
A remarkable solvent effect of fluorinated alcohols on transition metal catalysed C–H functionalizations
J.
Wencel-Delord
* and
F.
Colobert
Laboratoire de Chimie Moléculaire (UMR CNRS 7509), Université de Strasbourg, ECPM 25 Rue Becquerel, 67087, Strasbourg, France. E-mail: wenceldelord@unistra.fr; francoise.colobert@unistra.fr
First published on 2nd February 2016
Abstract
Fluorinated solvents like 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and trifluoroethanol (TFE) have recently emerged as a remarkable synthetic solution allowing challenging C–H activation reactions. A beneficial effect of these solvents on reactivity, as well as site- and stereoselectivity of various direct functionalization reactions has been observed. Moreover, several catalytic systems specifically require such fluorinated medium. This Highlight is devoted to showcasing the exceptional potential of fluorinated alcohols in the C–H activation field.
 Joanna Wencel-Delord | Joanna Wencel-Delord was educated in chemistry at the Ecole Nationale Supérieure de Chimie de Rennes, France and she received her PhD in 2010 from the University of Rennes 1, France (Dr C. Crévisy and Dr M. Mauduit). After postdoctoral studies with Prof. F. Glorius at the Westfälische Wilhelms-Universität Münster (Germany) and temporary assistant professor position (ATER) at the University of Strasbourg (Prof. P. Compain), she joined CNRS in 2013 as an associate researcher in the group of Prof. F. Colobert (University of Strasbourg, France). Her research focuses on the transition metal-catalyzed asymmetric C–H activation. |
Introduction
It is now about a decade since the C–H activation field has attracted an expanding interest from the scientific community, thus opening a new avenue in transition metal catalysis.1,2 Until recently, several major obstacles have however limited a broad and general utility of this modern strategy. Indeed, the vast majority of the catalytic systems not only required preinstallation on a substrate of a strongly coordinating directing group (DG)3 and harsh reaction conditions1i but was also almost exclusively limited to an ortho-functionalization of aromatic compounds.4 On the other hand, the direct functionalization of aliphatic molecules,5 the activation of remote C–H bonds and stereoselective transformations6 remained an almost uncovered research field. Nevertheless, since a few years, new synthetic solutions have been devised to overcome the above mentioned limitations largely expanding the portfolio of molecular scaffolds that can now be constructed via the C–H activation approach. Interestingly, a panel of solvents commonly employed in the C–H activation field, like alcohols, dichloroethane, aromatic solvents, dioxane and, maybe more sporadically, very polar media like DMF, DMSO or organic acids, was recently expanded to include polyfluorinated alcohols, such as 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and 1,1,1-trifluoroethanol (TFE). Owing to their unique properties, like their protic but non-nucleophilic character, high hydrogen bonding donor ability, high ionizing power, ability to stabilise cationic species and extreme polarity, these alcohols have been recently recognized as remarkable media for several types of transformations including oxidations, C–C and C–X bond formations, cycloadditions, etc.7 On the other hand, the beneficial effect of the acidic solvents to promote C–H activation is commonly acknowledged. Such media can indeed enhance an electrophilic metallation and accordingly various direct functionalization reactions occur in, or require an addition of strong organic acids such as acetic acid or trifluoroacetic acid. Besides, formation of cationic metallic species during the C–H transformations is frequently proposed. Finally, the DGs embedded within a C–H substrate (amides, carboxylates or nitriles for example), participate easily in hydrogen bonding. Taking into consideration all these points, it becomes quite obvious that polyfluorinated alcohols could advantageously impact the C–H activation reactions by means of various modes of action.To the best of our knowledge, one of the first to evidence a beneficial effect of TFE as a cosolvent for oxidative olefination was Zhangjie Shi. In 2007, while studying the direct functionalization of N,N-dimethylbenzylamines, he discovered that the most active catalytic system was obtained when TFE, together with the acetic acid additive was used.8 As this reaction was still efficient in other solvents (provided that an AcOH additive was employed), the origin of this TFE-induced reactivity enhancement was not elucidated. However, it can be hypothesized that when using TFE and AcOH, the amine DG is partially protonated yet modifying the features of the reactive species.
A more meaningful illustration of the unique potential of polyfluorinated alcohols to mediate C–H activation reactions was reported a few years later by Baran et al. In the context of a total synthesis of piperarborenine cores, the author noticed that the use of HFIP together with the pivalic acid additive was a critical parameter allowing for efficient arylation of the cyclobutane-derivatives (Scheme 1).9 Also in this case, selection of this particular solvent rather results from an empirical optimization than a foreseen reactivity. However, although there is a lack of rationalized explanation, this work progressively inspired numerous research groups to integrate fluorinated solvents into their “tool-box” and to screen them regularly during an optimization study. Rewardingly, many exciting and efficient transformations progressively emerged.
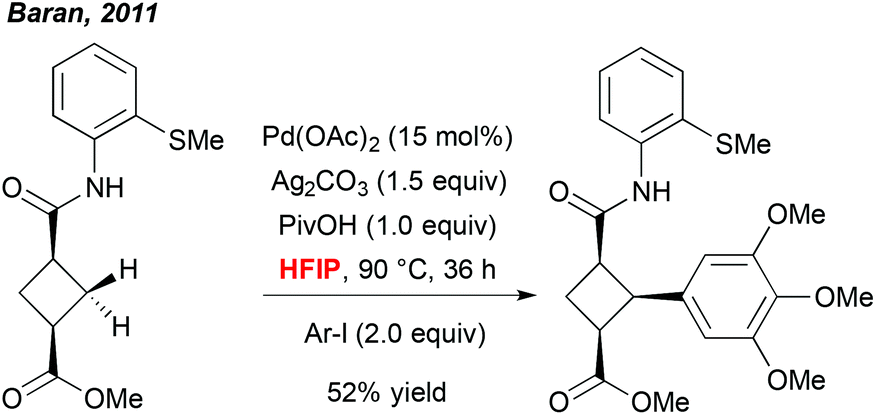 | ||
| Scheme 1 Diastereoselective direct arylation occurring in the HFIP medium. | ||
At this point we would like to highlight several important points. Although empirical observations showcase a particular role of HFIP and/or TFE in C–H activation reactions, their accurate role has not been studied in-depth and, if any, only very succinct comments are provided by the authors. Accordingly, the herein-presented explanations are rather hypothetical. Secondly, in some cases the authors mention that HFIP or TFE outcompetes other media but a lack of a detailed optimization study renders the analysis of such results illusive. Finally, it seems that in many cases the HFIP solvent was simply not tested. Accordingly, due to all these difficulties, the aim of this review is simple to highlight the different activation modes and beneficial effects that were observed when employing such a medium.
C–H activation of aliphatic substrates
Direct metallation of the C(sp3)–H bonds is widely recognized as highly challenging, and hence particular synthetic tools were needed to facilitate such transformations.5 Since 2006 Yu and colleagues have been particularly actively involved in this field, discovering several catalytic systems for direct C(sp3)–C and C(sp3)–X couplings.10,11 They, and others, observed that many of such transformations performed well in alcoholic solvents like tBuOH or tAmylOH or even in ether-type media.A major step forward in the C(sp3)–H activation field was achieved when the potential of bidentate DGs to enhance these couplings was evidenced. Such bidentate DG-induced couplings seem to tolerate well a wide range of solvents such as alcohols, DCE and toluene for example. However, an interesting HFIP-induced reactivity boost was observed independently by Fernández-Ibáñez and Carretero,12 Ma13 and Yu14 when alkyl substrates like the amino acid derivatives bearing a bidentate DG embedded either in the amine ((N-2-pyridyl)sulfonyl or 2-methoxyiminoacetyl auxiliaries (Scheme 2A)) or the carboxylate motif (OH-free dipeptide motif (Scheme 2B)) were subjected to the Pd-catalyzed direct arylation reaction. Truly efficient transformations could be achieved in HFIP while other standard solvents like toluene, non-fluorinated alcohols, dioxane, DCE, THF, etc. enabled the formation of the desired product in significantly lower yields. Although the authors did not comment on an exact role of HFIP in these reactions, the “peptide-like” structure of the substrates and strong hydrogen bonding properties of HFIP probably result in the formation of H-bonds between the substrate and the solvent, hence favouring an optimal geometry of the metallacyclic intermediates and/or positively influencing the electronic properties of the DG. More recently, HFIP has also been selected as an optimal solvent or a key additive in C(sp3)–H activation of simpler alkanes bearing monodentate auxiliaries like N-methoxyamide15 (Scheme 2C) or oxime ether.16 In the first case, the acidic character of this solvent is believed to prevent the decomposition of the starting material. In the second case, an increased efficiency in the presence of the extremely polar HFIP cosolvent (DCE/HFIP 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture) is attributed to an accelerated Ar group transfer from a diaryliodonium coupling partner to a palladacyclic intermediate. It is worth noting that when other commonly used bicoordinating DGs such as 8-aminoquinoline or 2-pyridin-2-ylisopropyl (PIP) moieties were installed on the amino acid scaffold, more standard solvents like hindered alcohols, DCE, acetonitrile or toluene were employed as optimal reaction media. However, to the best of our knowledge, HFIP was generally not tested in these cases (or at least the authors do not mention these tests).17,18
:1 mixture) is attributed to an accelerated Ar group transfer from a diaryliodonium coupling partner to a palladacyclic intermediate. It is worth noting that when other commonly used bicoordinating DGs such as 8-aminoquinoline or 2-pyridin-2-ylisopropyl (PIP) moieties were installed on the amino acid scaffold, more standard solvents like hindered alcohols, DCE, acetonitrile or toluene were employed as optimal reaction media. However, to the best of our knowledge, HFIP was generally not tested in these cases (or at least the authors do not mention these tests).17,18
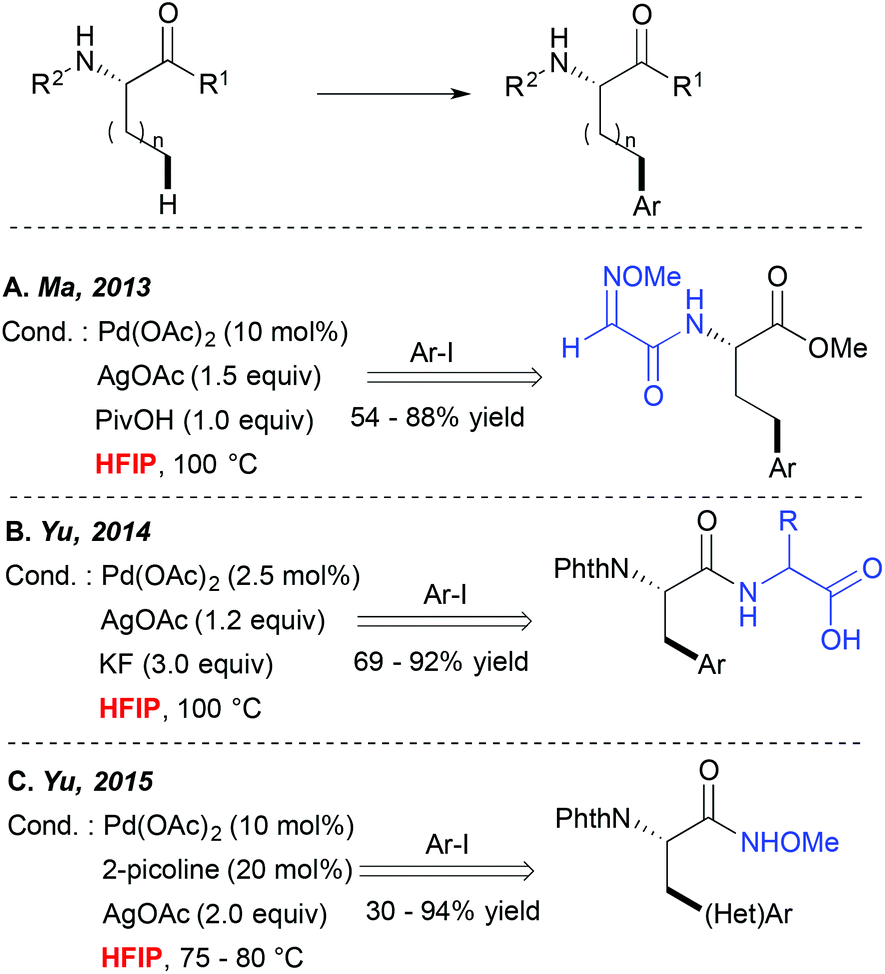 | ||
| Scheme 2 HFIP as a critical solvent for aliphatic C(sp3)–H bond activation. | ||
C–H activation using distal DG
For quite a long time the C–H activation field has been almost exclusively limited to the ortho-functionalization of the aromatic substrates bearing a coordinating group directly embedded in the aromatic ring and hence accommodating relatively stable 5-membered metallacyclic intermediates in the key C–H activation step. In contrast, the use of distal DGs and subsequent functionalization enabled by the generation of considerably larger metallacyclic intermediates represents a great synthetic challenge. Successful development of such transformations requires overcoming a high entropic barrier (due to the presence of a freely rotating sp3 carbon centre between the aromatic ring and the DG) necessary for assembling the desired transition state. Impressively, HFIP established itself as the solvent of choice to perform such distal C–H functionalizations (Fig. 1) as illustrated for the first time by Yu. The author discovered that the fluorinated alcohols (HFIP and TFE) are unique solvents supporting ortho-olefination of arenes bearing a distal, weakly coordinating ether DG.19 This challenging transformation totally failed when performed in other standard media like tert-amyl alcohol and DCE. Superior reactivity was observed in HFIP, permitting the isolation of the desired product in very high yields (even if a non-negligible amount of bifunctionalized products was generated). Importantly, further work of this research group revealed that other distal, weakly binding DGs such as Weinreb amides, esters and ketones may also be efficiently employed in combination with the HFIP solvent permitting direct C–C and C–O couplings.20 This solvent choice provoked also a drastic change of the reaction outcome when arylation of a mandelic acid was targeted; either any reactivity or only a trace amount of the desired product was generated in DCE, MeCN, toluene, DMF, AcOH and tert-amyl alcohol, whereas the use of HFIP, under otherwise identical reaction conditions, yielded the targeted scaffolds.21 Intriguingly, the authors noticed that the choice of the HFIP solvent is especially valuable if the targeted reaction occurs via a Pd(II)/Pd(IV) manifold (arylation, iodination, acetoxylation), whereas the oxidative Heck reaction following a Pd(II)/Pd(0) protocol works well in tert-amyl alcohol. In addition, HFIP stood out as the optimal medium when a sulfoxide DG is installed three carbons away from the aromatic ring, like in the case of phenylpropylsulfoxide.22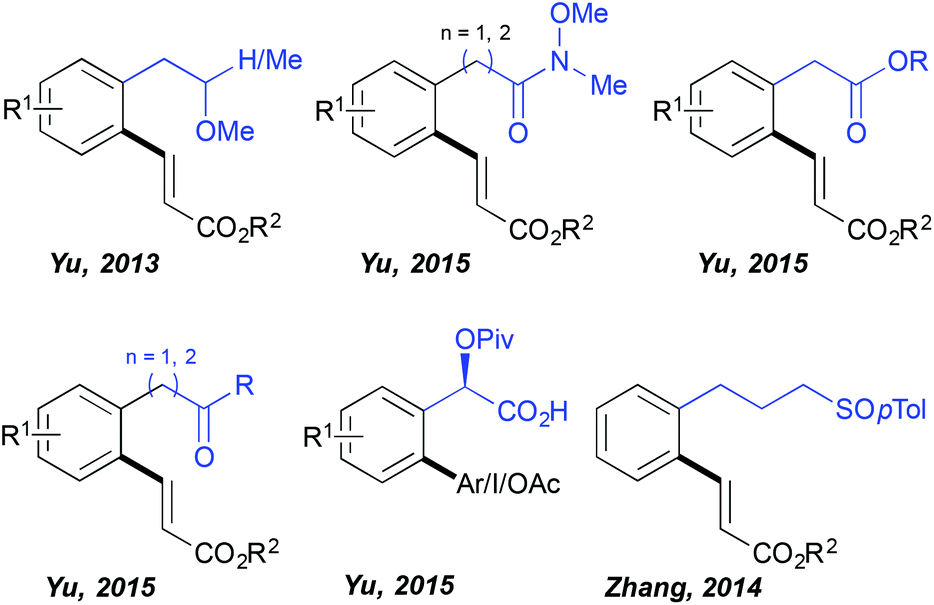 | ||
| Fig. 1 HFIP allowed direct C–H activation in the presence of a distal DG. | ||
Remote C–H activation
Expanding the scope of the C–H activation reactions beyond a proximity-driven reactivity (ie. ortho-functionalization) remained, until the beginning of this decade, an unsolved problem. Several different approaches have been imagined to achieve the meta-selective transformations4 and amongst them the design of an “end-on” template allowing the formation of larger metallacyclic intermediates and subsequent functionalization on distal positions has attracted particular attention (Fig. 2). In their seminal work, Yu et al. reported that a fine design of a removable template bearing a weakly coordinating nitrile group is the key to successfully deliver a metal catalyst to the vicinity of a tethered arene substrate, hence promoting the direct olefination of the meta-C–H bond.23 Rewardingly, when the template T1, considered as U-shaped tethered, was employed, a desired reactivity could be achieved with a high site-selectivity. The solvent selection had a major impact on both selectivity and efficiency of this reaction with HFIP outcompeting other media. Thereafter, the use of this T1 nitrile template paved also the way to the meta-arylation and alkylation of the aromatic substrates (using boronic acid derivatives).24 Following these seminal studies, several different “end-on” tethers (T2–T4) were designed to promote the remote functionalization of various synthetically useful cores such as aniline,25 phenol,26 phenyl acetic acid27 and benzylic alcohol28 derivatives as well as heterocyclic compounds like tetrahydroquinolines25 and indolines.29 Accordingly, the key factors governing the geometry of the metallacyclic intermediates and the resulting site-selectivity of the overall transformations could be identified. Remarkably, HFIP is still superior to other solvents. For instance only a trace amount of the desired product was observed when acetoxylation of the aniline-derivatives was conducted in DCE, tert-amyl alcohol or acetic acid.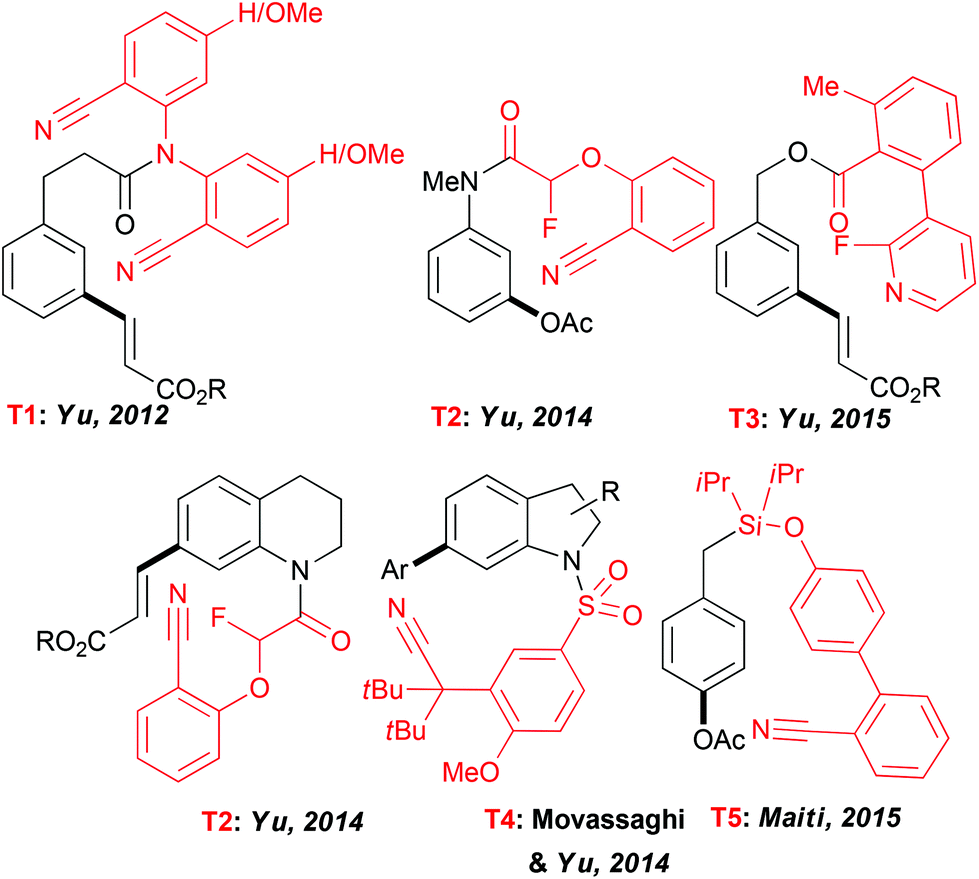 | ||
| Fig. 2 Examples of molecular scaffolds accessible via remote C–H activation occurring in HFIP. | ||
A further key achievement in the remote C–H activation was disclosed by Maiti who succeeded in designing a D-shaped template T5 directing C–H activation to the para-position.30 As reported previously, HFIP is crucial to ensure an excellent site-selectivity. Notwithstanding the lack of theoretical studies to elucidate the particular role of the fluorinated solvents in these exciting transformations, HFIP is believed to coordinate to the Pd catalyst, hence accommodating a desired pre-transition state and to enhance stabilisation of macrocyclic intermediates.
Besides, coordinative interactions between this fluorinated alcohol and Pd-catalyst could also account for an enhanced palladation step. Finally, formation of H-bonds between HFIP and the “end-on” nitrile moiety can also be reasonably expected, beneficially impacting both reactivity and selectivity of the overall transformation.
Aiming at a balanced presentation of the above-mentioned research axis, it is important to note that in some cases DCE may also be employed as a reaction medium, like in the case of meta-functionalization of benzylic ethers.31 Moreover, Tan devised a Si-tethered template attached to benzyl ethers and the corresponding remote meta-selective C–H activation occurs in DCE although addition of HFIP as an additive boosts the efficiency of this reaction and slightly improves its selectivity.32
Mild C–H activation
Achieving direct functionalization reactions under mild reaction conditions and in the absence of external oxidants offers the promise of designing truly synthetically useful transformations. Impressively, as illustrated very recently by Kuninobu and Kanai,33 (Scheme 3A) the choice of fluorinated alcohol as a medium may again be a privileged solution. In this case, Pd-catalyzed C–H activation, followed by coupling with oxiranes, afforded alkylated compounds under surprisingly mild reaction conditions and at room temperature. In this particular case, HFIP is expected to increase the stability of the oxirane coupling partner. In parallel, Zhao and Su34 discovered that picking the HFIP solvent is essential to perform high-yielding direct arylation of benzoic acid derivatives occurring at ambient temperature (Scheme 3B).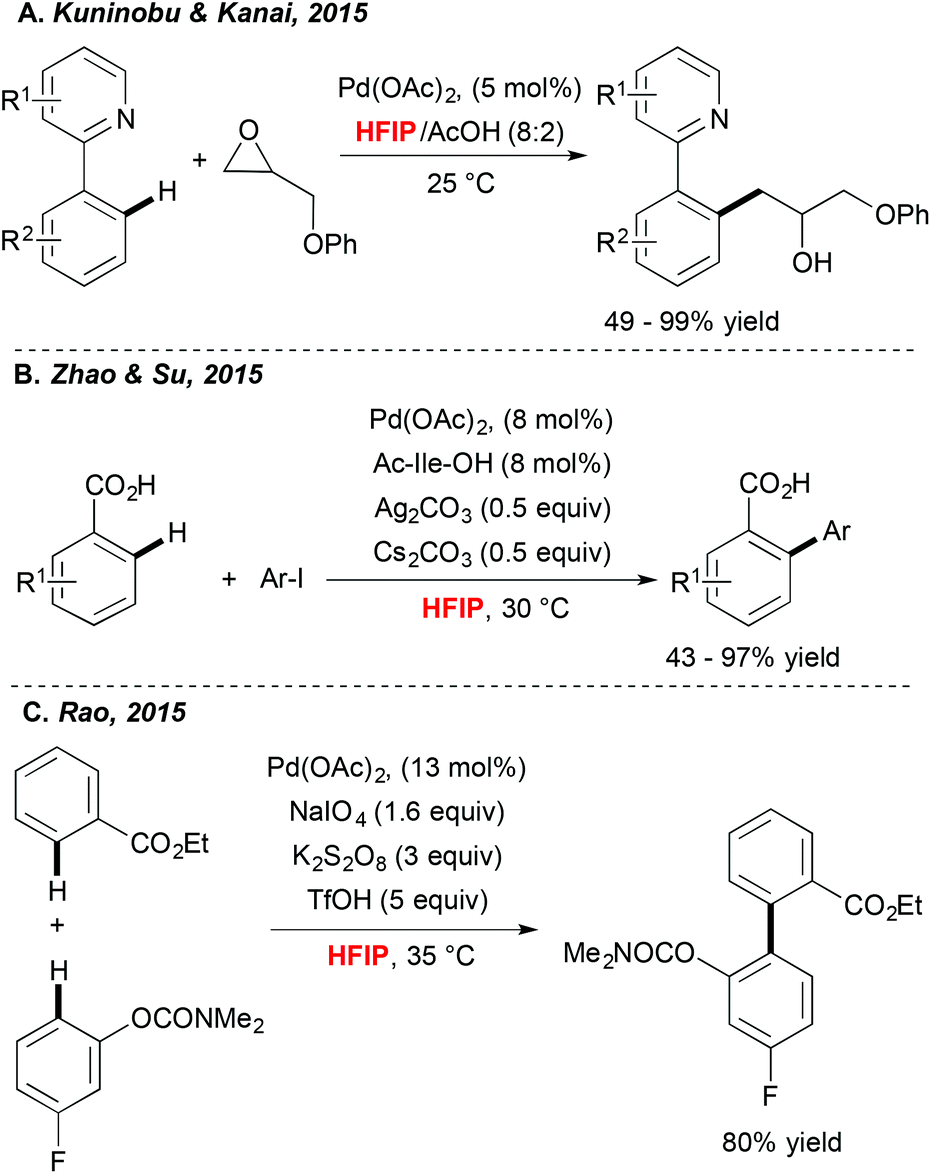 | ||
| Scheme 3 Mild Pd-catalyzed C–H activation occurring in HFIP. | ||
Another example of mild Pd-catalyzed and HFIP-mediated C–H activation has been disclosed very recently by Rao (Scheme 3C).35 The authors targeted the challenging two-fold C–H functionalization allowing for coupling of two aromatics bearing weakly coordinating DGs. Their extensive experimental work was rewarded by determination of an optimal catalytic system. Impressively, a choice of both an oxidant and a solvent, was a critical factor. A test reaction, i.e. homocoupling of benzophenone, was totally inefficient in DMSO, DMF and EtOH; moderate conversions were observed in DCE and TFE whereas the desired product was isolated in 83% yield when the reaction was performed in HFIP. Taking into consideration the choice of weakly coordinating DGs, the authors surmised that HFIP serves as a suitable ligand for palladium, stabilizing for example its high-oxidation state intermediates.
In addition to these Pd-catalyzed mild C–H activation reactions, Daugulis astutely employed TFE as a unique medium to perform cobalt-catalyzed olefination36 and carbonylation37 of aminoquinoline derivatives (Scheme 4). The superiority of the fluorinated alcohol over other solvents was likely attributed to increased solubility of the metal catalyst in this medium.
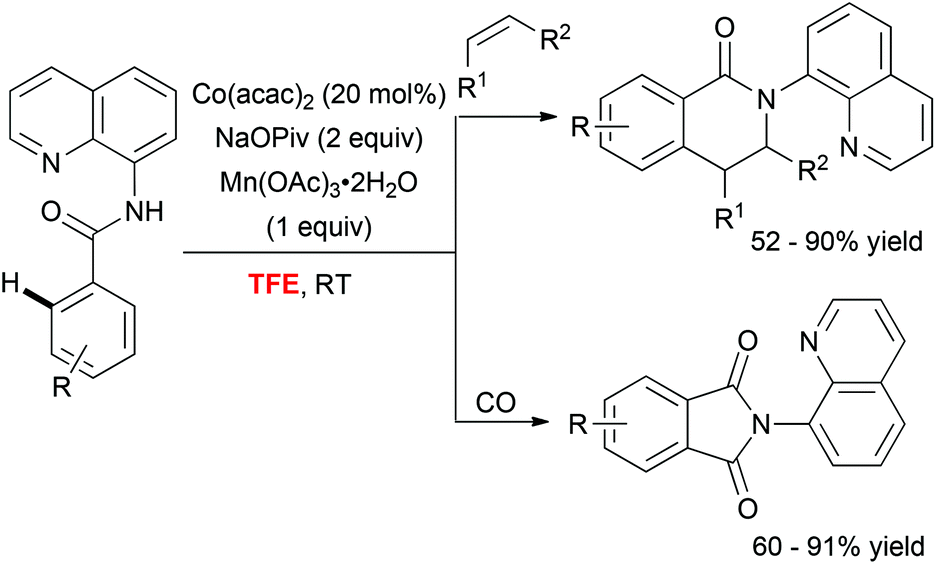 | ||
| Scheme 4 Cobalt catalysed mild C–H activation. | ||
Besides its compatibility with Pd and Co, Chang discovered recently that fluorinated alcohols are also a promising medium for Ir-catalyzed C–H transformation as illustrated by mild arylation of amide using aryldiazonium salts as an arylating agent.38 In this particular case, the lower Lewis basicity of this fluorinated alcohol compared to other conventional alcoholic solvents probably disfavours the coordination of the Ir-catalyst by the solvent, yet allowing for efficient binding of the diazonium coupling partner to the metal atom, expediting the overall transformation.
Notably, sporadic examples for the use of either HFIP or TFE solvents for Ru39 and Rh40 based transformations were also disclosed.
Stereoselective C–H activation
Taking into consideration the unique role of HFIP for both, the C–H activation step, which requires geometrically well-defined metallacyclic intermediates and mild transformations, the choice of this solvent to perform stereoselective transformations appears particularly attractive. This hypothesis is unambiguously confirmed by considering sulfoxide-directed atropodiastereoselective direct functionalization (Scheme 5).41 When biaryl substrates bearing a sulfoxide moiety as both DG and the chiral auxiliary were subjected to direct acetoxylation, iodination and olefination reactions, the use of this fluorinated solvent allowed not only to perform the desired transformations under very mild reaction conditions, but also to efficiently control the chiral induction during the overall transformation. Accordingly, the targeted, tri- and tetra-substituted biaryls were generated with excellent atroposelectivity. Notably, the mechanistic studies41b firstly proved a unique reactivity of this catalytic system in HFIP (use of other solvents such as DCE, AcOH, CHCl3 and iPrOH resulted in a reactivity shut down). Secondly, the efficiency and stereoselectivity enhancement observed in HFIP could be attributed to solvent-induced activation of the substrate by means of H-bond formation between the sulfoxide moiety and HFIP. Noteworthy, a closely related stereoselective transformation using biaryl substrates bearing a stereogenic phosphate DG has been recently reported and, once more, fluorinated solvent (TFE) was selected as an optimal medium.42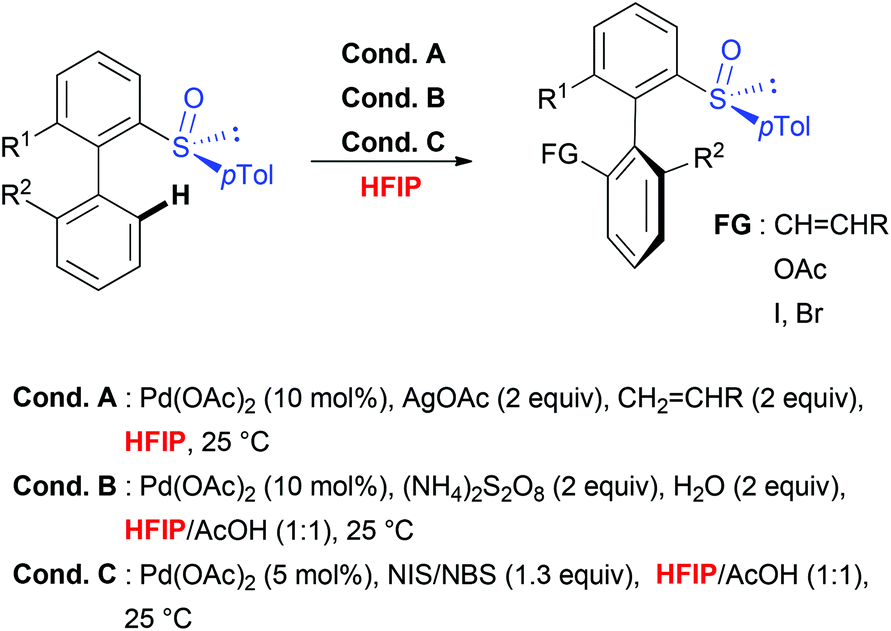 | ||
| Scheme 5 HFIP induced atropostereoselective C–H activation. | ||
Conclusions
Functionalization of the challenging C(sp3)–H bonds, remote and distal site-selectivity, high reactivity under surprisingly mild reaction conditions and excellent stereoselectivity are highly desirable features of the challenging C–H activation reactions which can be accomplished via numerous catalytic systems by employing fluorinated solvents such as HFIP and TFE. Although the particular mode of action of this medium is multiple and ambiguous, several hypotheses may be advanced. Firstly, thanks to the excellent H-bond donor ability of the fluorinated alcohols, these solvents are particularly suited to interact with DGs of the substrates yet accommodating their coordinating properties. Secondly, the coordinating character of this medium could facilitate controlled assembly of well-defined pretransition states, hence enhancing optimal site- and stereoselectivity. On the other hand, this solvent is also prompt to act as an ancillary ligand to coordinate Pd-species and for instance, stabilization of the electrophilic catalytic species in HFIP and TFE could also account for an improved overall efficiency.Nevertheless, although the benefits that may be expected when using HFIP, several disadvantages need to be taken into consideration. The volatility of HFIP (bp = 59 °C) renders it unsuitable for high temperature reactions and its elevated price may be prohibitive for its large-scale application (distillation of HFIP from a crude reaction mixture could however be considered). In this context, the use of HFIP and TFE rather as additives and activators than as solvents seems particularly tempting. Nonetheless, the unique potential of these solvents in the C–H activation field makes them a highly attractive tool to design unprecedented transformations and many exciting discoveries will certainly be reported in the near future.
Acknowledgements
We thank the CNRS (Centre National de la Recherche Scientifique), the “Ministere de l'Education Nationale et de la Recherche”, and ANR (Agence Nationale de la Recherche) France for financial support.Notes and references
- For selected general reviews see: (a) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (b) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS; (c) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed; (d) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (e) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (f) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (g) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (h) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (i) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (j) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- For selected reviews concerning application of the C–H activation see: (a) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (d) D. Y.-K. Chen and S. W. Youn, Chem. – Eur. J., 2012, 18, 9452 CrossRef CAS PubMed; (e) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- For selected reviews concerning weakly coordinating DG see: (a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (b) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef CAS.
- For selected reviews concerning distal C–H activation see: (a) R. Sharma, K. Thakur, R. Kumar, I. Kumar and U. Sharma, Catal. Rev.: Sci. Eng., 2015, 57, 345 CrossRef CAS; (b) J. Yang, Org. Biomol. Chem., 2015, 13, 1930 RSC; (c) I. Juliá-Hernández, M. Simonetti and I. Larossa, Angew. Chem., Int. Ed., 2013, 52, 11458 CrossRef PubMed.
- For selected reviews concerning C(sp3)–H activation see: (a) W. Zhang, N.-X. Wang and Y. Xing, Synlett, 2015, 26, 2088 CrossRef CAS; (b) R. K. Rit, M. R. Yadav, K. Ghosh and A. K. Sahoo, Tetrahedron, 2015, 71, 4450 CrossRef CAS; (c) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (d) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169 RSC.
- For recent reviews on asymmetric C–H activation see: (a) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC; (b) J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2013, 19, 14010 CrossRef CAS PubMed; (c) N. Cramer, Chimia, 2012, 66, 869 CrossRef CAS PubMed; (d) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (e) J. Wencel-Delord and F. Colobert, Synlett, 2015, 26, 2644 CrossRef CAS.
- For selected reviews see: (a) J.-P. Bégué, D. Bonnet-Delpon and B. Crousse, Synlett, 2004, 1, 18 Search PubMed; (b) I. A. Shuklov, N. V. Dubrovina and A. Börner, Synthesis, 2007, 19, 2925 CrossRef; (c) T. Sugiishi, M. Matsugi, H. Hamamoto and H. Amii, RSC Adv., 2015, 5, 17269 RSC.
- (a) G. Cai, Y. Fu, Y. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 7666 CrossRef CAS PubMed. For related work see: (b) Z. Liang, R. Feng, H. Yin and Y. Zhang, Org. Lett., 2013, 15, 4544 CrossRef CAS PubMed.
- W. R. Gutekunst and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 19076 CrossRef CAS PubMed.
- For selected examples see: (a) X. Chen, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 12634 CrossRef CAS PubMed; (b) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS PubMed; (c) D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190 CrossRef CAS PubMed; (d) M. Wasa, K. M. Engle, D. W. Lin, E. J. Yoo and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19598 CrossRef CAS PubMed; (e) K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Nat. Chem., 2014, 6, 146 CrossRef CAS PubMed.
- For an example of C(sp3)–H functionalization inefficient in HFIP see: K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138 CrossRef CAS PubMed.
- N. Rodríguez, J. A. Romero-Revilla, M. Á. Fernández-Ibáñez and J. C. Carretero, Chem. Sci., 2013, 4, 175 RSC.
- M. Fan and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12152 CrossRef CAS PubMed.
- W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940 CrossRef CAS PubMed.
- G. Chen, T. Shigenari, P. Jain, Z. Zhang, Z. Jin, J. He, S. Li, C. Mapelli, M. M. Miller, M. A. Poss, P. M. Scola, K.-S. Yeung and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 3338 CrossRef CAS PubMed.
- J. Peng, C. Chen and C. Xi, Chem. Sci., 2015 10.1039/C5SC03903G.
- For selected examples see: (a) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188 CrossRef CAS PubMed; (b) Q. Zhang, K. Chen, W. Rao, Y. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588 CrossRef CAS PubMed; (c) K. Chen, F. Hu, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 3906 RSC; (d) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135 CrossRef CAS PubMed.
- For an example of a related transformation totally ineffective in the HFIP medium see: K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950 CrossRef CAS PubMed.
- G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245 CrossRef CAS PubMed.
- G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391 CrossRef CAS PubMed.
- N. Dastbaravardeh, T. Toba, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 9877 CrossRef CAS PubMed.
- B. Wang, C. Shen, J. Yao, H. Yin and Y. Zhang, Org. Lett., 2014, 16, 46 CrossRef CAS PubMed.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518 Search PubMed.
- L. Wan, N. Dastbaravardeh, G. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 18056 CrossRef CAS PubMed.
- R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215 CrossRef CAS PubMed.
- H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567 CrossRef CAS PubMed.
- (a) Y. Deng and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 888 CrossRef CAS PubMed; (b) M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760 CrossRef CAS PubMed.
- L. Chu, M. Shang, K. Tanaka, Q. Chen, N. Pissarnitski, E. Streckfuss and J.-Q. Yu, ACS Cent. Sci., 2015, 1, 394 CrossRef CAS.
- G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R.-Y. Tang, M. Movassaghi and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807 CrossRef CAS PubMed.
- S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888 CrossRef CAS PubMed.
- See ref. 23; the use of HFIP during the optimization study of meta-functionalization of benzyl ethers is not mentioned.
- S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778 CrossRef CAS PubMed.
- (a) Z. Wang, Y. Kuninobu and M. Kanai, J. Am. Chem. Soc., 2015, 137, 6140 CrossRef CAS PubMed. For closely related work see: (b) G. Chen, T.-J. Li and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 10950 CrossRef PubMed.
- C. Zhu, Y. Zhang, J. Kan, H. Zhao and W. Su, Org. Lett., 2015, 17, 3418 CrossRef CAS PubMed.
- C. Zhang and Y. Rao, Org. Lett., 2015, 17, 4456 CrossRef CAS PubMed.
- L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4684 CrossRef CAS PubMed . For the early work see: L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209 CrossRef PubMed.
- L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4688 CrossRef CAS PubMed.
- K. Shin, S.-W. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 8584 CrossRef CAS PubMed.
- For an example see: B. Li, J. Ma, N. Wang, H. Feng, S. Xu and B. Wang, Org. Lett., 2012, 14, 736 CrossRef CAS PubMed.
- For an example see: J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735 CrossRef CAS PubMed.
- (a) C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871 CrossRef CAS PubMed; (b) Q. Dherbassy, G. Schwertz, M. Chessé, C. K. Hazra, J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2016, 22, 1735 CrossRef CAS PubMed; For an early example see: (c) T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139 CrossRef CAS.
- Y.-N. Ma, H.-Y. Zhang and S.-D. Yang, Org. Lett., 2015, 17, 2034 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2016 |