
Studies towards the synthesis of the functionalized C3–C14 decalin framework of alchivemycin A†
Kaiqing
Ma‡
ab,
Daohong
Liao‡
bc,
Shaoqiang
Yang
b,
Xiaofei
Li
c and
Xiaoguang
Lei
*bc
aModern Research Center for Traditional Chinese Medicine of Shanxi University, No. 92, Wucheng Road, Taiyuan 030006, Shanxi, China
bBeijing National Laboratory for Molecular Sciences, Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, Department of Chemical Biology, College of Chemistry and Molecular Engineering, Synthetic and Functional Biomolecules Center, and Peking-Tsinghua Center for Life Sciences, Peking University, Beijing 100871, China. E-mail: xglei@pku.edu.cn
cNational Institute of Biological Sciences (NIBS), Beijing 102206, China
First published on 21st December 2015
Abstract
We report our synthetic studies towards the synthesis of the C3–C14 fragment of alchivemycin A. The synthesis featured an asymmetric alkylation with excellent diastereoselectivity and a one-pot Julia–Kocienski olefination with excellent E-selectivity. An intramolecular Diels–Alder reaction was employed to construct the highly functionalized cis-decalin framework. Interestingly, the stereochemical outcome was not expected to generate the two stereoisomers 20 and 21 instead of the desired cis-decalin 5. The detailed mechanism of this transformation was discussed. These synthetic endeavors have offered us a number of crucial insights for the synthesis of the complex natural product alchivemycin A.
 Xiaoguang Lei | Prof. Dr Xiaoguang Lei was born and brought up in Beijing, China. He did his undergraduate studies at Peking University, and received a BSc degree in chemistry in 2001. He then moved to Boston and conducted his PhD research work from 2001 to 2006 under the supervision of Prof. John A. Porco at Boston University. After spending two years (2006–2008) as a postdoctoral fellow in the group of Prof. Samuel J. Danishefsky at Columbia University, Dr Lei joined the faculty of the National Institute of Biological Sciences (NIBS) as a Principal Investigator and Director of the Chemistry Center. He also held a joint professorship at Tianjin University. In early 2014, Dr Lei moved to Peking University and joined the faculty of the College of Chemistry and Molecular Engineering. His research program focuses on natural product total synthesis and chemical biology. |
Introduction
Alchivemycins A (1) and B (2) are two novel 17-membered polycyclic polyketides recently isolated from the culture extract of a plant-derived actinomycete Streptomyces sp. by Igarashi and co-workers in 20101 and 2013,2 respectively (Fig. 1). Alchivemycin A (1) shows promising biological activities with potent antimicrobial activity against Gram-positive Micrococcus luteus (MIC = 50 nM) and remarkable inhibitory effects against tumor cell invasion (IC50 = 0.34 μM) without showing cytotoxic effects. The structure and relative stereochemistry of alchivemycin A (1) were determined by spectroscopic analysis and X-ray crystallography.1 Altogether, the potent biological activity, combined with the unique structure containing an unprecedented 2H-tetrahydro-4,6-dioxo-1,2-oxazine heterocyclic ring as well as a highly functionalized cis-decalin (C3–C14 fragment), renders it an ideal target for total synthesis.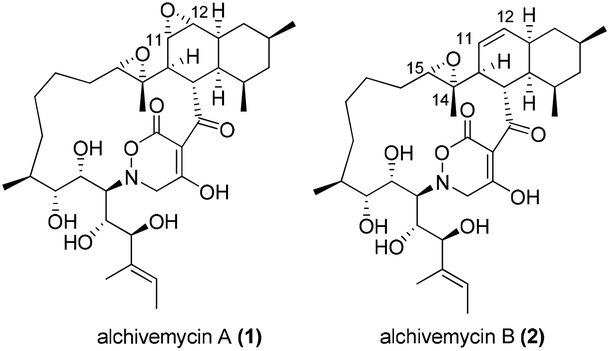 | ||
| Fig. 1 Structures of alchivemycins A (1) and B (2). | ||
To date, no total synthesis of alchivemycin A (1) or related synthetic studies towards its major fragment have been reported in the literature owing to the significant synthetic challenges underlying its complex structure.3 Herein, we disclose our synthetic endeavors towards the synthesis of alchivemycin A, focusing on the construction of the C3–C14 fragment containing the highly functionalized cis-decalin.
Results and discussion
From a synthetic perspective, alchivemycin A shows a considerable degree of structural complexity. Fifteen stereocenters are embedded within a 17-membered macrocycle featuring a novel 2H-tetrahydro-4,6-dioxo-1,2-oxazine heterocyclic ring and a highly functionalized cis-decalin framework. For maximal synthetic convergency and stereocontrol, we envisioned that alchivemycin A (1) could be forged from the C3–C14 ketone 4 and the C15–C30 phosphonate 3 (Fig. 2) through Horner–Wadsworth–Emmons (H–W–E)4 olefination and Dieckmann condensation.5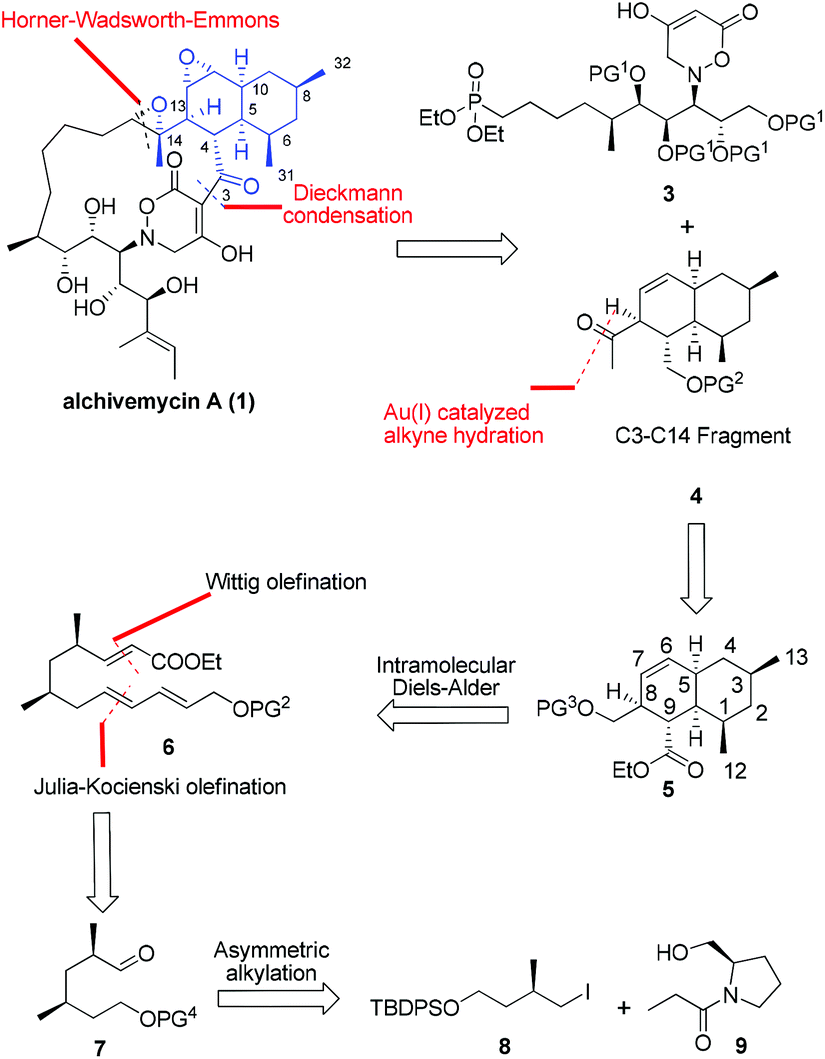 | ||
| Fig. 2 Retrosynthetic analysis of alchivemycin A. | ||
The proposed major C3–C14 fragment could be generated from compound 5 through a gold catalyzed alkyne hydration.6 Initially, a transannular Diels–Alder (TADA) strategy was considered for the synthesis of the cis-decalin system, however, this approach was not pursued because it unfortunately provided an undesired diastereomer containing the C8 and C9 syn-substituted motif on the decalin ring in compound 5.7 We thus decided to install the cis-decalin through an intramolecular Diels–Alder (IMDA) reaction8 from a linear precursor such as 6, favoring the exo-selective cycloaddition. The desired D–A substrate was to be assembled through one-pot Wittig and Julia–Kocienski (J–K) olefinations of 2,4-syn-dimethyl aldehyde 7,9 which in turn was planned to arise from the asymmetric alkylation of the known compound 9.
The synthesis began by recognizing that the 1,3-cis-dimethyl 13 (a potential precursor to 7) could be accessed from the diastereoselective methylation and subsequent reduction of compound 11, using Evans’ oxazolidinone auxiliary as the chiral source (Scheme 1). The amide 11 was prepared according to a literature procedure from the known acid 10.10 Unfortunately, under various conditions, the substrate modified with different chiral oxazolidinones and treated with methyl iodide failed to afford any useful level of diastereoselectivity, based on 1H-NMR analysis of the inseparable mixture.
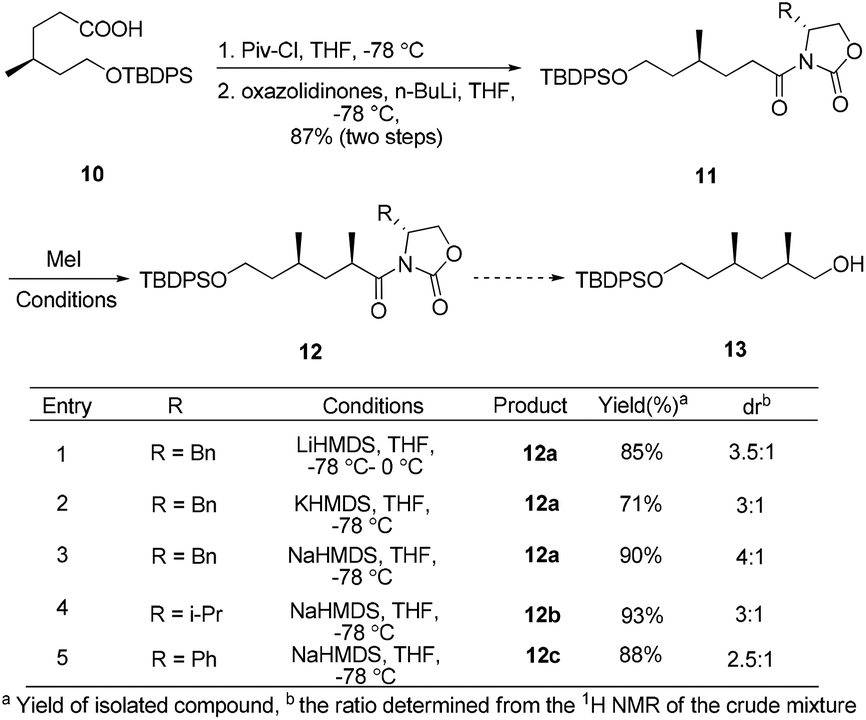 | ||
| Scheme 1 Attempted synthesis of the intermediate 13. | ||
In view of these results, a revised strategy was required for the stereocontrolled synthesis. Accordingly, a new approach was envisaged using the known iodide 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 and the L-prolinol N-propionamide 9,12 which were derived from (R)-methylsuccinic acid and L-prolinol, respectively. With 8 and 9 in hand, asymmetric alkylation was then re-investigated. To our delight, the reaction proceeded smoothly using Evans’ protocol13 to afford the desired product 14 in good yield and excellent diastereoselectivity (91:9 ratio of 2R- and 2S-diastereomers as determined by 1H-NMR) (Scheme 2). Conversion of the chiral auxiliary to the corresponding alcohol was then required. Excess LiEt3BH12 in THF led to epimerization of the substrate (5:1 dr). However, the chiral substituent was smoothly removed by reduction using LiH2NBH3 in THF at room temperature14 to provide the alcohol 13 without epimerization. Compound 13 was further subjected to a Swern oxidation/Wittig reaction sequence to afford the α,β-conjugated ester 15 in excellent yield (83%, over two steps).
11 and the L-prolinol N-propionamide 9,12 which were derived from (R)-methylsuccinic acid and L-prolinol, respectively. With 8 and 9 in hand, asymmetric alkylation was then re-investigated. To our delight, the reaction proceeded smoothly using Evans’ protocol13 to afford the desired product 14 in good yield and excellent diastereoselectivity (91:9 ratio of 2R- and 2S-diastereomers as determined by 1H-NMR) (Scheme 2). Conversion of the chiral auxiliary to the corresponding alcohol was then required. Excess LiEt3BH12 in THF led to epimerization of the substrate (5:1 dr). However, the chiral substituent was smoothly removed by reduction using LiH2NBH3 in THF at room temperature14 to provide the alcohol 13 without epimerization. Compound 13 was further subjected to a Swern oxidation/Wittig reaction sequence to afford the α,β-conjugated ester 15 in excellent yield (83%, over two steps).
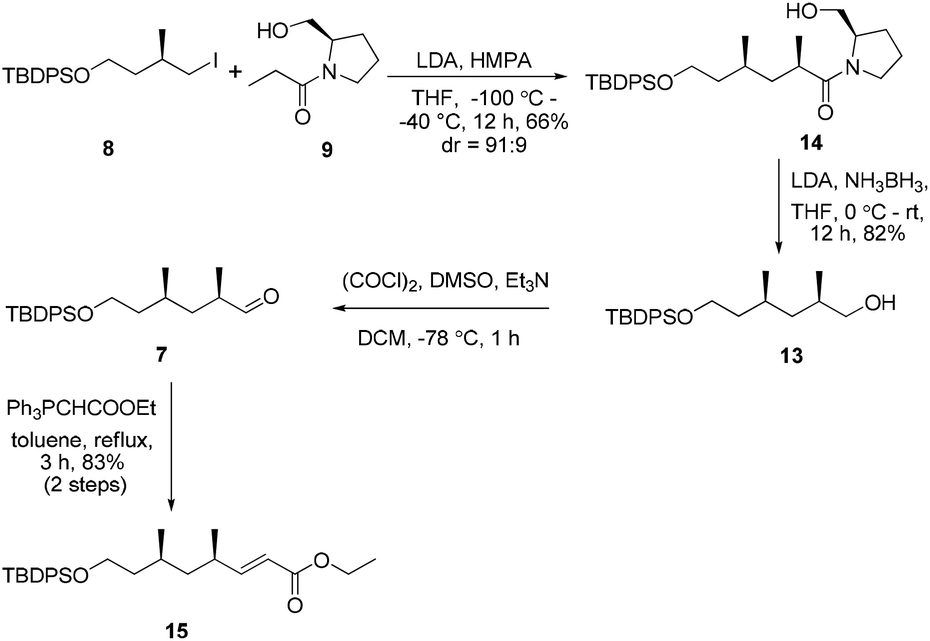 | ||
| Scheme 2 Synthesis of the intermediate 15. | ||
Our initial attempt at the synthesis of the Diels–Alder precursor began with ester 15. Desilylation of 15 with triethylamine and subsequent oxidation with Dess–Martin periodinane set the stage for the J–K olefination.15 Treatment of the aldehyde A with allylic sulfone B afforded a 1.5:1 inseparable mixture of E/Z isomers of C using the Barbier-type J–K protocol. The use of a more polar solvent such as DME led to the formation of an improved 3:1 mixture (Scheme 3).
 | ||
| Scheme 3 Attempt to synthesize the Diels–Alder precursor. | ||
Considering that the allylic sulfone B and the α-nonbranched aldehyde A may not be the optimal substrates for the E-selective olefination,16 we attempted the reaction with an alternative substrate pair:17 the known conjugated aldehyde 1918 and heteroarylsulfone 18 (PT = phenyltetrazolyl). The sulfone 18 was prepared from the primary alcohol 16 employing a sequence of Mitsunobu thioetherification and molybdenum-catalyzed sulfide oxidation. Gratifyingly, the coupling of 18 and 19 through one-pot J–K olefination furnished the desired (E/E/E)-triene substrate 6 in good yield and with excellent E/Z-selectivity (93:7) using the Barbier-type J–K protocol (Scheme 4).
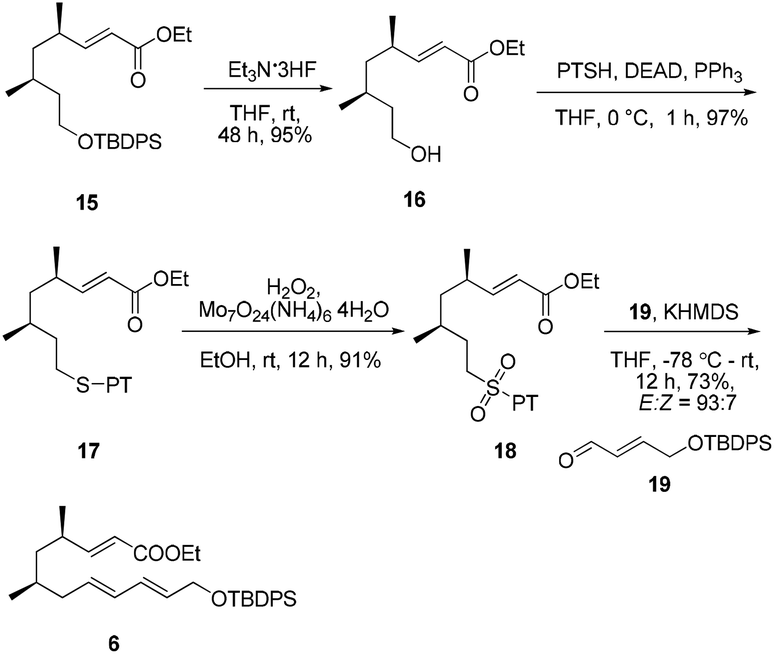 | ||
| Scheme 4 Synthesis of the Diels–Alder precursor 6. | ||
With triene 6 in hand, we began to further investigate the crucial IMDA reaction. The ester 6 underwent thermally promoted IMDA cycloaddition in toluene to afford mixtures of the adducts which could not be directly separated by chromatography, but they could be separated chromatographically upon desilylation with triethylamine trihydrofluoride to afford the lactone and the primary alcohol in 30% and 45% yield over two steps (Scheme 5). The lactones were identified as trans-fused bicyclic product 20 based on the cross peaks between H12–H9 and –H5 in the NOE experiments. The NOE experiment performed on another isomer revealed the cross peaks between H12–H9 and –H11 supporting the cis-fused primary alcohol 21 as the exo adduct, while the desired cis-fused decalin 5 required for alchivemycin A was not observed. From the IMDA reaction of triene 6, we expected four distinct decalin diastereomers from four possible transition states (Scheme 6). The cycloadducts 20 and 21 were generated through the equatorial state as the endo and exo products, respectively. The desired isomer 5 would form from the axial state with high energy which creates a severe 1,3-diaxial methyl interaction. The observed stereochemical outcome was consistent with Lee's findings.19 We envisioned that an organocatalytic process or an elevated reaction temperature could overcome the inherent substrate selectivity of this structure and favor the adduct 5 which was in the structure of alchivemycin A. The conditions of the IMDA reaction were extensively evaluated (Scheme 6). Treatment of ester 6 in dichlorobenzene at 220 °C affords 1:1.5 mixtures of compounds 23 and 24 (Scheme 6, entry 1). When the reaction solution was heated in trichlorobenzene at 240 °C,20 the ratio did not further improve, and in fact some decomposition of the substrate was observed (data not shown). The ethyl ester decomposed when the reaction was heated in DMSO (Scheme 6, entry 2). Bulky Lewis acids21 like aluminum tris(2,6-diphenylphenoxide) (ATPH) and chiral Lewis acids22 such as cationic oxazaborinane, known to promote the exo-selective D–A reaction, were also evaluated but led only to a 1:1 ratio of compounds 23 and 24 or compound 23 (Scheme 6, entries 3 and 4, respectively). The D–A reaction under neat conditions afforded the undesired endo adduct compound 23 as the sole product (Scheme 6, entry 5). Unfortunately, elevated reaction temperature or using a Lewis acid could not afford the desire adduct 5 based on the 1H-NMR of the crude mixture which was presumably due to the high energy of the transition state when both the methyls are at the axial position. In light of these results, a revised strategy was required to construct the cis-decalin 5. Based on the study by the Roush group,8 the Z dienophile facilitates the formation of the cis-decalin. It is a good strategy to synthesize the 9-epi-5 followed by epimerization under basic conditions to provide compound 5. However, in Lee's doctoral dissertation,19 they have changed the structure of 6 to (E,E,Z)-triene and examined the IMDA reaction. Unfortunately they observed that the reaction only afforded the undesired trans-decalin rather than the 9-epi-5. Eventually, the chiral auxiliary approach was proposed in which the Evans oxazolidinone was employed as a chiral auxiliary to obtain the desired exo-axial cycloadducts23 and the preparation of the precursor of the IMDA with the chiral auxiliary is ongoing.
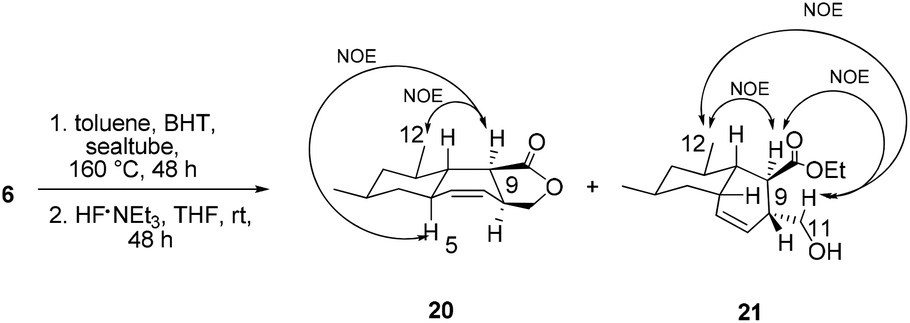 | ||
| Scheme 5 IMDA and deprotection reaction of 6. | ||
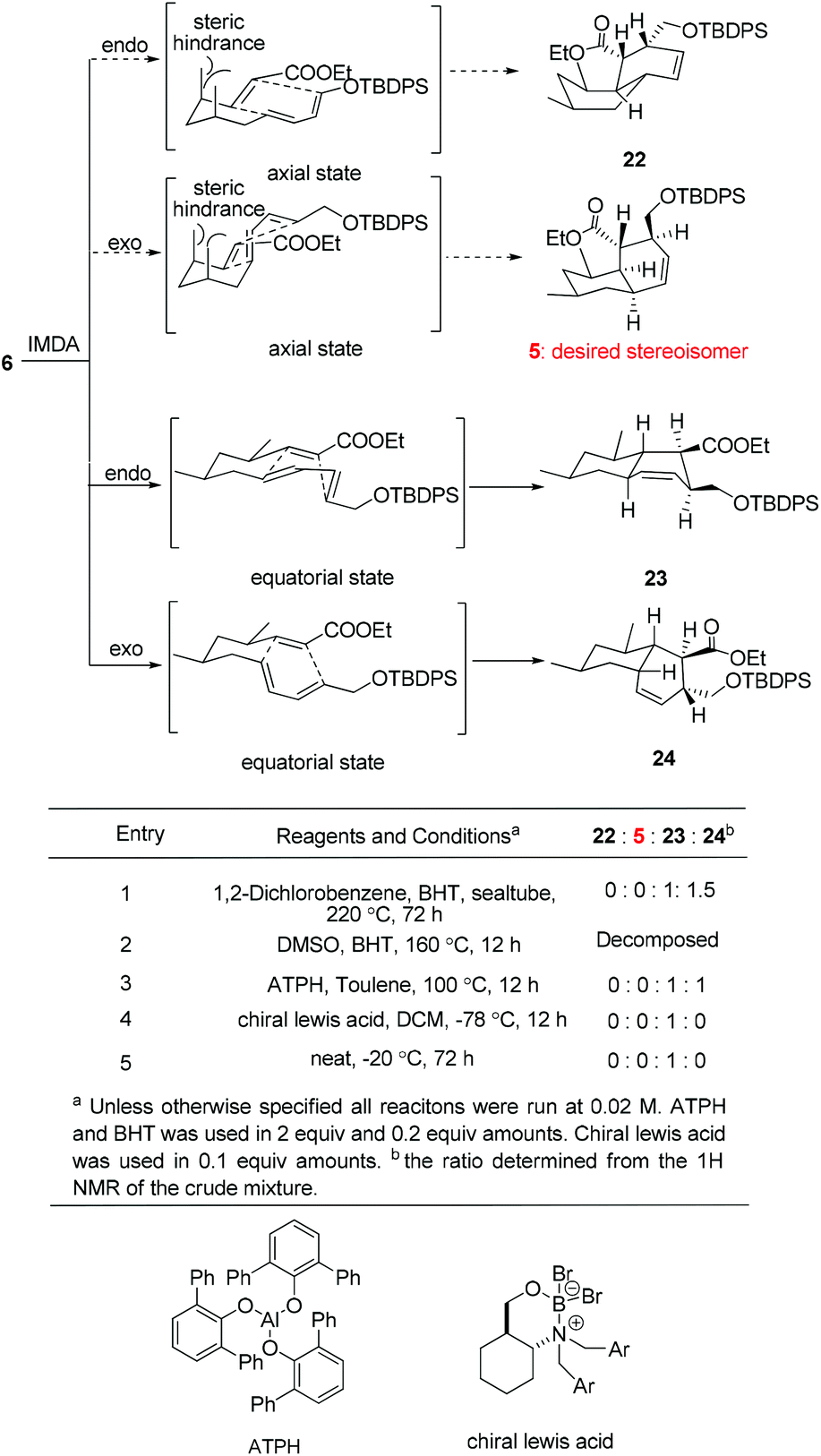 | ||
| Scheme 6 Possible transition state and condition evaluation of the IMDA reaction of 6. The terms axial and equatorial refer to the relative orientation of the methyl group at C1 and C3. | ||
Conclusions
We have reported our synthetic endeavors towards the synthesis of the C3–C14 fragment of alchivemycin A. The synthesis relies on an asymmetric alkylation and a one-pot E-selective J–K olefination. Although the decalin containing the relative stereochemistry required for alchivemycin A was not obtained under various conditions, ready access to compound 20, which is a common structural motif in the related polyketide, would facilitate synthetic endeavors toward other natural products.24 An alternative strategy to construct the cis-decalin of alchivemycin A is currently underway in our laboratory and will be reported in due course.Experimental section
General
1H NMR spectra were recorded on a Varian 400 MHz spectrometer at ambient temperature with CDCl3 as the solvent unless otherwise stated. 13C NMR spectra were recorded on a Varian 100 MHz spectrometer (with complete proton decoupling) at ambient temperature. Chemical shifts are reported in parts per million relative to chloroform (1H, δ 7.26; 13C, δ 77.00). Data for 1H NMR are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet) and coupling constants. Infrared spectra were recorded on a Thermo Fisher FT-IR200 spectrophotometer. High-resolution mass spectra were recorded at Peking University Mass Spectrometry Laboratory using a Bruker APEX Flash chromatograph. The samples were analyzed by HPLC/MS on a Waters Auto Purification LC/MS system (3100 Mass Detector, 2545 Binary Gradient Module, 2767 Sample Manager, and 2998 Photodiode Array (PDA) Detector). The system was equipped with a Waters C18 5 μm SunFire separation column (150 × 4.6 mm), equilibrated with HPLC grade water (solvent A) and HPLC grade methanol (solvent B) with a flow rate of 1.0 mL min−1 at room temperature. Analytical thin layer chromatography was performed using 0.25 mm silica gel 60-F plates. Silica gel column chromatography was performed using 200–400 mesh silica gel. Yields refer to chromatographically and spectroscopically pure materials, unless otherwise stated. All reagents were used as supplied by J&K, Sigma-Aldrich, and Alfa Aesar Chemicals. Toluene and methylene chloride were distilled from calcium hydride; tetrahydrofuran was distilled from sodium/benzophenone ketyl prior to use. All reactions were carried out in oven-dried glassware under an argon atmosphere unless otherwise noted.11 (0.20 g, 1.28 mmol) and HMPA (1.1 mL, 6.3 mmol) in anhydrous THF (1.2 mL). The resulting solution was warmed to room temperature and stirred for 1 h. The reaction mixture was cooled to −100 °C and the iodide 810b (0.38 g, 0.84 mmol) in THF (1.4 mL) was added to the solution dropwise. After being warmed to −40 °C in 1 h, the reaction mixture was stirred at this temperature for 11 h and quenched with saturated NH4Cl solution (5 mL). The resulting solution was extracted with EtOAc (20 mL × 3) and the combined extracts were washed with saturated NaCl solution (20 mL × 2), dried over anhydrous Na2SO4 and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EtOAc = 1/1) to afford 1,3-dimethyl compound 14 (0.27 g, 66%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.79–7.57 (m, 4H), 7.52–7.32 (m, 6H), 5.26 (dd, J = 7.7, 2.3 Hz, 1H), 4.19–4.25 (m, 1H), 3.78–3.61 (m, 3H), 3.60–3.51 (m, 2H), 3.43–3.50 (m, 1H), 2.63 (dd, J = 13.7, 6.8 Hz, 1H), 2.06–1.78 (m, 3H), 1.71–1.52 (m, 4H), 1.31–1.17 (m, 2H), 1.13 (d, J = 6.7 Hz, 3H), 1.04 (s, 9H), 0.83 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 178.4, 135.5, 134.0, 129.5, 127.6, 67.9, 61.9, 61.1, 47.8, 41.2, 39.3, 35.6, 28.3, 27.4, 26.8, 24.5, 20.0, 19.2, 18.0; IR (neat) νmax 3391, 1620, 1111, 739, 704 cm−1; HRMS (ESI) [M + H+] calculated for C29H44NO3Si: 482.30850, found: 482.30939; [α]20D −21.7 (c 0.7, CHCl3).
18 (0.32 g, 0.98 mmol) in THF (8 mL) was added KHMDS in toluene (0.7 M, 0.91 mL, 0.64 mmol) dropwise at −78 °C. After being stirred at this temperature for 30 min, the reaction mixture was warmed to room temperature and stirred for 12 h before being quenched by the addition of saturated NH4Cl solution at 0 °C and then warmed to room temperature. The solution was extracted with EtOAc (5 mL × 3). The combined extracts were washed with saturated NaCl solution (5 mL × 2), dried over anhydrous Na2SO4, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EtOAc = 30/1) to afford triene 6 (0.20 g, 73%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.85–7.56 (m, 4H), 7.54–7.31 (m, 6H), 6.81 (dd, J = 15.7, 8.3 Hz, 1H), 6.23 (dd, J = 15.2, 10.6 Hz, 1H), 6.02 (dd, J = 15.1, 10.7 Hz, 1H), 5.78 (d, J = 15.7 Hz, 1H), 5.71–5.53 (m, 2H), 4.23 (d, J = 5.0 Hz, 2H), 4.21–4.14 (m, 2H), 2.56–2.28 (m, 1H), 2.02–2.13 (m, 1H), 1.97–1.86 (m, 1H), 1.54–1.48 (m, 1H), 1.37–1.45 (m, 1H), 1.29 (t, J = 7.1 Hz, 3H), 1.18–1.10 (m, 1H), 1.06 (s, 9H), 1.05 (d, J = 7.2 Hz, 3H), 0.86 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.9, 154.4, 135.6, 133.7, 132.3, 131.3, 130.1, 130.0, 129.6, 127.6, 119.8, 64.3, 60.2, 43.3, 40.6, 34.3, 30.7, 29.7, 26.8, 20.4, 19.2, 19.2, 14.3; IR (neat) νmax 3071, 2959, 1721, 1652, 1113, 989, 704 cm−1; HRMS (ESI) [M + Na+] calculated for C32H44NaO3Si: 527.29519, found: 527.29622; [α]20D +11.2 (c 0.5, CHCl3).
Compound 20. 1 H NMR (400 MHz, CDCl3) δ 5.83 (dt, J = 9.3, 2.9 Hz, 1H), 5.67–5.59 (m, 1H), 4.41–4.33 (m, 1H), 4.23–4.26 (m, 1H), 2.92–2.99 (m, 1H), 2.71–2.62 (m, 1H), 1.96–1.84 (m, 1H), 1.77–1.65 (m, 2H), 1.59–1.48 (m, 1H), 1.46–1.35 (m, 1H), 1.09–1.16 (m, 1H), 1.05 (d, J = 6.6 Hz, 3H), 0.91 (d, J = 6.5 Hz, 3H), 0.90–0.78 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 179.6, 139.1, 127.7, 71.6, 45.9, 44.8, 43.8, 40.7, 39.6, 38.4, 36.7, 32.2, 22.3, 20.1; IR (neat) νmax 2965, 1756, 1441, 1203, 981 cm−1; HRMS (ESI) [M + Na+] calculated for C14H20NaO2: 243.13555, found: 243.13516; [α]20D −10.4 (c 0.9, CHCl3).
Compound 21. 1 H NMR (400 MHz, CDCl3) δ 5.54–5.64 (m, 2H), 4.30–4.02 (m, 2H), 3.69 (dd, J = 10.3, 6.5 Hz, 1H), 3.45–3.51 (m, 1H), 2.98 (t, J = 2.3 Hz, 1H), 2.71–2.79 (m, 1H), 2.52–2.26 (m, 1H), 1.75–1.64 (m, 2H), 1.62–1.44 (m, 4H), 1.25 (t, J = 7.1 Hz, 3H), 1.13–1.20 (m, 1H), 0.94 (d, J = 6.4 Hz, 3H), 0.85 (d, J = 6.5 Hz, 3H), 0.69 (q, J = 11.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 176.3, 133.1, 125.4, 65.4, 60.6, 44.4, 43.0, 41.2, 40.0, 38.7, 32.7, 30.1, 27.8, 22.5, 20.2, 14.3; IR (neat) νmax 3362, 1730, 1456, 1375, 1182, 1031 cm−1; HRMS (ESI) [M + H+] calculated for C16H27O3: 267.19547, found: 267.19504; [α]20D −8.9 (c 1.56, CHCl3).
Acknowledgements
We thank Dr Alexander Jones for helpful discussion, Ms Mingyan Zhao (NIBS) for NMR and HPLC-MS analyses, and Dr Jiang Zhou (Peking University) for HRMS analyses. We are also grateful for the financial support from the National High Technology Project 973 (2015CB856200 and 2012CB837400) and the NNSFC (21222209, 21472010, 91313303 and 21402111).References
- Y. Igarashi, Y. Kim, Y. In, T. Ishida, Y. Kan, T. Fujita, T. Iwashita, H. Tabata, H. Onaka and T. Furumai, Org. Lett., 2010, 12, 3402 CrossRef CAS PubMed.
- Y. Kim, Y. In, T. Ishida, H. Onaka and Y. Igarashi, Org. Lett., 2013, 15, 3514 CrossRef CAS PubMed.
- (a) G. Li, S. Kusari and M. Spiteller, Nat. Prod. Rep., 2014, 31, 1175 RSC; (b) S. Dhambri, S. Mohammad, O. N. Van Buu, G. Galvani, Y. Meyer, M. I. Lannou, G. Sorin and J. Ardisson, Nat. Prod. Rep., 2015, 32, 841 RSC.
- W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733 CrossRef CAS.
- B. Tse, J. Am. Chem. Soc., 1996, 118, 7094 CrossRef CAS.
- M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC.
- (a) E. Marsault, A. Toró, P. Nowak and P. Deslongchamps, Tetrahedron, 2001, 57, 4243 CrossRef CAS; (b) M. Tortosa, N. A. Yakelis and W. R. Roush, J. Am. Chem. Soc., 2008, 130, 2722 CrossRef CAS PubMed; (c) D. J. Mergott, S. A. Frank and W. R. Roush, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11955 CrossRef CAS PubMed.
- (a) W. R. Roush, J. A. Champoux and B. C. Peterson, Tetrahedron Lett., 1996, 37, 8989 CrossRef CAS; (b) T. A. Dineen and W. R. Roush, Org. Lett., 2005, 7, 1355 CrossRef CAS PubMed; (c) M. Tortosa, N. A. Yakelis and W. R. Roush, J. Org. Chem., 2008, 73, 9657 CrossRef CAS PubMed; (d) K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779 CrossRef CAS PubMed; (e) D. Craig, Chem. Soc. Rev., 1987, 16, 187 RSC; (f) M. Juhl and D. Tanner, Chem. Soc. Rev., 2009, 38, 2983 RSC.
- P. R. Blakemore, W. J. Cole, P. J. Kocieński and A. Morley, Synlett, 1998, 26 CrossRef CAS.
- (a) H. Mitome, M. Shinohara, H. Miyaoka and Y. Yamada, Chem. Pharm. Bull., 2003, 51, 640 CrossRef CAS PubMed; (b) D. A. Evans, M. Ennis and D. Mathre, J. Am. Chem. Soc., 1982, 104, 1737 CrossRef CAS; (c) J. S. Yadav, N. N. Yadav, T. S. Rao, B. V. S. Reddy and A. Ghamdi, Eur. J. Org. Chem., 2011, 4603 CrossRef CAS.
- (a) M. Lautens and T. A. Stammers, Synthesis, 2002, 1993 CrossRef CAS; (b) A. Francais, A. Leyva, G. Etxebarria-Jardi and S. V. Ley, Org. Lett., 2010, 12, 340 CrossRef CAS PubMed.
- D. A. Evans, R. L. Dow, T. L. Shih, J. M. Takacs and R. Zahler, J. Am. Chem. Soc., 1990, 112, 5290 CrossRef CAS.
- Y. Ujihara, K. Nakayama, T. Sengoku, M. Takahashi and H. Yoda, Org. Lett., 2012, 14, 5142 CrossRef CAS PubMed.
- A. G. Myers, B. H. Yang and K. J. David, Tetrahedron Lett., 1996, 37, 3623 CrossRef CAS.
- Q. Chen, D. Schweitzer, J. Kane, V. J. Davisson and P. Helquist, J. Org. Chem., 2011, 76, 5157 CrossRef CAS PubMed.
- F. Billard, R. Robiette and J. Pospisil, J. Org. Chem., 2012, 77, 6358 CrossRef CAS PubMed.
- M. Gärtner, D. Kossler, D. Pflästerer and G. Helmchen, J. Org. Chem., 2012, 77, 4491 CrossRef PubMed.
- Y. Mori, M. Asai, J.-i. Kawade and H. Furukawa, Tetrahedron, 1995, 51, 5315 CrossRef CAS.
- J. Lee, Part III. Studies towards the synthesis of alchivemycin A (Doctoral Dissertation), Stony Brook University, New York, USA, 2012 Search PubMed.
- M. Miyashita, M. Sasaki, I. Hattori, M. Sakai and K. Tanino, Science, 2004, 305, 495 CrossRef CAS PubMed.
- (a) K. Maruoka, H. Imoto and H. Yamamoto, J. Am. Chem. Soc., 1994, 116, 12115 CrossRef CAS; (b) E. L. Pearson, L. C. Kwan, C. I. Turner, G. A. Jones, A. C. Willis, M. N. Paddon-Row and M. S. Sherburn, J. Org. Chem., 2006, 71, 6099 CrossRef CAS PubMed.
- Y. Hayashi, J. J. Rohde and E. J. Corey, J. Am. Chem. Soc., 1996, 118, 5502 CrossRef CAS.
- (a) D. A. Evans and W. C. Black, J. Am. Chem. Soc., 1993, 115, 4497 CrossRef CAS; (b) D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1984, 106, 4261 CrossRef CAS; (c) D. A. Evans, K. T. Chapman and J. Bisaha, Tetrahedron Lett., 1984, 25, 4071 CrossRef CAS; (d) M. H. Lacoske, J. Xu, N. Mansour, C. Gao and E. A. Theodorakis, Org. Chem. Front., 2015, 2, 388 RSC.
- (a) S. W. Yang, R. Mierzwa, J. Terracciano, M. Patel, V. Gullo, N. Wagner, B. Baroudy, M. Puar, T. M. Chan, A. T. McPhail and M. Chu, J. Nat. Prod., 2006, 69, 1025 CrossRef CAS PubMed; (b) F. Cardoso-Martinez, J. M. de la Rosa, A. R. Diaz-Marrero, J. Darias, C. Cerella, M. Diederich and M. Cueto, Org. Biomol. Chem., 2015, 13, 7248 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00343a |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2016 |