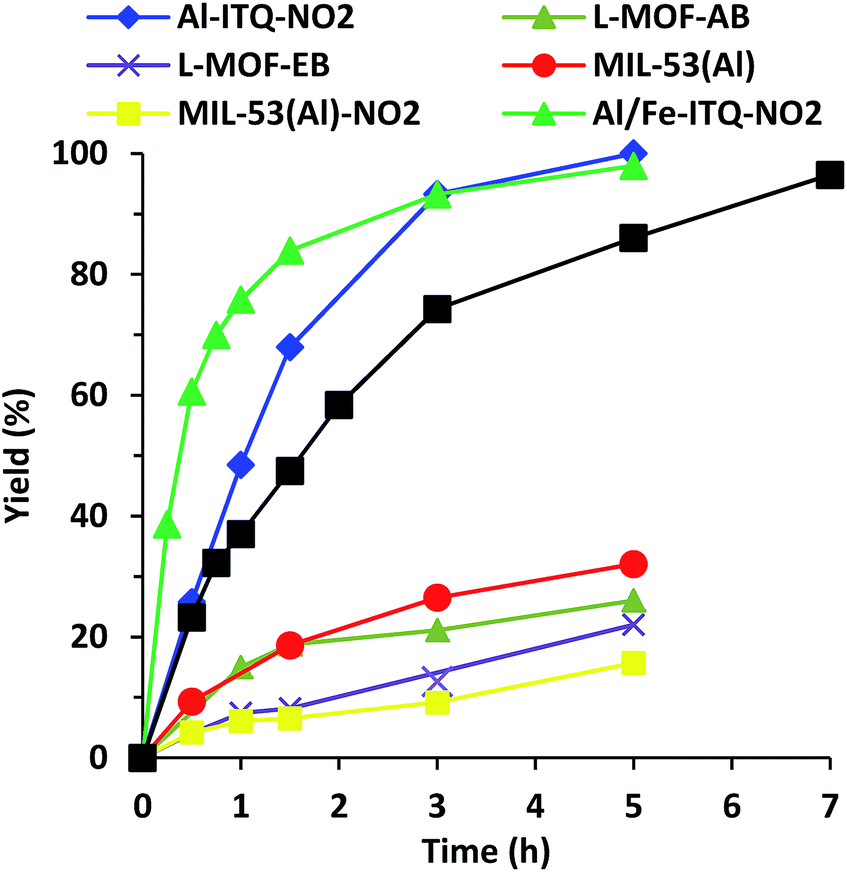 | ||
| Fig. 9 Kinetics for cyanohydrins production using Al-ITQ-NO2, Al/Fe-ITQ-NO2, Al-ITQ-Br, L-MOF-AB, L-MOF-EB, MIL-53(Al) and MIL-53(Al)–NO2 as solid catalysts (0.5 mol% Al). Blank test was performed, obtaining a yield of 24% at 5 h. | ||
| Samples | Yield (%) | r 0 (mol L−1 h−1) | TON | TOFrob (h−1) | TOFc (h−1) | TOFd (h−1) |
|---|---|---|---|---|---|---|
| a Reaction conditions: benzaldehyde (5 mmol), TMSCN (6 mmol), 0.5 mol% Al, at 30 °C, 5 h. b Calculated at the initial rate. c Calculated at 0.5 h. d Calculated at 3 h. | ||||||
| Al-ITQ-NO2 | 100 | 2.7 | 200 | 137 | 103 | 62 |
| Al/Fe-ITQ-NO2 | 98 | 8.0 | 154 | 410 | 242 | 62 |
| Al-ITQ-Br | 88 | 1.8 | 193 | 92 | 93 | 49 |
| L-MOF-AB | 26 | 0.6 | 52 | 30 | 36 | 14 |
| L-MOF-EB | 22 | 0.3 | 44 | 15 | 17 | 8 |
| MIL-53(Al) | 32 | 0.6 | 64 | 30 | 37 | 17 |
| MIL-53(Al)-NO2 | 15 | 0.2 | 30 | 10 | 17 | 6 |
| 4-Nitrobenzoic acid (0.5 mol%) | 24 | — | — | — | — | — |
| Blank | 24 | — | — | — | — | — |
Nevertheless, when the cyanosilylation was carried out in the presence of lamellar metal–organic materials, L-MOF-EB and L-MOF-AB, low yields close to 20% were reached after a reaction time of 5 hours. The poor catalytic performance of both 2D-metal–organic materials can be attributed not only to the lower Lewis acid strength of the generated CUS, because of the electron-donating character of the organic spacers, but also to the lamellar structuration of these materials. Indeed, in the interlamellar space, well-ordered ligands by intermolecular interactions via van der Waals forces and/or nitrogen bonds are present. This organization of the interlayer space conduces to reducing free porous volume (Fig. 8) and the material morphology is a key factor to perform the reaction since lamellar materials present great diffusional restrictions. The 3D materials are the most active catalysts because they combine higher accessibility to active sites and the most active Lewis acid sites because of the presence of electron-withdrawing ligands in their framework.
We compared the catalytic performances of the synthesised materials with monocarboxylate spacers with the corresponding one of 3D standard MIL-53(Al) which incorporates terephthalic acid linkers to show the benefit of the use of monodentate functional ligands. When, the cyanosilylation was carried out in the presence of 3D standard MIL-53(Al), a low 32% yield is obtained after 5 hours of reaction. Even MIL-53(Al) containing nitro groups in the bidentate organic linkers exhibited still lower yields and TON values than Al-ITQ-NO2 solids probably due the reduced number of CUS-type acid sites present in the structure of conventional 3D MOFs that exhibit high crystallinity (Fig. 9 and Table 2). This result showed that the 3D Al-ITQ-NO2 material exhibits higher catalytic properties not only because of the electron-withdrawing character of its ligand that contributes to increasing the Lewis acid strength, but also because of the use of monocarboxylate organic spacers, in the solvothermal process, that favour the formation of structural vacancies and a higher number of coordinatively unsaturated sites in the final hybrid framework.
To explore the stability of the Al-ITQ-NO2 structure under reaction conditions, and discarding the fact that active sites migrate from the solid to the liquid phase and could be responsible, at least in part, for the catalytic activity, a leaching test filtering off the solid catalyst after 1.5 h of reaction was carried out. After that, the reaction was continued and samples were taken until a reaction time of 5 hours. Fig. S5† shows that the yield did not increase substantially during this time. Furthermore, when the reaction was carried out with 0.5 mol% of 4-nitrobenzoic acid in order to study the capacity of the carboxylic acid to be active in the reaction, a yield of only 24% was achieved after 5 hours of reaction. Definitively, both experiments proved that the activity was due to Al-ITQ-NO2, confirming the heterogeneity of the reaction. In addition, a blank test without the solid catalyst was performed, and low yields (24%) were achieved with the thermal reaction, indicating the active role of the hybrid material Al-ITQ-NO2 to produce cyanohydrins (Table 2).
Another critical aspect of the use of solid catalysts is the stability of their catalytic properties and structure during recycles. Then, experiments on the recyclability of Al-ITQ-NO2 for cyanosilylation of benzaldehyde were performed and the results indicate that the activity was maintained for four consecutive runs, without significant deterioration (Fig. S6†), and the catalyst Al-ITQ-NO2 still maintains its structure, as can been observed from the XRD pattern (Fig. S7†). Finally, the scope of the cyanosilylation was determined using a series of aromatic aldehydes. The calculated TOF for molecules with the same functionality but different molecular dimensions, benzaldehyde and naphthaldehyde, was 103 and 134 after a reaction time of 0.5 h, indicating that no-diffusion limitations exist. Nevertheless, when an aldehyde with an electron-donating and deactivating group, OMe in the para position, was involved in the reaction, a slight decrease of the yield was observed. Even so, almost full conversion was reached in all cases (Fig. 10 and Table S2†).
 | ||
| Fig. 10 Kinetics for cyanosilylation of aromatic aldehydes catalyzed by Al-ITQ-NO2 (0.5 mol% Al). | ||
The catalytic comparison of this type of Al-ITQ-NO2 material, based on 1D organic–inorganic sub-domains, with other active conventional 3D MOFs for the preparation of cyanohydrins, under the same reaction conditions, showed that the yields, TOF and TON values are higher for the Al-ITQ-NO2 catalyst (Table S3†). Even the very active reported MOF catalyst for this reaction, MIL-101(Cr), exhibited a comparable activity, although being lower than that of In-MOFs which showed the highest activity for this reaction reported up to now.55 Interestingly, the Al-ITQ-NO2 catalyst showed higher activity for the 0.10–0.15% mol catalyst, the TOF being 300 h−1 and 405 h−1 for MIL-101(Cr) and Al-ITQ-NO2, respectively (Table S4†).57 Furthermore, the hybrid catalyst, Al-ITQ-NO2, based on 1D subdomains exhibited higher TOFs calculated at the initial reaction rates than zeolites and siliceous mesoporous materials, such as beta, M41S or SBA-15-type catalysts, which contained zirconium, erbium or organometallic complexes, respectively (see the comparison in Table S3†).30,49,58–60
As we have discussed before, the preparation of bi-metallic hybrid materials presents different advantages from the catalytic point of view. Then, with the aim of showing the versatility of the Al-ITQ-NO2 structure, the synthesis of bimetallic Al/Fe materials was performed. The incorporation of Fe should provide new functionalities, opening different applications as a heterogeneous catalyst since Fe can modify positively the catalytic properties of Al. Indeed, iron-based MOFs exhibit attractive catalytic properties for different reactions such as Friedel–Craft type-reactions, ring-opening of styrene oxide with methanol and aniline, aerobic oxidation of thiols to disulfides,61 synthesis of nitriles,62etc. In this paper, we have shown the successful incorporation of iron nodes in the framework when 4-nitrobenzoic acid was used as an organic spacer (Al/Fe-ITQ-NO2).
Specifically, this bimetallic organic–inorganic material was explored as a redox catalyst to perform aerobic oxidation of thiophenol to diphenylsulfide in acetonitrile as solvent. This reaction is an important process to obtain attractive chemical compounds for biological applications, using molecular oxygen as a sustainable oxidant to produce disulfides under more environmentally friendly reaction conditions.63–66 Thus, we explore the redox capacity of the bimetallic MOF-type material for thiophenol oxidation (Scheme 3). A high diphenylsulfide yield was reached in a few hours in the presence of Al/Fe-ITQ-NO2 (Fig. 11), whereas in the presence of Al-ITQ-NO2 only 32% yield was reached after 10 h of reaction. When 3D standard MIL-53(Al) was used as a catalyst in the oxidation procedure, 11% yield was achieved at the same reaction time. All these results confirmed the redox properties of iron nodes. Finally, the heterogeneity of the procedure was controlled by hot filtration of the catalyst after 0.5 h of reaction. The process was then continued for 10 h and no change in the yield was registered. This result confirms that no active species migrate from the solid to the liquid. On the other hand, the inability of the pure organic spacer to carry out the oxidation was clear since only 13% yield was achieved when the oxidation was performed in the presence of 10 mol% of 4-nitrobenzoic acid (Fig. S8 and Table S5†). All these results confirmed the heterogeneity of the process, showing the redox properties of the bimetallic Al/Fe hybrid material. Furthermore, the Al/Fe-ITQ-NO2 solid shows higher activity than well-known 3D Fe-MOFs, such as Fe(BTC) and MIL-100(Fe), exhibiting its potentiality as an alternative oxidation solid catalyst (see the comparison in Table S5†).67 Finally, a study of the recyclability of the Al/Fe-ITQ-NO2 hybrid material showed that the catalyst could be successfully reused for four consecutive cycles without observing a substantial activity loss, with the thiophenol conversion being almost complete (Fig. S9†). Furthermore, the catalyst kept its pristine structure after four uses, confirming the high stability of the material by XRD (Fig. S10†).
 | ||
| Scheme 3 Oxidation of thiophenol to diphenyldisulfide. | ||
 | ||
| Fig. 11 Kinetics for diphenyldisulfide production using Al/Fe-ITQ-NO2, Al-ITQ-NO2, MIL-53(Al) and MIL-53(Al)–NO2 as solid catalysts (10 mol% Fe). Blank test was performed, obtaining a yield of 3% at 10 h. | ||
At this stage of the study, the effect of the incorporation of a secondary metallic centre over the catalytic performance of the M-ITQ-NO2 hybrid material is still to be explored. When the cyanosilylation of benzaldehyde was performed in the presence of Al/Fe-ITQ-NO2, the initial rate of reaction was three-times higher, reaching 2.7 and 8.0 mol L−1 h−1 in the presence of Al-ITQ-NO2 and Al/Fe-ITQ-NO2, respectively (Table 2 and Fig. 9). Also, when the catalytic performance of Al/Fe-ITQ-NO2 was compared with reported catalysts and more especially with very active MOF catalyst, MIL-101(Cr), similar TOFs at the initial reaction rate and yields are achieved (382 h−1 and 409 h−1 for MIL-101(Cr) and Al/Fe-ITQ-NO2, respectively, see Table S4†). From these catalytic data, it can be concluded, as we supposed, that the incorporation of a secondary metallic centre has a beneficial cooperative effect established between metallic nodes of different nature, which modifies the final activity of Lewis acid active sites of the synthesized hybrid materials.
Conclusions
In this paper, the synthesis of five new metal–organic materials has been performed based on the assembly of 1D organic–inorganic sub-domains formed from octahedral metallic nodes (Al and Al/Fe) and functional monodentate benzylcarboxylate spacers with additional functions in the para-position. The importance of the choice of an appropriate organic linker, combined with optimal solvothermal synthesis conditions to control both the material structuration (2D or 3D) and the catalytic properties of the synthesised hybrid solids, has been shown. The different catalytic behaviour of the MOF-type materials for cyanosilylation of benzaldehydes clearly demonstrated the influence of the material morphology on the catalytic activity. The most active material was based on 3D type networks, incorporating electron-withdrawing (EWG) nitro groups. Moreover, a bimetallic Al/Fe metal–organic material containing iron nodes as secondary metallic structural nodes into the aluminium framework was successfully synthesized, and additional redox properties in these MOF-type materials for thiophenol oxidation were confirmed. Definitively, the use of functionalized substituted monodentate spacers and the cohabitation of metallic nodes with different nature open the possibilities to generate further families of metal–organic structures with different topologies, and at the same time it is also possible to modulate structural defects and vacancy concentration and, therefore, tune the acid strength of the hybrid solids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the Spanish Government by MAT2014-52085-C2-1-P, MAT2017-82288-C2-1-P and Severo Ochoa Excellence Program SEV-2016-0683. J. M. M. acknowledges Predoctoral Fellowships from MINECO for economical support. The authors thank the MULTY2HYCAT (EU-Horizon 2020 funded project under grant agreement no. 720783). The European Union is also acknowledged by ERC-AdG-2014-671093-SynCatMatch.References
- B. Li, H. M. Wen, H. Wang, H. Wu, M. Tyagi, T. Yildirim, W. Zhou and B. Chen, J. Am. Chem. Soc., 2014, 136, 6207 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., 2006, 118, 6120–6124 CrossRef.
- F. Y. Yi, D. Chen, M. K. Wu, L. Han and H.-L. Jiang, ChemPlusChem, 2016, 81, 675 CrossRef CAS.
- R. B. Lin, S. Y. Liu, J. W. Ye, X. Y. Li and J. P. Zhang, Adv. Sci., 2016, 3, 1500434 CrossRef PubMed.
- E. Reale, A. Leyva, A. Corma, C. Martínez, H. García and F. Rey, J. Mater. Chem., 2005, 15, 1742 RSC.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS.
- S. Inagaki, S. Guan, T. Ohsuna and O. Terasaki, Nature, 2002, 416, 304 CrossRef CAS PubMed.
- U. Díaz and A. Corma, Coord. Chem. Rev., 2016, 311, 85–124 CrossRef.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. García, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. LLabrés i Xamena, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem.–Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- C. Volkringer, H. Leclerc, J. C. Lavalley, T. Loiseau, G. Férey, M. Daturi and A. Vimont, J. Phys. Chem. C, 2012, 116, 5710–5719 CrossRef CAS.
- N. Klein, H. C. Hoffmann, A. Cadiau, J. Getzschmann, M. R. Lohe, S. Paasch, T. Heydenreich, K. Adil, I. Senkovska, E. Brunner and S. Kaskel, J. Mater. Chem., 2012, 22, 10303 RSC.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700–7702 RSC.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449 RSC.
- B. Zhao, X. Y. Chen, P. Cheng, D. Z. Liao, S. P. Yan and Z. H. Jiang, J. Am. Chem. Soc., 2004, 126, 15394–15395 CrossRef CAS PubMed.
- A. N. Khlobystov, A. J. Blake, N. R. Champness, D. A. Lemenovskii, A. G. Majouga, N. V. Zyk and M. Schröder, Coord. Chem. Rev., 2001, 222, 155–192 CrossRef CAS.
- A. Michaelides, M. Aravia, M. G. Siskos and S. Skoulika, CrystEngComm, 2015, 17, 124 RSC.
- S. Inoue and S. Fujihara, Inorg. Chem., 2011, 50, 3605–3612 CrossRef CAS PubMed.
- T. Ródenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés i Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef PubMed.
- P. García-García, J. M. Moreno, U. Díaz, M. Bruix and A. Corma, Nat. Commun., 2016, 7, 10835 CrossRef PubMed.
- J. M. Moreno, I. Navarro, U. Díaz, J. Primo and A. Corma, Angew. Chem., 2016, 55, 11026–11030 CrossRef CAS PubMed.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81–88 CrossRef CAS.
- X. Cui, M. C. Xu, L. J. Zhang, R. X. Yao and X. M. Zhang, Dalton Trans., 2015, 44, 12711 RSC.
- K. Mo, Y. Yang and Y. Cui, J. Am. Chem. Soc., 2014, 136, 1746–1749 CrossRef CAS PubMed.
- G. E. Gómez, A. M. Kaczmarek, R. Van Deun, E. V. Brusau, G. E. Narda, D. Vega, M. Iglesias, E. Gutiérrez-Puebla and M. A. Monge, Eur. J. Inorg. Chem., 2016, 1577–1588 CrossRef.
- S. Telalovic and U. Hanefeld, Catal. Commun., 2011, 12, 493–496 CrossRef CAS.
- K. Iwanami, J. C. Choi, B. Lu, T. Sakakura and H. Yasuda, Chem. Commun., 2008, 1002–1004 RSC.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed.
- P. W. Siu, Z. J. Brown, O. K. Farha, J. T. Hupp and K. A. Scheidt, Chem. Commun., 2013, 49, 10920–10922 RSC.
- S. Biswas, T. Ahnfeldt and N. Stock, Inorg. Chem., 2011, 50, 9518–9526 CrossRef CAS PubMed.
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS PubMed; A. Santiago-Portillo, S. Navalón, P. Concepción, M. Álvaro and H. García, ChemCatChem, 2017, 9, 2506–2511 CrossRef.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- A. M. Rasero-Almansa, M. Iglesias and F. Sánchez, RSC Adv., 2016, 6, 106790–106797 RSC.
- X. Yang and Q. Xu, Cryst. Growth Des., 2017, 17, 1450–1455 CrossRef CAS.
- Q. Sun, M. Liu, K. Li, Y. Han, Y. Zuo, F. Chai, C. Song, G. Zhang and X. Guo, Inorg. Chem. Front., 2017, 4, 144 RSC.
- E. A. Dolgopolova, A. J. Brandt, O. A. Ejegbavwo, A. S. Duke, T. D. Maddumapatabandi, R. P. Galhenage, B. W. Larson, O. G. Reid, S. C. Ammal, A. Heyden, M. Chandrashekhar, V. Stavila, D. A. Chen and N. B. Shustova, J. Am. Chem. Soc., 2017, 139, 5201–5209 CrossRef CAS PubMed.
- R. Zou, P.-Z. Li, Y.-F. Zeng, J. Liu, R. Zhao, H. Duan, Z. Luo, J.-G. Wang, R. Zou and Y. Zhao, Small, 2016, 12(17), 2334–2343 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. Llabres i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed; M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef PubMed.
- F. Vermoortele, R. Ameloot, L. Alaerts, R. Matthesen, J. Gascon, F. Kapteijn and D. E. De Vos, J. Mater. Chem., 2012, 22, 10313 RSC.
- U. Ravon, M. Savonnet, S. Aguado, M. E. Domine, E. Janneau and D. Farrusseng, Microporous Mesoporous Mater., 2010, 129, 319 CrossRef CAS.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- C. Volkringer, M. Meddouri, T. Loiseau, N. Guillou, J. Marrot, G. Férey, M. Haouas, F. Taulelle, N. Audebrand and M. Latroche, Inorg. Chem., 2008, 47, 11892–11901 CrossRef CAS PubMed.
- I. Syozi, Prog. Theor. Phys., 1951, 6, 306–308 CrossRef.
- J. Frunzke, T. Hansen, A. Harrison, J. S. Lord, G. S. Oakley, D. Visserc and A. S. Willsa, J. Mater. Chem., 2001, 11, 179–185 RSC.
- J. J. Delpuech, M. R. Khaddar, A. A. Peguy and P. R. Rubini, J. Am. Chem. Soc., 1975, 97, 3373–3379 CrossRef CAS.
- A. Vimont, J. M. Goupil, J. C. Lavalley, M. Daturi, S. Surble, C. Serre, F. Millange, G. Férey and N. Audebrand, J. Am. Chem. Soc., 2006, 128, 3218–3227 CrossRef CAS PubMed.
- Z. Fang, J. P. Dürholt, M. Kauer, W. Zhang, Ch. Lochenie, B. Jee, B. Albada, N. Metzler-Nolte, A. Pöppl, B. Weber, M. Muhler, Y. Wang, R. Schmid and R. A. Fischer, J. Am. Chem. Soc., 2014, 136, 9627–9636 CrossRef CAS PubMed.
- P. García-García, M. Müller and A. Corma, Chem. Sci., 2014, 5, 2979–3007 RSC.
- L. Alaerts, E. Séguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem.–Eur. J., 2006, 12, 7353–7363 CrossRef CAS PubMed; N. B. Pathan, A. M. Rahatgaonkar and M. S. Chorghade, Catal. Commun., 2011, 12, 1170–1176 CrossRef.
- R. J. H. Gregory, Chem. Rev., 1999, 99, 3649–3682 CrossRef CAS PubMed.
- J. M. Brunel and I. P. Holmes, Angew. Chem., Int. Ed., 2004, 43, 2752–2778 CrossRef CAS PubMed.
- L. M. Aguirre-Díaz, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. A. Monge, CrystEngComm, 2013, 15, 9562–9571 RSC; L. M. Aguirre-Díaz, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. A. Monge, RSC Adv., 2015, 5, 7058–7065 RSC.
- M. A. Lacour, N. J. Rahier and M. Taillefer, Chem.–Eur. J., 2011, 17, 12276–12279 CrossRef CAS PubMed.
- T. Yang, A. Bartoszewicz, J. Ju, J. Sun, Z. Liu, X. Zou, Y. Wang, G. Li, F. Liao, B. Martín-Matute and J. Lin, Angew. Chem., Int. Ed., 2011, 50, 12555 CrossRef CAS PubMed; Y. Ogasawara, S. Uchida, K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2009, 15, 4343 CrossRef PubMed.
- J. M. De Simone, Science, 2002, 297, 799–803 CrossRef CAS PubMed.
- Z. Zhang, J. Chen, Z. Bao, G. Chang, H. Xing and Q. Ren, RSC Adv., 2015, 5, 79355–79360 RSC.
- A. Procopio, G. Das, M. Nardi, M. Oliverio and L. Pasqua, ChemSusChem, 2008, 1, 916–919 CrossRef CAS PubMed.
- S. Zhang, B. Zhang, H. Liang, Y. Liu, Y. Qiao and Y. Qin, Angew. Chem., Int. Ed., 2018, 57, 1091–1095 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2010, 46, 6476–6647 RSC.
- A. Rapeyko, M. J. Climent, A. Corma, P. Concepcion and S. Iborra, ChemSusChem, 2015, 8, 3270–3282 CrossRef CAS PubMed.
- A. Saxena, A. Kumar and S. Mozumdar, J. Mol. Catal. A: Chem., 2007, 269, 35–40 CrossRef CAS.
- M. Oba, K. Tanaka, K. Nishiyama and W. Ando, J. Org. Chem., 2011, 76, 4173–4177 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Alvaro and H. García, Chem. Commun., 2010, 46, 6476–6478 RSC.
- A. Dhakshinamoorthy, M. Alvaro, P. Horcajada, E. Gibson, M. Vishnuvarthan, A. Vimont, J.-M. Grenèche, C. Serre, M. Daturi and H. García, ACS Catal., 2012, 2, 2060–2065 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization details of metal–organic hybrid materials (TG and DTA curves and FTIR spectra after CO adsorption), Hammett plot for cyanosilylation of benzaldehyde, kinetics and additional catalytic results are shown. See DOI: 10.1039/c8sc04372h |
| This journal is © The Royal Society of Chemistry 2019 |