 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
VOC emission rates over London and South East England obtained by airborne eddy covariance†
Adam R.
Vaughan
 a,
James D.
Lee
*b,
Marvin D.
Shaw
b,
Pawel K.
Misztal
c,
Stefan
Metzger
de,
Massimo
Vieno
f,
Brian
Davison
g,
Thomas G.
Karl
h,
Lucy J.
Carpenter
b,
Alastair C.
Lewis
b,
Ruth M.
Purvis
b,
Allen H.
Goldstein
c and
C. Nicholas
Hewitt
g
a,
James D.
Lee
*b,
Marvin D.
Shaw
b,
Pawel K.
Misztal
c,
Stefan
Metzger
de,
Massimo
Vieno
f,
Brian
Davison
g,
Thomas G.
Karl
h,
Lucy J.
Carpenter
b,
Alastair C.
Lewis
b,
Ruth M.
Purvis
b,
Allen H.
Goldstein
c and
C. Nicholas
Hewitt
g
aDepartment of Chemistry, University of York, York, UK
bNational Centre for Atmospheric Science, University of York, York, UK. E-mail: james.lee@york.ac.uk
cUniversity of California, Berkeley, USA
dNational Ecological Observatory Network, Boulder, USA
eUniversity of Colorado, Boulder, USA
fNatural Environment Research Council, Centre for Ecology & Hydrology, Penicuik, UK
gLancaster Environment Centre, Lancaster University, Lancaster, UK
hInstitute of Atmospheric and Cryospheric Sciences, University of Innsbruck, Innsbruck, Austria
First published on 3rd February 2017
Abstract
Volatile organic compounds (VOCs) originate from a variety of sources, and play an intrinsic role in influencing air quality. Some VOCs, including benzene, are carcinogens and so directly affect human health, while others, such as isoprene, are very reactive in the atmosphere and play an important role in the formation of secondary pollutants such as ozone and particles. Here we report spatially-resolved measurements of the surface-to-atmosphere fluxes of VOCs across London and SE England made in 2013 and 2014. High-frequency 3-D wind velocities and VOC volume mixing ratios (made by proton transfer reaction – mass spectrometry) were obtained from a low-flying aircraft and used to calculate fluxes using the technique of eddy covariance. A footprint model was then used to quantify the flux contribution from the ground surface at spatial resolution of 100 m, averaged to 1 km. Measured fluxes of benzene over Greater London showed positive agreement with the UK’s National Atmospheric Emissions Inventory, with the highest fluxes originating from central London. Comparison of MTBE and toluene fluxes suggest that petroleum evaporation is an important emission source of toluene in central London. Outside London, increased isoprene emissions were observed over wooded areas, at rates greater than those predicted by a UK regional application of the European Monitoring and Evaluation Programme model (EMEP4UK). This work demonstrates the applicability of the airborne eddy covariance method to the determination of anthropogenic and biogenic VOC fluxes and the possibility of validating emission inventories through measurements.
1. Introduction
Non-methane volatile organic compounds (VOCs) are a class of organic molecules that are sufficiently volatile to enter the atmosphere. Some, including benzene, are directly toxic to humans, while others are reactive in the atmosphere producing secondary pollutants such as ozone and particles, and hence impact air quality.1 Globally, most VOCs originate from the terrestrial biosphere, but in cities and other areas of high anthropogenic emissions, pollution sources usually dominate.Emissions of VOCs in cities such as London have been well studied, with vehicles recognised as a significant source,2–4 either via emission in the exhaust gas or by the evaporation of unburnt or partially burnt fuel.5–7 Na et al. (2005) found that 58% of aromatic VOC emissions in Seoul, Korea, originated from vehicle exhausts.6 Measurements made during summer 2001 in Sacramento by Rubin et al. (2006) found fuel evaporative emissions could contribute up to 29% of total vehicular VOC emissions.7 Quantifying the emission rates of individual VOCs is a prerequisite to their cost-effective and successful control and this is routinely attempted by the construction of bottom-up emission inventories such as the UK’s National Atmospheric Emissions Inventory (NAEI).8 In the case of VOCs of biogenic origin, the most widely used models of emissions are derived from that of Guenther et al. (1995).9
The validation of emission inventory estimates is difficult as they are built up from many emission factors and activity rates. Measurements of atmospheric volume mixing ratios do not allow their direct validation but require a model of atmospheric chemistry and transport to infer emission rates. Micrometeorologically-based surface-to-atmosphere flux measurements can allow direct validation, but normally only at one point on the surface, which is not representative of an entire city. For example, Langford et al. (2010) assessed VOC emission fluxes from central London by making flux measurements at the BT Tower. They found good agreement for benzene, toluene and C2 alkyl-benzenes fluxes to NAEI emission estimates generated using a spatial footprint model2 but the study was limited by confined spatiality.
Measurements from aircraft allow for larger spatial assessment compared to tower measurements. Due to the high speed at which aircraft move, coupling aircraft measurements to the surface has different challenges compared to measuring at a tower site. Small aircraft which can fly low and slow have previously been used to study atmospheric turbulent structure through airborne eddy-covariance.10,11 One such approach employs disjunct eddy-covariance (DEC), as first described by Karl et al. (2002).12 DEC allows for sample processing time to be slower than the air sampling rate (<1 s) causing data to become discontinuous, while still being able to fully capture the turbulent statistics required for eddy-covariance.12–14 Karl successfully implemented DEC on-board an aircraft, to assess city-wide emissions of toluene and benzene from Mexico City, and found that sampling rates above ≥2 Hz captured the majority of eddy flux contributions.13 Misztal et al. (2014) also investigated VOC emissions from an aircraft via DEC. High spatiality isoprene fluxes were quantified over a large region of California. The study gave direct assessment of a statewide emission inventory used in predicting ozone concentrations, which previously had not been possible on such a large scale.15
Shaw et al. 2015 assessed the temporo-spatial distributions of benzene, toluene NOx mixing ratios over London using an aircraft in 2013. The strong correlation between aircraft and ground site measurements highlighted the applicability for using aircraft to assess ground level pollution sources from London, with vehicles predicted to be a major source for measured VOCs over central London.16 During the same flights, Vaughan et al. (2016) measured fluxes of nitrogen oxides (NOx) over London and found that the NAEI greatly underestimated NOx fluxes, mainly because road vehicle sources were underpredicted by the Inventory.17 This was the first attempt to validate the NAEI at the city scale, and the present study continues this by directly assessing the NAEI, and an inventory of biogenic VOC emissions, to provide a direct validation of the current ability of emissions inventories to predict VOC emissions in SE England. The work has important policy implications as it highlights current weaknesses in the ability to estimate VOC emission rates to the atmosphere.
2. Methodology
The Ozone Precursor Fluxes in an Urban Environment (OPFUE) campaigns ran during July 2013 and July 2014. The strategy for both campaigns was to determine the highly spatially resolved emissions of a range of ozone precursor species from both anthropogenic and biogenic sources, over Greater London and SE England.162.1. Flight strategy
The OPFUE campaign (24th June–9th July 2013) consisted of 12 research flights aboard the Natural Environment Research Council’s Airborne Research and Survey Facility (ARSF) Dornier-228 aircraft, based at Gloucestershire Airport. Each flight involved replicated legs across Greater London and the rural area to its south (described as South Sussex below). Fig. 1a shows all flight transects across Greater London (∼50 km length). Fig. 1c shows all flight transects across the rural South Sussex region (∼50 km length). At the end of every flight a profile ascent was conducted to high-altitude (up to 2400 m) allowing an assessment of the boundary layer height and for instrument calibrations to be conducted in clean free tropospheric air. | ||
| Fig. 1 (a) RF transects over London during July 2013, and (b) July 2014. (c) RF biogenic transects over the South Sussex region during July 2013, and (d) July 2014. | ||
In July 2014 (9th–16th July), similar flight paths were used over London, with extra legs added to form an incomplete figure of 8 (Fig. 1b) with each transect ∼120 km long. The expanded flight path allowed for greater spatial coverage, passing over important emission areas, including the industrialised area around the Dartford crossing over the River Thames. Flights over the South Sussex region followed the same flight path as used in July 2013, shown in Fig. 1d.
In summary, total spatial coverage over Greater London during 2013 was ∼400 km2 and ∼700 km2 during 2014. Over rural South Sussex, total spatial coverage of ∼170 km2 was achieved during 2013 and ∼180 km2 during 2014.
2.2. Scientific payload
VOC measurements were obtained at a sampling rate of 5 Hz and a repetition rate of ∼2 Hz. The target protonated masses and likely contributing compounds were m/z 57 (methyl tert-butyl ether, MTBE), m/z 69 (isoprene), m/z 71 (methyl vinyl ketone/methacrolein, MVK/MACR), m/z 79 (benzene), m/z 93 (toluene), m/z 107 (C2 alkyl-benzenes ethylbenzene/benzaldehyde/xylene isomers) and m/z 121 (C3 alkyl-benzene isomers). Additionally, both the primary ion m/z 21 (H3O18+), its first water cluster (H3O18 H2O16+) at m/z 39 and O2+ at m/z 32 were determined. PTR-MS drift tube pressure, temperature and voltage were held constant at 2.0 mbar, 40 °C and 480 V respectively, maintaining an E/N ratio of approximately 110 Td. For flights at ∼360 m a.g.l, the m/z 21 primary ion count ranged between (4–7) × 107 ion counts per second (cps) with an average of 6 × 107. Ion counts of m/z 32 ranged between (0.8–3) × 106 cps, with an average of 2 × 106 cps, which represented 3% of the primary ion signal. Ion counts of m/z 39 ranged between (1–5) × 106 cps with an average of 3 × 106 cps, which represented 6% of the primary ion signal. Due to O2+ and NO+ impurities in PTR-MS (∼1–5%), elevated concentrations of butane and higher alkanes are going to be detected at the most abundant ion fragments such as at m/z 57 and 71, which will contribute to the ion signals of MTBE and MVK/MACR.22
In PTR-MS several major vehicle exhaust emission components are expected to contribute to the ion signal at m/z 57, including MTBE and C4H8 (butene) isomers. The butenes react to form protonated molecular ions at this mass while greater than 95% of the MTBE proton transfer reaction products fragment to the mass 57 ion.23 It is now recognized that the concentrations derived from ion intensity measurements at m/z 57 involving petroleum vehicular emission predominately reflect the sum of the butene isomers and MTBE. However, Rogers et al. (2006) concluded that m/z 57 ion receives significant intensity from a wide variety of neutral components including the isomeric butenes, acrolein, higher order alkenes and alkanes particularly abundant in vehicular diesel exhaust.24 The ion signal at m/z 57 (MTBE‡) was therefore assumed to represent the vehicular emission source across Greater London with mixing ratios calculated assuming kcMTBE‡ of 2 × 10−9 cm3 s−1.
During flights, ambient air was sampled from the forward facing isokinetic inlet along a heated (70 °C) 5 m 1/4′′ Teflon (PFA) tube pumped by a stainless-steel diaphragm pump (Millipore) at a flow-rate of 22 L min−1. A portion of this ambient air (∼300 sccm) was diverted into the pressure controlled inlet of the PTR-MS instrument such that the overall delay time was <3 s. To determine blank VOC mixing ratios, the remaining ambient air was purged into a custom built zero air generator, which consisted of a 3/8′′ stainless steel tube packed with 1 g of platinum coated quartz wool (Elemental Microanalysis) which efficiently removes VOCs.26 The zero-air generator was operated at 350 °C and 30 psi for the duration of the flights to maintain optimal operating conditions. The catalytic converter does not remove water vapour from the sample stream, which is of importance as background impurities may depend upon sample air humidity. Zero air was periodically back-flushed through the inlet system to determine the instrument background signal.
2.3. Disjunct eddy covariance
Eddy covariance is a well-defined technique for quantifying trace gases emissions from the surface to the atmosphere. Here we use a moving aircraft to gain greater spatial coverage than possible from a tower, hence continuous wavelet transform (CWT) was chosen to generate fluxes with high time (and hence spatial) resolution. CWT gives both frequency and time-resolved flux information, compared to frequency resolved information only from the more conventional Fast Fourier Transform (FFT) approach. CWT gives advantages over conventional FFT, with stationarity not needing to be conserved and time resolved spectral contributions quantified. All calculated mixing ratios from the PTR-MS are dry mixing ratios, with fluxes calculated using this methodology not requiring water vapour content correction. Prior to calculating the flux, lag time correction via cumulative covariance was carried out.Using identical methodology as proven successful in previous studies,15,17,27,28 we conduct CWT viaeqn (1), which defines the transformation of the discrete sequence of data x(n) (being either the instantaneous change of concentration or vertical wind speed data from its mean), using the daughter scaled Morlet wavelet:29
 | (1) |
For flux data quality control, conventional FFT flux analysis was run at the same time as the CWT analysis. A comparison was then made between the two. By analysing the co- and cumulative-co-spectra, we are able assess the need for any frequency correction. Fig. 2 shows the CWT and FFT flux spectra across all frequencies for heat and C2 alkyl-benzenes fluxes for one run over London. For all analysed runs, we found spectra followed the general trend shown, with all major flux contributions below 0.1 Hz, inferring that high frequency correction for flux loss was not needed. Any major deviations found between FFT and CWT spectras would result in data being discarded. Good fluxes were deemed to display similar spectra to that of the standard FFT, showing no high frequency loss, and a ratio of FFT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CWT of 0.7–1.3. The calculated FFT flux data would still be affected by nonstationarity and inhomogeneity.15 However, this comparison still points to whether the CWT flux analysis had been successful.
:CWT of 0.7–1.3. The calculated FFT flux data would still be affected by nonstationarity and inhomogeneity.15 However, this comparison still points to whether the CWT flux analysis had been successful.
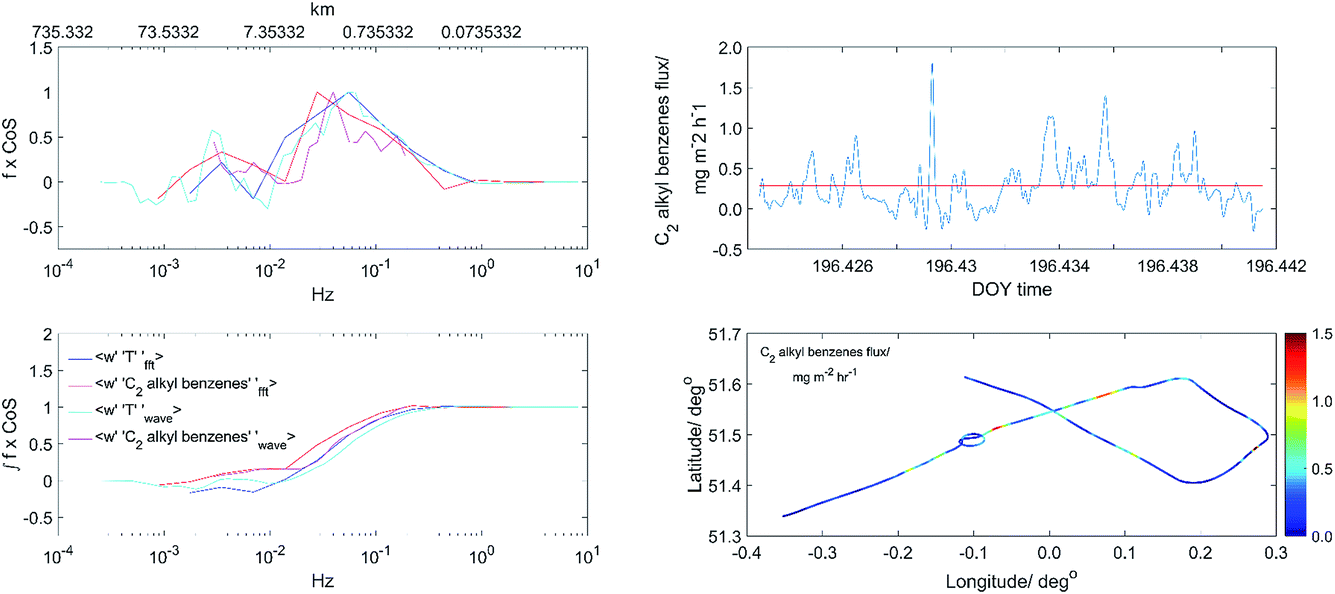 | ||
| Fig. 2 Co- and cumulative co-spectra of wavelet (wT) and FFT C2 alkyl benzenes and temperature fluxes. | ||
A final data quality process involved the removal of any flux contributions which sat outside of the cone of influence (COI). The COI is the area of the wavelet cross-spectrum which is free from edge effects, with the area outside it being of lower flux quality.30 Edge effects were found to be greatest at the beginning and end of each flight run. To help reduce this, the beginning and end of each run was padded with instrument zeros before conducting CWT analysis; however, fluxes calculated at the beginning and end of each run will still be prone to greater uncertainties than fluxes from the more central parts of each run.
The calculated error associated with each flux transect considers random, systematic and disjunct errors as an average across the transect, using the methods of Lenschow et al. (1994) and Karl et al. (2013).28,31 It should be noted that the error associated with instantaneous fluxes was much greater than the average error across each flight track, with calculated errors increasing to 100–300%. A full summary of calculated flux errors as an average over each transect is shown in Table 1.
| Compound | Systematic error/% | Random error/% | Disjunct error/% |
|---|---|---|---|
| C2 alkyl-benzenes | 7.1 | 25.1 | 0.8 |
| Toluene | 19.1 | 35.0 | 1.0 |
| Benzene | 25.3 | 47.0 | 1.1 |
| C3 alkyl-benzenes | 5.0 | 12.0 | 0.7 |
| MTBE | 2.4 | 18.0 | 1.3 |
| Isoprene | 9.5 | 30.1 | 2.2 |
| MVK/MACR | 2.1 | 16.6 | 1.6 |
| Monoterpenes | 2.3 | 17.7 | 1.9 |
2.4. Footprint calculation
A footprint model was used to calculate the origin of the emissions as measured by the aircraft: the scalars of interest are mixed and transported downwind during the interval from the time of emission from the surface to their registration at the aircraft position. The footprint model permits calculating an ensemble trajectory for each aircraft observation, and thus to spatially attribute the measured emissions to surface sources in a probabilistic framework. Here, a computationally efficient parameterization for along-wind dispersion (Kljun et al. 2004) is coupled with a cross-wind dispersion function to permit 2-dimensional source area definition. Metzger et al. (2012) provide an in-depth description of this coupled parameterisation alongside a comparison with alternative footprint models.34,35The parameterisation depends upon friction velocity u*, measurement height z, standard deviation of the vertical wind σw, standard deviation of the crosswind wind σv, aerodynamic roughness length z0, and the boundary layer depth zi, and is valid in the range −200 ≤ z/L ≤ 1, u* ≥ 0.2 m s−1, and 1 m ≤ z ≤ zi. As previously used in Vaughan et al. (2016), z0 values for London were taken from the Drew et al. (2013) study at 1 km2 resolution.17,36 Time of flight zi estimates for London were obtained via Li-DAR sounding from Heathrow airport in West London. For flights over South Sussex region, zi estimates were obtained from vertical profiles at the beginning and end of each flight. z0 values for the South Sussex region were calculated according to Monin–Obukhov theory.36–38 The remaining variables are directly available from the aircraft measurements.
For the aircraft flux observations, each 1 km along the flight track, the model calculates a surface weighting matrix at 1 km resolution, which is the same as the emission inventory grid resolution. The surface weights for each observation sum to unity, which permits cell-wise multiplication of the surface weights with the emission inventory, and subsequent aggregation over the entire grid extent. This results in the estimate of the emission strength that should be detected by the aircraft, based on the emission inventory.35,39 Additional methodological detail is provided in Metzger et al. (2013).
2.5. NAEI comparison
Measured anthropogenic fluxes were compared to emission estimates of benzene and total non-methane hydrocarbons (NMHCs) from 11 source sectors made by the UK’s National Atmospheric Emissions Inventory at 1 km2 spatial resolution.40 The source sectors used in the NAEI include solvent use, road transport, agriculture and industrial processes. NAEI road transport estimates follow the COPERT 4 emission factor model, which can be found in the European Monitoring and Evaluation Programme/European Economic Area (EMEP/EEA) air pollutant emission inventory guidebook.41 A full description of NAEI model methodology can be found in Bush et al. (2008).8 Comparison to measured fluxes using the NAEI was done using year-specific inventory data for either 2013 or 2014.Emission estimates at 1 km2 resolution from each source sector were quantified using the footprint model described above. As NAEI emission estimates are annual averages, month-of-the-year, day-of-the-week and time-of-day scaling is required to produce hourly average emission estimates for the specific days of the year on which flux measurements were made. Each source sector was scaled separately, with each scaling factor accounting for temporal variation in emissions for each month, day and hour.40 Scaled estimates for each source sector were then summed up for every 1 km along the flight track, giving time-of-day emission estimates along the flight track.
Measured fluxes of toluene and C2 alkyl-benzenes were compared to NMHCs estimates from the NAEI. As before, annual estimates were scaled to time-of-day using the described scaling factors, giving temporally-resolved hour of the day estimates. To extract specific VOC emission estimates from the inventory, the Passant (2002) study was used.42 This gives detailed characterisation for each NAEI source sector, with percentage contribution of each VOC quantified. Each source sector was scaled using the relevant percentage contribution, with all sectors summed up every 1 km along the flight track, giving time-of-day emission estimates for toluene and C2 alkyl-benzenes.
2.6. EMEP4UK comparison
Emission measurements of biogenically-derived isoprene were compared with emission estimates made using the European Monitoring and Evaluation Programme model (EMEP MSc-W) for the United Kingdom (EMEP4UK). The model gives 5 km2 emission estimates of isoprene for the whole of the UK with hourly resolution using near-surface air temperature and photosynthetically active radiation (PAR) flux.43–46 A detailed description of EMEP4UK biogenic isoprene emissions can be found in Simpson et al. (2012).47 Using the described footprint model, time-of-day estimates of isoprene emission rates were quantified using interpolated 1 km2 EMEP4UK estimates. Comparison using the EMEP4UK model to measured isoprene fluxes was done using year-specific inventory data either 2013 or 2014.3. Airborne measurements
3.1. Anthropogenic VOC fluxes over London
Measurements conducted during the 2013 flights over London focused on two anthropogenic VOCs, benzene and toluene. Both compounds are highly volatile with a wide range of previously identified emission sources in London.2,16 Meteorological conditions during the flights are summarised in Table S1,† with prevailing wind directions predominately from the SW and temperatures above 20 °C. Inverse distance weighing (obtained using ArcGIS) at 500 m, was used to interpolate all measured fluxes from all RF transects, generating spatially-average benzene and toluene fluxes, as shown in Fig. 3a. Approximately 4 hours of benzene fluxes were obtained from 5 RFs, covering a total area of ∼400 km2 over London. These fluxes highlight the spatial heterogeneity of VOC emission over central London, with maximum emission rates of 0.20 mg m−2 h−1. Higher benzene fluxes were also observed around the M25 ring road around Greater London and industrial areas of London, as shown in Fig. 3a. The mean benzene flux from all RFs was 0.051 ± 1 mg m−2 h−1.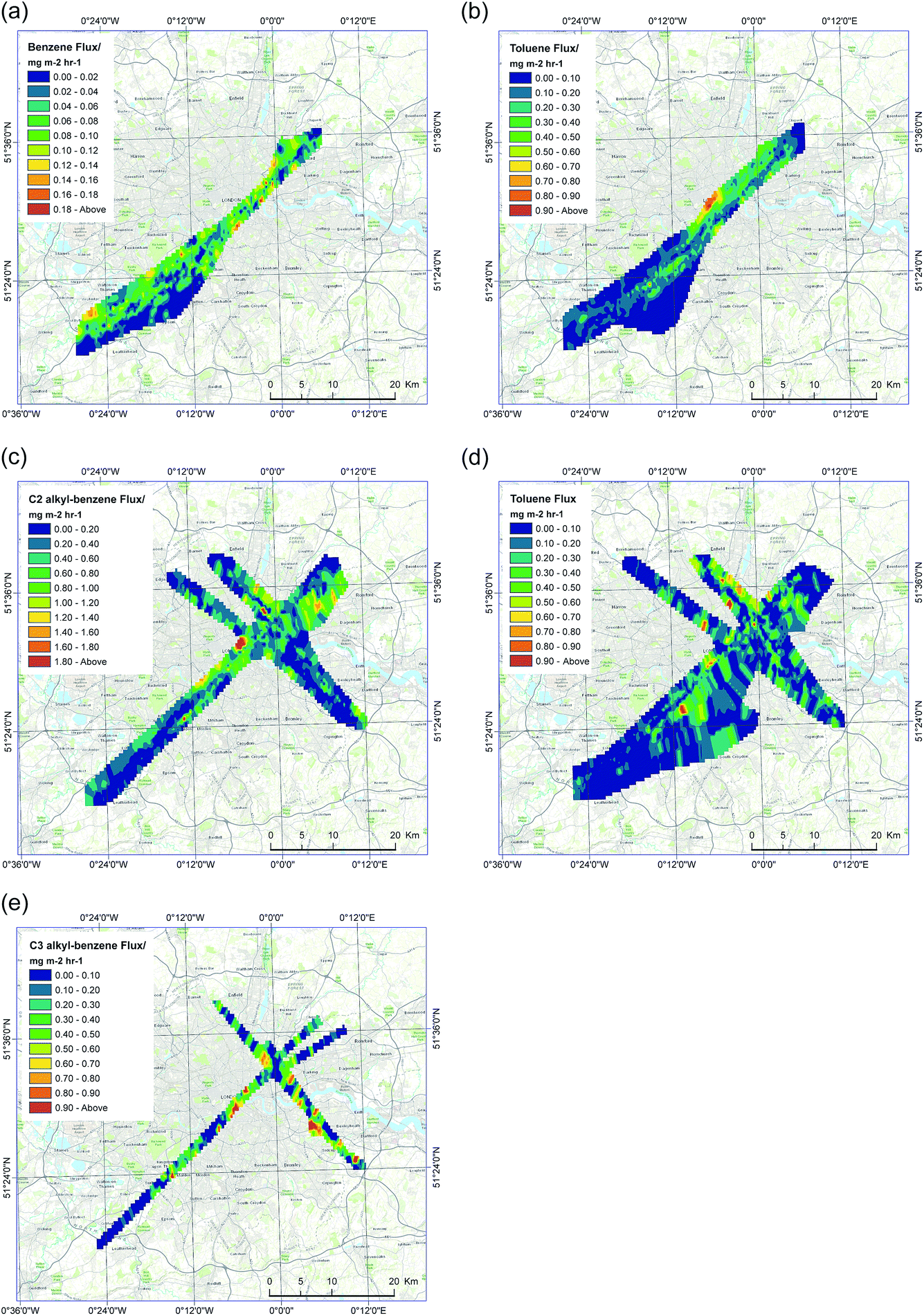 | ||
| Fig. 3 Measured instantaneous anthropogenic fluxes of (a) benzene (2013), (b) toluene (2013), (c) C2 alkyl-benzenes (2014), (d) toluene (2014) and (e) C3 alkyl-benzenes (2014), interpolated at 500 m using inverse distance weighting from all RF transects. | ||
Toluene fluxes were quantified during from 33 flight flux transects in 6 RFs, giving ∼5.5 hours of flux data with total coverage over London of ∼400 km2. The observed structure in toluene emissions was comparable to that of benzene, with highest averaged emissions of 0.30 ± 1 mg m−2 h−1 found over central London. Toluene emissions across all of London were always higher than those of benzene, with a mean toluene flux from all RFs of 0.18 ± 1 mg m−2 h−1. Emission rates exceeding 1.0 mg m−2 h−1 of toluene were observed in central London (Fig. 3b). In 2014, toluene fluxes were obtained from 5 RFs, with >75% of transects generating fluxes. A total of ∼3 hours of toluene fluxes with coverage of ∼700 km2 over London were obtained. The emissions profile was comparable to the previous year’s data, with the highest measure toluene flux of 0.30 mg m−2 h−1. Fig. 3d shows interpolated toluene fluxes from all transects across London. No benzene flux data were available from the 2014 flights due to reduced instrument performance.
As discussed by Langford et al. (2009), benzene/toluene ratios (b/t) can help evaluate the emission source of measured fluxes. Ratios in the range of 0.41–0.83 indicate vehicle exhaust emissions as dominant.14,48 To investigate the spatial emission distribution over London, the flight path was split into 4 zones: Suburban London latitudes of 51.30–51.35°; Southwest Greater London with latitudes of 51.35–51.42° containing mainly residential areas; Central London latitudes of 51.42–51.56° containing the most densely populated areas of London; and Northeast Greater London, at latitudes of 51.56–51.60°, containing a mix of residential and industrial areas.
Suburban London showed an average b/t ratio of 0.43, indicative of vehicular exhaust emissions as the main VOC source. Within the suburban region of London is the M25 motorway ring road, which encompasses the entire Greater London area. The highest b/t ratio observed within this area was 0.82, indicating direct VOC emissions coming from the motorway. As discussed by Rogers, higher b/t ratios can also be attributed to correct operation of vehicle emission control systems, i.e. the catalytic converter. Ratios will however depend on the performance of the converter and the fuel used.23
For Southwest Greater London, the average b/t ratio was lower at 0.28. Ratios below 0.41 are indicative of other sources being dominant.13,23 The average b/t ratio for central London was also 0.28, suggesting similar sources are responsible for these two regions’ VOC emissions. Northeast Greater London showed the lowest average b/t ratio of 0.20, which as discussed by Karl et al. (2009), is indicative of more industrial processes and evaporation. Karl et al. (2009) observed a ratio of 0.13 over Mexico City, with a multitude of emission sources predicted to be responsible.13 Langford et al. (2010) observed similar b/t flux ratios from the BT tower in central London of 0.21.2 The similarities between their findings and aircraft calculated ratios indicate the measured flux in this study can capture ground level emission sources successfully.
Measurements made during the 2014 flights over the London focused on a wider range of VOCs. Meteorological conditions during all 2014 flights (RFs 3–7) are shown in Table S1.† Flux measurements were not possible during RFs 1 and 2 due to adverse weather conditions and instrument problems. Prevailing wind directions during many of the RFs over London were from the SW, with high summer air temperatures. The higher order benzenoid compounds, C2 alkyl-benzenes and C3 alkyl-benzenes, were measured during the 2014 flights. Fluxes of C2 alkyl-benzenes were measured during 5 RFs, giving 1.5 hours of flux data and a total spatial coverage of ∼450 km2. High emissions were observed over central London and industrial areas of the Greater London region (Fig. 3c), with a maximum flux of 1.00 mg m−2 h−1. Measurements of C3 alkyl-benzenes fluxes were obtained during 2 RFs, giving ∼30 min of data, over a total area of 150 km2. C3 alkyl-benzenes such as 1,2,4-trimethylbenzenes are a product of petroleum refining and have industrial applications, including as a blending additive in petroleum fuels.49,50 Maximum observed emissions of C3 alkyl-benzenes were found to be ∼1.00 mg m−2 h−1, observed over central London. Fig. 3e shows interpolated instantaneous C3 alkyl-benzenes fluxes from all RF transects.
MTBE‡ fluxes were measured during two (2014) RFs, giving total spatial coverage of ∼100 km2. Methyl tert-butyl ether (MTBE) is a key additive in petrol fuel, acting as an anti-knocking agent.50 The highest measured emissions of MTBE‡ were observed over areas with similarly high toluene emissions, with up to 1.00 mg m−2 h−1 of MTBE‡ observed. Fig. 4 shows latitude averages of all flight transects (1 km resolved) of MTBE and toluene emission, with both in good agreement. The average MTBE‡ flux for Central London was calculated to be 0.21 mg m−2 h−1, compared to 0.20 mg m−2 h−1 for toluene, suggesting an approximate 1:1 ratio. In 2008 in the UK, the average MTBE concentration in petroleum was 3.4% (v/v).51 As MTBE‡ is a good indicator of the relative importance of vehicular emissions, the observed toluene emissions from central London can be attributed mainly to vehicle emissions.
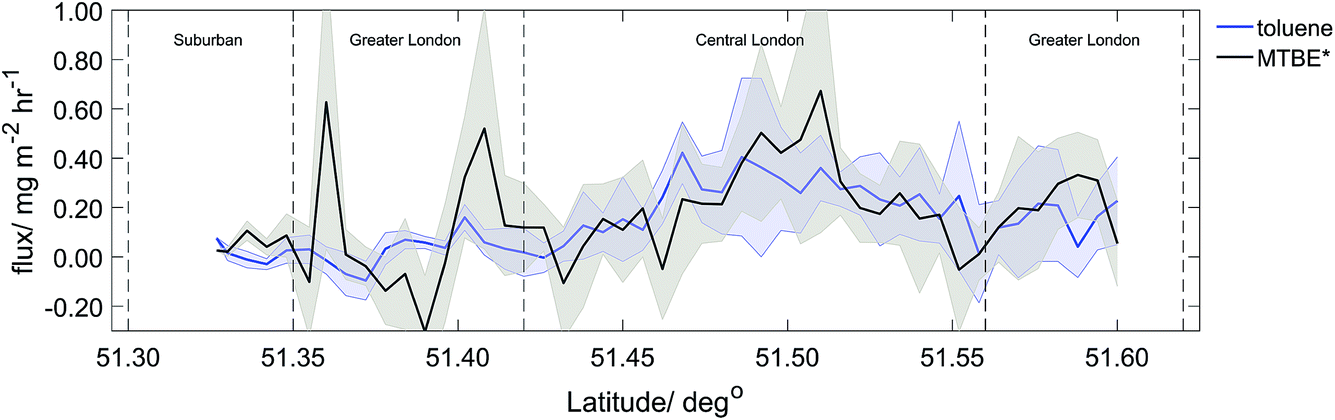 | ||
| Fig. 4 Latitude averaged (1 km) MTBE‡ (grey) and toluene flux (blue) with standard deviation (shaded area). | ||
3.2. NAEI comparison
Benzene fluxes quantified for all RFs in 2013 were compared to 1 km2 footprint estimates of benzene from the NAEI, scaled to time-of-day as described above. The lateral displacement of the footprint from the flight track due to the horizontal boundary layer wind extended 4–12 km upwind and is shown in Fig. 5. Fig. 6a shows 1 km latitude averages of measured benzene fluxes and NAEI benzene emission estimates with the standard deviation shaded. Good agreement between measurements and NAEI emission estimates was found for across most of London. Due to the significant uncertainty of benzene flux at 1 km2 resolution (100–300%), benzene flux and NAEI estimates were averaged across the entire flight path, reducing uncertainty. The average measured benzene flux (0.051 mg m−2 h−1) and the average NAEI estimate (0.037 mg m−2 h−1) agree within the bounds of measurement uncertainty, with a ratio of 1.40. To investigate the spatial emission distribution over London, the flight path was split into four zones as previously discussed, reducing the overall error associated with 1 km averages. The suburban London region was found to have the largest difference between averaged benzene flux and NAEI. The average flux was found to be 3 times higher than predicted NAEI estimates. Greater and Central London areas showed better agreement, with ratios of average flux/NAEI below 1.70.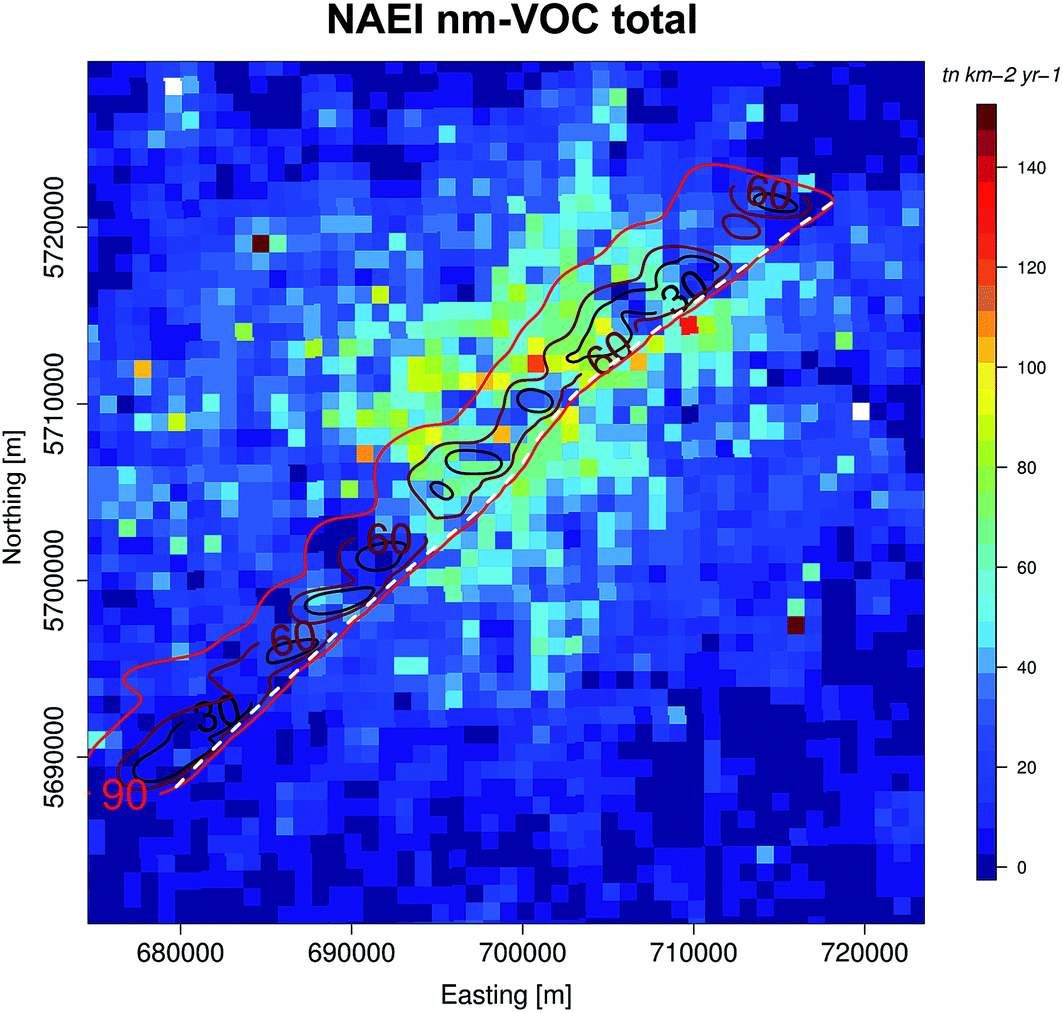 | ||
| Fig. 5 Example footprint for 2014 RF transect over London showing the 30, 60, 90% footprint influence areas from the flight track (white), overlaid onto 1 km2 NAEI total NMHC emission estimates. | ||
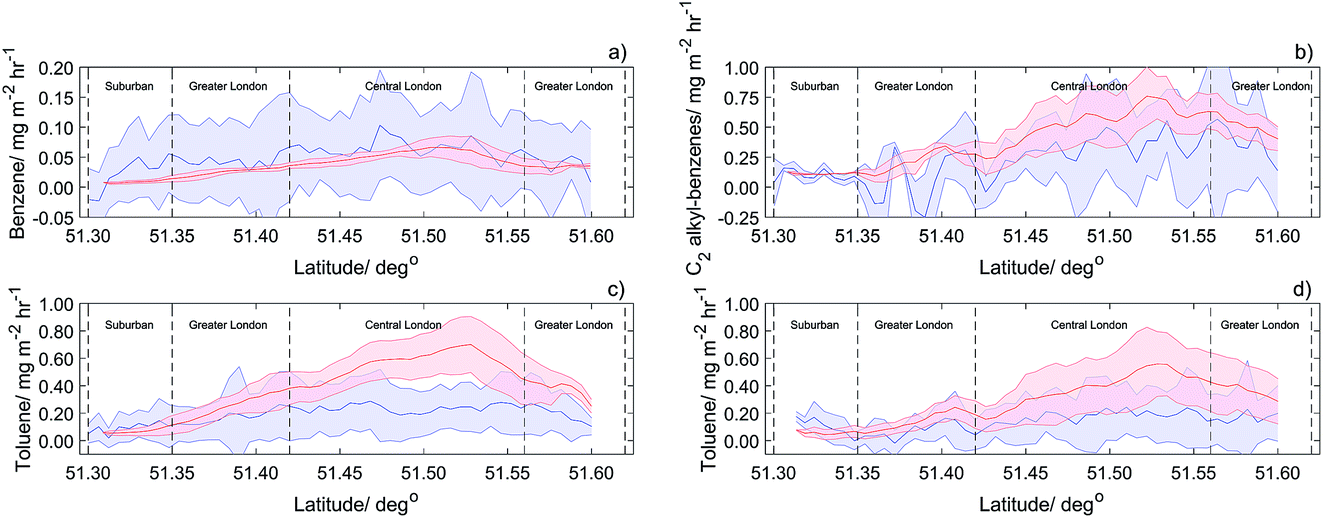 | ||
| Fig. 6 Latitude averages (1 km) for (a) benzene flux (2013), (b) C2 alkyl benzenes flux (2014), (c) toluene flux (2013) and (d) toluene flux (2014) shown in blue, with footprint NAEI estimates in red. Shaded areas are the calculated standard deviation of all RF transects. | ||
Toluene fluxes measured in 2013 were also compared to the NAEI emission estimates. Fig. 6c shows 1 km2 latitude averages of toluene flux and NAEI estimates with standard deviation shaded. In suburban areas of London, measured fluxes are a factor of 1.4 higher than NAEI estimates. In the other zones, measured fluxes are 30 to 50% lower than estimates. When averaged across the whole of London, the NAEI emission estimates for toluene were a factor of two higher than the average measured flux (0.39 mg m−2 h−1 from the NAEI compared with the observed flux of 0.18 mg m−2 h−1).
Comparison of 1 km2 latitude average toluene flux (2014) and NAEI estimates are shown in Fig. 6d, with good agreement for outer London. Again, higher NAEI emission estimates are observed for most of London. Average NAEI toluene estimates are a factor of 2 higher compared to measured fluxes, with average NAEI estimate of 0.28 mg m−2 h−1, compared to the average measured value of 0.14 mg m−2 h−1. With stricter legislation now restricting emissions from sources such as road transport, solvent emissions are now predicted to be the main source for NMHCs in London.52 As observed in 2013, Greater and Central London areas show measured fluxes to be 50% less than predicted by the NAEI, further implying the need for refinement of emission sources for toluene within the NAEI.
C2 alkyl-benzenes compared to 1 km2 NAEI estimates (Fig. 6b) show good agreement across London. Average NAEI estimates for C2 alkyl-benzenes are higher than the average measured C2 alkyl-benzenes flux. This overestimation is likely due to errors in the source sector factors for C2 alkyl-benzenes emissions. Average suburban measured C2 alkyl-benzenes flux agreed well with the NAEI estimates, compared to the average Southwest Greater London average flux which was 70% lower than NAEI estimates. Table 2 gives an in-depth review of measured fluxes and NAEI footprint estimates for both years’ flights, giving averages and ratios for each of the four defined London zones.
| Average flux/mg m−1 h−1 | Suburban London/mg m−1 h−1 | Southwest Greater London/mg m−1 h−1 | Central London/mg m−1 h−1 | Northeast Greater London/mg m−1 h−1 | |
|---|---|---|---|---|---|
| Benzene (2013) | 0.052 | 0.035 | 0.044 | 0.065 | 0.039 |
| Benzene NAEI (2013) | 0.037 | 0.009 | 0.026 | 0.052 | 0.034 |
| Toluene (2013) | 0.180 | 0.098 | 0.180 | 0.240 | 0.190 |
| Toluene NAEI (2013) | 0.390 | 0.069 | 0.260 | 0.560 | 0.380 |
| Toluene (2014) | 0.140 | 0.104 | 0.076 | 0.170 | 0.160 |
| Toluene NAEI (2014) | 0.280 | 0.058 | 0.140 | 0.380 | 0.370 |
| C2 alkyl-benzenes (2014) | 0.320 | 0.107 | 0.064 | 0.320 | 0.380 |
| C2 alkyl-benzenes (2014) NAEI | 0.490 | 0.110 | 0.220 | 0.540 | 0.510 |
| Average flux/NAEI | Suburban London/ratio | Southwest Greater London/ratio | Central London/ratio | Northeast Greater London/ratio | |
|---|---|---|---|---|---|
| Benzene (2013) | 1.40 | 3.70 | 1.70 | 1.30 | 1.10 |
| Benzene NAEI (2013) | |||||
| Toluene (2013) | 0.46 | 1.40 | 0.70 | 0.40 | 0.50 |
| Toluene NAEI (2013) | |||||
| Toluene (2014) | 0.50 | 1.80 | 0.50 | 0.50 | 0.40 |
| Toluene NAEI (2014) | |||||
| C2 alkyl-benzenes (2014) | 0.64 | 0.90 | 0.30 | 0.60 | 0.70 |
| C2 alkyl-benzenes (2014) NAEI |
3.3. Biogenic VOC fluxes
Measurements of several VOCs of predominantly biogenic origin were made over the South Sussex region of Southern England. Fig. 1c and d show all flight transects conducted during the two campaign periods. Fig. 7 shows the calculated footprint extent from the flight track overlaid onto the UK’s National Land Classification map at 25 m2 resolution. This shows that for a typical flight over the region, areas of mixed broadleaved woodland containing Quercus and other tree species known to emit isoprene occur within the footprint.53–55 | ||
| Fig. 7 RF flight track during a 2013 flight over the South Sussex region. (Left) Calculated footprint extent 30, 60, and 90% from flight track, overlaid onto EMEP4UK isoprene emission grid. (Right) Footprint extent 30, 60, and 90% from flight track, overlaid onto UK National Land Class Inventory 2012 at 25 m2 resolution. | ||
Measured isoprene fluxes varied from 0.20 to 2.00 mg m−2 h−1 (Fig. 8a and b). In general, the temperature and the flux of photosynthetically active radiation (PAR) were higher during the 2013 flights than during the 2014 flights. Fig. 9 shows mean isoprene emissions from identical flight transects conducted on three consecutive days (7–9th July 2013), each flight consisting of 1.5 hours and ∼40 km repeated flight legs. Mean measured isoprene emission rates in areas of broadleaved woodland displayed a dependence on both PAR and ground temperature. Maximum isoprene emissions measured during both campaigns were found to be 2.0 mg m−2 h−1 and 1.0 mg m−2 h−1 for July 2013 and July 2014, respectively. This difference can be explained by differences in weather conditions (temperature and cloudiness) during the two measurement periods.
 | ||
| Fig. 8 Measured biogenic instantaneous fluxes of (a) isoprene (2013), (b) isoprene (2014), (c) monoterpenes (2013) and (d) monoterpenes (2014), interpolated at 500 m using inverse distance weighting from all RF transects. | ||
 | ||
| Fig. 9 Mean isoprene emissions from 3 OPFUE (July 2013) identical flight transects conducted on 3 consecutive days (7/7/13–9/7/13), each flight consisting of 1.5 hours of data and ∼40 km repeated flight legs. Mean isoprene emission in the dense broadleaved woodland area displayed a dependence on photo-synthetically active radiation (PAR) (black) and ground temperature (grey). | ||
MVK/MACR fluxes were quantified during two RFs in 2014. Overall, one hour of flux data was obtained, giving spatial coverage of ∼180 km2. Maximum fluxes of ∼0.20 mg m−2 h−1 were observed between latitudes 51.10–51.20°, with high isoprene fluxes observed over the same area. NO mixing ratios measured during all flights were found to be ≤1 ppbv. Under these conditions, the MCM 3.1.1 predicts that 70% of the measured flux will be MVK and 30% MACR.
Emissions of monoterpenes were also quantified during both campaigns. PTR-MS intensities at m/z 137 were assumed to give a measure of the total monoterpene flux, since no separation of individual species was possible. In 2013, 1 hour of monoterpene flux data was obtained from 2 RFs and ∼160 km2 of spatial coverage. Monoterpenes measured during 2014 generated 1.25 hours of flux data, with overall spatial cover of ∼180 km2. Both campaigns showed similar emission characteristics, with highest emissions found at latitudes 51.00–50.90°. Maximum emissions were found to be 1.00 mg m−2 h−1 during 2013 and 0.80 mg m−2 h−1 during 2014. Fig. 8c and d show interpolated monoterpene fluxes during both years. Table S2† gives a full statistical breakdown of all measured biogenic VOCs during 2013 and 2014.
3.4. EMEP model comparison
The measured isoprene fluxes from 2013 and 2014 were compared to hourly-average EMEP4UK isoprene estimates at 5 km2 resolution. Because EMEP4UK estimates only account for biogenic sources of isoprene, the NMHCs NAEI was used to account for any anthropogenic contribution. Isoprene contribution for each of the 11 source sectors within the NAEI was accounted for in remaining unspeciated contributions, with no separation.42 Total anthropogenic contributions of isoprene for the region were found to be less than 1% of EMEP4UK estimates, highlighting biogenic sources as dominant.The flux footprint represented an area 5–10 km upwind from the flight tracks (Fig. 7 left). Latitude averages of isoprene flux and EMEP4UK time-of-day estimates for 2013 (Fig. 10 left) show good agreement from latitudes of 51.15–51.30°. The EMEP4UK model however fails to capture the high isoprene fluxes observed at latitudes between 51.05–51.15°. The model predicts isoprene emissions of <0.20 mg m−2 h−1, with measured isoprene fluxes ranging from 1.00–2.00 mg m−2 h−1. This discrepancy may be due to the relatively coarse (5 km2) land classification used by the emission model (Fig. 7 left) or too low normalized (‘base’) emission rates used in the model.
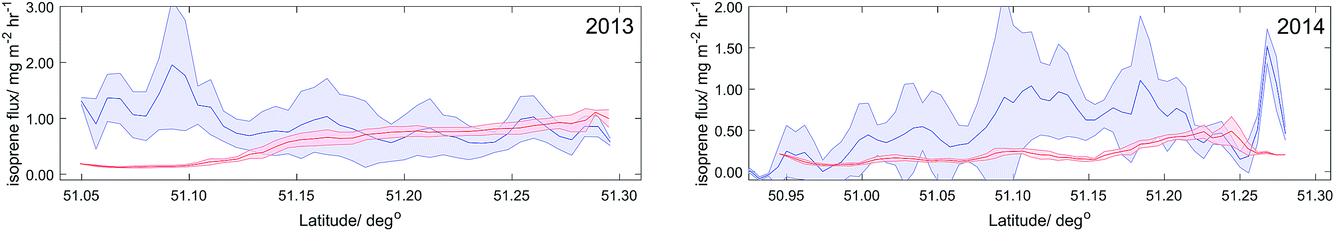 | ||
| Fig. 10 Latitude averages (1 km) of measured isoprene flux (blue) and EMEP4UK isoprene emission (red) with shaded standard deviation for 2013 and 2014. | ||
Fig. 10 (right) shows latitude-averaged isoprene fluxes compared to the EMEP4UK estimates for 2014. The degree of agreement between the measurement and the inventory is poor, with measured flux consistently higher than EMEP4UK estimates. Measured isoprene fluxes range from 0.10–1.00 mg m−2 h−1, compared to the EMEP4UK estimates of ≤0.40 mg m−2 h−1. Again, the discrepancy may be due to land classification resolution or unrealistic normalized emission rates used in the emissions model.
The highly spatially resolved (1 km2) flux measurements have significant uncertainty (100–300%). To overcome this, overall average fluxes obtained from all the measurement data were compared with the corresponding EMEP4UK emissions estimate. A full statistical description is given in Table 3 for average comparison between measured isoprene flux and EMEP4UK estimates. In 2013, the average measured isoprene flux was 60% higher than the EMEP4UK estimate. In 2014, the average measured isoprene flux was 140% higher than the EMEP4UK estimate.
| Flux to EMEP/ratio | Flux mean/mg m−1 h−1 | Flux median/mg m−1 h−1 | Standard deviation | Standard error | 95th percentile | |
|---|---|---|---|---|---|---|
| Isoprene (2013) | 1.60 | 0.902 | 0.820 | 0.320 | 0.032 | 1.50 |
| EMEP4UK (2013) | 0.570 | 0.650 | 0.301 | 0.030 | 0.95 | |
| Isoprene (2014) | 2.40 | 0.480 | 0.470 | 0.310 | 0.025 | 0.960 |
| EMEP4UK (2014) | 0.201 | 0.170 | 0.100 | 0.008 | 0.402 |
4. Conclusions
This study has allowed the first direct measurements of VOC emission rates over Greater London and SE England. A total of 11 flights during July 2013 and 2014 gave 16 hours of high spatial resolution flux measurements over London for a range of anthropogenic NMHCs, corresponding to emission measurements from ∼4500 grid cells of 1 km2 each. Measured benzene fluxes over London displayed high spatial heterogeneity and compared relatively well to NAEI estimates. Measured toluene and C2 alkyl-benzenes fluxes showed high emissions emanating from central and industrial regions of London, although these were overestimated by the NAEI. It is clear that further refinement of the NAEI, both in terms of the speciation of its NMHC inventory and in terms of individual VOC emission rates, is required.Isoprene and monoterpene fluxes, presumably predominantly from biogenic sources, were measured over rural SE England. A total of 5 flights over SE England gave 6.5 hours of highly spatially resolved fluxes covering ∼800 km2. Measured isoprene fluxes showed relatively higher emissions over areas containing Quercus and other broadleaf tree species known to emit isoprene. The EMEP4UK inventory significantly underestimated the measured isoprene fluxes. This may be due in part to the failure of the model to capture regions of elevated isoprene flux due to its use of land classification at 5 km2 resolution when much of the woodland in SE England is smaller than this. Improvements in the spatial resolution of land use classification could help improve model performance with respect to isoprene and monoterpene emissions. This is important as it is possible that biogenic VOCs (especially isoprene) could become of increasing importance to secondary pollutant formation in the UK and Northern Europe in a future warming climate, with increased prevalence of high temperature events, and as anthropogenic VOC emissions continue to decline with improved regulation and control technologies.
Overall this work demonstrates the suitability of using a low-flying aircraft to determine anthropogenic and biogenic VOC fluxes by eddy covariance and the possibility of validating emission inventories with these measurements. Further evaluation and improvements to the emission inventories of anthropogenic and biogenic VOCs used for regulatory, policy and research purposes is clearly an urgent requirement, especially as some current air quality policies are based on a flawed understanding of VOC emission rates.
Author contribution statement
CNH, ACL, JL and RP conceived the study and obtained funding. AV, JL and MS wrote the paper. AV, SM, PM, AG, TK and LC carried out the flux data analysis, footprint modelling and interpretation the results. JL, MS, ACL, CNH, RP and BD made the measurements. MV provided the EMEP4UK isoprene emissions inventory model, as well as help interpret the results. All authors contributed to the discussion and commented on the manuscript.Acknowledgements
We would like to thank Captain Carl Joseph and co-pilot James Johnson of the Natural Environment Research Council (NERC) Airborne Research and Survey Facility for their contribution to the OPFUE campaign during 2013, through flight planning and flying of the Dornier 228 research aircraft. We would also like to thank NERC (grant NE/J00779X/1) and the UK Department of Environment, Food and Rural Affairs for funding. The National Ecological Observatory Network is a project sponsored by the National Science Foundation and managed under cooperative agreement by Battelle Ecology, Inc. This material is based upon work supported by the National Science Foundation under the grant DBI-0752017. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.References
- A. L. Robinson, N. M. Donahue, M. K. Shrivastava, E. A. Weitkamp, A. M. Sage, A. P. Grieshop, T. E. Lane, J. R. Pierce and S. N. Pandis, Science, 2007, 315, 1259–1262 CrossRef CAS PubMed
.
- B. Langford, E. Nemitz, E. House, G. Phillips, D. Famulari, B. Davison, J. Hopkins, A. Lewis and C. N. Hewitt, Atmos. Chem. Phys., 2010, 10, 627–645 CrossRef CAS
- A. Kansal, J. Hazard. Mater., 2009, 166, 17–26 CrossRef CAS PubMed
- K. Na, Y. P. Kim, I. Moon and K.-C. Moon, Chemosphere, 2004, 55, 585–594 CrossRef CAS PubMed
- A. Srivastava, A. Joseph, A. More and S. Patil, Environ. Monit. Assess., 2005, 109, 227–242 CrossRef CAS PubMed
- K. Na, K.-C. Moon and Y. P. Kim, Atmos. Environ., 2005, 39, 5517–5524 CrossRef CAS
- J. I. Rubin, A. J. Kean, R. A. Harley, D. B. Millet and A. H. Goldstein, J. Geophys. Res.: Atmos., 2006, 111(D3) CrossRef CAS
-
T. Bush, I. Tsagatakis, K. King and N. Passant, NAEI UK emission mapping methodology 2006, 2008 Search PubMed
- A. Guenther, C. N. Hewitt, D. Erickson, R. Fall, C. Geron, T. Graedel, P. Harley, L. Klinger, M. Lerdau and W. McKay, J. Geophys. Res.: Atmos., 1995, 100, 8873–8892 CrossRef CAS
- C. Thomas and T. Foken, Theor. Appl. Climatol., 2005, 80, 91–104 CrossRef
- M. Mauder, R. L. Desjardins and I. MacPherson, J. Geophys. Res.: Atmos., 2007, 112(D13112) CAS
- T. Karl, C. Spirig, J. Rinne, C. Stroud, P. Prevost, J. Greenberg, R. Fall and A. Guenther, Atmos. Chem. Phys., 2002, 2, 279–291 CrossRef CAS
- T. Karl, E. Apel, A. Hodzic, D. Riemer, D. Blake and C. Wiedinmyer, Atmos. Chem. Phys., 2009, 9, 271–285 CrossRef CAS
- B. Langford, B. Davison, E. Nemitz and C. N. Hewitt, Atmos. Chem. Phys., 2009, 9, 1971–1987 CrossRef CAS
- P. K. Misztal, T. Karl, R. Weber, H. H. Jonsson, A. B. Guenther and A. H. Goldstein, Atmos. Chem. Phys., 2014, 14, 10631–10647 Search PubMed
- M. D. Shaw, J. D. Lee, B. Davison, A. Vaughan, R. M. Purvis, A. Harvey, A. C. Lewis and C. N. Hewitt, Atmos. Chem. Phys., 2015, 15, 5083–5097 CAS
- A. R. Vaughan, J. D. Lee, P. K. Misztal, S. Metzger, M. D. Shaw, A. C. Lewis, R. M. Purvis, D. C. Carslaw, A. H. Goldstein, C. N. Hewitt, B. Davison, S. D. Beevers and T. G. Karl, Faraday Discuss., 2016, 189, 455–472 RSC
- K. M. Beswick, M. W. Gallagher, A. R. Webb, E. G. Norton and F. Perry, Atmos. Chem. Phys., 2008, 8, 5449–5463 CrossRef CAS
- J. de Gouw and C. Warneke, Mass Spectrom. Rev., 2007, 26, 223–257 CrossRef CAS PubMed
- C. N. Hewitt, S. Hayward and A. Tani, J. Environ. Monit., 2003, 5, 1–7 RSC
- W. Lindinger and A. Hansel, Plasma Sources Sci. Technol., 1997, 6, 111 CrossRef CAS
- O. Amador-Muñoz, P. K. Misztal, R. Weber, D. R. Worton, H. Zhang, G. Drozd and A. H. Goldstein, Atmos. Meas. Tech., 2016, 9, 5315–5329 Search PubMed
- J. J. Schauer, M. J. Kleeman, G. R. Cass and B. R. Simoneit, Environ. Sci. Technol., 1999, 33, 1578–1587 CrossRef CAS
- T. Rogers, E. Grimsrud, S. Herndon, J. Jayne, C. E. Kolb, E. Allwine, H. Westberg, B. Lamb, M. Zavala and L. Molina, Int. J. Mass Spectrom., 2006, 252, 26–37 CrossRef CAS
- R. Taipale, T. Ruuskanen, J. Rinne, M. Kajos, H. Hakola, T. Pohja and M. Kulmala, Atmos. Chem. Phys., 2008, 8, 6681–6698 CrossRef CAS
- J. de Gouw, C. Warneke, R. Holzinger, T. Klüpfel and J. Williams, Int. J. Mass Spectrom., 2004, 239, 129–137 CrossRef CAS
- T. Karl, E. Apel, A. Hodzic, D. D. Riemer, D. R. Blake and C. Wiedinmyer, Atmos. Chem. Phys., 2009, 9, 271–285 CrossRef CAS
- T. Karl, P. K. Misztal, H. H. Jonsson, S. Shertz, A. H. Goldstein and A. B. Guenther, J. Atmos. Sci., 2013, 70, 3277–3287 CrossRef
- B. Yuan, L. Kaser, T. Karl, M. Graus, J. Peischl, T. L. Campos, S. Shertz, E. C. Apel, R. S. Hornbrook, A. Hills, J. B. Gilman, B. M. Lerner, C. Warneke, F. M. Flocke, T. B. Ryerson, A. B. Guenther and J. A. de Gouw, J. Geophys. Res.: Atmos., 2015, 120, 6271–6289 CAS
- C. Torrence and G. P. Compo, Bull. Am. Meteorol. Soc., 1998, 79, 61–78 CrossRef
- D. Lenschow, J. Mann and L. Kristensen, J. Atmos. Ocean. Tech., 1994, 11, 661–673 CrossRef
- T. Karl, A. Guenther, A. Turnipseed, G. Tyndall, P. Artaxo and S. Martin, Atmos. Chem. Phys., 2009, 9, 7753–7767 CrossRef CAS
- M. E. Jenkin, J. C. Young and A. R. Rickard, Atmos. Chem. Phys., 2015, 15, 11433–11459 CAS
- N. Kljun, P. Calanca, M. W. Rotach and H. P. Schmid, Bound.-Layer. Meteor., 2004, 112, 503–523 CrossRef
- S. Metzger, W. Junkermann, M. Mauder, F. Beyrich, K. Butterbach-Bahl, H. P. Schmid and T. Foken, Atmos. Meas. Tech., 2012, 5, 1699–1717 CrossRef
- D. R. Drew, J. F. Barlow and S. E. Lane, J. Wind Eng. Ind. Aerod., 2013, 121, 98–105 CrossRef
- J. A. Businger, J. C. Wyngaard, Y. Izumi and E. F. Bradley, J. Atmos. Sci., 1971, 28, 181–189 CrossRef
- T. Foken, Bound.-Layer. Meteor., 2006, 119, 431–447 CrossRef
- S. Metzger, W. Junkermann, M. Mauder, K. Butterbach-Bahl, B. T. Y. Widemann, F. Neidl, K. Schafer, S. Wieneke, X. H. Zheng, H. P. Schmid and T. Foken, Biogeosciences, 2013, 10, 2193–2217 CrossRef
- Defra and DECC, NAEI data, http://naei.defra.gov.uk/.
-
EEA, EMEP/EEA air pollutant emission inventory guidebook 2013. Technical guidance to prepare national emission inventories, Report 12/2013, 2013 Search PubMed
-
N. Passant, AEA Technology Report ENV-0545, Culham, Abingdon, United Kingdom, 2002 Search PubMed
- M. Vieno, A. J. Dore, D. S. Stevenson, R. Doherty, M. R. Heal, S. Reis, S. Hallsworth, L. Tarrason, P. Wind, D. Fowler, D. Simpson and M. A. Sutton, Atmos. Chem. Phys., 2010, 10, 7963–7978 CrossRef CAS
- M. Vieno, M. R. Heal, S. Hallsworth, D. Famulari, R. M. Doherty, A. J. Dore, Y. S. Tang, C. F. Braban, D. Leaver, M. A. Sutton and S. Reis, Atmos. Chem. Phys., 2014, 14, 8435–8447 CrossRef
- M. Vieno, M. R. Heal, M. M. Twigg, I. A. MacKenzie, C. F. Braban, J. J. N. Lingard, S. Ritchie, R. C. Beck, A. Móring, R. Ots, C. F. D. Marco, E. Nemitz, M. A. Sutton and S. Reis, Environ. Res. Lett., 2016, 11, 044004 CrossRef
- M. Vieno, M. R. Heal, M. L. Williams, E. J. Carnell, E. Nemitz, J. R. Stedman and S. Reis, Atmos. Chem. Phys., 2016, 16, 265–276 CrossRef CAS
- D. Simpson, A. Benedictow, H. Berge, R. Bergström, L. D. Emberson, H. Fagerli, C. R. Flechard, G. D. Hayman, M. Gauss, J. E. Jonson, M. E. Jenkin, A. Nyíri, C. Richter, V. S. Semeena, S. Tsyro, J. P. Tuovinen, Á. Valdebenito and P. Wind, Atmos. Chem. Phys., 2012, 12, 7825–7865 CAS
- N. V. Heeb, A.-M. Forss, C. Bach, S. Reimann, A. Herzog and H. W. Jäckle, Atmos. Environ., 2000, 34, 3103–3116 CrossRef CAS
- I. R. Kaplan, Y. Galperin, H. Alimi, R. P. Lee and S. T. Lu, Groundwater Monit. Rem., 1996, 16, 113–124 CrossRef CAS
- W. Xiong, R. Bernesky, R. Bechard, G. Michaud and J. Lang, Sci. Total Environ., 2014, 487, 452–462 CrossRef CAS PubMed
-
Environment Agency, Motor fuel components: environmental occurrence and potential future risks, Science Summary SC040087/SS, 2008 Search PubMed
-
R. Bailey, R. Claxton, L. Jones, E. Kilroy, T. Misselbrook, Y. Pang, N. Passant, E. Salisbury, H. Smith, G. Thistlethwaite, D. Wakeling and C. Walker, Air Quality Pollutant Inventories for England, Scotland, Wales and Northern Ireland: 1990-2014, 2016 Search PubMed
- C. Guenther, Atmos. Chem. Phys., 2006, 6, 3181–3210 CrossRef
- A. Arneth, G. Schurgers, J. Lathiere, T. Duhl, D. Beerling, C. Hewitt, M. Martin and A. Guenther, Atmos. Chem. Phys., 2011, 11, 8037–8052 CrossRef CAS
- P. K. Misztal, J. C. Avise, H. H. Jonsson, A. B. Guenther and A. H. Goldstein, Atmos. Chem. Phys., 2016, 16, 9611 CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7fd00002b |
| ‡ Potential contributions from butene isomers, C4 and higher alkenes and alkanes. |
| This journal is © The Royal Society of Chemistry 2017 |