
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total chemical synthesis of ester-linked ubiquitinated proteins unravels their behavior with deubiquitinases†
Hao
Sun
,
Roman
Meledin
,
Sachitanand M.
Mali
and
Ashraf
Brik
 *
*
Schulich Faculty of Chemistry, Technion Israel Institute of Technology, Haifa, 3200008, Israel. E-mail: abrik@technion.ac.il
First published on 11th January 2018
Abstract
Ester-linked ubiquitinated proteins have been reported by several groups to be involved in ubiquitin signalling. However, due to the lack of the suitable tools to homogeneously produce such conjugates, their exact physiological roles and biochemical behavior remain enigmatic. Here, we report for the first time on the development of a novel synthetic strategy based on total chemical synthesis of proteins to construct ubiquitinated proteins, where ubiquitin is linked to the substrate via an ester bond. In this study, we prepared ester- and isopeptide-linked ubiquitinated α-globin and examined their relative behaviors with various deubiquitinases. We found that deubiquitinases are able to cleave the ester linkage with different efficiency relative to the isopeptide-linked substrate. These results may indicate that ester-linked ubiquitinated proteins are natural substrates for deubiquitinases.
Introduction
Ubiquitination is a posttranslational modification (PTM) which targets proteins for a variety of processes, such as proteasomal degradation, ER-associated degradation and intracellular trafficking.1 The ubiquitin (Ub) monomers form a chain in which its C-terminal Gly can be linked to any of the seven available Lys side chains or the N-terminus of the preceding Ub, yielding a large diversity of polyUb chains.2 Ub or any of the polyUb chains can be linked to a protein substrate through a Lys residue of a protein via an isopeptide bond. In addition, unusual ester or thioester linkages through Ser/Thr and Cys, respectively, can be formed (Fig. 1).3 In these processes, Ub conjugates are assembled by three enzymatic steps including the activating enzyme E1, the conjugating enzyme E2 and the E3 ligase.4 Once being installed, the Ub modifier is recognized by Ub binding domains based on location, topology and conformation of the Ub chain5 to initiate a specific signal according to the type of ubiquitination. Like many PTMs, ubiquitination is a reversible process where deubiquitinases (DUBs) disassemble the Ub chains or the ubiquitinated proteins, therefore playing crucial roles in Ub signalling.6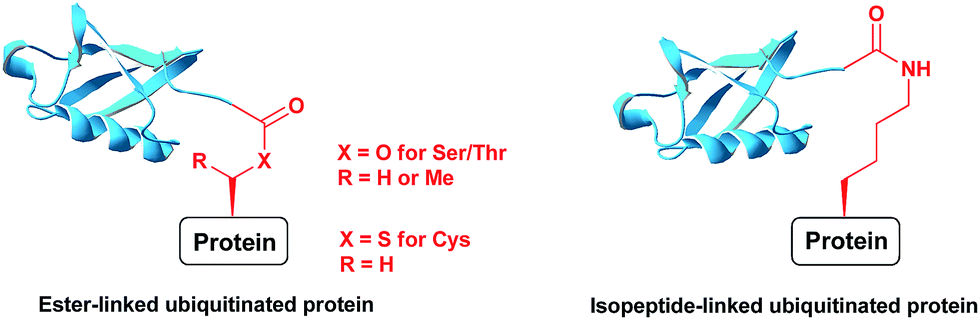 | ||
| Fig. 1 Ubiquitinated proteins with different linkages. | ||
Despite the fact that most proteins are linked to Ub or polyUb via their Lys residue there is growing evidence about the existence and physiological relevance of the “non-canonical” ubiquitination.7,8 Indeed, it was reported by several groups that proteins lacking Lys or those, where the Lys residues were mutated to Arg can still be ubiquitinated and targeted for the proteasomal or ER-associated degradation.9–11 For example, the heavy chain of the major histocompatibility complex-I (MHC-I HC) was shown to undergo ubiquitination through Ser, Thr and Lys residues by the viral E3 ligase mK3.12 This ubiquitination subsequently stimulates the ER-associated degradation of the MHC-I and thus prevents the immune response against viral infected cells. Notably, in this example and for unclear reason ubiquitination through Ser or Thr residues was found to be preferentially formed than via conventional isopeptide linkage. Another important aspect of protein ubiquitination through the Thr/Ser side chains is the nature of the ester linkage. In addition to having a different number of atoms, ester also provides a higher degree of rotational freedom than the corresponding amide bond. These properties could confer unusual surface interactions of the proximal anchored Ub with the Ub binding proteins that might lead to the specific outcomes.13 Indeed, Hansen and colleagues reported that the ubiquitination of HC MHC-I via its Ser in contrast to Lys leads to subsequent polyUb chain build up, exclusively through the K48 linkage type.14
Despite these emerging observations, the field of non-canonical protein ubiquitination is still in its infancy. Here, most reported studies rely heavily on mutagenic approaches, which enable indirect observations of the ubiquitination type. While chemical biologists have already developed powerful toolboxes for the preparation of isopeptide bond based ubiquitinated proteins,15–19 strategies to assemble ester-linked equivalents are still lacking. Filling this gap would enable the preparation of highly pure and homogenous ester-linked ubiquitinated proteins to investigate the interplay between these conjugates and their modulating enzymes, such as DUBs and E2/E3 ligases, including functional, structural and interactome analyses. In addition, it will increase our knowledge about their exact roles in Ub signalling such as in proteasomal degradation and will enable to explain the differences between canonical versus non-canonical types of ubiquitination. Finally, such conjugates could be useful as templates for the development of antibodies, which will open the door for a variety of imaging and diagnostics studies.
In order to prepare desired conjugates by conventional enzymatic approaches, one should identify a specific set of E2/E3 ligases, which in turn have to possess a high selectivity towards the specific location and type of ubiquitination. Since there is a shortage of knowledge about these ligases for the ester-linked ubiquitination, biochemical strategies remain significantly restricted. Chemistry, however, offers an alternative approach for the preparation of various complex conjugates based on the total chemical synthesis of proteins.20 Regarding the incorporation of an ester unit in proteins there are only a few examples that were reported. For instance, Dawson and colleagues replaced four backbone amide bonds with the ester in chymotrypsin inhibitor II protein to study its folding and stability.21 The Liu group applied the solubilizing tag of polyarginine attached through its C terminus carboxylic acid to the threonine side chain of Autophagosomal Marker Protein LC3-II.22 Kent and co-workers prepared proinsulin analogues having an ester linkage between threonine and glutamic acid side chains.23 Despite the existence of these examples, there are no reported studies about the chemical preparation of a highly complex ester-linked conjugates such as ubiquitinated proteins. Here, for the first time, we report on the total chemical synthesis of ubiquitinated protein, where ubiquitin is linked to a substrate via an ester bond to shed light on the behavior of these conjugates with known DUBs.
Results and discussion
To examine the properties of an ester-linked ubiquitinated protein compared to the isopeptide analogue, we examined first the synthesis of a model peptide containing a branched ester bond (Scheme 1). Such synthesis would enable us to test the stability of the ester bond under different conditions often used in chemical protein synthesis. In this synthesis, the first five amino acids of the 28-mer peptide (1) were coupled on the solid support, using standard solid phase peptide synthesis (SPPS).24 We then coupled N-terminal Fmoc-Thr-OH with unprotected hydroxyl group using benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) and hydroxybenzotriazole (HOBT) in presence of N-methylmorpholine (NMM).25 Next, the N-terminal Fmoc protecting group was removed and phenylalanine was coupled to give 2. Subsequently, allyloxycarbonyl (Alloc) protected Gly was coupled to the free hydroxyl group of the Thr using N,N-diisopropylcarbodiimide (DIC) and 4-dimethylaminopyridine (DMAP) to form the ester linkage (3). The ester bond could be formed only after the phenylalanine coupling to prevent a five-member ring induced intramolecular attack of the Thr free amine on the ester bond. Next, we completed the synthesis of the backbone peptide and introduced N-terminal thiazolidine (Thz) as the last amino acid to afford 4. Subsequently, the Alloc protecting group on the branched Gly was removed by Pd(PPh3)4, followed by coupling the dipeptide Arg–Gly to prevent intramolecular diketopiperazine formation. The branched model sequence was accomplished using standard SPPS to give peptide 6 after the cleavage the deprotection step using TFA. Finally, Thz was deprotected by [Pd(Allyl)Cl]2 (ref. 26) to afford peptide 7, following by native chemical ligation with peptide-thioester27 and desulfurization28,29 to give peptide 8. Importantly, through all these synthetic steps we did not observe any hydrolysis of the ester linkage (Fig. S1†).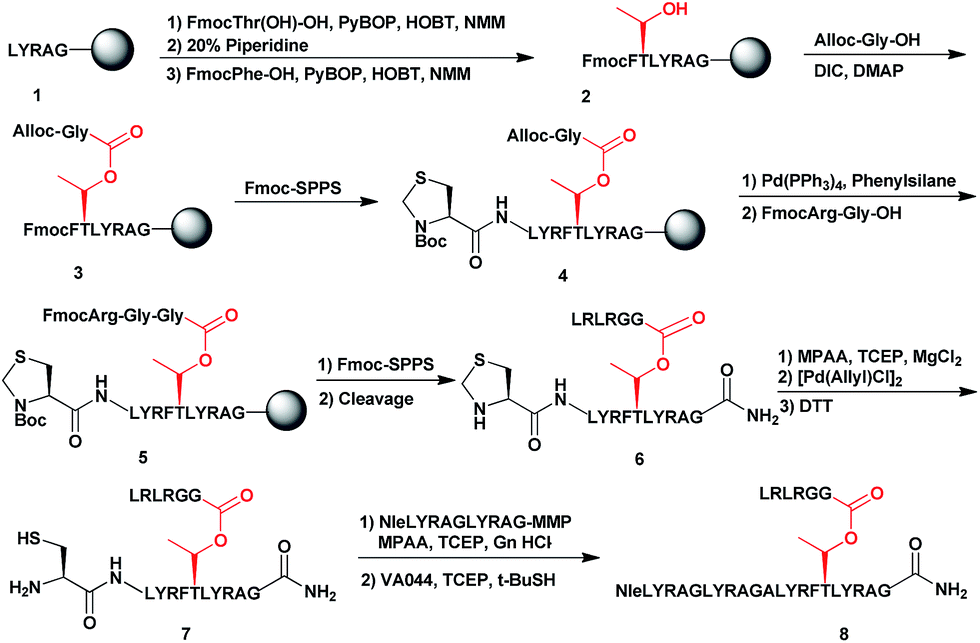 | ||
| Scheme 1 Synthetic strategy for the model peptide containing the ester linkage. | ||
Having succeeded with the model peptide, we then turned our attention to prepare ester-linked ubiquitinated protein. We chose α-globin as the Ub acceptor protein (Scheme 2A), since in addition to its naturally being ubiquitinated,30 it has been also shown to behave as an excellent model for DUB's and proteasomal degradation studies.31,32 To compare the properties of the ester- and the isopeptide-linked ubiquitinated α-globin, we designed synthetic schemes for both variants. As the anchoring location of Ub we chose Thr127 because of its surface exposure. In addition, α-globin has been reported to undergo ubiquitination at different positions, including the Thr region.33 To prepare desired the ester- and isopeptide-linked ubiquitinated HA-α-globin we divided the ester analogue 19 (Scheme 3A) to four fragments: 10, 13, 14 and 15 (Scheme 2B), while for the isopeptide analogue 20 (Scheme 3A) we used the four fragments 12, 13, 14 and 15 (Scheme 2B). Fragment 10, Ub(46–76)-α-globin(120–150), which contains an ester linkage via the Thr127 side chain was prepared in a similar manner to the model peptide 8 (Scheme 2C & Fig. S2†). On the other hand, fragment 12 bearing the isopeptide bond was synthesized using Alloc protected Lys to enable side chain elongation of the Ub peptide(46–76) at position127 (Scheme 2D & Fig. S3†). This peptide, like 10 has also N-terminal Thz. Next, upon Alloc removal by Pd(PPh3)4 on resin, SPPS of the Ub sequence was accomplished.34 HA-α-globin(1–61)-MMP, 13, and Ub(1–45)-MMP, 15, were prepared on the solid support using N-acyl-N′-methyl-benzimidazolinone (MeNbz) chemistry.35 On the other hand, Cys-α-globin(63–119)-NHNH214 was prepared with N-acyl-benzimidazolinone (Nbz) approach.36 This was followed by switching 13 and 15 to the thioester precursors and of 14 to hydrazide functionality.37 During the cyclization step of fragment 14 in DCM we obtained only ∼30% conversion, while cyclization in DMF according to our previous reports led to a full conversion.38 Interestingly, we found that we could obtain the same efficiency of conversion without the need of the extra step of treatment with the base N,N-diisopropylethylamine.36 The N-terminal amino acids in the fragments 10, 12 and 14 were temporarily changed to Cys to enable subsequent ligation steps, which at later stage will be converted back to Ala via desulfurization.
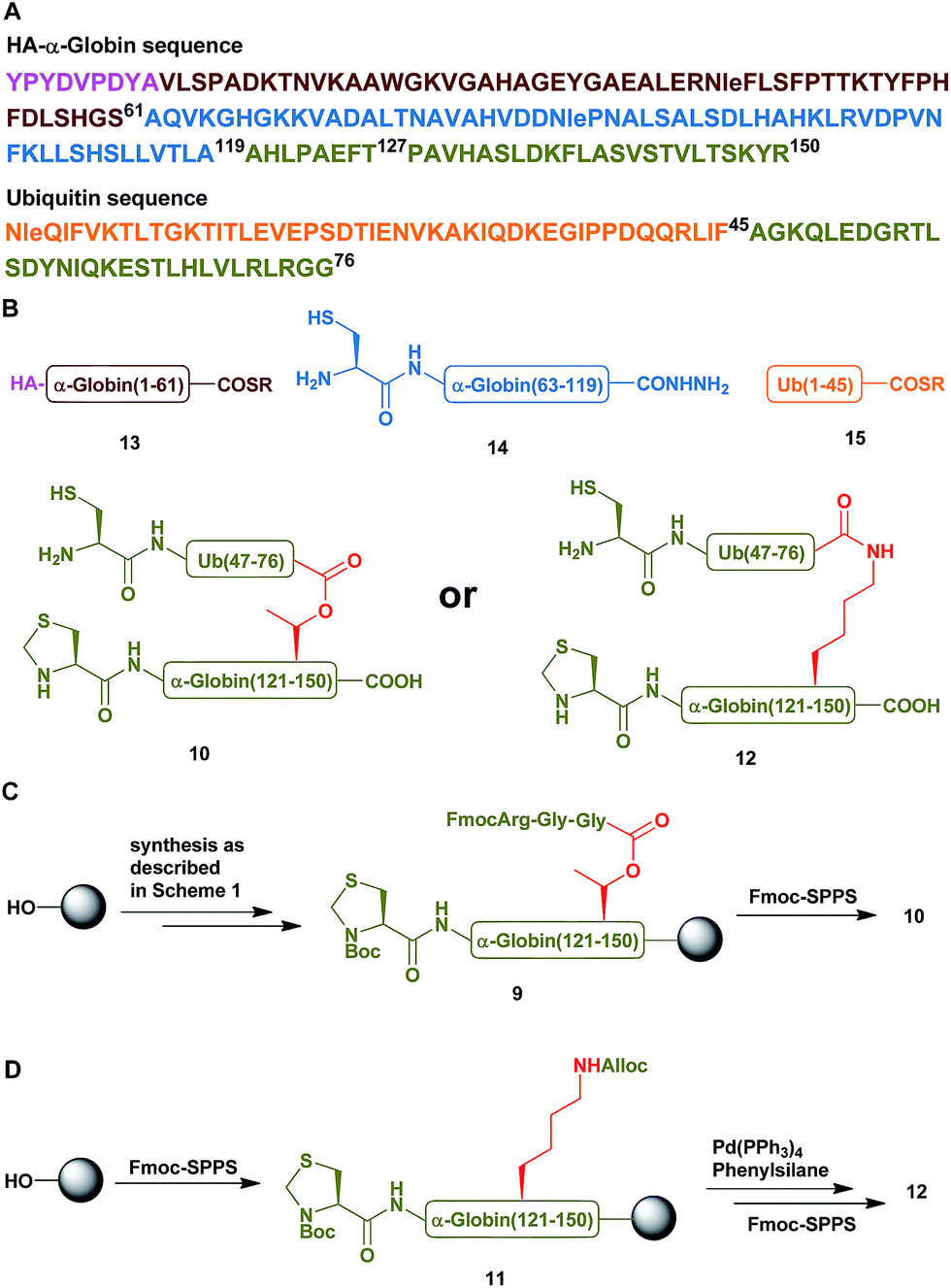 | ||
| Scheme 2 (A) The sequences of HA-α-globin and Ub. (B) The peptide building blocks for the ubiquitinated α-globin analogues. (C & D) Synthetic strategies for the preparation of fragment 10 with the ester linkage and of fragment 12 with the isopeptide linkage, respectively. | ||
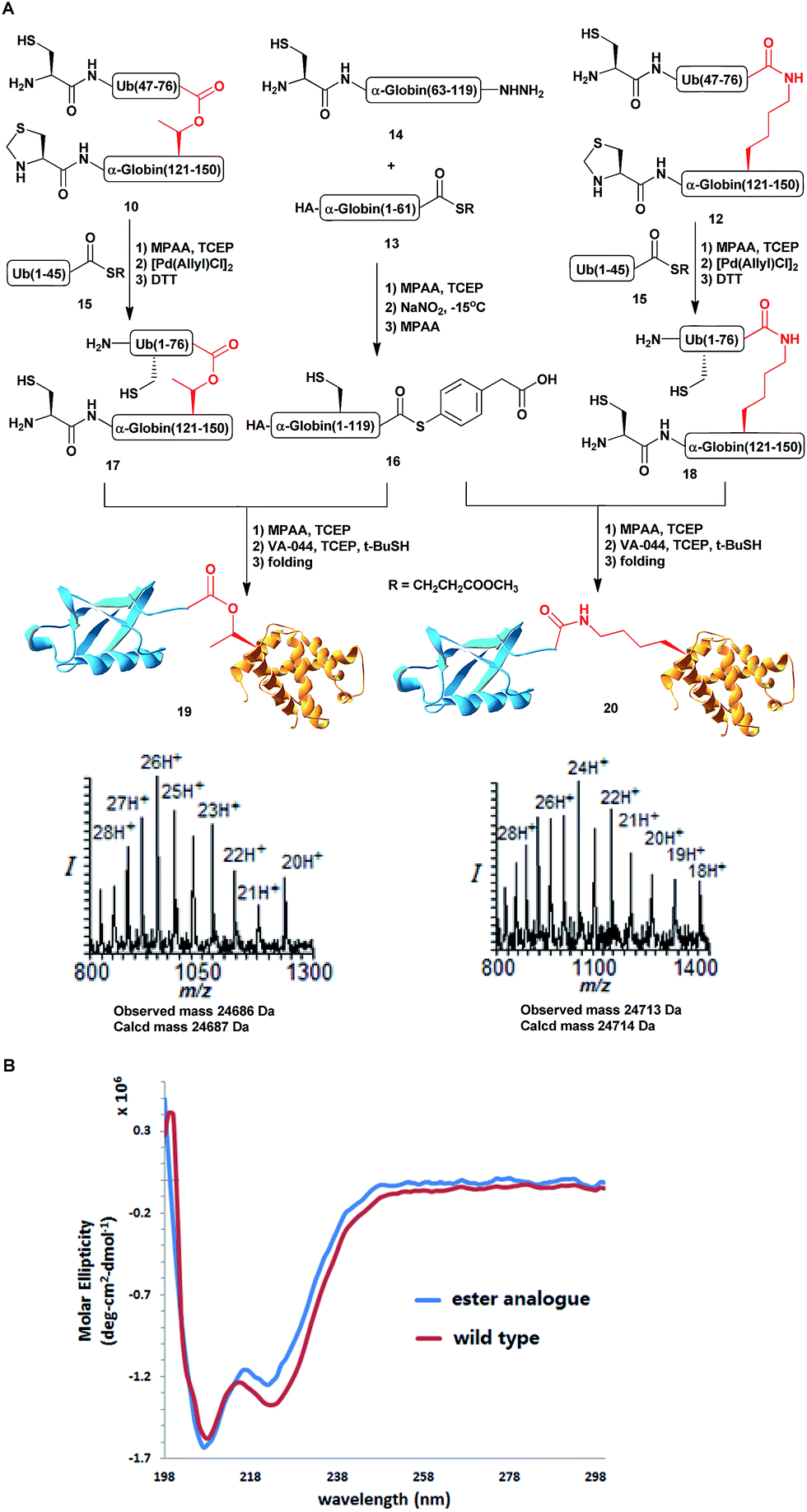 | ||
Scheme 3 (A) Syntheses of HA-α-globin-Ub having the ester or isopeptide linkages with the observed masses of 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 686 Da (calcd 24687 Da) and 24713 Da (calcd 24714 Da), respectively. (B) CD analysis of the HA-α-globin-Ub analogues. 686 Da (calcd 24687 Da) and 24713 Da (calcd 24714 Da), respectively. (B) CD analysis of the HA-α-globin-Ub analogues. | ||
With all fragments in hand, we performed stepwise synthesis of the two variants of HA-α-globin-Ub (Scheme 3A). First, fragment 13 was ligated with fragment 14, followed by switching of the ligated product to thioester 16. In parallel, fragment 10 or 12 was ligated with 15, followed by Thz deprotection to give 17 or 18, respectively. Finally, fragments 16 was ligated with 17 or 18 and the product was desulfurized in one-pot manner to give the final HA-α-globin-Ub analogue.39 The secondary structures of these ubiquitinated proteins were verified by circular dichroism (CD), supporting correct folding (Scheme 3B).
Next, we turned our attention to biochemical characterization of the ester- and isopeptide-linked conjugates using DUBs cleavage assay. The nature of the isopeptide bond is considered chemically more stable than the ester, because of higher stability of the amide's resonance form. However, due to the variation in length, conformation and the structure of the linkages, their recognition by DUBs might vary, which could lead to a difference in cleavage efficiency and DUBs specificities. To examine this, a panel of purified DUBs was incubated with each of the HA-α-globin-Ub variants at 37 °C for 5 minutes, followed by western blot analysis to evaluate the cleavage efficiency (Fig. 2 & S11†). Our results show that USP2, USP7 and USP15 cleave both ester- and isopeptide-linked conjugates with different efficiency. Interestingly, the cleavage of each analogue by USP2 is weaker than by USP7 and USP15. To compare the cleavage efficiency of isopeptide versus ester-linked ubiquitinated HA-α-globin we chose to focus on USP2, which was the slowest in the previous experiment. By incubating these analogues with USP2 at different time points we found that the isopeptide-linked HA-α-globin-Ub undergoes faster cleavage than the ester-linked analogue (Fig. 3 & S12†). For example, after 7.5 minutes of incubation, the ester-linked HA-α-globin-Ub exhibited around 30% cleavage, while the isopeptide analogue underwent over 70% (Fig. 4). To examine if this observation depends on the type of DUB we performed similar analysis with USP15, which exhibited with both α-globin-Ub variants faster cleavage efficiencies compared to USP2. Similar to the USP2 case, we found comparable differences in the cleavage of the two analogues, highlighting the effect of the linkage type on the enzymatic cleavage (ESI, Fig. S13†).
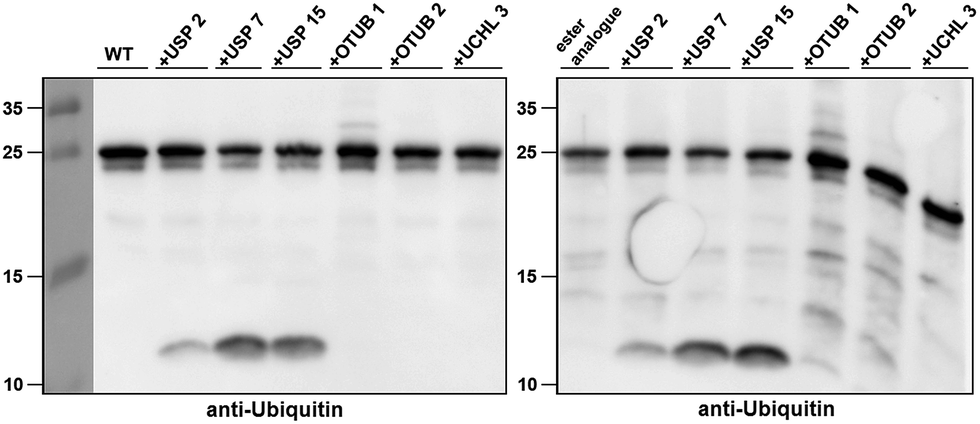 | ||
| Fig. 2 Western blot analysis of ester- and isopeptide-linked HA-α-globin-Ub treated with panel of DUBs. The analogues were treated with 1:10 enzyme to substrate molar ratio. | ||
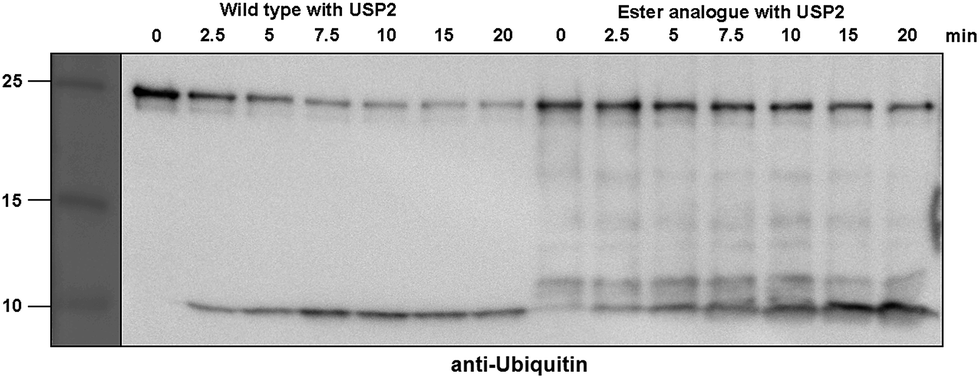 | ||
| Fig. 3 Representative western blot analysis of ester-linked HA-α-globin-Ub (right gel) and wild type (left gel). The analogues were treated with USP2 at 1:50 (enzyme:substrate) molar ratio for different time points. Untreated analogues were used as a control. | ||
 | ||
| Fig. 4 Summary of wild type and ester analogue deubiquitination by USP2, quantified from three independent western blot analyses. | ||
These findings are interesting because it shows for the first time the ability of DUBs to cleave an ester bond between Ub and its acceptor protein. Our preliminary analysis shows that the cleavage with two different DUBs is slower compared to the isopeptide bond counterpart. We cannot exclude, however, the possible effect of the natural protein substrate, which dictates conformation and exposure of the ester linkage toward DUBs, on this process. As it is known that proteins labeled for the degradation by Ub, may be rescued by the action of DUBs, it is tempting to assume that difference in the removal of Ub could tune the degradation rate by the proteasome.32,40 Hence, the more stable Ub-substrate the less chance to be rescued from degradation. In this context, it has been shown that lysine-less mutant of NS-1 immunoglobulin κ LC, where ubiquitination occurs via the ester linkage, degrades faster compared to the wild type protein.41 Further support for these hypotheses requires detailed studies, including comprehensive kinetic characterizations of their cleavage by DUBs. In addition, the effect of Ub location as well as specific type of ester bond (Ser vs. Thr) should also be examined.
Conclusions
In summary, we have developed for the first time a new synthetic strategy to chemically prepare ubiquitinated proteins where Ub is linked to a protein substrate via an ester bond. This allowed us to examine the activity of these types of conjugates with various DUBs. Our study demonstrated that ester-linked ubiquitinated protein could serve as substrates for DUBs, albeit being cleaved at slower rate compared to the isopeptide ubiquitinated protein counterpart. This novel platform opens the door to investigate in more detailed analysis the effect of the ester connectivity on various aspects of Ub signalling and whether such a linkage could be another layer of regulation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Brik is a Neubauer Professor and a Taub Fellow, Supported by the Taub Foundations. H. S. is supported in part at the Technion by a Technion-Guangdong Fellowship.References
- M. H. Glickman and A. Ciechanover, Physiol. Rev., 2002, 82, 373–428 CrossRef CAS PubMed.
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef CAS PubMed.
- Y. Kravtsova-Ivantsiv, T. Sommer and A. Ciechanover, Angew. Chem., Int. Ed. Engl., 2013, 52, 192–198 CrossRef CAS PubMed.
- C. M. Pickart, Annu. Rev. Biochem., 2001, 70, 503–533 CrossRef CAS PubMed.
- I. Dikic, S. Wakatsuki and K. J. Walters, Nat. Rev. Mol. Cell Biol., 2009, 10, 659–671 CrossRef CAS PubMed.
- F. E. Reyes-Turcu and K. D. Wilkinson, Chem. Rev., 2009, 109, 1495–1508 CrossRef CAS PubMed.
- Y. Kravtsova-Ivantsiv and A. Ciechanover, J. Cell Sci., 2012, 125, 539–548 CrossRef CAS PubMed.
- G. S. McDowell and A. Philpott, Int. J. Biochem. Cell Biol., 2013, 45, 1833–1842 CrossRef CAS PubMed.
- K. Cadwell and L. Coscoy, Science, 2005, 309, 127–130 CrossRef CAS PubMed.
- S. Ishikura, A. M. Weissman and J. S. Bonifacino, J. Biol. Chem., 2010, 285, 23916–23924 CrossRef CAS PubMed.
- J. M. Vosper, G. S. McDowell, C. J. Hindley, C. S. Fiore-Heriche, R. Kucerova, I. Horan and A. Philpott, J. Biol. Chem., 2009, 284, 15458–15468 CrossRef CAS PubMed.
- X. Wang, R. A. Herr, W. J. Chua, L. Lybarger, E. J. Wiertz and T. H. Hansen, J. Cell Biol., 2007, 177, 613–624 CrossRef CAS PubMed.
- X. Wang, R. A. Herr and T. H. Hansen, Traffic, 2012, 13, 19–24 CrossRef CAS PubMed.
- X. Wang, R. A. Herr, M. Rabelink, R. C. Hoeben, E. J. Wiertz and T. H. Hansen, J. Cell Biol., 2009, 187, 655–668 CrossRef CAS PubMed.
- C. E. Weller, M. E. Pilkerton and C. Chatterjee, Biopolymers, 2014, 101, 144–155 CrossRef CAS PubMed.
- G. B. van Tilburg, A. F. Elhebieshy and H. Ovaa, Curr. Opin. Struct. Biol., 2016, 38, 92–101 CrossRef CAS PubMed.
- G. H. Pham and E. R. Strieter, Curr. Opin. Chem. Biol., 2015, 28, 57–65 CrossRef CAS PubMed.
- R. Meledin, S. M. Mali and A. Brik, Chem. Rec., 2016, 16, 509–519 CrossRef CAS PubMed.
- S. M. Mali, S. K. Singh, E. Eid and A. Brik, J. Am. Chem. Soc., 2017, 139, 4971–4986 CrossRef CAS PubMed.
- S. Bondalapati, M. Jbara and A. Brik, Nat. Chem., 2016, 8, 407–418 CrossRef CAS PubMed.
- G. S. Beligere and P. E. Dawson, J. Am. Chem. Soc., 2000, 122, 12079–12082 CrossRef CAS.
- Y. C. Huang, Y. M. Li, Y. Chen, M. Pan, Y. T. Li, L. Yu, Q. X. Guo and L. Liu, Angew. Chem., Int. Ed. Engl., 2013, 52, 4858–4862 CrossRef CAS PubMed.
- Y. Sohma, Q. X. Hua, J. Whittaker, M. A. Weiss and S. B. Kent, Angew. Chem., Int. Ed. Engl., 2010, 49, 5489–5493 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- P. M. Fischer, K. V. Retson, M. I. Tyler and M. E. Howden, Int. J. Pept. Protein Res., 1991, 38, 491–493 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity, M. Seenaiah and A. Brik, J. Am. Chem. Soc., 2016, 138, 5069–5075 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CAS.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed. Engl., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- J. R. Shaeffer, J. Biol. Chem., 1983, 258, 13172–13177 CAS.
- H. P. Hemantha, S. N. Bavikar, Y. Herman-Bachinsky, N. Haj-Yahya, S. Bondalapati, A. Ciechanover and A. Brik, J. Am. Chem. Soc., 2014, 136, 2665–2673 CrossRef CAS PubMed.
- S. K. Singh, I. Sahu, S. M. Mali, H. P. Hemantha, O. Kleifeld, M. H. Glickman and A. Brik, J. Am. Chem. Soc., 2016, 138, 16004–16015 CrossRef CAS PubMed.
- J. R. Shaeffer, J. Biol. Chem., 1994, 269, 29530–29536 CAS.
- K. S. Kumar, L. Spasser, S. Ohayon, L. A. Erlich and A. Brik, Bioconjugate Chem., 2011, 22, 137–143 CrossRef CAS PubMed.
- J. B. Blanco-Canosa, B. Nardone, F. Albericio and P. E. Dawson, J. Am. Chem. Soc., 2015, 137, 7197–7209 CrossRef CAS PubMed.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed. Engl., 2008, 47, 6851–6855 CrossRef CAS PubMed.
- G. M. Fang, Y. M. Li, F. Shen, Y. C. Huang, J. B. Li, Y. Lin, H. K. Cui and L. Liu, Angew. Chem., Int. Ed. Engl., 2011, 50, 7645–7649 CrossRef CAS PubMed.
- P. Siman, O. Blatt, T. Moyal, T. Danieli, M. Lebendiker, H. A. Lashuel, A. Friedler and A. Brik, ChemBioChem, 2011, 12, 1097–1104 CrossRef CAS PubMed.
- T. Moyal, H. P. Hemantha, P. Siman, M. Refua and A. Brik, Chem. Sci., 2013, 4, 2496–2501 RSC.
- D. Komander, M. J. Clague and S. Urbe, Nat. Rev. Mol. Cell Biol., 2009, 10, 550–563 CrossRef CAS PubMed.
- Y. Shimizu, Y. Okuda-Shimizu and L. M. Hendershot, Mol. Cell, 2010, 40, 917–926 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04518b |
| This journal is © The Royal Society of Chemistry 2018 |