
Chirality transfer in metal-catalysed intermolecular addition reactions involving allenes
José Miguel
Alonso
,
María Teresa
Quirós
and
María Paz
Muñoz
*
School of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: m.munoz-herranz@uea.ac.uk
First published on 5th July 2016
Abstract
Allene chemistry in the presence of transition metal complexes is nowadays a very important topic that underpins many challenges and advances in organic synthesis. The number of research articles covering new transformations of allenes is vast and the development of enantioselective reactions involving allenes has flourished in the last 10–15 years. In this review we cover three important topics in allene chemistry that we feel are timely and appropriate for this special issue celebrating the work of Prof. Trost: the metal-catalysed reactions involving chirality transfer from chiral allenes to products; the analysis of the possible racemization processes that have been observed in the interaction of some metals with allenes; and chirality transfer using racemic allenes in reactions catalysed by metal complexes bearing chiral ligands to produce enantioenriched products. We have focussed the review on intermolecular addition reactions as they are still much less explored than the intramolecular version.
 José Miguel Alonso | Dr Alonso received his B.Sc. and Ph.D. from the Universidad Complutense de Madrid, Spain, under the supervision of Prof. Benito Alcaide and Dr Pedro Almendros (2003). Besides a diverse experience in industry (environmental science, polymer chemistry), he worked as a Postdoctoral Researcher in Prof. Enrique Guitian's group at Universidade de Santiago de Compostela (Spain, 2009 and 2013–2015), and in the group of Benito Alcaide and Pedro Almendros at UCM (Spain, 2010–2013). He is currently working as Senior Research Associate at the University of East Anglia (UK, 2015) in Dr María Paz Muñoz's research group. |
 María Teresa Quirós | Dr Quirós received her B.Sc. and M.Sc. in Organic Chemistry from the Universidad Complutense de Madrid (Spain), where she carried out her Ph.D. under the supervision of Prof. Benito Alcaide and Dr Pedro Almendros (2014). During her Ph.D. she completed a short stay at the University of Heidelberg in the group of Prof. A. Stephen K. Hashmi (November 2012–January 2013). From March 2014, she joined Dr María Paz Muñoz's research group at the University of East Anglia (UK), where she was first a Senior Postdoctoral Research Associate (March 2014–March 2015) and then a Marie Sklodowska-Curie Research Fellow (April 2015–April 2016). |
 María Paz Muñoz | Dr Muñoz received her B.Sc. and M.Sc. in Organic Chemistry, from the Universidad Autónoma de Madrid, Spain, where she carried out her Ph.D. under the supervision of Prof. Antonio M. Echavarren. From November 2004, she joined the group of Prof. Guy C. Lloyd-Jones (University of Bristol, UK), where she held the positions of Research Assistant (LigBank Project), Post-Doctoral Fellow (MEC), and Research Associate (EPSRC). In May 2009, she joined the Instituto de Química Orgánica General (IQOG-CSIC) in Madrid, with a prestigious Ramón y Cajal Award. In September 2011 she became Lecturer in Chemistry at the University of East Anglia, UK. |
1 Introduction
The chemistry of allenes has experienced great advances in the last 20 years on all fronts, from the synthesis of these fascinating structures in achiral and chiral versions, to their application as very versatile building blocks in organic and organometallic chemistry for the synthesis of complex molecules in a straightforward manner.The structure of allenes consists of a linear 3-carbon skeleton formed by two π-bonds, orthogonal to each other joined by a sp-hybridised carbon. Due to this orbital arrangement, if the two terminal sp2-hybridised carbons bear different substituents, these species are provided with a rare and interesting property, axial chirality,1 that can be exploited in asymmetric synthesis.
Amongst the most important reactions involving allenes are the transition metal-catalysed additions of nucleophiles or electrophiles to one or the other double bond of a cumulene. Intramolecular reactions are very important strategies for the synthesis of carbo- or heterocycles in an atom economical manner.2 However, the intermolecular reaction to form allyl derivatives is also a very useful and versatile strategy to build molecular complexity, and many examples have appeared in the literature with different outcomes and mechanisms depending on the nature of the metal and nucleophile employed.3
However, despite all the advances in the field, the axial-to-central chirality transfer4 from enantioenriched allenes to products in the presence of transition metal complexes still remains a challenge. Therefore, in recent years, efforts towards the development of chiral catalysts capable of creating a chiral environment that allows the facial selectivity around the allene, introducing enantioselection in the process have been made.
In this review we will summarise the chirality transfer challenges in metal-catalysed reactions involving addition of different partners to allenes. We will start with the reported attempts of axial-to-central chirality transfer from chiral enantioenriched allenes to the reaction products. Then, the possible racemization processes that have been observed in the interaction of some metals with allenes will be described. Finally, we will examine the examples on chirality transfer using racemic allenes in reactions catalysed by metal complexes bearing chiral ligands to produce enantioenriched products.
We have focussed the review on intermolecular addition reactions of nucleophiles and other partners, such as carbonyl groups, alkynes or silaboranes. Intermolecular cycloadditions5 and intramolecular reactions involving allenes, including the enantioselective version,2 have been much more reviewed recently and are not covered in this review.
2 Metal-catalysed reactions involving chirality transfer from chiral allenes to products
In the last few years the number of methodologies for the synthesis of chiral allenes has increased exponentially, opening a new pool of organic chiral precursors for enantioselective synthesis.6–8 With this broad range of chiral starting materials in hand it was a matter of time before the development of methodologies taking advantage of the axial-to-central chirality transfer offered by allenes for the synthesis of chiral molecules would arise. Thus, a number of metal-free enantioselective synthesis based on chiral allenes have been developed in the last few years.9,10 However, the number of metal-catalysed reactions of chiral allenes is scarce. The reason why these processes remain underexplored is the tendency of allenes to racemize under the catalytic conditions. So, under metal catalysis, two competitive processes could occur, the reaction of the allene to give the desired product in an enantioselective fashion, or the racemization, prior to the reaction, converting the chiral allene in a racemic mixture which would then react to give the desired product with decreased or no enantiomeric excess.In this section, we will discuss reactions in which the racemization issue has been overcome (totally or partially) to form products with good enantiomeric excesses. In section 3, we will explore the racemization process in more detail and the mechanisms that have been proposed to explain it.
2.1 Chirality transfer in metal-catalysed addition of heteronucleophiles to chiral allenes
Metal-catalysed nucleophilic additions to allenes are possibly amongst the most studied methodologies involving these substrates. Particularly, the intramolecular version has proven its importance in the synthesis of carbo- and heterocycles.2 Less studied has been the intermolecular version, although its importance has been recently discussed in different reviews.3The reported examples of metal-catalysed nucleophilic additions to chiral allenes are mostly limited to alcohols11 and amines12 as nucleophiles, and mainly using gold catalysts.
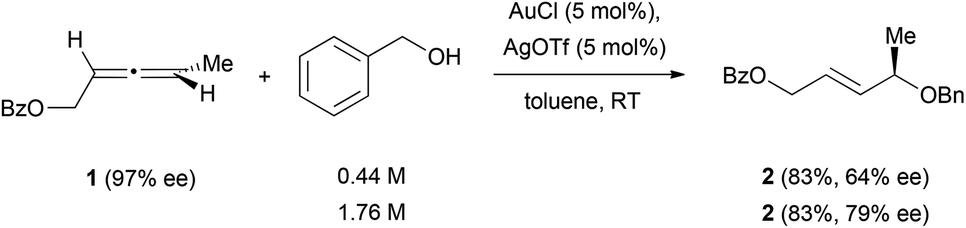
The intermolecular reaction of chiral allenes with alcohols leads in almost every case to racemic allylic alcohols.13 The only example in which a partially successful chirality transfer was observed has been reported by Widenhoefer et al. in the presence of a cationic gold catalyst (Scheme 1).14 In their work, they reported that treatment of the chiral allene 1 with a mixture of AuCl and AgOTf in toluene at room temperature afforded the enantioenriched product 2, in which the enantiomeric excess was lower than the one of the initial allenes. An increase in the concentration of alcohol (from 0.44 M to 1.76 M) also increased the efficiency in chirality transfer.
 | ||
| Scheme 1 | ||
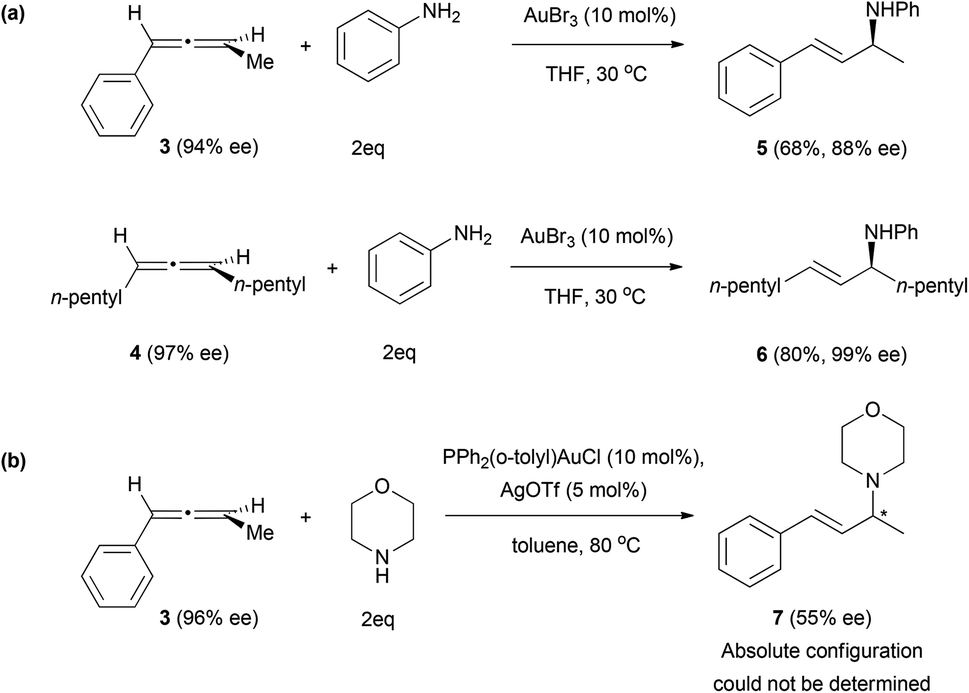
Better results have been obtained when amines are used as nucleophiles. The group of Yamamoto explored the nucleophilic addition of amines to chiral allenes, also under gold catalysis.13,15 The addition of aromatic amines to allenes 3 and 4 gave high values of enantiomeric excess on the final allyl amines 5 and 6 (Scheme 2a). In the case of aliphatic amines such as morpholine, the chirality transfer was poorer, only obtaining a 55% ee from allene 3 (Scheme 2b). A possible explanation could lie in the difference in the catalyst and harder conditions of the reaction, which was carried out at 80 °C instead of 30 °C.
 | ||
| Scheme 2 | ||
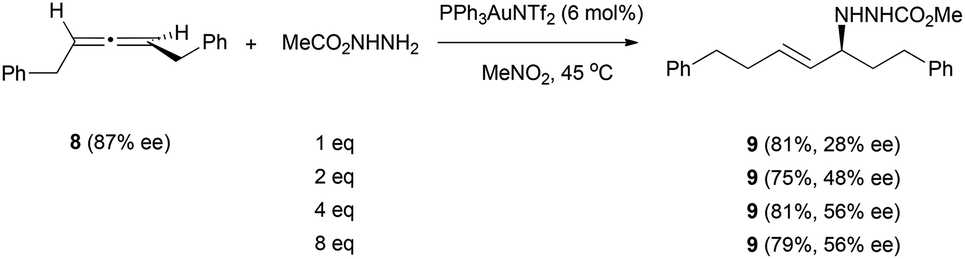
The scope of nitrogen nucleophile addition to chiral allenes was expanded by Toste et al. on their studies of the gold-catalysed reaction of allene 8 with carbazate nucleophiles (Scheme 3).16 They observed that the efficiency of chirality transfer was also improved when increasing the amount of nucleophile, with the best results obtained with more than 4 equivalents of the nucleophile.
 | ||
| Scheme 3 | ||
It is worth mentioning that, in the examples described here, racemization of the chiral allene has been detected under gold-catalysis in the absence of nucleophiles. These results suggest that racemization of the chiral allene and nucleophilic attack at the coordinated allene are competing processes and that, although the enantioselectivity of the nucleophilic attack is dependent on the nucleophile concentration, the racemization is independent of it.
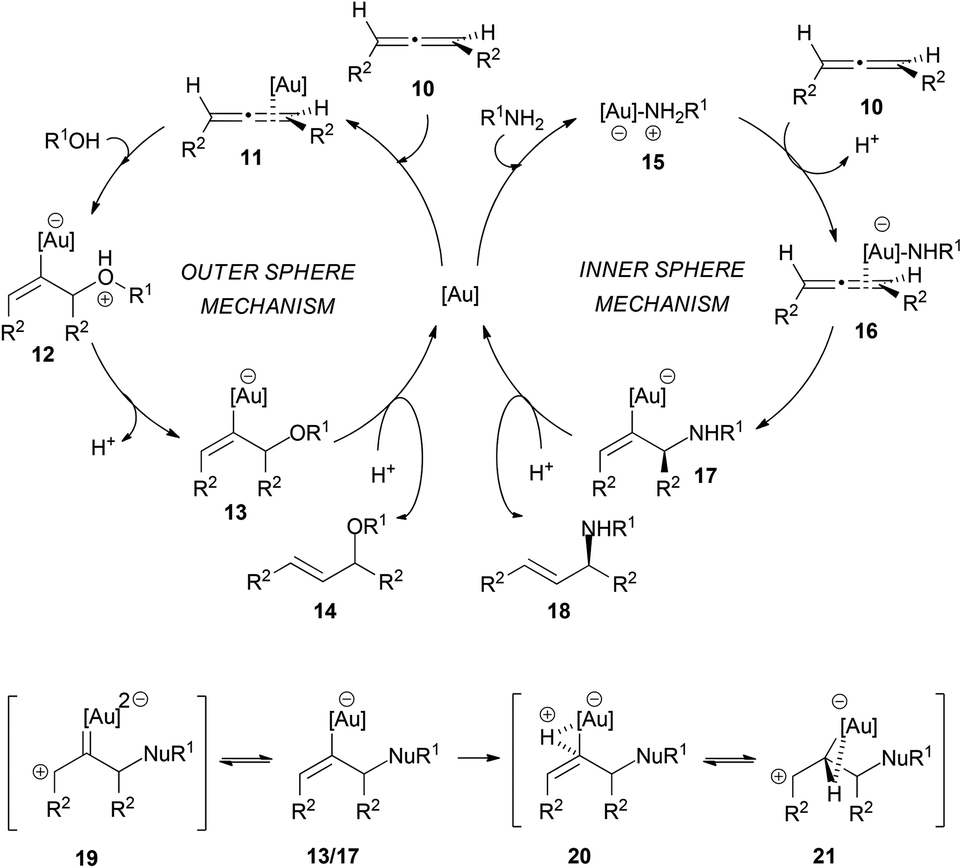
The mechanism of these transformations is quite controversial and different authors have proposed different pathways depending on the nucleophile employed, to explain the differences in the degree of chirality transfer in the attack of amines and alcohols. Yamamoto et al. explained the difference between alcohols and amines on the basis of outer and inner sphere mechanisms, proposing a better chirality transfer when nitrogen nucleophiles are used due to their ability to react through an inner sphere mechanism (Scheme 4). Thus, the intermolecular hydroalkoxylation of allenes would proceed via an outer sphere mechanism, where the alcohol attacks at the coordinated metal–allene complex 11. In contrast, hydroamination would occur through an inner sphere mechanism, in which a metal–amine complex 15 would form first and the nucleophilic attack would occur from the gold coordinating sphere.14,15,17 The gold–amine complex would coordinate to the less hindered face of the allene, and therefore the amine attack would also occur in that face. Furthermore, the formation of this gold–amine complex is supported by the fact that there is an induction period in the hydroamination process and also because the yield decreased when the gold catalyst was not exposed to the amine before the addition of allene. The formation of the E-allyl product was initially explained through an equilibrium between the vinyl-gold intermediates 13/17 and a gold carbene 19, which would allow double bond isomerization. However, afterwards, the authors proposed an alternative explanation via a three-centred intermediate 21. The outer sphere mechanism for the intermolecular attack of alcohols has also been proposed by the group of Widenhoefer.14,18
 | ||
| Scheme 4 | ||
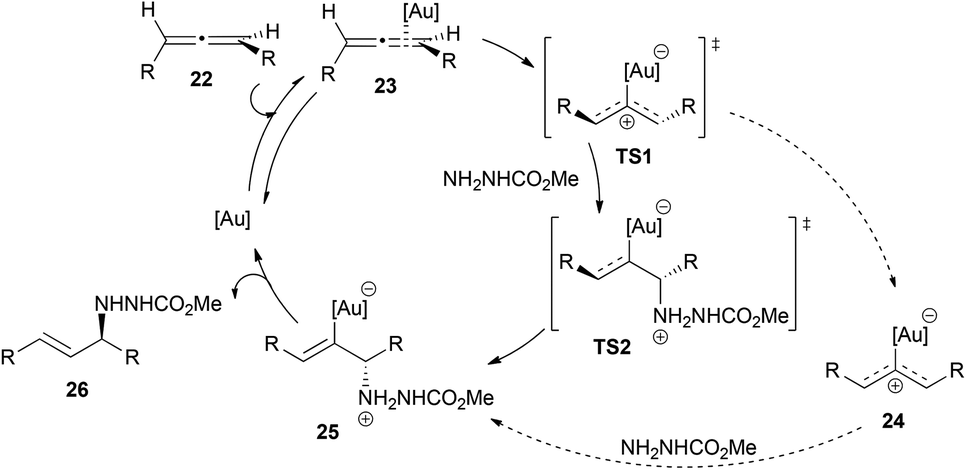
Toste et al. performed an in-depth study of the mechanism of the intermolecular reaction of allenes with methyl carbazate shown in Scheme 3.16 They carried out different experiments, including kinetics, Hammett analysis, NMR studies, competition experiments and DFT calculations. On the basis of the results obtained, and in contrast to the previously proposed inner sphere mechanism with nitrogen nucleophiles, they proposed an outer sphere mechanism, in which the first step would be the formation of a gold–allene complex 23 (Scheme 5). This complex could evolve to a bent-allene–gold complex transition state TS1, which is proposed to be the rate-determining transition state.19 After that, two possible pathways are possible: the first one could explain the chirality transfer and would be the direct conversion of TS1 to TS2 without the intervention of a reaction intermediate in a two-step, no intermediate process. This might be the main mechanism at high concentrations of nucleophiles, leading to higher enantiomeric excess in the final product; the second pathway would account for the racemization of the chiral starting allene, and implies the conversion of TS1 to a planar, cationic gold complex 24 that then suffers the nucleophilic attack, through a traditional two-step outer sphere process. This second route could be the one occurring at low concentrations of the nucleophile and it accounts for the less efficient chirality transfer observed.
 | ||
| Scheme 5 | ||
2.2 Chirality transfer in metal-catalysed addition of carbon-based nucleophiles to chiral allenes
The only example to date in the literature of gold-catalysed addition of carbon nucleophiles to chiral allenes has been reported by Che et al. During their investigations on the enantioselective hydroarylation of allenes with indoles, they observed that the reaction of allene 27 with N-methyl indole in the presence of different gold catalysts led to the formation of the desired product 28 as a racemic mixture (Scheme 6).20,21 Although chirality transfer from the allene was unsuccessful in this case, they reported the enantioselective hydroarylation of racemic allenes using chiral binuclear gold(I) phosphine complexes, and this will be discussed in section 4 of this review.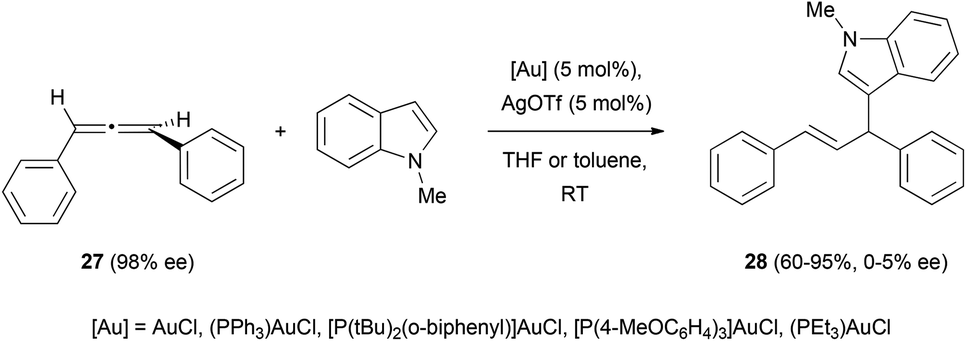 | ||
| Scheme 6 | ||
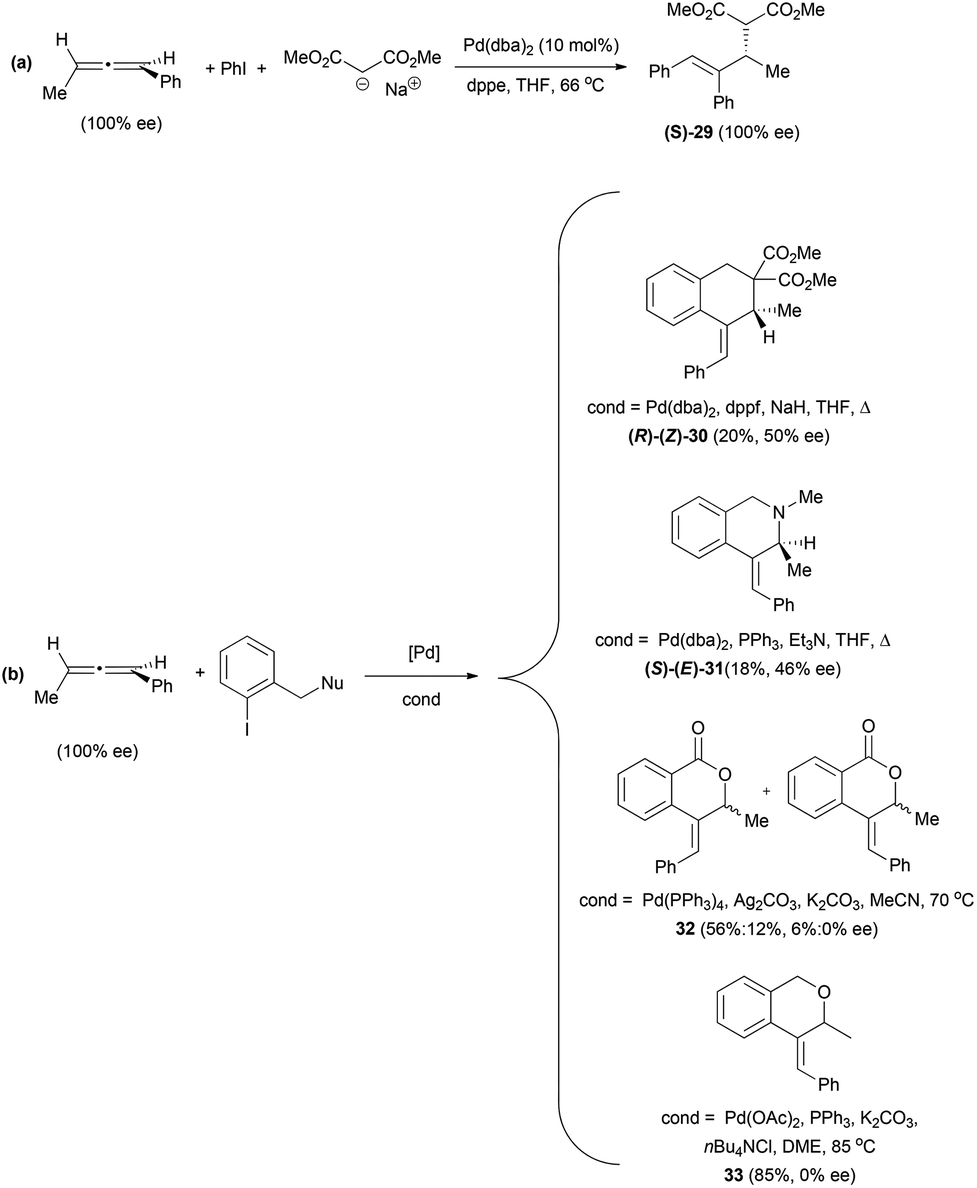
Related to the carbon-based nucleophilic addition processes, Hiroi et al. have described an asymmetric palladium-catalysed reaction of allenes with iodobenzene and sodium malonate.22,23 The reaction proceeds though a carbopalladation in the central carbon of the allene, followed by nucleophilic addition to a terminal carbon of the π-allyl-palladium intermediate (vide infra). Thus, the reaction of 1,3-disubstituted allenes allowed the formation of product (S)-29 with complete stereospecificity (Scheme 7a). The authors exploited this methodology for the synthesis of carbo- and heterocycles using iodobenzenes with a pending nucleophile (Scheme 7b). The best enantioselectivities were achieved under different conditions depending on the nucleophile, although in the case of oxygen nucleophiles the chirality was lost during the reaction. However, the best conditions for the enantiomeric excess were not always the ones generating higher yields.
 | ||
| Scheme 7 | ||
The way the chirality transfer is explained implies that the carbopalladation occurs regioselectively (benzene attack at the central carbon) and with positional selectivity at the double bond of the allene that is closer to the phenyl group. This addition would occur preferentially from the top face of the allene, which is less sterically hindered since the methyl group is on the other face of the allene. The π-allyl palladium complex formed 34 would present a 1,3-allylic strain and will therefore isomerize to the more stable π-allyl palladium complex 35, which will be the one suffering the nucleophilic attack (Scheme 8). If the carbopalladation occurred at the double bond of the allene closer to the methyl group, the product formed would have been the enantiomer of the one actually obtained.
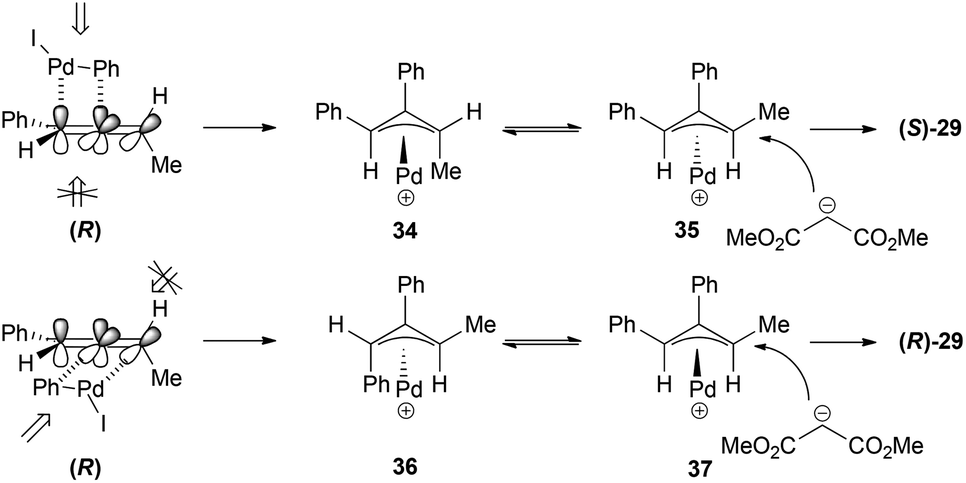 | ||
| Scheme 8 | ||
The formation of the cyclic products 30–33 (Scheme 7) could be explained in a similar way through initial selective carbopalladation. In this case, though, the nucleophilic attack would occur intramolecularly. In contrast with the selective intermolecular process, in this case the two pathways described in Scheme 8 could occur in a competitive manner, accounting for the lower or non-existent transfer of chirality (Scheme 9). It is interesting to note that the enantiomer formed preferentially in the case of the malonate is the opposite of the one formed in the case of the nitrogen nucleophile. The explanation given was that the lower reactivity of the nitrogen nucleophile allows time for the isomerization of the π-allyl palladium complex 38 into 40, in which there is a steric interaction between both aromatic rings. Complex 41 would then be the one reacting to form the major product (S)–(E)-31.
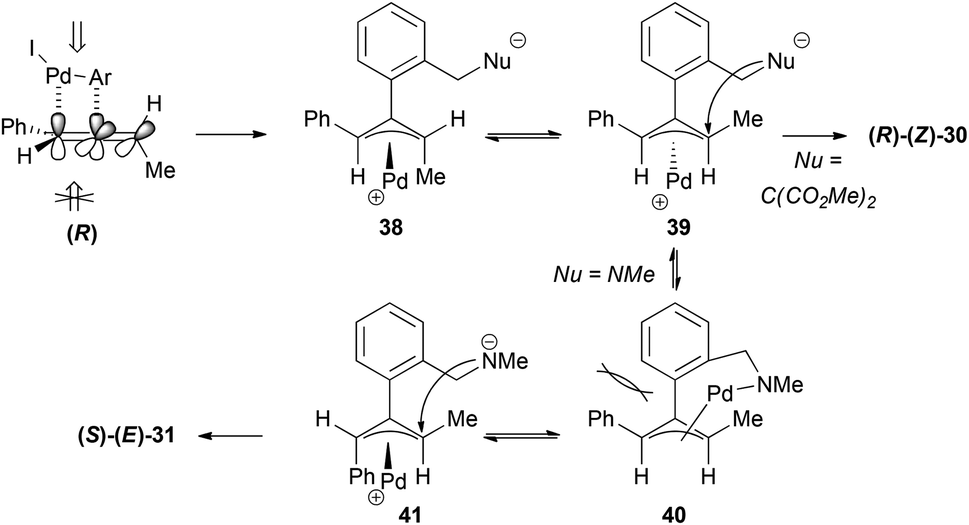 | ||
| Scheme 9 | ||
In a different approach, the reaction of chiral chloroallenes with organometallic reagents has been explored by the group of Alexakis.24 The treatment of allenes 42 with a Grignard reagent under copper catalysis gave the substituted allenes 43 with complete retention of the enantiomeric excess when an aryl group was introduced (Scheme 10). In contrast, the introduction of an aliphatic group led to the formation of the terminal alkynes 44. The change of the solvent from THF to CH2Cl2 was also necessary, since the THF afforded the dechlorinated non-chiral allenes.
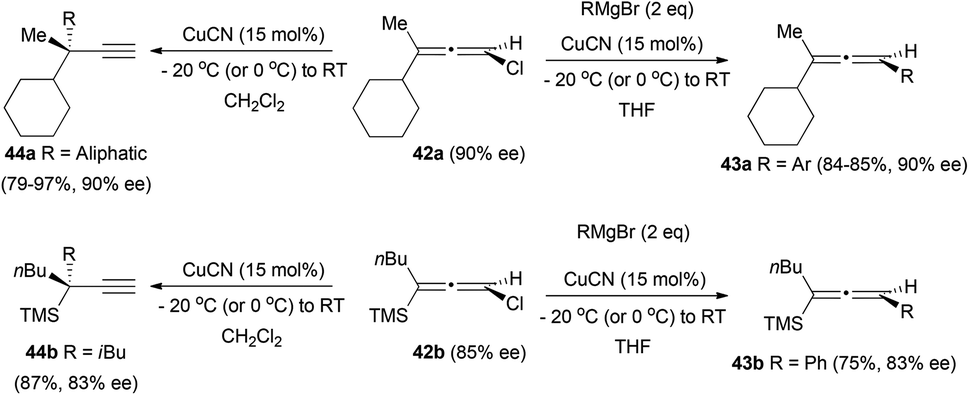 | ||
| Scheme 10 | ||
Finally, a C–H functionalization of benzamides involving chiral allenes has been described by Ma et al.25 The reaction of the chiral allenols 45 with N-methoxy benzamide 46 proceeded smoothly to give lactones 47, with moderate yields but with a high degree of chirality transfer (Scheme 11). The transformation would start with the formation of rhodacycle 48 from the reaction of the benzamide 46 with the rhodium catalyst. This rhodium complex could coordinate with the allenol in its less substituted double bond to form intermediate 49, which would then undergo an insertion of the double bond to form rhodacycle 50. Protonolysis regenerates the catalyst and forms compound 51, which after lactonization forms lactones 47. The stereoselectivity of the double bond and the chirality transfer could be explained by the steric interaction between the methoxy group and the biggest substituent of the allene in the Rh–allene complex 49′.
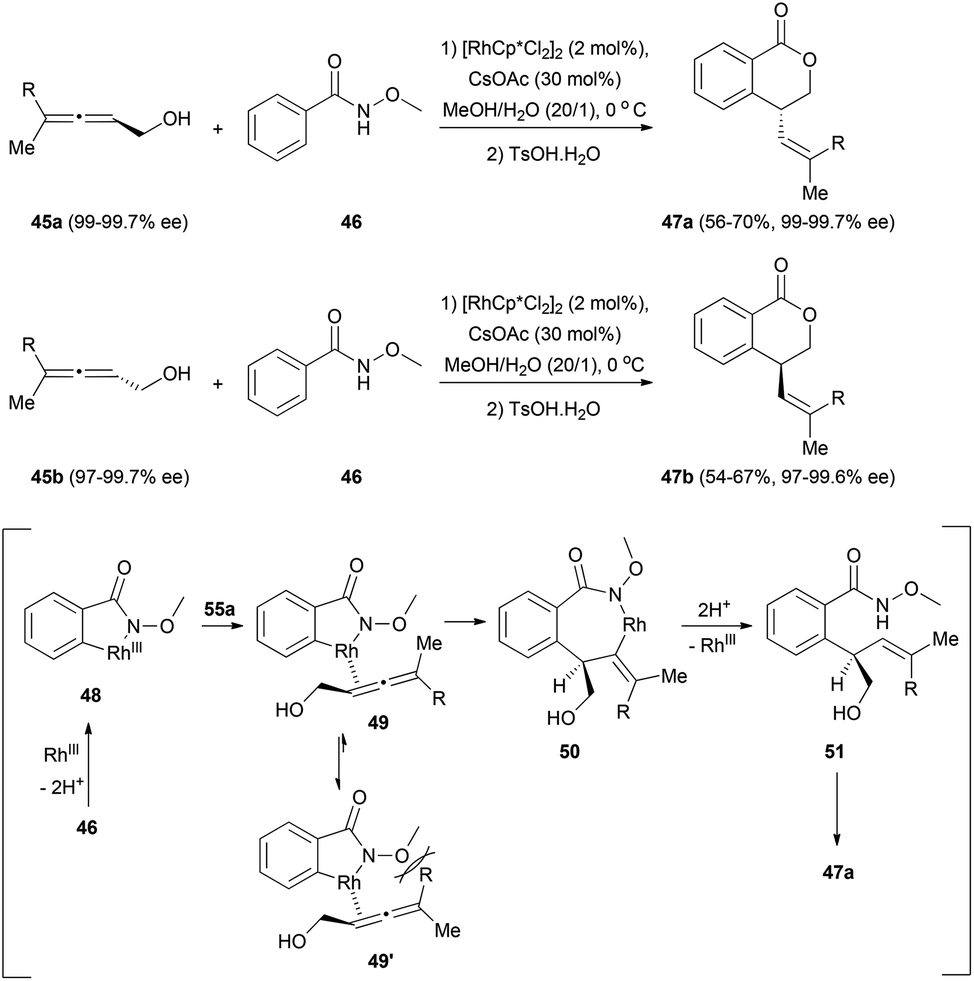 | ||
| Scheme 11 | ||
2.3 Chirality transfer in diverse metal-catalysed additions to chiral allenes
Reactions of chiral allenes with aldehydes have been explored in their non-catalytic version, usually using titanium salts to form a metallated allene that then adds to the carbonyl group.26 However, some interesting examples of metal catalysed reactions of chiral allenes with aldehydes have appeared in the literature.The group of Jamison described a multicomponent coupling of chiral allenes 52, aromatic aldehydes and silanes catalysed by nickel salts, which furnished silyl-protected allylic alcohols 53 with good yields and total chirality transfer when a NHC carbene (IPr = 1,3-bis-(2,6-diisopropylphenyl)imidazol-2-ylidene) was added as an additive to the reaction mixture (Scheme 12).27,28 One of the disadvantages of the method is that a mixture of various homoallylic alcohols was obtained as subproducts in the reaction. The reaction mechanism would start with the selective formation of nickel complex 54. From the four possible nickel complexes, this would be the only one forming since the metal is on the less hindered allene face and in the less hindered double bond. The aromatic aldehyde would then coordinate to the nickel from the opposite face of the metal group and with the phenyl group pointing to the front. The next step would be a reorganization to allow overlap between a C–Ni bond and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and oxidative addition to give metallacycle 55. This metallacycle would evolve through a σ-bond metathesis with Et3SiH to give η3-allyl-Ni complex 56. Finally, reductive elimination with retention could lead to the observed final product 53a, in which the alkyne has a Z configuration.
O, and oxidative addition to give metallacycle 55. This metallacycle would evolve through a σ-bond metathesis with Et3SiH to give η3-allyl-Ni complex 56. Finally, reductive elimination with retention could lead to the observed final product 53a, in which the alkyne has a Z configuration.
 | ||
| Scheme 12 | ||
In a related example, Woerpel et al. prepared alkylidenesilacyclopropanes from chiral allenes and then studied their reactivity with aldehydes.29 In the case of chiral allene 57, the formation of the silacyclopropane 58 was performed in situ in the presence of the aldehyde to form oxasilacyclopentane 59 in good yield and with good enantiomeric excess (Scheme 13). The stereochemistry of the obtained product suggests that the formation of the new carbon–carbon bond in the reaction with the aldehyde occurs in the face opposite to the substituent on a three-membered ring.
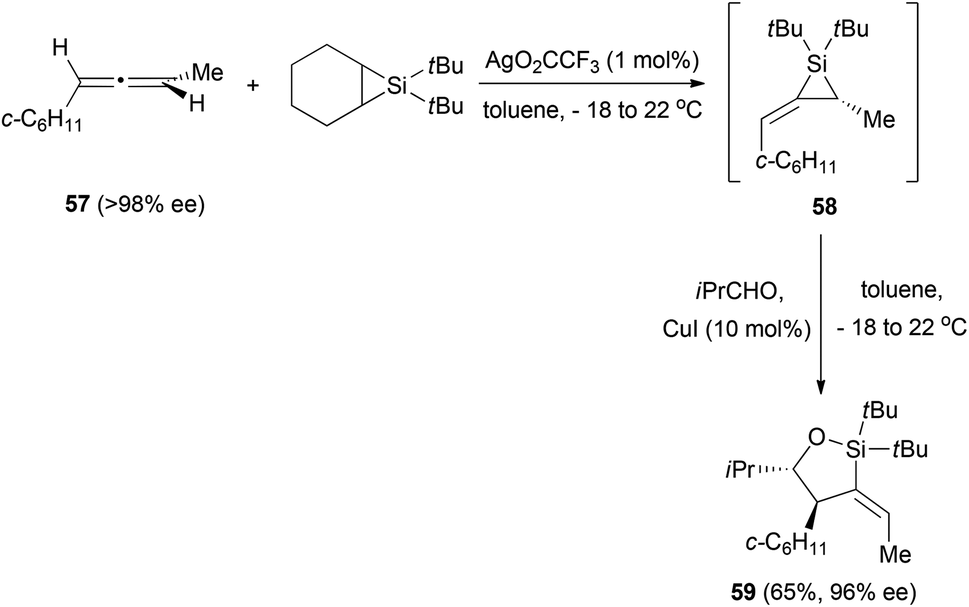 | ||
| Scheme 13 | ||
Similar reactivity was observed by the group of Zhou in a reaction of chiral allenes with hydrogen peroxide catalysed by methyltrioxorhenium (MTO).30 The allenes chosen to study this transformation were chiral 1,2-allenylic diphenyl phosphine oxides 60, whose reactions led to the formation of β-carbonyl-γ-hydroxyl diphenyl phosphine oxides 61 with good regioselectivity, since the reaction only occurs at the electron poor double bond, the one non-substituted with the diphenyl phosphinyl group (Scheme 14). Chirality transfer is also good in this transformation, from 97% ee of the starting material to 93% ee in the final product. The mechanism of this process would occur through the formation of monoperoxo- and bisperoxo-rhenium complexes, which would catalyse the epoxidation of the allenes to form methylene oxiranes 62. These intermediates could react to form the five membered cycles 63, which would then suffer a ring opening via the nucleophilic attack of a water molecule and a final tautomerization of 64 affords the final products 61. This mechanism is in accordance with the stereochemistry of the final products. However, the slight decrease of the enantiomeric excess suggest that other minor reaction pathways could be operating, which has also been suggested by 18O labelling experiments.
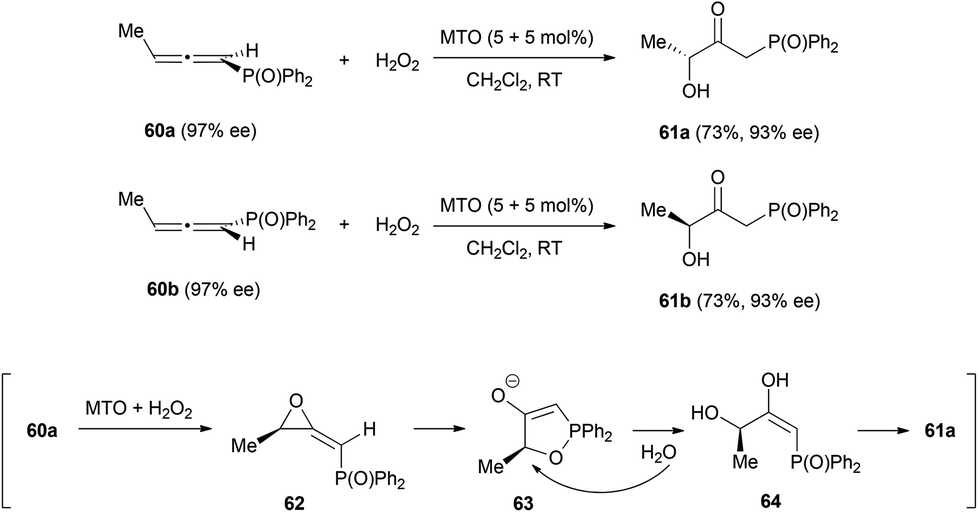 | ||
| Scheme 14 | ||
3 Racemization processes in metal-catalysed reactions of chiral allenes
As has already been mentioned, one of the reasons why not many examples of axial-to-central chirality transfer from chiral allenes have been reported so far is the fact that the majority of the most used metal catalysts in allene chemistry promote the racemization of optically active allenes. Apart from the examples mentioned above, racemization of chiral allenes in the presence of metal catalysts has been observed on more than one occasion in the literature, mainly in the presence of gold catalysts,19b,31–35 but also with palladium36–38 and copper,39 among others.40–42 Moreover, 1,3-dialkyl allenes have been found to racemize thermally with no catalyst,43 and in the presence of photosensitizers.44To understand the causes of the metal catalysed racemization of allenes, first the modes of coordination of the metal to the allene need to be understood. The possible types of metal–allene coordination can be summarized in two coordination modes: η2 and η1. The η2-coordination involves one of the orthogonal double bonds of the allene and different structures could be depicted depending on the contribution of each of the carbons of the double bond to the coordination (65–67, Scheme 15). The slippage of the metal centre towards one or the other carbon would depend on the substituents of the allene and the ligands of the metal. The second possibility is the η1-coordination, in which the metal would coordinate exclusively to the central carbon of the allene. This type of coordination can be seen as a η1-coordinated bent allene 68, as a σ-allylic cation 69 or as a zwitterionic carbene 70.45,46 While in the η2-coordination the allene retains its chirality, the σ-allylic cation form 69 in the η1-coordination is a planar structure which would lose the chiral information of the allene, and therefore could explain the racemization. It is worth mentioning that the η1-coordinated bent allene 68 still retains the stereochemistry of the initial allene, so reactions in which this structure participates as an intermediate could still have high degrees of chirality transfer.
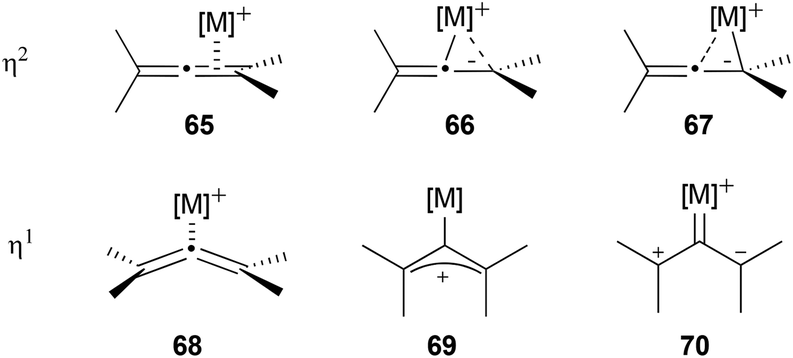 | ||
| Scheme 15 | ||
Not many studies have been performed on the racemization process itself, but several racemization mechanisms have been proposed depending on the metal, additives and reaction conditions employed. In the case of palladium catalysis, Bäckwall et al. described a method for the racemization of chiral allenes catalysed by Pd(OAc)2 and LiBr.37 With this catalytic system the authors investigated the racemization of different α-substituted allenes. The proposed mechanism would occur via a bromopalladation–debromopalladation sequence, through a π-allylpalladium complex 73 as the intermediate responsible for the racemization (Scheme 16).
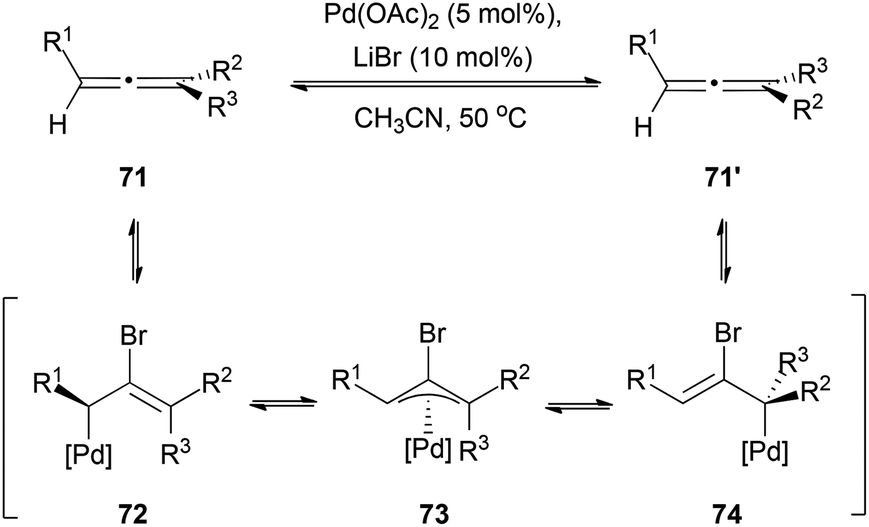 | ||
| Scheme 16 | ||
The case of gold has been more deeply investigated.47,48 Gandon, Fensterbank, Malacria et al. studied the type of coordination and the racemization processes in different gold allene complexes using computational chemistry.45 When they studied (R)-1,3-dimethylallene with different gold fragments they encountered that in all the cases the ground state presented an η2 coordination. In Scheme 17 the possible interconversion pathways between the different gold–allene complexes are shown for [Au] = Au(PMe3) with the relative enthalpies obtained in the calculations. Only one η1 structure (77) could be optimized as an intermediate, the others were all transition states. There are two possible racemization pathways in which the transition state is a σ-allylic cation. The chirality can also be lost during the formation of 77, through a bent allene transition state.
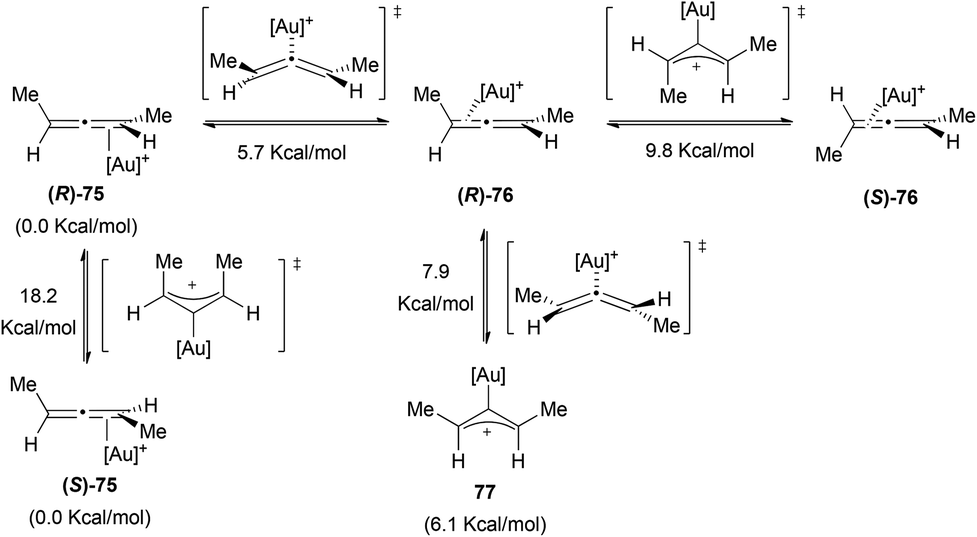 | ||
| Scheme 17 | ||
When these authors performed the study with allenes bearing an electron donating group, the trend changed and in those cases the allylic cations of type 69 were found as the ground state structures. Thus, it seems that the formation of η1-coordinating structures and, therefore, the racemization is more favoured if the allene has electron donating substituents. Similarly, the more the substitution on the allene, the better the degree of chirality transfer that can be achieved.
A more experimental study was performed by the group of Widenhoefer on the kinetics of racemization of aromatic 1,3-disubstituted allenes.49 Based on their experimental information the authors proposed a mechanism in which the first step would be the rapid and reversible formation of a η2-gold–allene complex 79, followed by its conversion to σ-allylic cation 80via a bent allene complex transition state TS3, with this last step being the turnover limiting step (Scheme 18). The rate law for the racemization process is second order with first order in allene and first order in the catalyst [(Ph3P)AuOTf]. Hammett analysis for both, the allene and the phosphine, suggests a decrease in the electron density on the terminal carbons of the allene and an accumulation of the electron density in the phosphine ligands in the rate limiting transition state, which accounts for the high reactivity of cationic gold complexes in the racemization process.
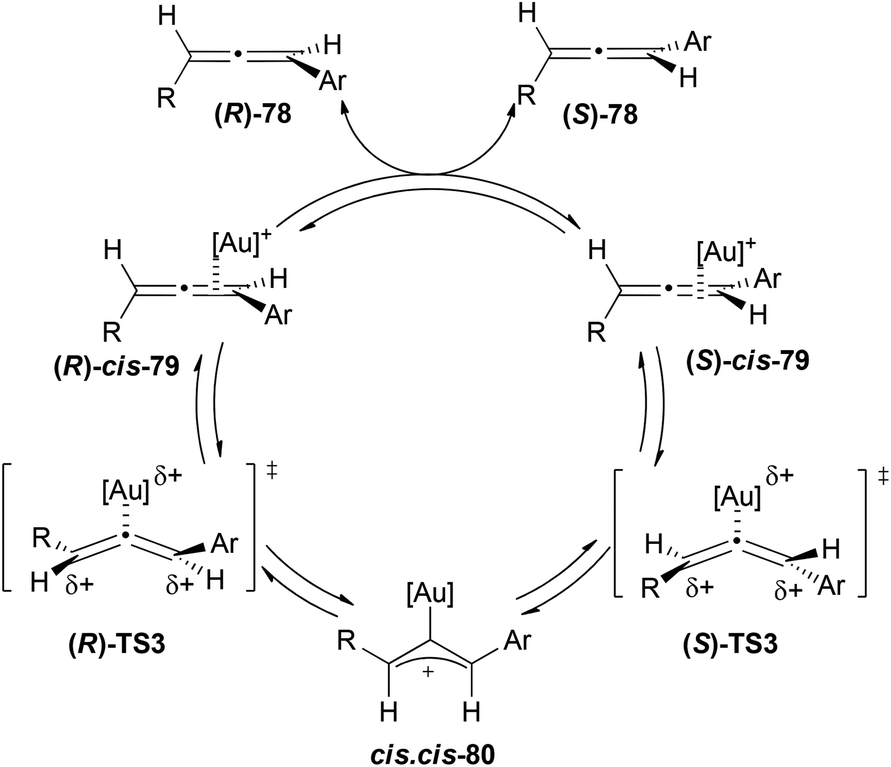 | ||
| Scheme 18 | ||
While there is not much data available for comparison, an important point when discussing the racemization process is the substitution of the allene. As just presented in the previous examples, disubstituted allenes tend to racemize very easily because the formation of an allylic cation is not disfavoured by allylic strain. However, tri- and even tetrasubstituted allenes seem to be more resistant to racemization, as shown by Mouriés-Mansuy and Fensterbank in an example of almost total chirality transfer in a cyclisation process from a tetrasubstituted chiral allene in the presence of cationic gold complexes.50
Although the proposed mechanisms for the racemization of chiral allenes with gold proposed by Widenhoefer, Gandon, Fensterbank and Malacria seem to agree, it is important to mention that different manifolds could also occur depending on the substitution of the allene. An interesting example has been recently reported by Liu and Feng et al.51 The authors explain the racemization of allenic esters 81via the formation of 3-alkynyl-2-enolates 83 (Scheme 19). More details about this reaction will be covered in the next section.
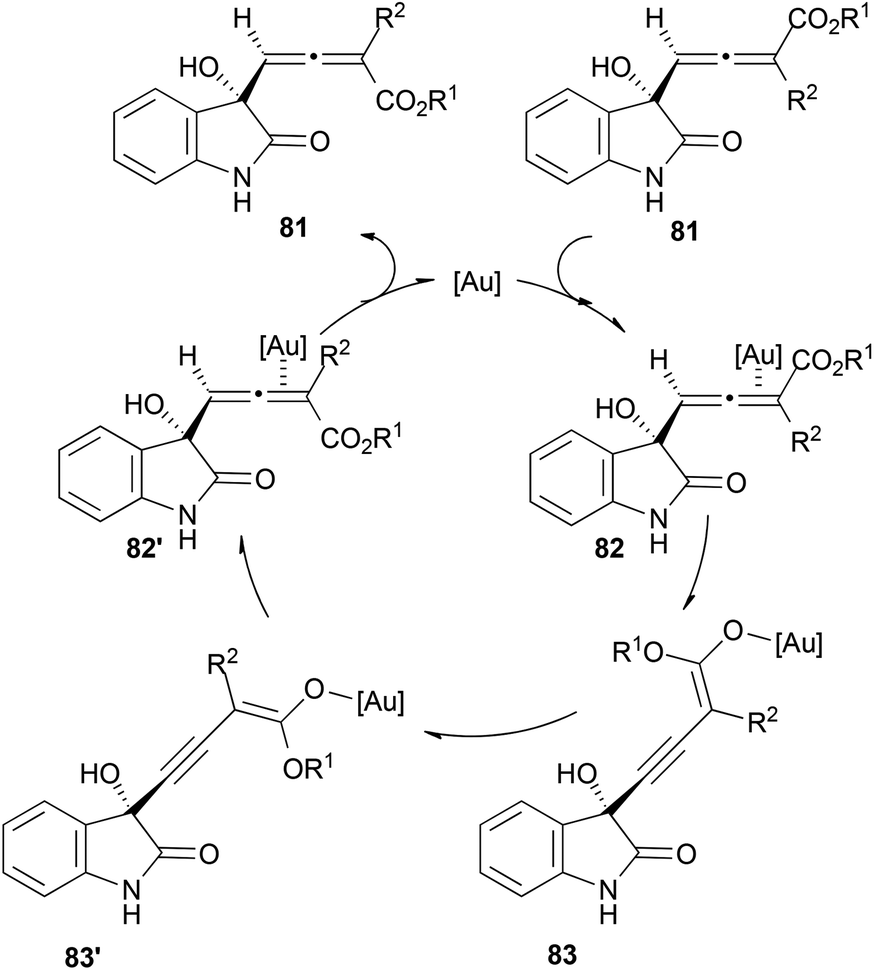 | ||
| Scheme 19 | ||
The difficulty in finding reaction conditions for the successful chirality transfer in optically active allenes avoiding the racemization process is possibly one of the reasons why the interest of chemists has moved towards the development of asymmetric methodologies which allow the creation of chiral centres from racemic allenes. The next section will cover recent developments in this area in asymmetric metal-catalysed intermolecular addition reactions involving allenes.
4 Chirality transfer using racemic allenes in reactions catalysed by metal complexes bearing chiral ligands to produce enantioenriched products
Chirality transfer on addition reactions to allene systems has witnessed a remarkable and growing interest in the last few years. Some examples can be found dealing with the use of chiral reagents, both the allene system and its counterpart,52 or with the use of chiral organocatalysts, such as different amines or acids,53 and specially phosphine derivatives.54 Nevertheless, the use of chiral ligands in metal-catalysed reactions has been probably the most attractive strategy for organic chemists, and the main part of the reported work within the field of chirality transfer using allenes. Thus, in this section we will focus on the last efforts in chirality transfer, starting from racemic allenes, and using different ligands as asymmetric inductors in metal catalysed addition reactions.4.1 Asymmetric metal-catalysed reactions of allenes with heteronucleophiles
The transition metal-catalysed enantioselective version of the addition of heteronucleophiles to allenes has experienced great progress in the last few years, taking advantage of the inherent nucleophilic nature of certain heteroatoms, such as oxygen, nitrogen or sulphur. The challenge on these reactions is not only the enantioselectivity, but also the regioselectivity, as the attack of the nucleophile at the terminal carbon in monosubstituted allenes will lead to an achiral product. Different strategies have been developed to overcome this problem, taking advantage of the different reaction mechanisms operating depending on the metal and the nucleophile used, similarly to the examples reported in the previous section.Breit's research group is one of the pioneers in this area and has contributed with many remarkable examples exploring rhodium catalysis on terminal allenes, to yield branched allylic products regio- and enantioselectively. In this area, they developed the first direct hydrothiolation of terminal allenes 84 in a racemic (Scheme 20, left)55 and enantioselective (Scheme 20, right) manner, using the much less explored sulfones as nucleophiles, giving access to chiral allylic sulfones 85, recurring motifs in bioactive and pharmacological compounds.56 The reported work comprises a screening of different phosphines as ligands, finding (R)-difluorophos L2 as the best chiral inductor, providing high enantioselectivity, in addition to an almost complete regioselectivity (Scheme 20).
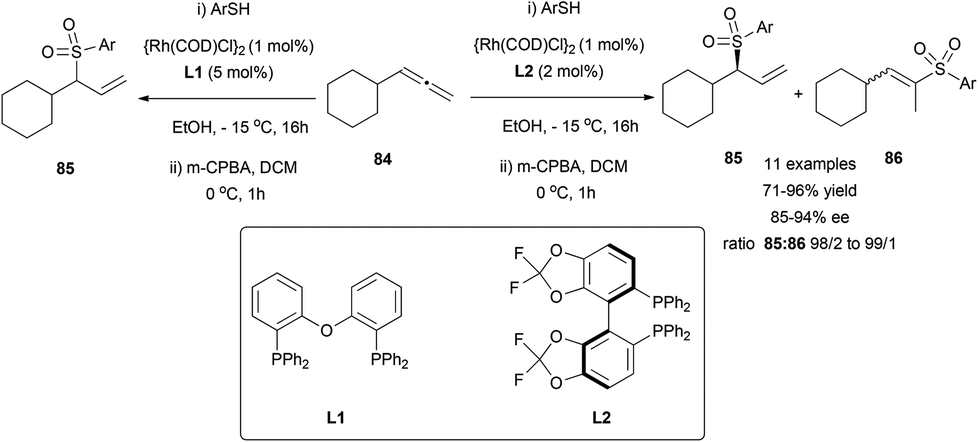 | ||
| Scheme 20 | ||
The methodology was extended to both aromatic and aliphatic thioethers and different terminal allenes, and further transformations of the resulting allylic compounds make the reported strategy a versatile tool for building a wide variety of structures, like sulfone-based aldehydes, epoxides, alcohols or halogen derivatives (Scheme 21).
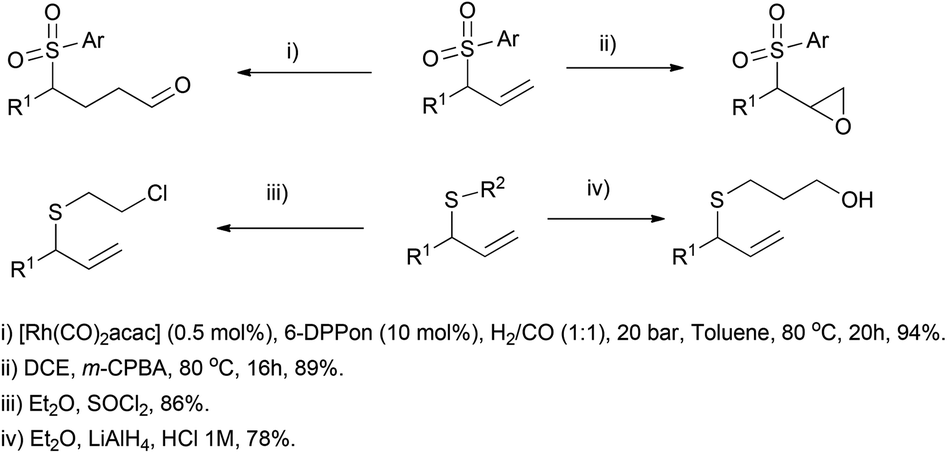 | ||
| Scheme 21 | ||
The more commonly used nitrogen nucleophiles have also been widely tested on rhodium-catalysed additions to allenes, yielding an extended family of allylic branched alkaloids. Thus, different nucleophiles such as primary amines,57 pyridines,58 imidazoles,59 imines,60 pyrazoles,61 tetrazoles,62 or hydrazines63 have been investigated, finding in every case the appropriate chiral ligand to induce excellent regio- and enantioselectivities as summarised in Scheme 22. The ferrocenyl and biphenyl/binaphthyl derived diphosphines, highlighted in the scheme, were the ligands that gave the best results.
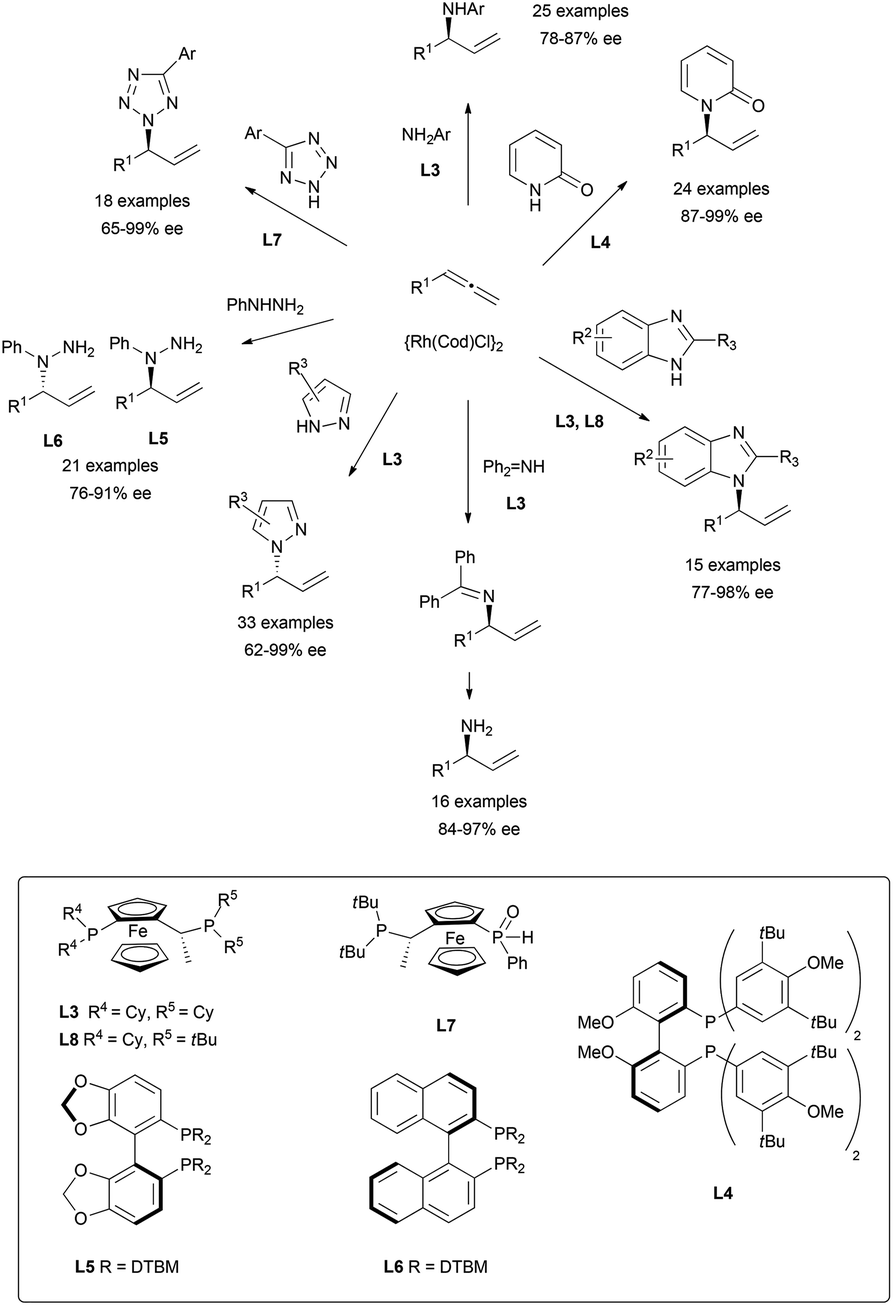 | ||
| Scheme 22 | ||
Examples of gold-catalysed enantioselective addition of nucleophiles to allenes are scarce. As mentioned in the previous section, once the gold is coordinated to one of the double bonds of the allene, the mechanisms proposed for these transformations could involve a planar achiral gold intermediate. Creating a chiral environment by modulating the ligands in the gold complex, could allow the facial selectivity on the nucleophilic addition, and the introduction of enantioselection in the process. The only example reported to date of enantioselective hydroamination of allenes, is the gold-catalysed reaction of 1-aryl-1,2-butadienes 87 with carbazates and carbamates reported by Widenhoefer (Scheme 23).35 In this reaction, regioselectivity was controlled by the substitution in the allene, where attack of the nitrogen nucleophile always occurs at the carbon opposite to the aromatic group to form chiral allyl derivatives 88, with enantioselectivities up to 92% ee when using the (S)-DTBM-MeOBIPHEP ligand (6,6′-dimethoxybiphenyl-2,2′-diyl)bis[bis(3,5-di-tert-butyl-4-methoxyphenyl)phosphine], L4) that forms di-gold complexes.
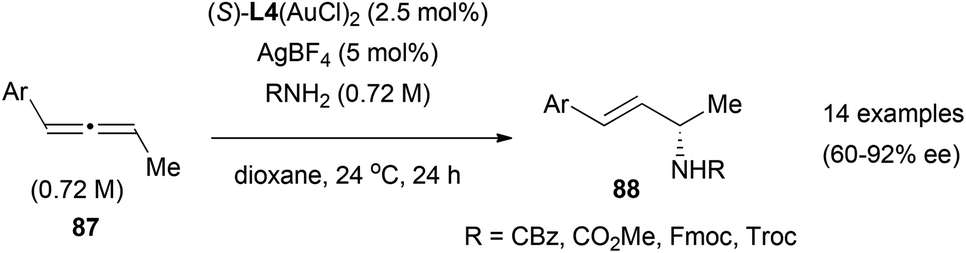 | ||
| Scheme 23 | ||
It is interesting to mention here that, Widenhoefer et al. observed in these reactions two racemization processes: the racemization of the chiral allenes in the presence of gold complexes, and also a decrease in enantioselectivity of the allyl derivative product with increasing conversion of the intermolecular hydroamination process. They proposed an outer sphere mechanism in which one of the gold centres in the chiral di-gold complex participates as the catalytically active site, while the other is a spectator that can ligate the allene, the carbamate or either enantiomer of the allyl product, affecting the efficiency of the enantioselectivity of the reaction. This suggests that the nature of the catalytic species and the chiral pocket change during the reaction with increased concentration of the product, but the process is not yet fully understood. Further studies towards the development of ligand-modulated enantioselective gold catalysts are still needed.
The gold-catalysed enantioselective addition of oxygen nucleophiles to allenes is still unknown. However, Breit and co-workers have described the use of carboxylic acids as oxygen-based nucleophiles for the enantioselective addition to racemic terminal allenes using rhodium catalysts.64 A wide scope of allyl esters 89 is accessible through their reported strategy, including the formation of ester 90 bearing quaternary stereocentres, and the application of the methodology for the synthesis of γ-butyrolactone 91, without loss of chirality (Scheme 24).
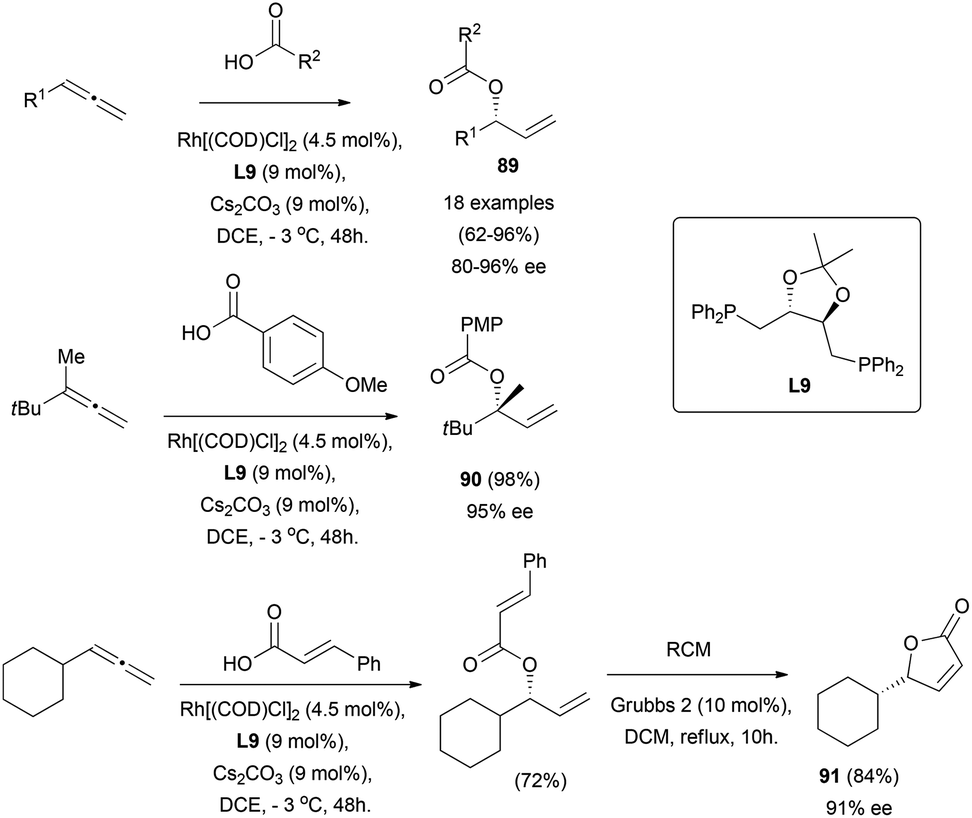 | ||
| Scheme 24 | ||
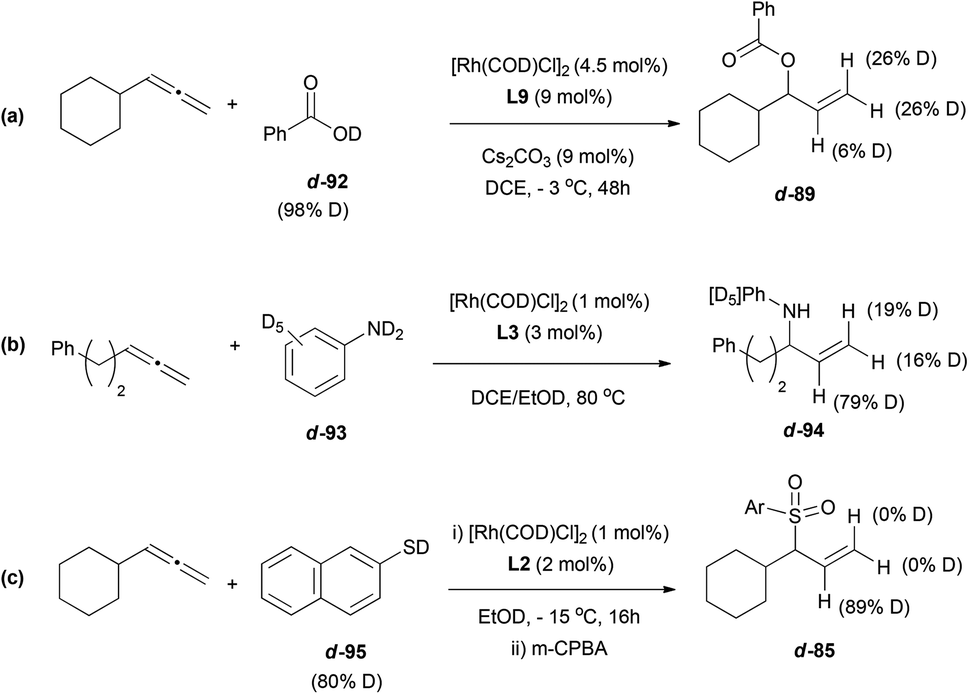
Interestingly, deuterium-labelling experiments support the idea of different mechanistic pathways for the rhodium-catalysed reaction depending on the nucleophile. Addition of deuterated carboxylic acids 92, or deuterated anilines 93, resulted in deuterium labelled products, 89 and 94, both at terminal positions and at the central carbon of the allyl moiety (Scheme 25a and b). In contrast, when the reaction was performed with deuterated thiol 95, the final product 85 showed only deuterium incorporation at the central position (c, Scheme 25).
 | ||
| Scheme 25 | ||
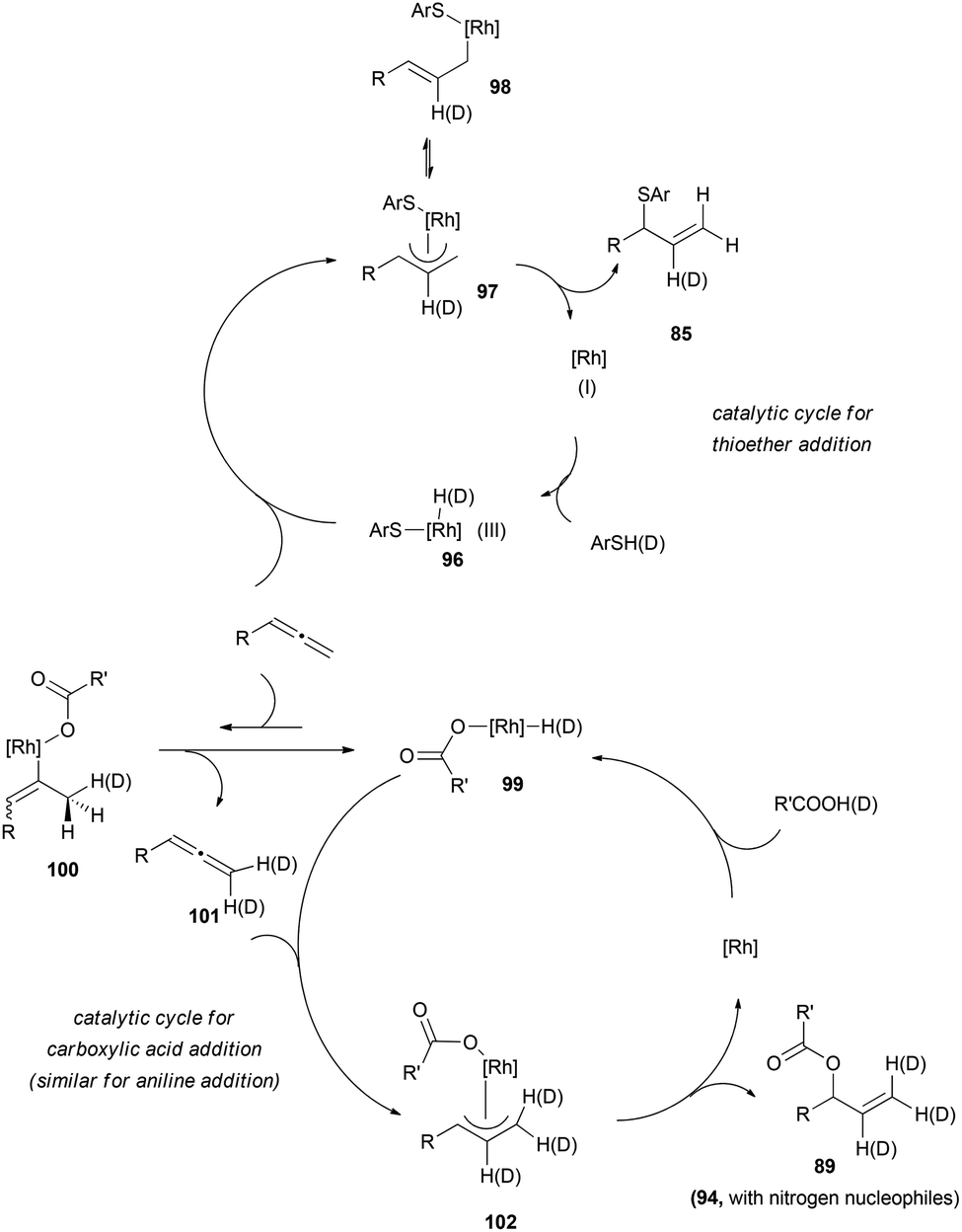
These results could be explained through formation of nucleophile-rhodium-hydrides 96 and exclusive hydrometalation of the more substituted allenic carbon, leading to the π-allyl complex 97 for the sulphur-based nucleophile, while reactions with carboxylic acids (and anilines) could proceed through a previous and fast reversible hydrometalation of the less substituted allenic carbon, furnishing the σ-complex 100. β-Hydrogen elimination of these species would explain the incorporation of deuterium at the terminal positions, generating deuterated allene 101. Then, a slower hydrometalation of the substituted carbon would provide the corresponding π-complex 102, which could yield the observed allylic products 89 (or 94) after enantioselective reductive elimination with the nucleophile always attached to the more substituted terminus of the allyl rhodium (Scheme 26). The regioselectivity of this step is in accordance with the ones reported for Rh-π-allyl complexes, and has been tentatively ascribed to an increased electrophilicity of the Rh-allyl intermediate, which electronically promotes alkylation at the more substituted terminus of the allyl fragment.65
 | ||
| Scheme 26 | ||
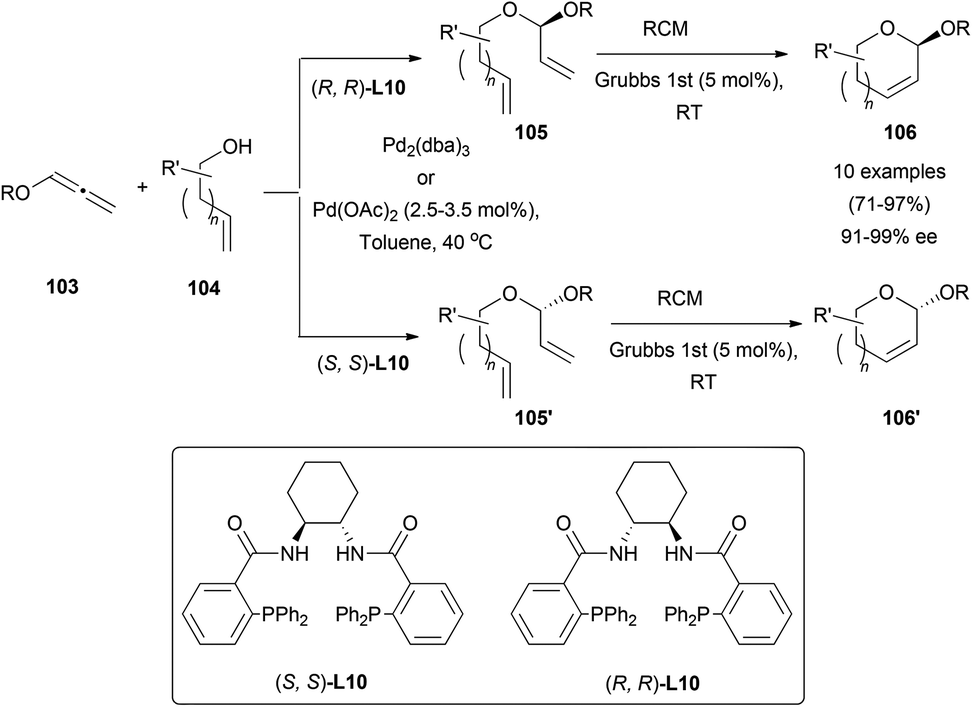
Rhee's research group has recently developed a smart approach to cyclic acetals 106 by a palladium-catalysed asymmetric hydroalkoxylation reaction of hydroxyallenes 103.66 The well-known poor nucleophilicity of primary alcohols 104 in π-allyl chemistry,67 compared with carbon or nitrogen-based reagents was solved after an extensive screening of reaction conditions. The best examples were obtained when the reaction was performed using Pd(OAc)2 or Pd2(dba)3 as a catalyst, and Trost's ligand (L10) as an asymmetric inductor, yielding acetals 105 and 105′, controlling the regioselectivity of the addition to the carbon containing the BnO-group, and the configuration of the new chiral centres by selecting the right configuration in the chiral ligand. Subsequent ring-closing-metathesis afforded cyclic acetals 106 and 106′. This unique strategy to control the configuration of the anomeric carbon allows the synthesis of mono-saccharide precursors, avoiding long multiple step routes and activating group chemistry (Scheme 27).
 | ||
| Scheme 27 | ||
4.2 Asymmetric metal-catalysed reactions of allenes with carbon-based nucleophiles
Trost and co-workers’ contribution to the enantioselective addition of carbon-based pronucleophiles to allenes has probably set the basis for all the later reports in this field. In an early work, they described the reaction of benzyloxyallene 103a with Meldrum's acid 107 or azlactones 109, yielding the corresponding allylic branched structures 108 and 110, respectively.68 It was found that the catalytic species formed by palladium trifluoroacetate and Trost's ligand (S,S)-L10, allowed the allylic alkylation of enolates in good yields and excellent enantioselectivities. It's worth noting that, the addition of acid additives in the reaction with Meldrum's acid notably improved the efficiency of the process, while the addition of bases as potassium tert-butoxide enhanced the azlactone alkylation. This result is presumed by the authors as a possible consequence of the different pKa of Meldrum's reagent (around 5) and azlactone (around 9), with the enol form being the active tautomer for the reported reaction with Meldrum's acid (Scheme 28). In both cases, attack at the more substituted external carbon of the allene was observed due to the presence of the BnO-group.69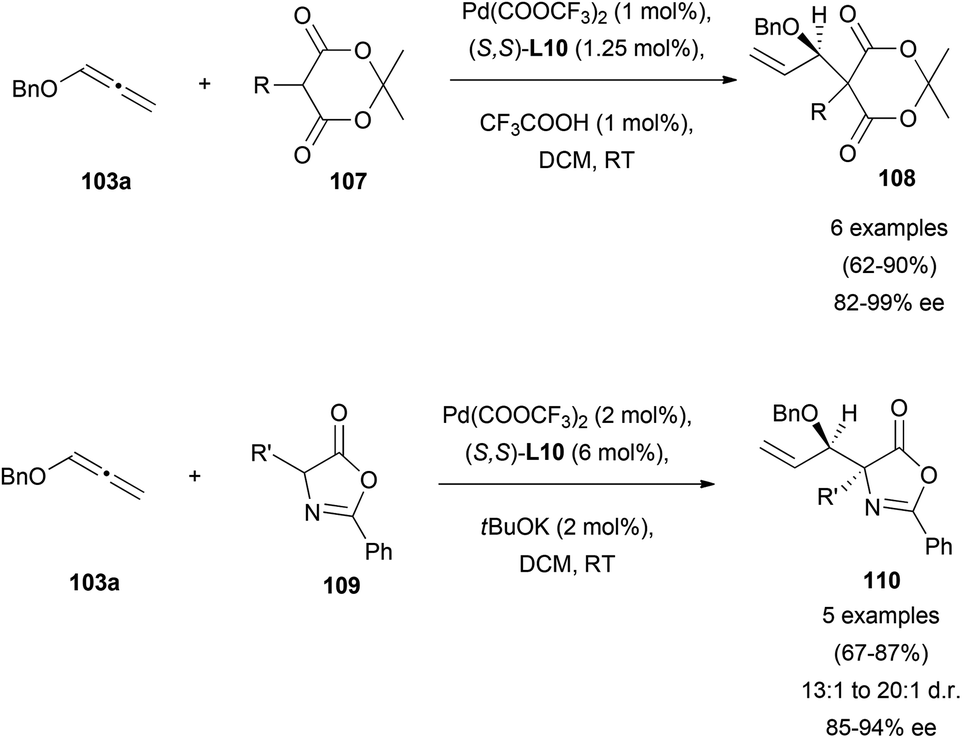 | ||
| Scheme 28 | ||
Electron-rich aromatics are common carbon-based nucleophiles in the metal-catalysed reaction with allenes.21 However, there are not many examples in the enantioselective version. In this area, the gold-catalysed enantioselective hydroarylation of 1,3-diarylallenes 27 with N-methyl indoles has been reported by Che et al.20 Interestingly, the transfer of chirality from the allene under these conditions was not successful, as mentioned in the previous section (Scheme 4). However, the chiral induction using chiral di-gold phosphine complexes with (S)-MeOBIPHEP (L11, Scheme 29) as a ligand gave the corresponding allyl indoles 28 with moderate enantioselectivities up to 63% ee. Deuteration experiments and DFT calculations on the process lead to propose an outer sphere mechanism with attack of the indole from the opposite site of the gold–allene complex, and with gold–gold interactions being beneficial for the enantiodiscrimination.
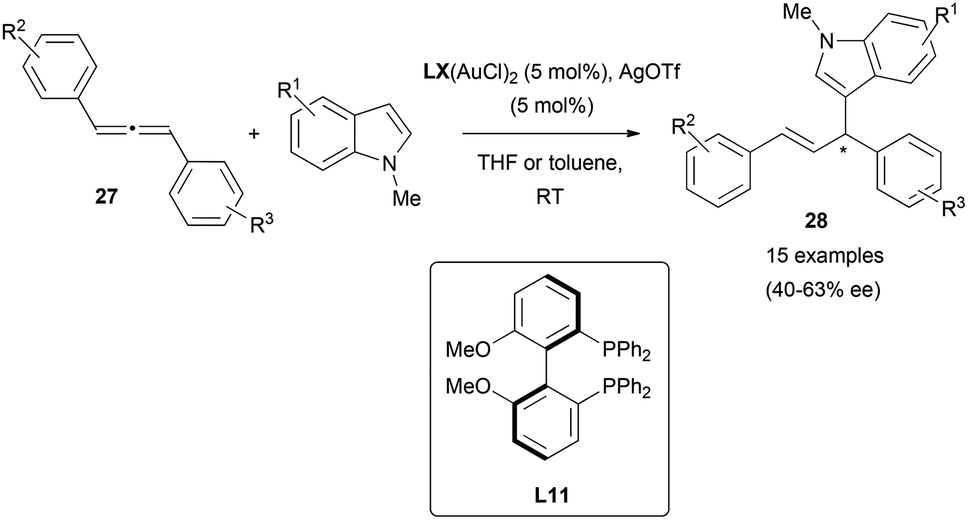 | ||
| Scheme 29 | ||
In a recent report, tryptamines 112 have been used as carbon-based nucleophiles for the addition reaction to terminal allenes 111, towards the synthesis of pyrroloindoline skeletons 113.70 In this case, gold catalysis promoted the addition to the less substituted allenic carbon, and further in situ cyclization led to the mentioned alkaloid motif. In the asymmetric version, the use of a chiral phosphine L12 allowed the synthesis of the indicated structures in good to excellent yields, and enantioselectivities up to 99% (Scheme 30, right). Interestingly, an alternative strategy based on palladium catalysis yielded related pyrroloindoline systems. In the route to naturally occurring gliocladin C (116), it has been reported that the asymmetric addition of oxindoles 114 to terminal allenes is the key step of the synthesis.71 In these examples, the catalytic pair Pd2(dba)3 and Trost's ligand (R,R)-L10, together with benzoic acid as a co-catalyst, provided the best results regarding regio- and enantioselectivity. An exhaustive screening of reaction conditions including catalyst concentration, solvent, and different substituted oxindoles and allenes was also reported, leading to the desired indole-type systems 115 in good to excellent yields and selectivities up to 93% ee (Scheme 30, left).
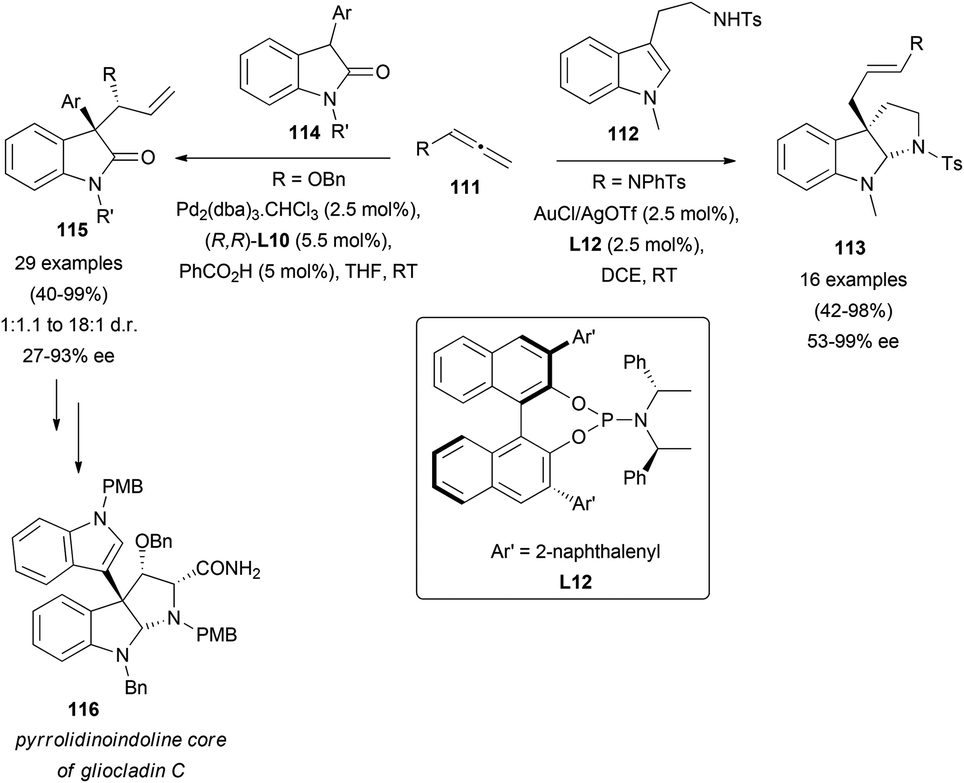 | ||
| Scheme 30 | ||
In addition, mechanistic insights and reaction selectivity were investigated in the reaction with the palladium system. Hydropalladation of the allene from in situ generated palladium-hydrides would form allyl complex syn-118. The stereochemical outcome of the nucleophilic addition can be rationalized through the attack at the more substituted carbon with an approach of oxindoles 114 from the Si-face onto syn-118, yielding major product 115. Attack of the nucleophile from Re-face to furnish the diastereomer 115′, would be therefore less favoured due to steric interaction between the oxindole and the bulkier side of the chiral ligand (Scheme 31).
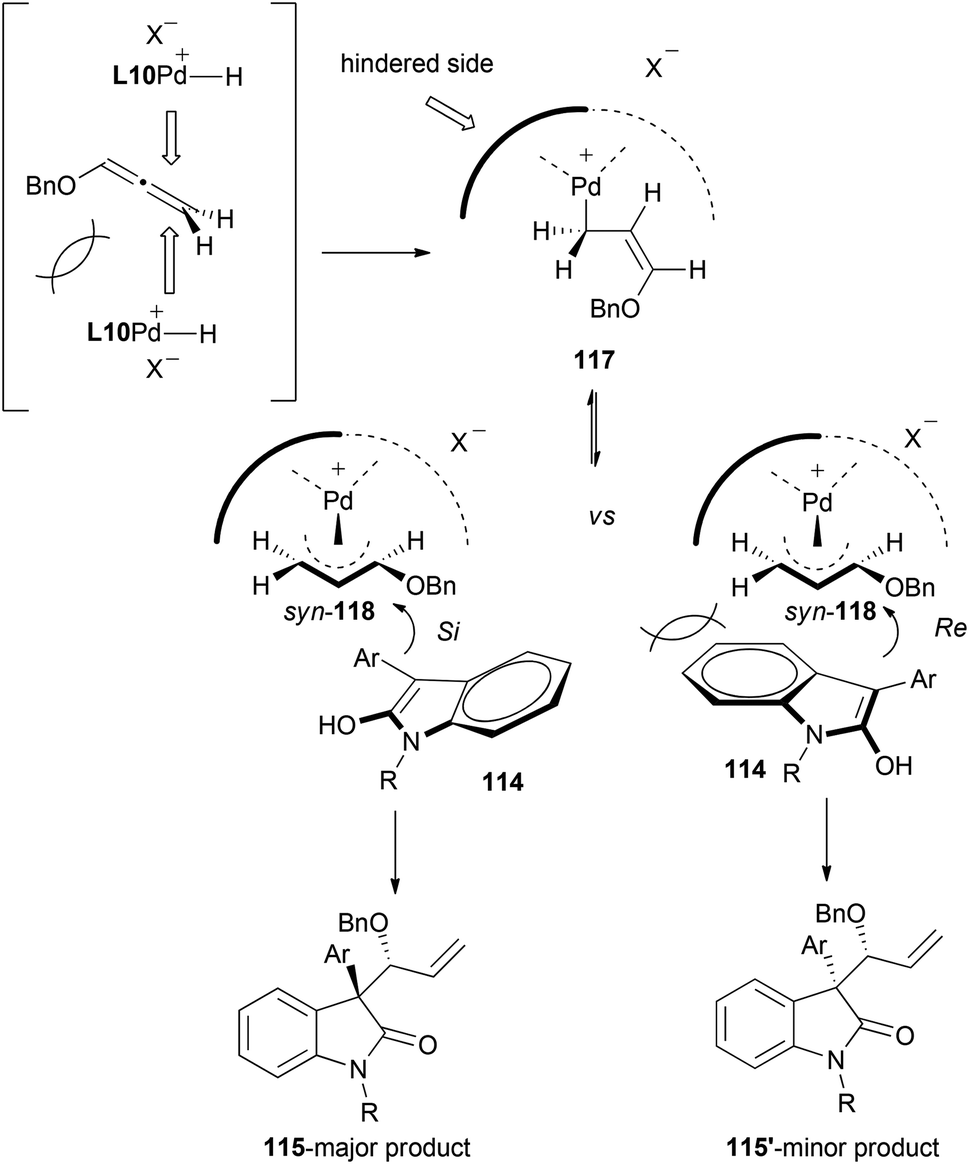 | ||
| Scheme 31 | ||
Despite the general use of terminal alkynes in addition reactions to polar multiple bonds,72 the number of reports dealing with addition to allenes is scarce. In this context, cobalt-catalysed asymmetric addition of terminal alkynes 120 to allenes 119 has been described, giving access to useful conjugated internal enynes 121 in excellent yields and enantiomeric excess up to 99.5% using ferrocenyl diphosphine derivatives as chiral ligands (L13, Scheme 32).73
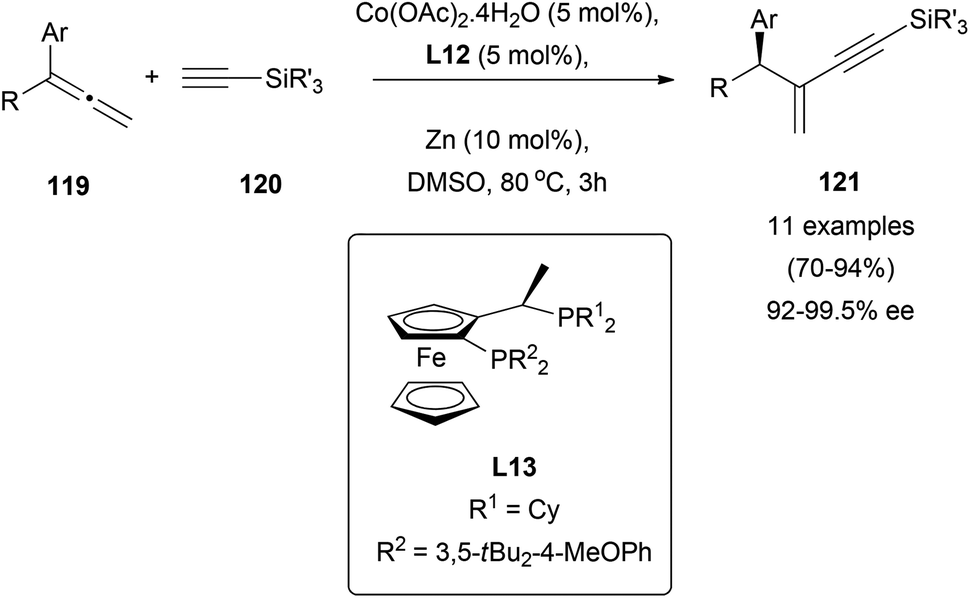 | ||
| Scheme 32 | ||
4.3 Asymmetric metal-catalysed silaboration and diboration reactions of allenes
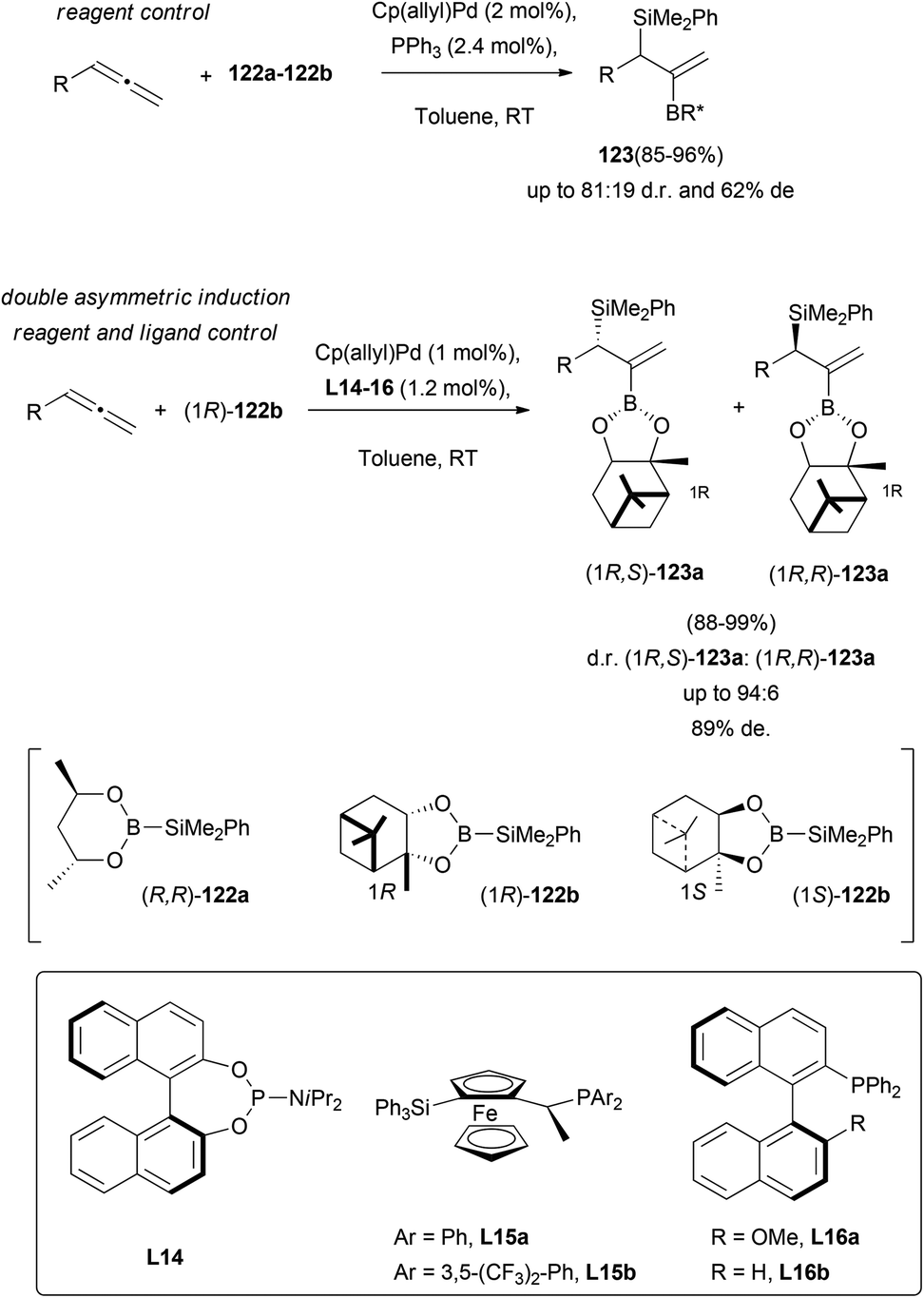
Allyl silanes are considered synthetic intermediates of great interest for the construction of complex organic molecules. Stability, reactivity and their tolerance to different functional groups, make them a useful tool in modern organic chemistry.74 In addition, chiral allyl silanes allow the asymmetric synthesis of many optically active structures, with highly efficient chirality transfer. One of the described methodologies to prepare this building block deals with the metal catalysed enantioselective addition of silaboranes to allenes. This strategy provides functionalized allyl silanes, difficult to obtain through any other known methodology, and easily converted into many attractive organic structures. Suginome and co-workers have achieved a notable advance in this field, exploring the efficiency of the chirality transfer in palladium-based allene silaboration.75 In first reports, a general reaction condition screening showed that the catalytic pair Cp(allyl)Pd/tertiary phosphine in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, promoted the desired process through considerably milder conditions and higher yields, compared to related previous studies.76 Then, a double asymmetric induction was tested, using different chiral monophosphine ligands L14–L16, and optically active boranes. It was found that the combination of silaborane (1R)-122b and phosphine L16b gave the best results, yielding the expected allyl-silanes 123 in excellent yields and 89% de (Scheme 33).
:1 ratio, promoted the desired process through considerably milder conditions and higher yields, compared to related previous studies.76 Then, a double asymmetric induction was tested, using different chiral monophosphine ligands L14–L16, and optically active boranes. It was found that the combination of silaborane (1R)-122b and phosphine L16b gave the best results, yielding the expected allyl-silanes 123 in excellent yields and 89% de (Scheme 33).
 | ||
| Scheme 33 | ||
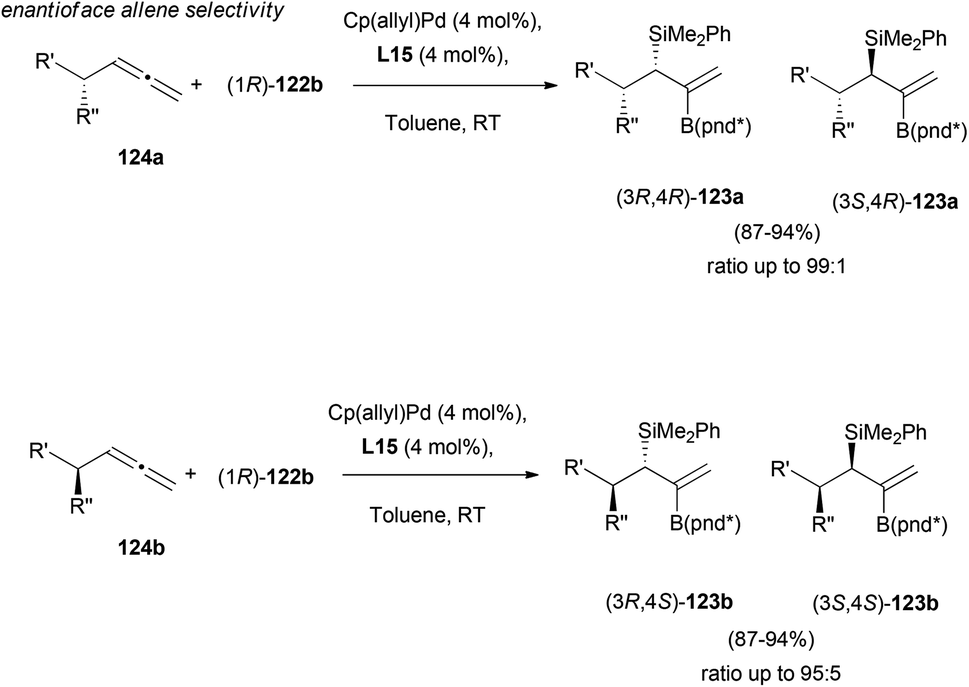
Enantioface-selectivity was then explored, in the reaction on α-chiral allene substrates 124. Interestingly, the reagent-controlled asymmetric addition provided excellent results both for the matched and mismatched combinations, showing selectivities up to 99:1 in final adducts 123 (Scheme 34).
 | ||
| Scheme 34 | ||
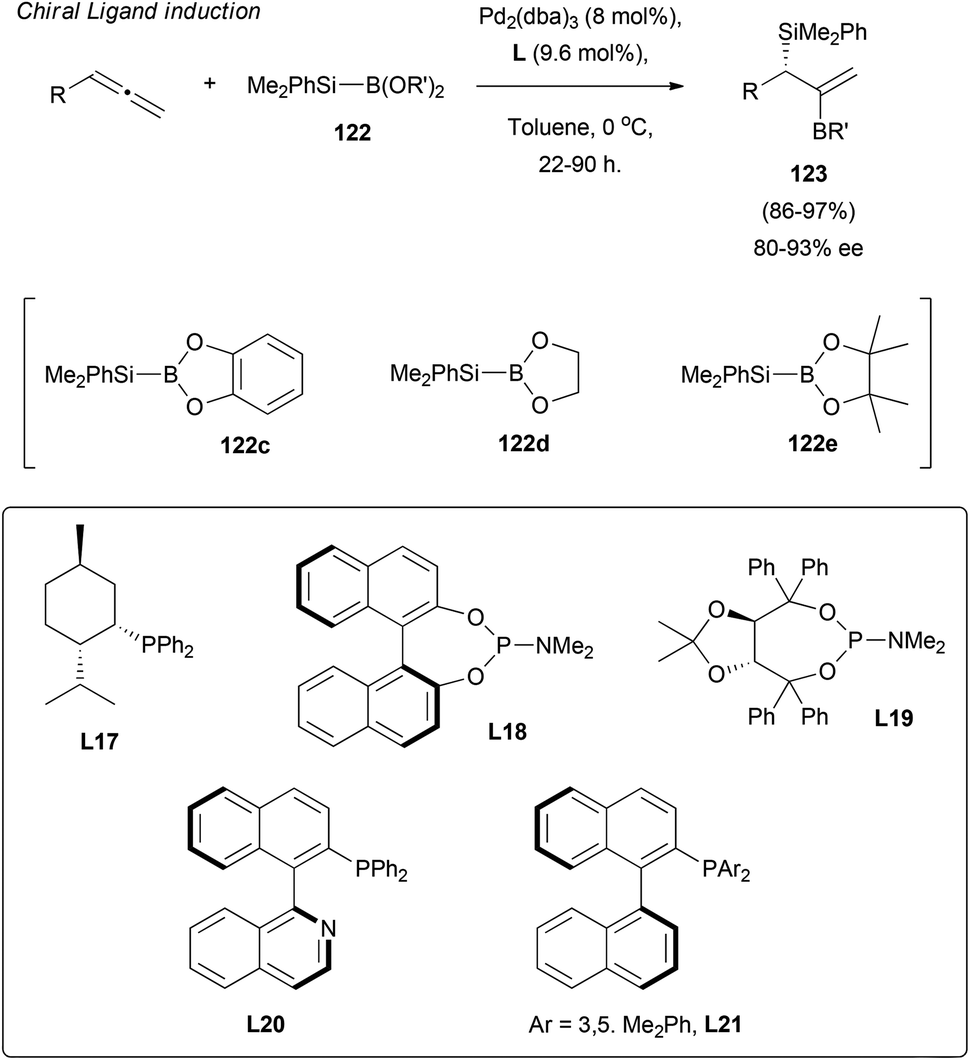
Finally, the efforts of the group moved to explore optically active ligands as the sole chirality inductors in allene silaborations.77 Thus, after the first moderate results using achiral boranes 122c and 122d and ligands L17–21, enantioselectivity was considerably improved by replacing the mentioned boranes with sterically hindered 122e. Excellent yields and selectivity around 80–93% ee were reported (Scheme 35).
 | ||
| Scheme 35 | ||
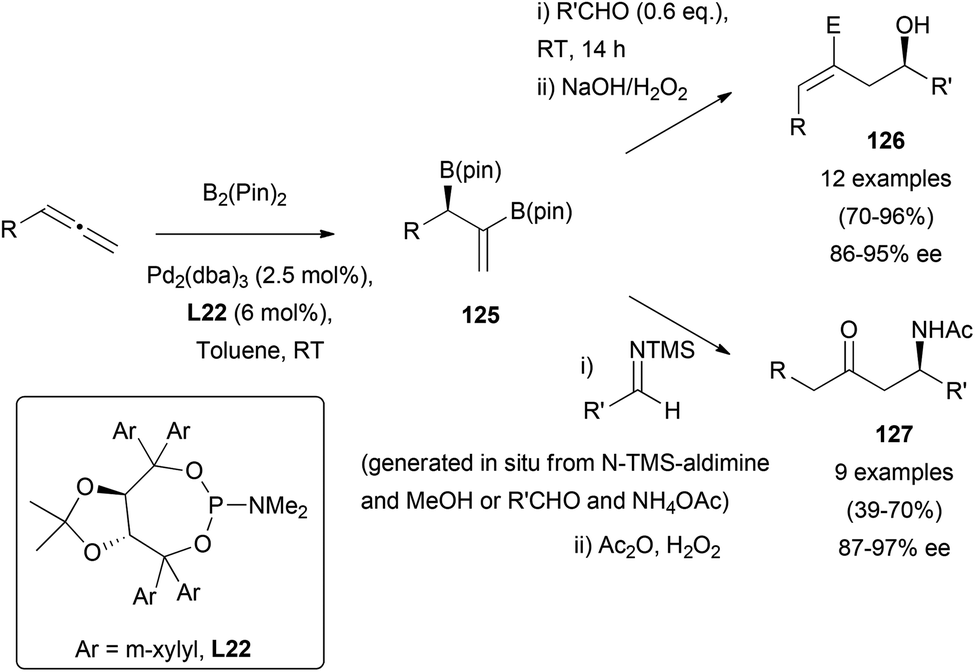
The catalytic asymmetric diboration of allenes78 has also been described as a useful strategy for the preparation of optically active amines,79 or alcohols.80 Morken's research group has reported the use of Pd2(dba)3, and TADDOL (α,α,α′,α′-tetraaryl-2,2-disubstituted 1,3-dioxolane-4,5-dimethanol)-derived phosphoramidite ligand L22, as a chiral inductor for the reaction of racemic allenes and pinacol-based diboranes. Resulting vinyl diborates 125 were directly subjected to allylation in the presence of carbonyl groups or imines, providing functionalized alcohols 126 or keto-amines 127 in moderate to excellent yields (Scheme 36).
 | ||
| Scheme 36 | ||
4.4 Asymmetric metal-catalysed reaction of allenes with carbonyl groups
As mentioned in the previous section, some interesting examples of metal catalysed reactions of chiral allenes with carbonyl groups have been reported. There are also a few examples of metal-catalysed enantioselective reactions of allenes with carbonyl groups.Although aldol reactions of carbonyl groups and allenoates are widely studied,81 its asymmetric metal-catalysed version remains rarely explored. Recently, a reaction between achiral or racemic allenoates 128 and different isatins 129 has been described, yielding allenols 81 through a selective γ-addition in a diastereo- and enantioselective manner (Scheme 37).51 The process takes place in the presence of catalytic amounts of gold or platinum salts, and using dioxides L23 as chiral transfer agents. Moreover, the presence of this non-phosphorous ligand is crucial, as it has been stated that the reaction does not take place in its absence.
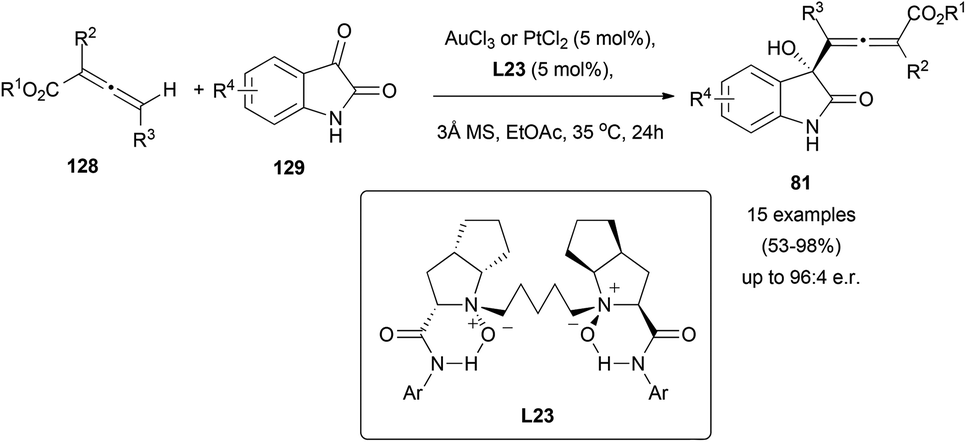 | ||
| Scheme 37 | ||
Interestingly, when the reaction was performed with chiral allenoates 128, the recovered starting material showed a 89% ee, meaning that kinetic resolution of the allenoate was taking place during the process. To explain the observed results, the authors proposed a mechanistic pathway for this reaction where first γ-deprotonation of the gold-coordinated allenoate 130 should take place leading to the active species 131, which could react as the nucleophile with isatins 129, building complex 133. Protonation and metal elimination would then yield the corresponding allenols 81 (Scheme 38).
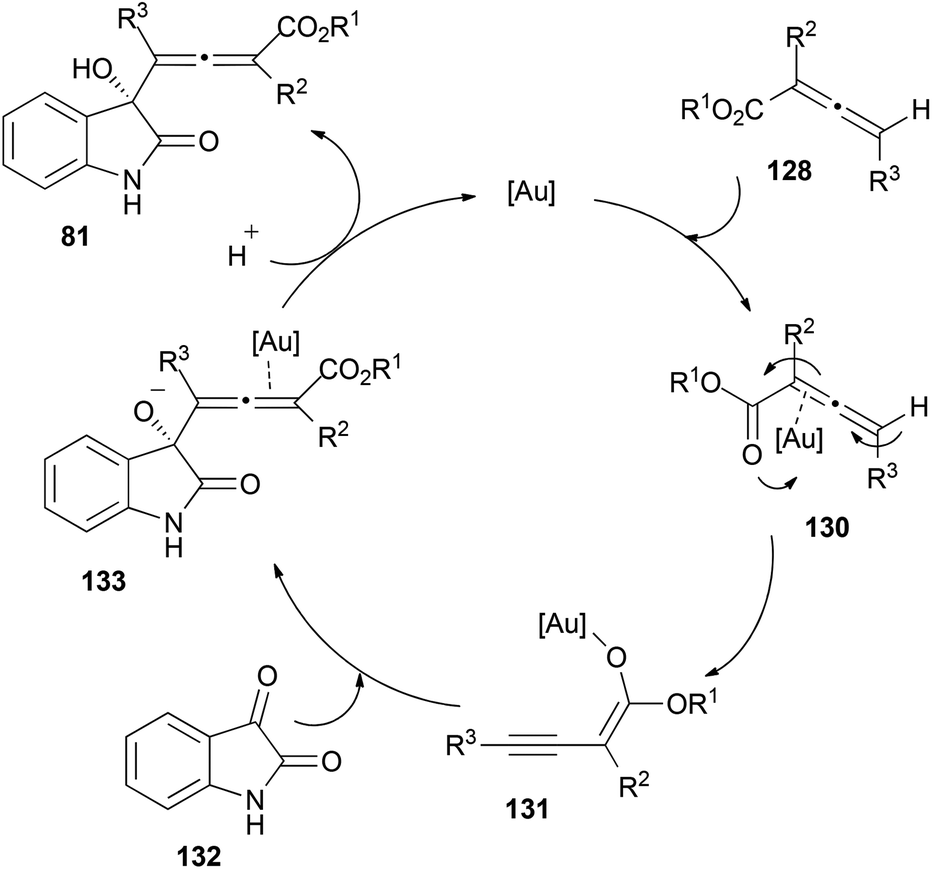 | ||
| Scheme 38 | ||
As mentioned in the previous section about racemization, allenols 81 bearing hydrogen atoms in the R3 substituents showed epimerization in the allene. This result could be explained through a proposed complementary catalytic cycle, where γ-deprotonation of metal-activated species 82 would lead to internal alkyne 83, with a free rotation axis (Scheme 19, vide supra).
Different reactivity regarding the aldol addition was observed using a copper–zinc catalytic pair. Starting from related allenoates 135 and unactivated ketones 134, Shibasaki's research group has described an asymmetric alkylative aldol coupling, by using alkylzinc derivatives 136, and (R)-difluorophos ligand L2 as a chiral inductor.82 This rare example of a multicomponent asymmetric reaction showed an excellent γ-selectivity in the aldol addition, and furnished a quaternary chiral centre in final lactones 137 (Scheme 39).
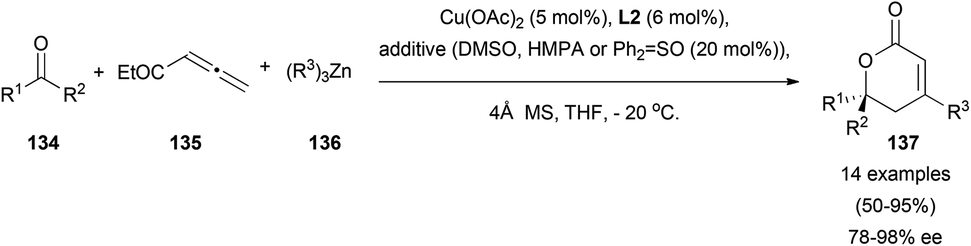 | ||
| Scheme 39 | ||
In the mechanistic proposal, a divergent pathway leading to both γ- and α-adducts is stated. A first reduction of Cu(II) to Cu(I) in the presence of alkylzinc could produce the alkylcopper–phosphine complex 138. Subsequent conjugate addition of this species to allenoate 135 could afford the enolate 139, which is likely to be the active intermediate for the next aldol reactions. Thus, two different aldol products could be formed, α-140 and γ-140. Selectivity between both species seemed to be dependent on substrates and conditions. Further reaction with alkylzinc 136 would furnish aldolate 141 and regenerate alkylcopper 138. The addition of a Lewis base to destabilize aldol adduct α-140, and promote therefore the retro-aldol process, proved the reversibility of the α-cycle. On the other hand, irreversibility of the lactonization step from γ-140 aldolate could result in the total conversion of allenoates and ketones into the final lactones 137 (Scheme 40).
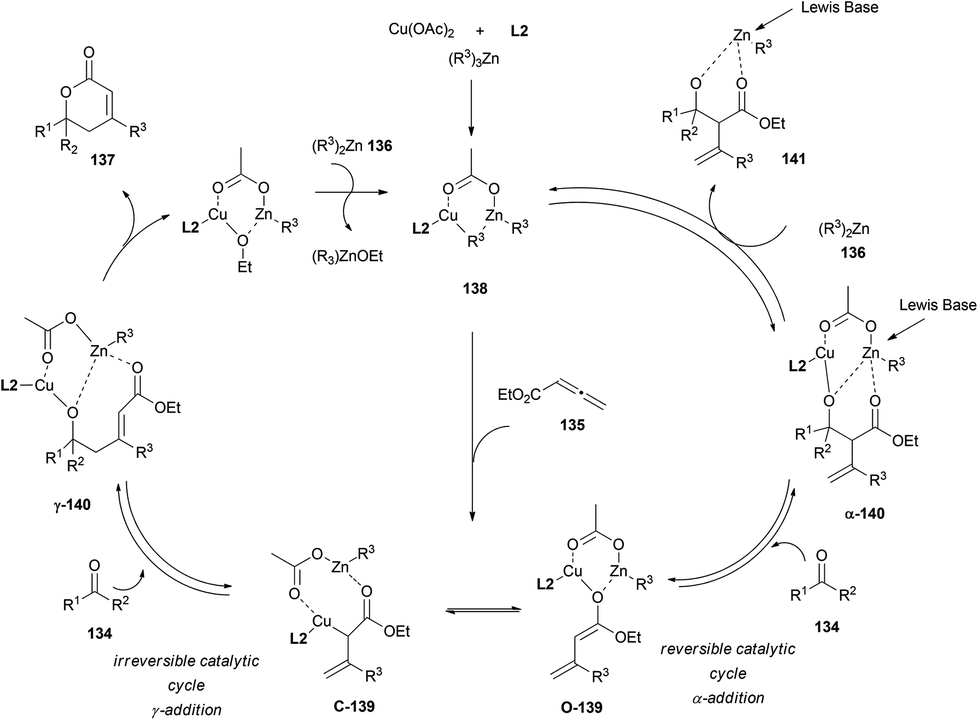 | ||
| Scheme 40 | ||
An interesting example has been reported by Lu et al., where they described the palladium-catalysed tandem annulation of 2-formylboronic acids 142 with allenoates 143 to form indenol derivatives 144 diastereoselectively and with enantioselectivities up to 84% ee when using BINAP ((±)-2,2′-bis(diphenylphosphino)-1,1′-binaphthalene)-type ligands (L5 and L6) and cationic palladium complexes (Scheme 41).83
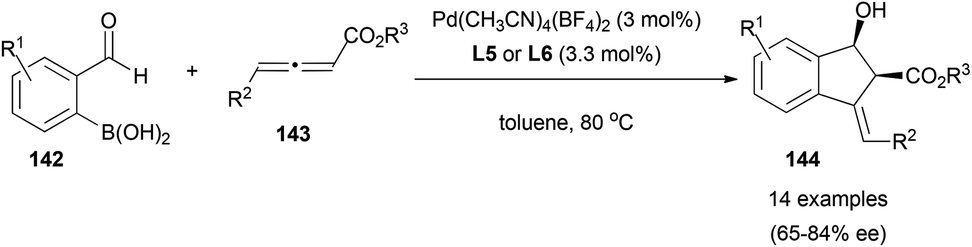 | ||
| Scheme 41 | ||
The proposed mechanism would start with transmetallation of the cationic palladium complex with boronic acid, and formation of a η1-allyl palladium species 145 after coordination with the allenoate. This intermediate could undergo an intramolecular 1,2-addition with the carbonyl group activated by the palladium centre (which avoids formation of η3-allyl palladium intermediates, controlling the selectivity of the reaction). Subsequent hydrolysis of 146 would complete the catalytic cycle (Scheme 42).
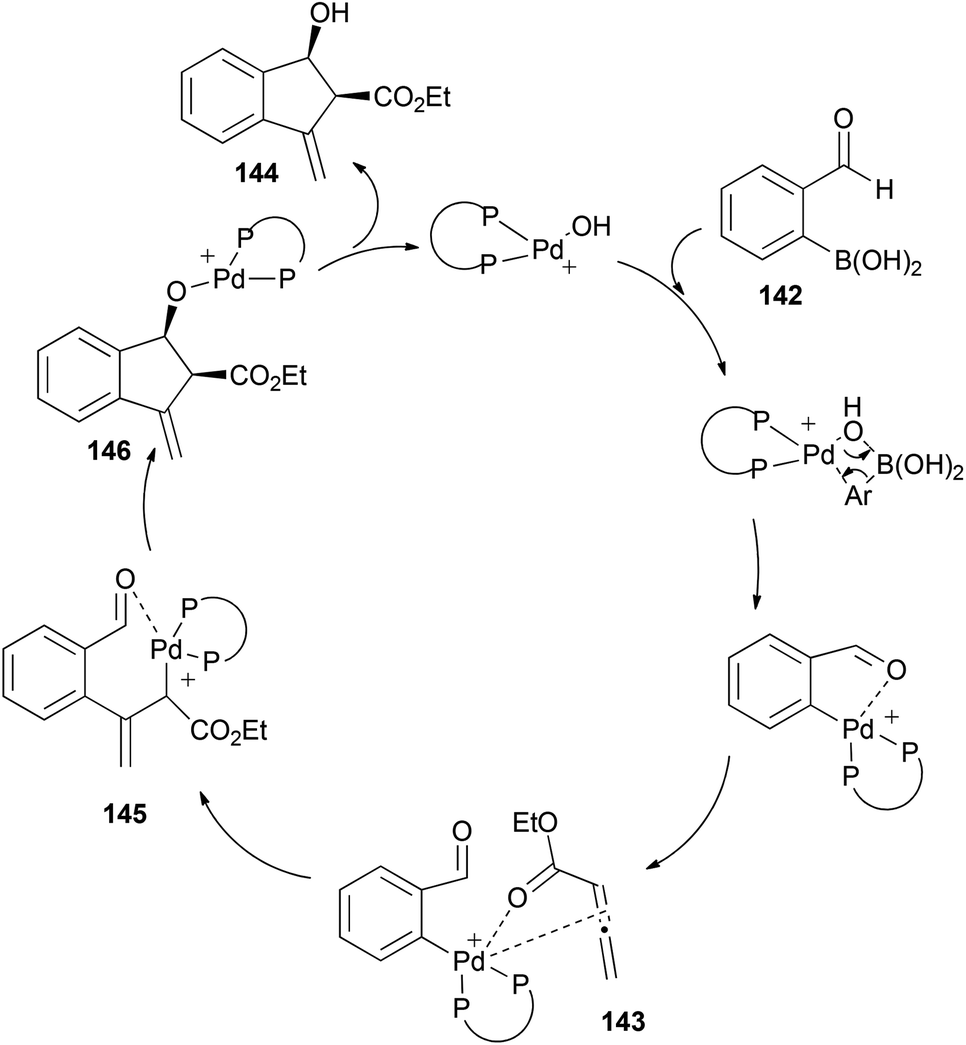 | ||
| Scheme 42 | ||
Finally, in a different approach, efforts have also been directed to the metal-catalysed enantioselective hydroacylation reaction of allenes using chiral ligands. In 2008, Willis et al. reported the first enantioselective rhodium-catalysed intermolecular hydroacylation of 1,3-disubstituted allenes 147 with β-S-aldehydes 148, using Me-DuPhos (1,2-bis[(2R,5R)-2,5-dimethylphospholano]benzene, L24, Scheme 43) as the chiral inductor.84 The hydroacylation process to yield derivatives 149 occurred regioselectively and with enantioselectivities up to 96% ee, by activation of the C–H bond of the aldehyde group by the rhodium complex, followed by insertion into one of the double bonds of the allene.
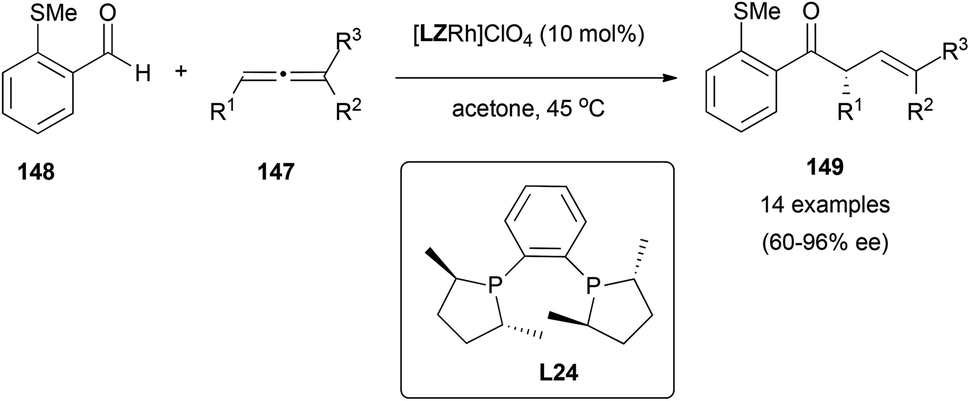 | ||
| Scheme 43 | ||
Interestingly, when they used an enantiomerically enriched chiral allene in the presence of both enantiomers of the catalysts, they observed catalyst control in both cases (they observed the same ee obtained with the racemic allene), and also partial racemization of the recovered allenes (from 99% ee to 31% ee with the (R,R)-ligand), suggesting that a dynamic kinetic asymmetric transformation is in operation in the reaction, in accordance with the studies with other metals as reported in the previous section of this review.
5 Conclusions
In this review we summarise the efforts made in recent years to develop intermolecular enantioselective reactions of allenes under metal catalysis. Whether starting from chiral or racemic allenes, the metal–allene coordination seems to involve a dynamic transformation where competitive processes of allene and product racemization and the desired reaction can occur.Several different mechanisms have been proposed to understand and explain the enantioselectivity of the reactions. For example, reactions catalysed by palladium and rhodium complexes have been proposed to occur mainly through metal-hydride and π-allyl metal intermediates. In these reactions, regioselectivity is controlled and high chirality transfer or enantioselective processed are observed.
In contrast, reactions catalysed by gold occur mainly via η2-gold–allene intermediates, and outer or inner sphere mechanisms are proposed depending on the external partner in the reaction. Reactions proceeding via this η2-metal allene intermediate or via the η1-coordinated bent allene retain the allene chirality, and can be performed in an enantioselective manner, but they are still limited in the nucleophile scope. However, reactions proceeding through the planar σ-allylic cation form in the η1-coordination, lose the chiral information of the allene, which explains racemization processes.
The chiral ligands that have shown better results in the reported enantioselective processes are generally phosphines derived from biphenyl or ferrocenyl derivatives, with the Trost's ligands playing an important role in the enantioselective transformations using palladium chemistry.
It seems clear that understanding the interaction of allene–metal has been crucial for the successful development of strategies for the axial-to-central chirality transfer from enantioenriched allenes and the design of chiral catalysts for the asymmetric induction in processes involving racemic allenes. However, the enantioselectivities obtained in the methodologies developed so far in each variant, can still be improved. In particular, the design of new asymmetric ligands with special pockets for enantioselection and their application in catalysis with less explored metals, such as platinum, will open new avenues for research in the near future.
In this respect, incorporation of the allene framework as part of the ligand scaffold for asymmetric catalysis has rarely been reported. Chiral allenes have been reported to induce asymmetry in the enantioselective addition of diisopropyl zinc to pyrimidine carbaldehyde85 and in the asymmetric ring opening of meso-epoxides.86 More recently, allenic bipyridines87 and allenic bisphosphines88,89 have been reported as axial chiral ligands for transition metal complexes with some promising results, but their full exploitation in asymmetric synthesis has not yet been achieved and further advances in this area, in which imagination and creativity will be crucial, are yet to come.
Acknowledgements
Funding by the University of East Anglia, the EPSRC (EP/L012855/1), the EU (H2020-MSCA-IF-2014-EF-ST-658172) (MTQ) and the Leverhulme Trust (RPG-2015-143) (JMA) is gratefully acknowledged.References
- (a) G. Lowe, Chem. Commun., 1965, 411–413 RSC; (b) J.-C. Damiano, J.-L. Luche and P. Crabbé, Tetrahedron, 1978, 34, 3137–3140 CrossRef CAS; (c) G. P. Moss, Pure Appl. Chem., 1996, 68, 2193–2222 CrossRef CAS.
- For selected reviews on intramolecular transition metal-catalysed reactions of allenes with nucleophiles, see: (a) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2000, 39, 3590–3593 CrossRef CAS; (b) R. W. Bates and V. Satcharoen, Chem. Soc. Rev., 2002, 31, 12–21 RSC; (c) S. Ma, Acc. Chem. Res., 2003, 36, 701–712 CrossRef CAS PubMed; (d) A. S. K. Hashmi, in Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley, 2004, ch. 15, vol. 2, pp. 877–923 Search PubMed; (e) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed; (f) N. Bonger and N. Krause, Angew. Chem., Int. Ed., 2008, 47, 2178–2181 CrossRef PubMed; (g) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS PubMed; (h) J. Muzart, Tetrahedron, 2008, 64, 5815–5849 CrossRef CAS; (i) A. S. K. Hashmi and M. Buhrle, Aldrichimica Acta, 2010, 43, 27–33N CAS; (j) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994–2009 CrossRef CAS PubMed; (k) B. Alcaide and P. Almendros, Adv. Synth. Catal., 2011, 353, 2561–2576 CrossRef CAS; (l) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954–1993 CrossRef CAS PubMed.
- For selected reviews covering intermolecular transition metal-catalysed reactions of allenes with nucleophiles, see: (a) M. P. Muñoz, Org. Biomol. Chem., 2012, 10, 3584–3594 RSC; (b) M. P. Muñoz, Chem. Soc. Rev., 2014, 43, 3164–3183 RSC.
- For a review on axial to central chirality transfer in cyclization processes, see: D. Campolo, S. Gastaldi, C. Roussel, M. P. Bertrand and M. Nechab, Chem. Soc. Rev., 2013, 42, 8434 RSC.
- For selected reviews on cycloaddition reactions with allenes, see: (a) F. López and J. L. Mascareñas, Chem. – Eur. J., 2011, 17, 418–428 CrossRef PubMed; (b) K. M. Brummond and J. M. Osbourn, Beilstein J. Org. Chem., 2011, 7, 601–605 CrossRef CAS PubMed; (c) S. Kitagaki, F. Inagaki and C. Mukai, Chem. Soc. Rev., 2014, 43, 2956–2978 RSC; (d) F. López and J. L. Mascareñas, Chem. Soc. Rev., 2014, 43, 2904–2915 RSC.
- J. Ye and S. Ma, Org. Chem. Front., 2014, 1, 1210–1224 RSC.
- R. K. Neff and D. E. Frantz, ACS Catal., 2014, 4, 519–528 CrossRef CAS.
- M. Ogasawara, Tetrahedron: Asymmetry, 2009, 20, 259–271 CrossRef CAS.
- R. K. Neff and D. E. Frantz, Tetrahedron, 2015, 71, 7–18 CrossRef CAS.
- S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS PubMed.
- For selected examples of non-enantioselective intermolecular metal-catalysed addition of alcohols to allenes, see: (a) S. S. Kinderman, R. Doodeman, J. W. Van Beijma, J. C. Russcher, K. C. M. F. Tjen, T. M. Kooistra, H. Mohaselzadeh, J. H. Van Maarseveen, H. Hiemstra, H. E. Schoemaker and F. P. J. T. Rutjes, Adv. Synth. Catal., 2002, 344, 736–748 CrossRef CAS; (b) R. Doodeman, F. P. J. T. Rutjes and H. Hiemstra, Tetrahedron Lett., 2000, 41, 5979–5983 CrossRef CAS; (c) Z. Zhang and R. A. Widenhoefer, Org. Lett., 2008, 10, 2079–2081 CrossRef CAS PubMed; (d) N. Nishinaa and Y. Yamamoto, Tetrahedron Lett., 2008, 49, 4908–4911 CrossRef; (e) Z. Zhang, S. D. Lee, A. S. Fisher and R. A. Widenhoefer, Tetrahedron, 2009, 65, 1794–1798 CrossRef CAS PubMed; (f) R. S. Paton and F. Maseras, Org. Lett., 2009, 11, 2237–2240 CrossRef CAS PubMed; (g) D. M. Cui, Z.-L. Zheng and C. Zhang, J. Org. Chem., 2009, 74, 1426 CrossRef CAS PubMed; (h) M. S. Hadfield and A.-L. Lee, Org. Lett., 2010, 12, 484–487 CrossRef CAS PubMed; (i) M. P. Muñoz, M. C. de la Torre and M. A. Sierra, Adv. Synth. Catal., 2010, 352, 2189–2194 CrossRef . For the first gold-catalysed reaction of allenes, see: A. S. K. Hashmi, L. Schwarz, J.-H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285–2288 Search PubMed.
- For selected examples of non-enantioselective intermolecular metal-catalysed addition of amines to allenes, see: (a) R. A. Widenhoefer and X. Han, Eur. J. Org. Chem., 2006, 4555–4563 CrossRef CAS; (b) N. Nishina and Y. Yamamoto, Synlett, 2007, 1767–1770 CAS; (c) R. E. Kinder, Z. Zhang and R. A. Widenhoefer, Org. Lett., 2008, 10, 3157–3159 CrossRef CAS PubMed; (d) A. W. Hill, M. R. J. Elsegood and M. C. Kimber, J. Org. Chem., 2010, 75, 5406–5409 CrossRef CAS PubMed; (e) K. L. Toups and R. A. Widenhoefer, Chem. Commun., 2010, 46, 1712–1714 RSC; (f) R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2011, 50, 5560–5563 CrossRef CAS PubMed.
- N. Nishina and Y. Yamamoto, Tetrahedron, 2009, 65, 1799–1808 CrossRef CAS.
- Z. Zhang and R. A. Widenhoefer, Org. Lett., 2008, 10, 2079–2081 CrossRef CAS PubMed.
- N. Nishina and Y. Yamamoto, Angew. Chem., Int. Ed., 2006, 45, 3314–3317 CrossRef CAS PubMed.
- Z. J. Wang, D. Benitez, E. Tkatchouk, W. A. Goddard III and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 13064–13071 CrossRef CAS PubMed.
- N. Nishina and Y. Yamamoto, Tetrahedron Lett., 2008, 49, 4908–4911 CrossRef CAS.
- For a paper on proven intermediates in gold catalysis, see: A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232–5241 CrossRef CAS PubMed.
- For papers supporting the notion of bent-allenes and their isolation of complexes characterised by X-ray, see: (a) C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206–3209 CrossRef CAS PubMed; (b) A. Fürstner, M. Alcarazo, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 3210–3214 CrossRef PubMed . For a review on activation of allenes by gold complexes, see: M. Malacria, L. Fensterbank and V. Gandon, Top. Curr. Chem., 2011, 302, 157–182 Search PubMed.
- M.-Z. Wang, C.-Y. Zhou, Z. Guo, E. L.-M. Wong, M.-K. Wong and C.-M. Che, Chem. – Asian J., 2011, 6, 812–824 CrossRef CAS PubMed.
- For selected examples of non-enantioselective intermolecular metal-catalysed of allenes with electron-rich aromatics, see: (a) M. A. Tarselli, A. Liu and M. R. Gagné, Tetrahedron, 2009, 65, 1785–1789 CrossRef CAS PubMed; (b) K.- L.- Toups, G. T. Liu and R. A. Widenhoefer, J. Organomet. Chem., 2009, 694, 571–575 CrossRef CAS PubMed; (c) W. Kong, J. Cui, Y. Yu, G. Chen, C. Fu and S. Ma, Org. Lett., 2009, 11, 1213–1216 CrossRef CAS PubMed; (d) K. L. Toups, G. T. Liu and R. A. Widenhoefer, J. Organomet. Chem., 2009, 694, 571–575 CrossRef CAS PubMed; (e) R. R. Suresh and K. C. K. Swamy, J. Org. Chem., 2012, 77, 6959–6969 CrossRef CAS PubMed; (f) M. P. Muñoz, M. de la Torre and M. A. Sierra, Chem. – Eur. J., 2012, 18, 4499–4504 CrossRef PubMed.
- F. Kato and K. Hiroi, Chem. Pharm. Bull., 2004, 52, 95–103 CrossRef CAS PubMed.
- For selected reviews, see: (a) Ref. 2c; ; (b) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989–1000 CrossRef CAS PubMed; (c) J. L. Bras and J. Muzart, Chem. Soc. Rev., 2014, 43, 3003–3040 RSC.
- H. Li, D. Müller, L. Guénée and A. Alexakis, Org. Lett., 2012, 14, 5880–5883 CrossRef CAS PubMed.
- R. Zeng, C. Fu and S. Ma, J. Am. Chem. Soc., 2012, 134, 9597–9600 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Ogasawara, A. Okada, V. Subbarayan, S. Sorgel and T. Takahashi, Org. Lett., 2010, 12, 5736–5739 CrossRef CAS PubMed; (b) M. Ogasawara, Y. Ge, A. Okada and T. Takahashi, Eur. J. Org. Chem., 2012, 1656–1663 CrossRef CAS.
- S.-S. Ng and T. F. Jamison, Tetrahedron, 2005, 61, 11405–11417 CrossRef CAS.
- S.-S. Ng and T. F. Jamison, J. Am. Chem. Soc., 2005, 127, 7320–7321 CrossRef CAS PubMed.
- K. M. Buchner, T. B. Clark, J. M. N. Loy, T. X. Nguyen and K. A. Woerpel, Org. Lett., 2009, 11, 2173–2175 CrossRef CAS PubMed.
- J. Hou, Y. Chen, D. Ma, B. Cordes, J. Wang, X. Wang, F. E. Kuhn, H. Guo and M. Zhou, Chem. Commun., 2015, 51, 7439–7442 RSC.
- Z. Zhang, C. F. Bender and R. A. Widenhoefer, J. Am. Chem. Soc., 2007, 129, 14148–14149 CrossRef CAS PubMed.
- J. H. Lee and F. D. Toste, Angew. Chem., Int. Ed., 2007, 46, 912–914 CrossRef CAS PubMed.
- C. Deutsch, B. Gockel, A. Hoffmann-Röder and N. Krause, Synlett, 2007, 1790–1794 CAS.
- B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 15978–15979 CrossRef CAS PubMed.
- K. L. Butler, M. Tragni and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2012, 51, 5175–5178 CrossRef CAS PubMed.
- H. E. Burks, S. Liu and J. P. Morken, J. Am. Chem. Soc., 2007, 129, 8766–8773 CrossRef CAS PubMed.
- A. Horvath and J.-E. Backvall, Chem. Commun., 2004, 964–965 RSC.
- G. A. Molander, E. M. Sommers and S. R. Baker, J. Org. Chem., 2006, 71, 1563–1568 CrossRef CAS PubMed.
- A. Claesson and L.-I. Olsson, J. Chem. Soc., Chem. Commun., 1979, 524–525 RSC.
- D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 10630–10634 CrossRef CAS PubMed.
- S. M. Oon and W. M. Jones, Organometallics, 1988, 7, 2172–2177 CrossRef CAS.
- J. D. Osborne, H. E. Randell-Sly, G. S. Currie, A. R. Cowley and M. C. Willis, J. Am. Chem. Soc., 2008, 130, 17232–17233 CrossRef CAS PubMed.
- W. R. Roth, G. Ruf and P. W. Ford, Chem. Ber., 1974, 107, 48–52 CrossRef CAS.
- O. Rodriguez and H. Morrison, J. Chem. Soc. D, 1971, 679–679 RSC.
- V. Gandon, G. Lemière, A. Hours, L. Fensterbank and M. Malacria, Angew. Chem., Int. Ed., 2008, 47, 7534–7538 CrossRef CAS PubMed.
- E. Soriano and I. Fernandez, Chem. Soc. Rev., 2014, 43, 3041–3105 RSC.
- T. Cañeque, F. M. Truscott, R. Rodriguez, G. Maestri and M. Malacria, Chem. Soc. Rev., 2014, 43, 2916–2926 RSC.
- W. Yang and A. S. Hashmi, Chem. Soc. Rev., 2014, 43, 2941–2955 RSC.
- R. J. Harris, K. Nakafuku and R. A. Widenhoefer, Chem. – Eur. J., 2014, 20, 12245–12254 CrossRef CAS PubMed . During the preparation of this manuscript, a new paper with the kinetics and mechanism of gold-catalysed allene racemization appeared: H. Li, R. J. Harris, K. Nakafuku and R. A. Widenhoefer, Organometallics, 2016, 35(13), 2242–2248 CrossRef.
- A. Vanitcha, G. Gontard, N. Vanthuyne, E. Derat, V. Mouriés-Mansuy and L. Fensterbank, Adv. Synth. Catal., 2015, 357, 2213–2218 CrossRef CAS.
- G. Wang, X. Liu, Y. Chen, J. Yang, J. Li, L. Lin and X. Feng, ACS Catal., 2016, 6, 2482–2486 CrossRef CAS.
- (a) M. Sugimone, T. Ohmura, Y. Miyake, S. Mitani, Y. Ito and M. Murakami, J. Am. Chem. Soc., 2003, 125, 11174–11175 CrossRef PubMed; (b) F. Ferreira, M. Audouin and F. Chemla, Chem. – Eur. J., 2005, 11, 5269–5278 CrossRef CAS PubMed; (c) X. Ariza, J. Cornellá, M. Font-Bardia, J. Garcia, J. Ortiz, C. Sánchez and X. Solans, Angew. Chem., Int. Ed., 2009, 48, 4202–4205 CrossRef CAS PubMed; (d) M. Curtis-Long and Y. Aye, Chem. – Eur. J., 2009, 15, 5402–5416 CrossRef CAS PubMed; (e) J. Louvel, F. Chemla, E. Demont, F. Ferreira, A. Pérez-Luna and A. Voituriez, Adv. Synth. Catal., 2011, 353, 2137–1251 CrossRef CAS; (f) M. Yoshida, T. Kasai, T. Mizuguchi and K. Namba, Synlett, 2014, 1160–1162 CrossRef.
- (a) P. Elsner, L. Bernardi, G. Dela Salla, J. Overgaard and K. A. Jorgensen, J. Am. Chem. Soc., 2008, 130, 4897–4905 CrossRef CAS PubMed; (b) D. Barber, A. Duris, A. Thompson, H. Sanganee and D. J. Dixon, ACS Catal., 2014, 4, 634–638 CrossRef CAS PubMed; (c) S. Zhang, Y.-C. Luo, X.-Q. Hu, Z.-Y. Wang, Y.-M. Liang and P.-F. Xu, J. Org. Chem., 2015, 80, 7288–7294 CrossRef CAS PubMed; (d) Y. Huang, Z. Lv, C. Cai, C. Kai, Y. Pei and Y. Feng, Angew. Chem., Int. Ed., 2015, 54, 7299–7302 CrossRef CAS PubMed; (e) X. Yang and D. Toste, Chem. Sci., 2016, 7, 2653–2656 RSC.
- For recent reviews, see: (a) Q. Y. Zhao, Z. Lian, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 1724–1732 RSC; (b) Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927–2940 RSC. For selected examples, see: (c) R. J. Lundgren, A. Wilsily, N. Marion, C. Ma, Y. K. Chung and G. C. Fu, Angew. Chem., Int. Ed., 2013, 52, 2525–2528 CrossRef CAS PubMed; (d) J. Chen, Y. Cai and G. Zhao, Adv. Synth. Catal., 2014, 356, 359–363 CrossRef CAS; (e) T.-H. Nguyen, M. Toffano, C. Bournard and G. Vo-Thanh, Tetrahedron Lett., 2014, 55, 6377–6380 CrossRef CAS; (f) M. Kalek and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 9438–9442 CrossRef CAS PubMed; (g) T. Wang, Z. Yu, D. L. Hoon, K. W. Huang, Y. Lan and Y. Lu, Chem. Sci., 2015, 6, 4912–4922 RSC.
- For an example of a non-enantioselective gold-catalysed thiolation of allenes, see: M. Menggenbateer, G. Narsireddya, N. Ferrara, T. J. Nishina and Y. Yamamoto, Tetrahedron Lett., 2010, 51, 4627–4629 CrossRef CAS.
- A. B. Pritzius and B. Breit, Angew. Chem., Int. Ed., 2015, 54, 3121–3125 CrossRef CAS PubMed.
- M. L. Cooke, K. Xu and B. Breit, Angew. Chem., Int. Ed., 2012, 51, 10876–10879 CrossRef CAS PubMed.
- C. Li, M. Kähny and B. Breit, Angew. Chem., Int. Ed., 2014, 53, 13780–13784 CrossRef CAS PubMed.
- K. Xu, N. Thieme and B. Breit, Angew. Chem., Int. Ed., 2014, 53, 2162–2165 CrossRef CAS PubMed.
- K. Xu, Y. H. Wang, V. Khakyzadeh and B. Breit, Chem. Sci., 2016, 7, 3313–3316 RSC.
- A. Haydl, K. Xu and B. Breit, Angew. Chem., Int. Ed., 2015, 54, 7149–7153 CrossRef CAS PubMed.
- K. Xu, W. Raimondi, T. Bury and B. Breit, Chem. Commun., 2015, 51, 10861–10863 RSC.
- K. Xu, T. Gilles and B. Breit, Nat. Commun., 2015, 6, 7616 CrossRef PubMed.
- P. Koschker, A. Lumbroso and B. Breit, J. Am. Chem. Soc., 2011, 133, 20746–20749 CrossRef CAS PubMed . During the preparation of this manuscript, a new paper describing the rhodium-catalysed enantioselective hydroalkoxylation of allenes has been published: Z. Liu and B. Breit, Angew. Chem., Int. Ed., 2016, 55, 8440–8443 CrossRef PubMed.
- P. A. Evans and L. J. Kennedy, J. Am. Chem. Soc., 2001, 123, 1234–1235 CrossRef CAS PubMed.
- W. Lim, J. Kim and Y. H. Rhee, J. Am. Chem. Soc., 2014, 136, 13618–13621 CrossRef CAS PubMed.
- For selected examples of metal-catalysed asymmetric allylic substitution reaction using aliphatic alcohols as a nucleophile, see: (a) P. A. Evans, D. K. Leahy, W. J. Andrews and D. Uraguchi, Angew. Chem., Int. Ed., 2004, 43, 4788–4791 CrossRef CAS PubMed; (b) C. Shu and J. F. Hartwig, Angew. Chem., Int. Ed., 2004, 43, 4794–4797 CrossRef CAS PubMed; (c) F. L. Lam, T. T.-L. Au-Yeung, F. Y. Kwong, Z. Zhou, K. Y. Wong and A. S. C. Chan, Angew. Chem., Int. Ed., 2008, 47, 1280–1283 CrossRef CAS PubMed; (d) M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5568–5571 CrossRef CAS PubMed.
- (a) B. M. Trost, C. Jäkel and B. Plietker, J. Am. Chem. Soc., 2003, 125, 4438–4439 CrossRef CAS PubMed; (b) B. M. Trost, A. B. C. Simas, B. Plietker, C. Jäkel and J. Xie, Chem. – Eur. J., 2005, 11, 7075–7082 CrossRef CAS PubMed.
- For a review of chemistry of alkoxyallenes, see: R. Zimmer and H.-U. Reissig, Chem. Soc. Rev., 2014, 43, 2888–2903 RSC.
- Z. Q. Shen, X. X. Li, J. W. Shi, B. L. Chen and Z. Chen, Tetrahedron Lett., 2015, 56, 4080–4083 CrossRef CAS.
- B. M. Trost, J. Xie and J. D. Sieber, J. Am. Chem. Soc., 2011, 133, 20611–20622 CrossRef CAS PubMed.
- For reviews on reaction of alkynes and multiple bonds, see: (a) D. E. Frantz, R. Fässler, C. S. Tomooka and E. M. Carreira, Acc. Chem. Res., 2000, 33, 373–381 CrossRef CAS PubMed; (b) L. Pu, Tetrahedron, 2003, 59, 9873–9886 CrossRef CAS; (c) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2004, 4095–4105 CrossRef CAS; (d) G. Lu, Y.-M. Li, X.-S. Li and A. S. C. Chan, Coord. Chem. Rev., 2005, 249, 1736–1744 CrossRef CAS; (e) K. Yamada and K. Tomioka, Chem. Rev., 2008, 108, 2874–2886 CrossRef CAS PubMed; (f) L. Zani and C. Bolm, Chem. Commun., 2006, 4263–4275 RSC; (g) B. M. Trost and A. H. Weiss, Adv. Synth. Catal., 2009, 351, 963–983 CrossRef CAS PubMed; (h) C.-J. Li, Acc. Chem. Res., 2010, 43, 581–590 CrossRef CAS PubMed.
- T. Sawano, K. Ou, T. Nishimura and T. Hayashi, J. Org. Chem., 2013, 78, 8986–8993 CrossRef CAS PubMed.
- For selected examples of chemistry and importance of allyl silanes, see: (a) T. Hayashi, M. Konishi and M. Kumada, J. Am. Chem. Soc., 1982, 104, 4963–4965 CrossRef CAS; (b) I. Fleming, J. Dunogues and R. Smithers, Org. React., 1989, 37, 57 CAS; (c) C. E. Masse and J. S. Panek, Chem. Rev., 1995, 95, 1293–1316 CrossRef CAS; (d) T. K. Sarkar, in Science of Synthesis, ed. I. Fleming, Georg Thieme Verlag, Stuttgart, 2002, vol. 4, p. 837 Search PubMed; (e) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2794 CrossRef CAS PubMed.
- (a) M. Suginome, T. Ohmura, Y. Miyake, S. Mitani, Y. Ito and M. Murakami, J. Am. Chem. Soc., 2003, 125, 11174–11175 CrossRef CAS PubMed; (b) T. Ohmura and M. Suginome, Org. Lett., 2006, 8, 2503–2506 CrossRef CAS PubMed.
- (a) S. Onozawa, Y. Hatanaka and M. Tanaka, Chem. Commun., 1999, 1863–1864 RSC; (b) M. Suginome, Y. Ohmori and Y. Ito, J. Organomet. Chem., 2000, 611, 403–413 CrossRef CAS; (c) M. Suginome, Y. Ohmori and Y. Ito, J. Am. Chem. Soc., 2001, 123, 4601–4602 CrossRef CAS PubMed.
- T. Ohmura, H. Taniguchi and M. Suginome, J. Am. Chem. Soc., 2006, 128, 13682–13683 CrossRef CAS PubMed . For the mechanistic study and regioselectivity of the reactions, see: Y. Abe, K. Kuramoto, M. Ehara, H. Nakatsuji, M. Sugimore, M. Murakami and Y. Ito, Organometallics, 2008, 27, 1736–1742 CrossRef.
- (a) N. F. Pelz, A. R. Woodward, H. E. Burks, J. D. Sieber and J. P. Morken, J. Am. Chem. Soc., 2004, 126, 16328–16329 CrossRef CAS PubMed; (b) See ref. 36.
- J. D. Sieber and J. P. Morken, J. Am. Chem. Soc., 2006, 128, 74–75 CrossRef CAS PubMed.
- A. R. Woodward, H. E. Burks, L. M. Chan and J. P. Morken, Org. Lett., 2005, 7, 5505–5507 CrossRef CAS PubMed.
- (a) B. Xu and G. B. Hammond, Angew. Chem., Int. Ed., 2008, 47, 689–692 CrossRef CAS PubMed; (b) J. C. Aponte, G. B. Hammond and B. Xu, J. Org. Chem., 2009, 74, 4623–4625 CrossRef CAS PubMed; (c) Z. Qin, R. Ma, S. Xu and Z. He, Tetrahedron, 2013, 69, 10424–10430 CrossRef CAS; (d) E. Lee, J. Bang, J. Kwon and C. M. Yu, J. Org. Chem., 2015, 80, 10359–10363 CrossRef CAS PubMed; (e) J. Bang, H. Kim, J. Kim and C. M. Yu, Org. Lett., 2015, 17, 1573–1576 CrossRef CAS PubMed.
- K. Oisaki, D. Zhao, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 7439–7443 CrossRef CAS PubMed.
- X. Yu and X. Lu, Org. Lett., 2009, 11, 4366–4369 CrossRef CAS PubMed.
- J. D. Osborne, H. E. Randell-Sly, G. S. Currie, A. R. Cowley and M. C. Willis, J. Am. Chem. Soc., 2008, 130, 17232–17233 CrossRef CAS PubMed . For a non-enantioselective example, see: K. Kokubo, K. Matsumasa, Y. Nishinaka, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1999, 72, 303–311 CrossRef.
- I. Sato, Y. Matsueda, K. Kadowaki, S. Yonekubo, T. Shibata and K. Soai, Helv. Chim. Acta, 2002, 85, 3383 CrossRef CAS.
- X. Pu, X. Qi and J. M. Ready, J. Am. Chem. Soc., 2009, 131, 10364 CrossRef CAS PubMed.
- S. Löhr, J. Averbeck, M. Schürmann and N. Krause, Eur. J. Org. Chem., 2008, 552 CrossRef.
- F. Cai, X. Pu, X. Qi, V. Lynch, A. Radha and J. M. Ready, J. Am. Chem. Soc., 2011, 133, 18066 CrossRef CAS PubMed.
- A. Vanitcha, C. Damelincourt, G. Gontard, N. Vanthuyne, V. Mouriés-Mansuy and L. Fensterbank, Chem. Commun., 2016, 52, 6785–6788 RSC.
| This journal is © the Partner Organisations 2016 |