
Palladium-catalyzed decarboxylative alkoxycarbonylation of potassium aryltrifluoroborates with potassium oxalate monoesters†
Jinmin
Miao
,
Ping
Fang
,
Sahota
Jagdeep
and
Haibo
Ge
*
Department of Chemistry and Chemical biology, Indiana University Purdue University Indianapolis, 402 N Blackford Street, Indianapolis, IN 46202, USA. E-mail: geh@iupui.edu; Fax: +1 317-274-4701; Tel: +1 317-274-6876
First published on 11th December 2015
Abstract
Palladium-catalyzed decarboxylative alkoxycarbonylation of potassium aryltrifluoroborates with potassium oxalate monoesters in the presence of potassium persulfate was performed under mild conditions. A number of benzoyl esters with a wide variety of substituents at different positions were efficiently synthesized with this method. Mechanism of the palladium-catalyzed decarboxylative carbonylation of aryltrifluoroborates was studied, and a radical-mediated Pd(II)/Pd(IV) catalytic cycle was proposed.
 Haibo Ge | Haibo Ge received his PhD degree in Medicinal Chemistry from the University of Kansas in 2006 with Professor Gunda Georg. He then moved to The Scripps Research Institute for his postdoctoral study with Professor Dale Boger. In 2009, he began his independent academic career at the Department of Chemistry and Chemical Biology at Indiana University Purdue University Indianapolis (IUPUI). Research in his group is mainly focused on the development of novel methods towards carbon–carbon and carbon–heteroatom bond formation through transition metal catalyzed C–H functionalization. |
Introduction
During the past decade, transition metal-catalyzed cross coupling has been extensively studied as a powerful synthetic tool for selective carbon–carbon (C–C) bond formation.1 In particular, transition metal-catalyzed decarboxylative coupling has recently attracted more and more attention.2 Compared with the traditional coupling reactions, this transformation is more environmentally friendly since stoichiometric organometallic waste is replaced by the innocuous CO2 gas. In addition, as the coupling partners, carboxylic acids are readily available at low cost, fairly stable and easy to handle and store in laboratory. However, although the first decarboxylative cross-coupling reaction was realized with unsatisfactory yield in 1960s,3 it remained unelaborated until recent years. In 2002, Myers reported the silver-mediated decarboxylation of benzoic acid derivatives, followed by a palladium-catalyzed Heck reaction with alkenes.4 This discovery opened the door to a new area of synthetic methodology. Later on, the milestone discoveries were reported by Goossen and co-workers,5 who developed the synthesis of biaryls via palladium/copper-catalyzed decarboxylative coupling of aryl carboxylic acids and aryl halides. Furthermore, alkyl, alkenyl, and alkynyl carboxylic acids were also demonstrated as effective substrates in decarboxylative cross-coupling reactions, turning the methods into highly valuable alternatives to classical reactions for the C–C bond formation.In 2008, α-oxocarboxylic acids were first utilized as coupling partners by Goossen's group in a Cu/Pd-catalyzed acylation reaction of aryl halides.6 Alkoxycarbonylation of aryl halides via decarboxylation of oxalate monoesters was later realized by Liu and co-workers (Scheme 1, eqn (1)).7 However, high temperature was required for the decarboxylation process in these reports, which limits the substrate scope of these reactions.
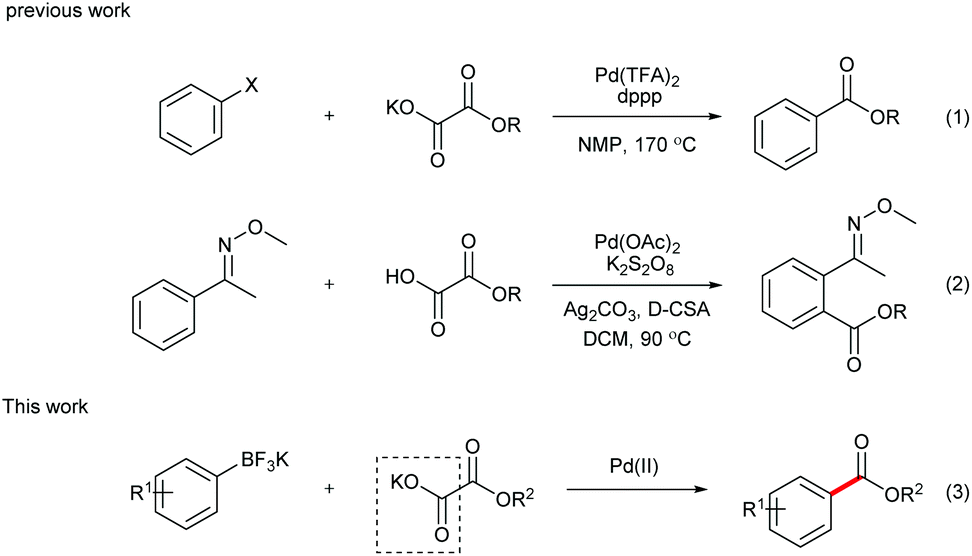 | ||
| Scheme 1 Synthesis of aryl esters with Pd-catalyzed decarboxylative coupling. | ||
Inspired by Minisci's work on peroxydisulfate,8 we discovered that decarboxylative ortho-carbonylation of acetanilides with α-oxocarboxylic acids can be realized at room temperature in the presence of a persulfate.9 Recently, Wang's group developed the Pd-catalyzed ortho-ethoxycarbonylation of O-methylketoximes with potassium oxalate monoester using Ag2CO3 and K2S2O8 as oxidants (eqn (2)).10 However, these reactions can be performed only on substrates with a directing group, which limits the potential application of this approach. We envisioned that boronic acids or their derivatives may be utilized in the coupling reactions to broaden the product range of the method. Arylboronic acids and their derivatives are staple substrates in Suzuki–Miyaura coupling reactions.11 However, the most common pathway of direct transformation from arylboronic acids to carbonyl compounds, the insertion of carbon monoxide,12 suffers from the use of high pressure of the toxic and flammable CO gas, which diminishes its practical utility. To provide an alternative access to aryl ketones, Goossen and Yamamoto developed the Pd-catalyzed decarboxylative cross-coupling reactions of arylboronic acids with anhydrides, carboxylic acids, and α-oxocarboxylic acids, in which the requirement for high temperature or stoichiometrical expensive metal reagents or strong bases is avoided.13 Inspired by these results, our group realized the decarboxylative acylation and aminocarbonylation of potassium aryltrifluoroborates with α-oxocarboxylic acids and oxamic acids.14 Herein, as a supplement to the previous methods, we report the Pd-catalyzed decarboxylative alkoxycarbonylation of potassium aryltrifluoroborates with potassium oxalate monoesters under mild conditions.
Results and discussion
On the basis of our success on decarboxylative cross coupling of aryltrifluoroborates with α-oxocarboxylic acids and oxamic acids,14 we investigated the decarboxylative coupling reaction between potassium phenyltrifluoroborate and potassium 2-ethoxy-2-oxoacetate (Table 1). The ethyl benzoate was obtained with 10 mol% Pd(OAc)2 and 2 equiv. K2S2O8 in a mixture of DMSO and water at room temperature (entry 1). Further screening of solvent showed that the mixture of MeCN/DMSO/H2O was the best (entries 2–5). Although (NH4)2S2O8 was also effective, K2S2O8 was found to be the optimal oxidant (entries 6 and 7). Gratifyingly, the product yield was improved when the reaction was heated at 70 °C for 5 min and then cooled to room temperature (entry 8). Finally, a high yield was acquired by increasing the amount of the K2S2O8 to 3 equiv. (entry 9). It was noted that Pd(OAc)2 was the most efficient catalyst in this reaction (entries 9–12). Furthermore, the desired coupling product was not observed in the absence of a palladium catalyst (entry 13).|
|
||||
|---|---|---|---|---|
| Entry | Pd catalyst | Oxidant | Solvant (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v) :v) |
Yieldb (%) |
| a Conditions: 1a (0.3 mmol), 2a (0.6 mmol), PdX2, oxidant (0.6 mmol), 6 mL solvent, rt, overnight. b Yields and conversions are based on 1a, determined by 1H-NMR using dibromomethane as the internal standard. Isolated yield is in parenthesis. c Preheated at 70 °C for 5 min, and then rt for 1 h. d With 3.0 eq. (0.9 mmol) K2S2O8. | ||||
| 1 | Pd(OAc)2 | K2S2O8 | DMSO/H2O (4:1) |
21 |
| 2 | Pd(OAc)2 | K2S2O8 | MeCN/H2O (4:1) |
<5 |
| 3 | Pd(OAc)2 | K2S2O8 | DME/H2O (4:1) |
0 |
| 4 | Pd(OAc)2 | K2S2O8 | diglyme/H2O (4:1) |
0 |
| 5 | Pd(OAc)2 | K2S2O8 | MeCN/DMSO/H2O (2:2:1) |
30 |
| 6 | Pd(OAc)2 | (NH4)2S2O8 | MeCN/DMSO/H2O (2:2:1) |
18 |
| 7 | Pd(OAc)2 | H2O2 | MeCN/DMSO/H2O (2:2:1) |
0 |
| 8c | Pd(OAc)2 | K2S2O8 | MeCN/DMSO/H2O (2:2:1) |
72 |
| 9c,d | Pd(OAc)2 | K2S2O8 | MeCN/DMSO/H2O (2:2:1) |
84(82) |
| 10c,d | Pd(TFA)2 | K2S2O8 | MeCN/DMSO/H2O (2:2:1) |
60 |
| 11c,d | Pd(acac)2 | K2S2O8 | MeCN/DMSO/H2O (2:2:1) |
39 |
| 12c,d | Pd(MeCN)2(BF4)2 | K2S2O8 | MeCN/DMSO/H2O (2:2:1) |
68 |
| 13c,d | — | K2S2O8 | MeCN/DMSO/H2O (2:2:1) |
0 |
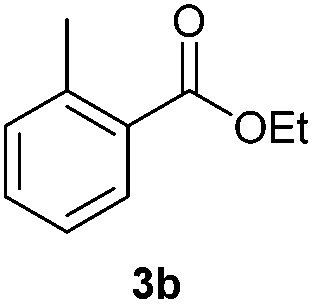
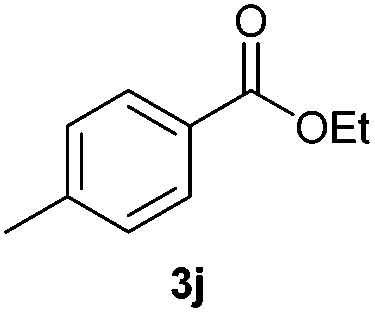
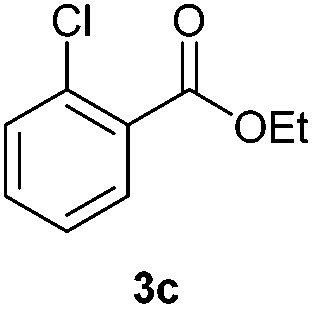
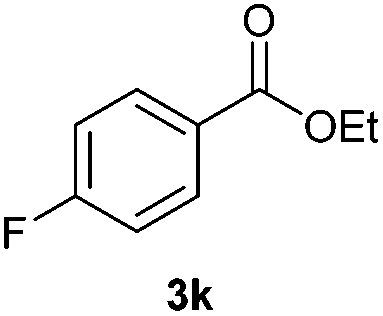
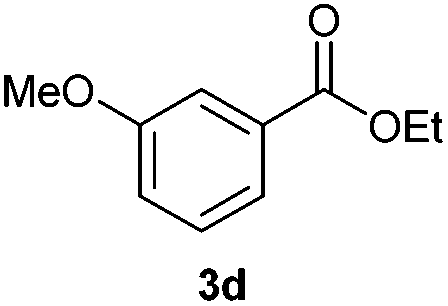
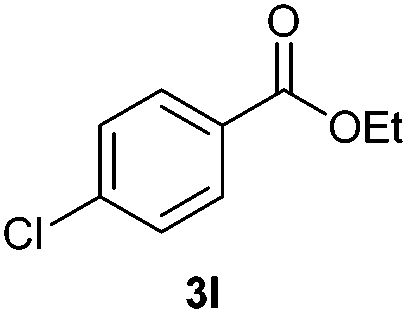
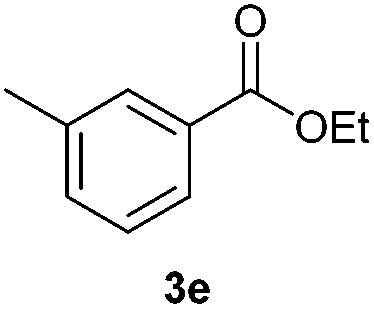
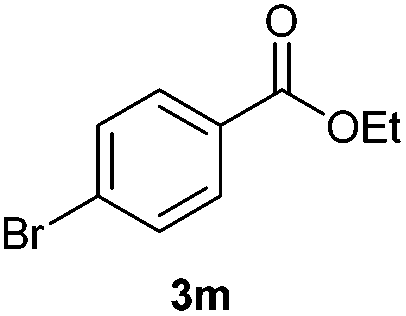
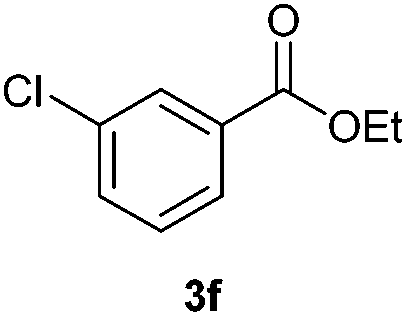
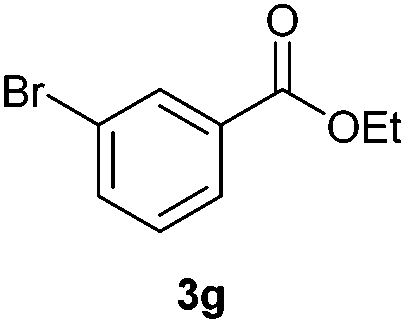
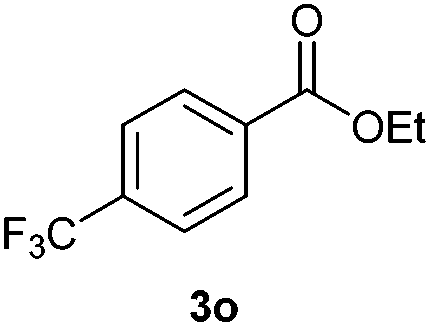
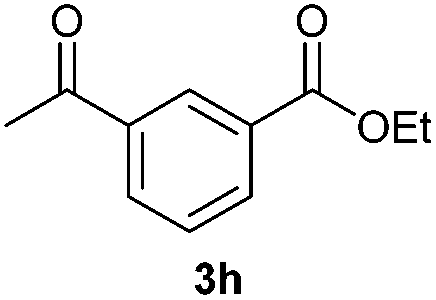
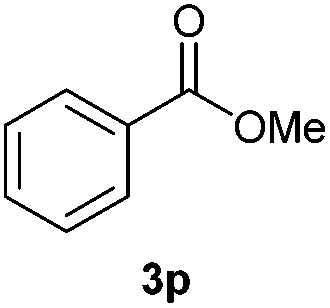
With the optimized conditions in hand, we then investigated the substrate scope of the alkoxycaronylation reaction. As shown in Table 2, a variety of aryl esters were synthesized under the standard conditions. Potassium phenyltrifluoroborates with a methyl group at the meta- or para-position of the phenyl ring (Table 2, 3e and 3j) gave comparable yields to that of 3a. The ortho-methyl phenyltrifluoroborate gave a lower yield than those of the counterparts with a meta- or para- methyl group (3bvs. 3e, 3j), presumably due to the steric effect. Ester 3k, which has a fluoro group at the para-postion of the phenyl ring, could be produced in a good yield from the corresponding substrate (3k). Methoxy-, chloro-, and bromo-substituted phenyltrifluoroborates gave moderate yields (3c, 3d, 3f, 3g, 3i, 3l and 3m), while lower yields were observed with the electron-withdrawing groups on the phenyl ring (3h, 3n, 3o). In addition, potassium 2-methoxy-2-oxoacetate (7b) was demonstrated as a feasible substrate, affording a satisfying yield (3p).
|
|
|||
|---|---|---|---|
| Product | Yieldb (%) | Product | Yieldb (%) |
|
a Conditions: 1 (0.3 mmol), 2a or 2b (0.6 mmol, 2.0 equiv.), Pd(OAc)2 (0.030 mmol, 10 mol%), K2S2O8 (0.9 mmol, 3.0 equiv.), DMSO/MeCN/H2O (4:4:2, v/v/v, 6 mL), preheated at 70 °C for 5 min, then at rt for 1 h.
b Isolated yields based on 1.
|
|||
|
82 |
|
44 |
|
52 |
|
76 |
|
60 |
|
79 |
|
43 |
|
67 |
|
81 |
|
63 |
|
58 |

|
35 |
|
63 |
|
35 |
|
57 |
|
76 |
Mechanistic study
As mentioned above and in our previous reports,14 the alkoxycarbonylation and aminocarbonylation reactions require heating at the beginning of the reaction. To illustrate the function of increased temperature and thereby to provide some insights of the catalytic process, time courses for acylation, aminocarbonylation, and alkoxycarbonylation of potassium phenyltrifluoroborate were examined (Fig. 1 and 2). Not surprisingly, the reaction rates for production of the ketone and the amide at room temperature were comparatively high at the beginning, as most of the product was accumulated in the first 2 hours (Fig. 2, red and purple). In the case of heated reactions, the early reaction rates were extraordinarily fast (Fig. 2, blue and green). Remarkably, the alkoxycarbonylation process was almost completed within 2 min at 70 °C (Fig. 1, pink).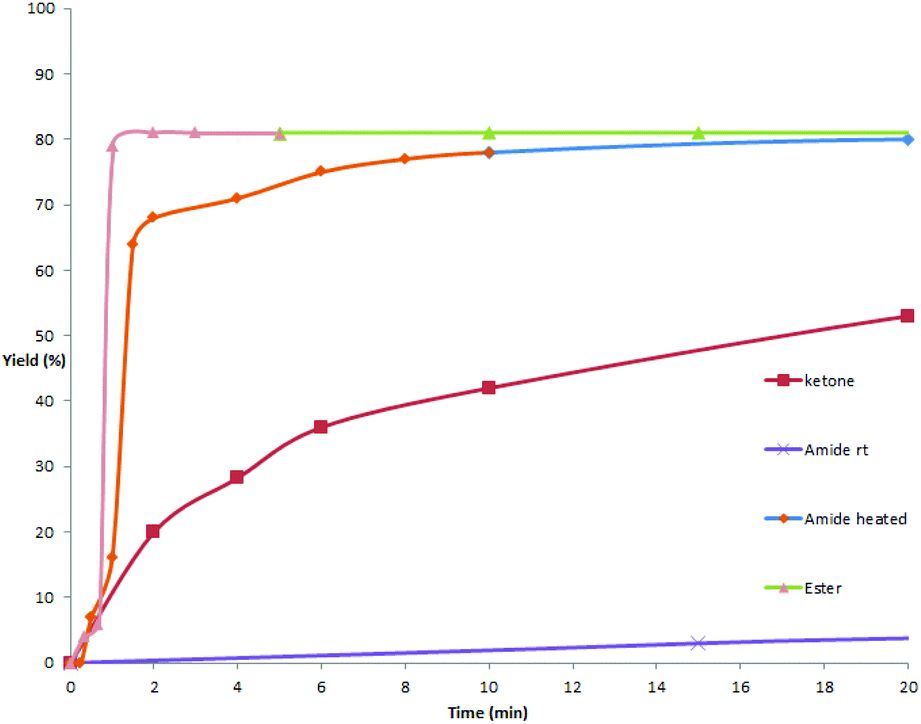 | ||
| Fig. 1 Time courses of the cross-coupling reactions. 0–20 min. | ||
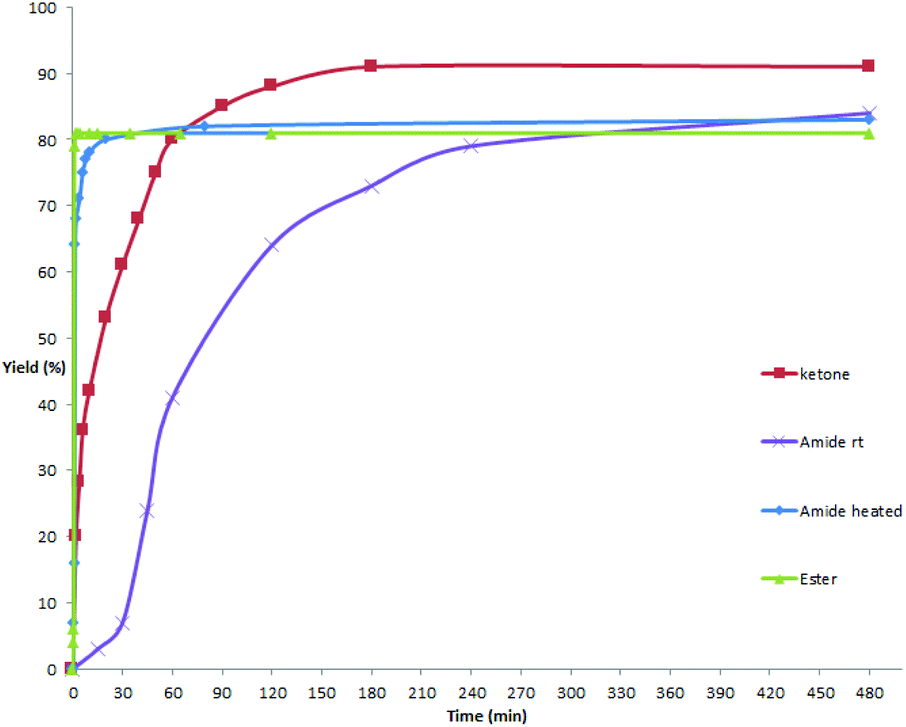 | ||
| Fig. 2 Time courses of the cross-coupling reactions. 0–480 min. | ||
To further study the mechanistic aspects of the catalytic decarboxylative cross-coupling, TEMPO was introduced to the reactions under standard conditions as a radical trapping reagent (Scheme 2). It was found that the formation of the decarboxylative coupling products was suppressed, while TEMPO- aldehyde, amide, and ester adducts were detected by LC-MS respectively. The decarboxylative coupling reactions were not completely inhibited, which is consistent with the reaction rate study since the coupling reactions were so fast at the beginning that they could overwhelm the competing reactions. In addition, the yields of the coupling products were reduced by increasing the amount of TEMPO in the reaction systems. Furthermore, the TEMPO adducts were isolated from the control experiments under the similar conditions in the absence of PhBF3K and Pd(OAc)2 (see Experimental section). Thus, all the results suggest that radical intermediates are likely formed and involved in these coupling reactions, which indicates a different reaction pathway from the previously proposed ligand exchange process of direct decarboxylative acylation of acetanlides.9
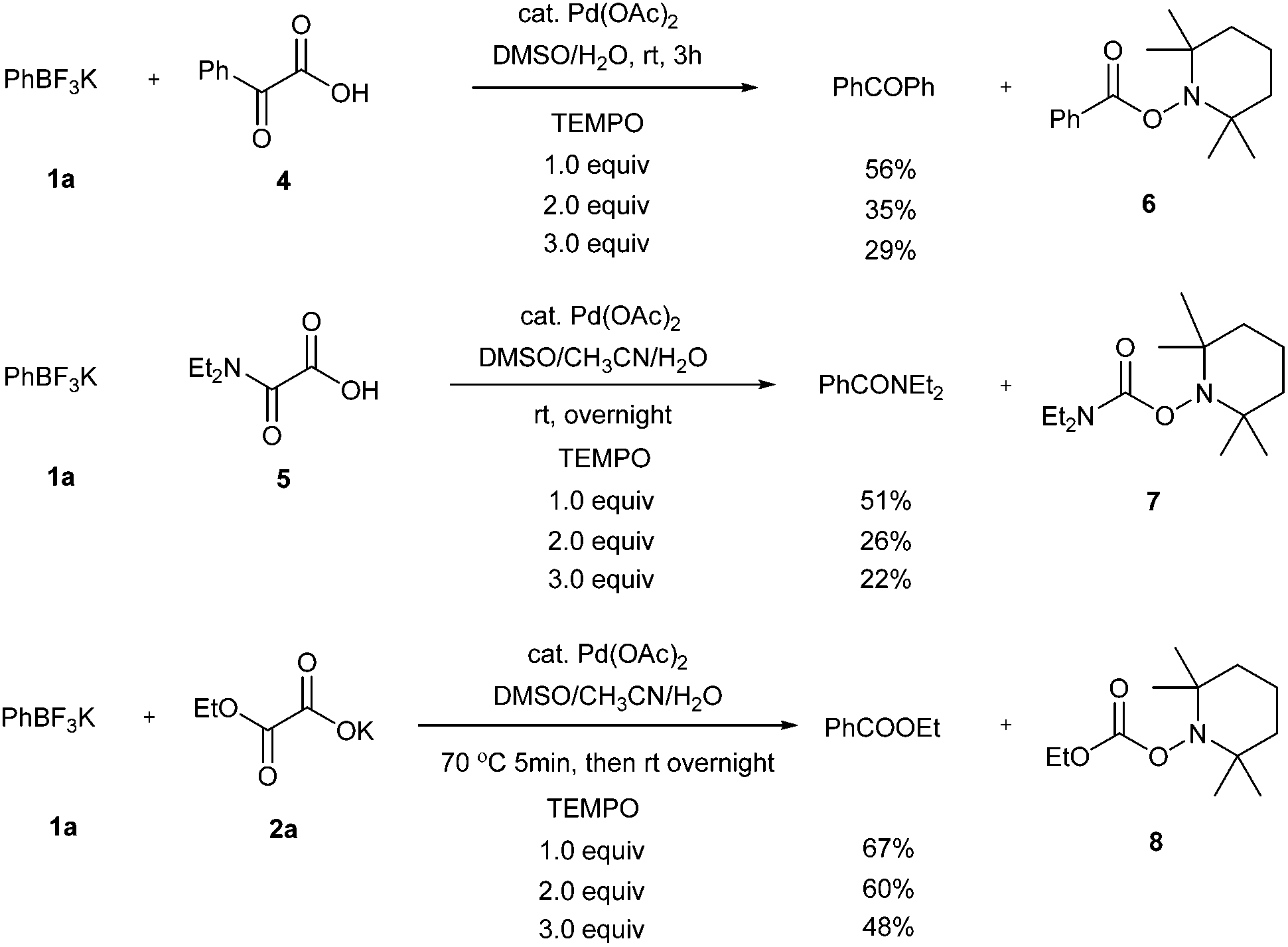 | ||
| Scheme 2 Control experiments with TEMPO. | ||
Based on the above observations and previous literature reports,15 a tentative mechanism for the cross coupling is proposed (Fig. 3). The reaction is initiated by the transmetallation between the Pd(II) catalyst and the boronic intermediates derived from hydrolysis of the aryltrifluoroborate to afford the Pd(II) intermediate A. Oxidation of A in the presence of the carbonyl radical C, which is formed by the decarboxylation of an α-oxocarboxylic or oxamic acid, generates the Pd(IV) intermediate B. The desired carbonyl product is then produced via reductive elimination of B, while the Pd(II) species is reproduced. It's noteworthy that a dimeric Pd(III) mechanism cannot be excluded.16
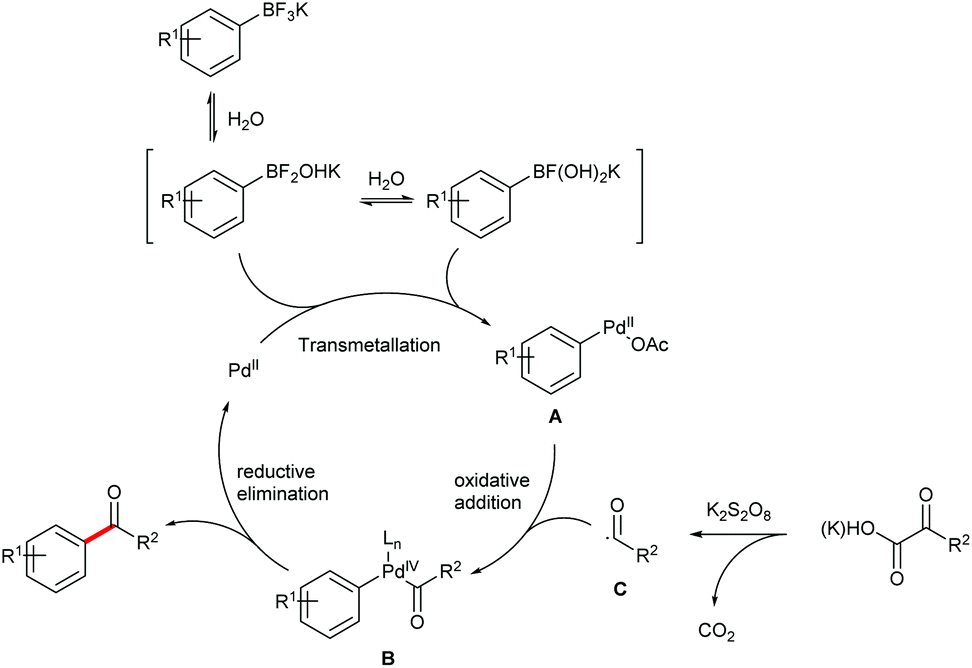 | ||
| Fig. 3 Plausible reaction mechanism. L = Ligand. | ||
Conclusions
In summary, we have demonstrated that a handy and efficient palladium-catalyzed alkoxycarbonylation of potassium aryltrifluoroborates with the decarboxylation of potassium oxalate monoester could be performed under mild and compatible conditions. This unprecedented reaction provides a promising pathway towards a variety of aryl esters. Additionally, the mechanistic study suggests that radicals should be involved in this process, constituting the possible Pd(II)/Pd(IV) catalytic cycle.Experimental
General methods
All reactions were carried out in oven-dried glassware. Pd(II) catalysts, Ag2CO3, K2S2O8 and (NH4)2S2O8 were purchased and used directly. All other solvents and commercially available reagents (boronic acids, KHF2, amines and potassium oxalate monoester) were purchased and used directly. For TLC analysis, precoated plates (0.25 mm thick) were used; for air-flashed column chromatography, flash silica gel (32–63 μm) was used. 1H NMR and 13C NMR spectra were measured on a 500 MHz spectrometer (1H at 500 MHz, 13C at 125 MHz), using CDCl3 as solvent with tetramethylsilane (TMS) as an internal standard at room temperature. 1H NMR data was reported as: chemical shift (δ ppm), multiplicity, coupling constant (Hz), integration. 13C NMR data was reported in terms of chemical shift (δ ppm).Starting materials
Potassium aryltrifluoroborates (1a, 1i, 1j and 1l) and potassium oxalate monoesters (2a, 2b) were purchased and used directly. Other potassium aryltrifluoroborates were prepared from boronic acids with KHF2 according to the reported procedure.17N,N-Diethyloxamic acid (5) was prepared from diethyl oxalate with N,N-diethylamine according to the reported procedure.18General procedure for the synthesis of aryl esters (3)
An 8 mL vial was charged with magnetic bar, ArBF3K (1, 0.3 mmol), potassium oxalic acid monoesters (2, 0.6 mmol, 2.0 equiv.), K2S2O8 (0.9 mmol, 3.0 equiv.), followed by Pd(OAc)2 (DMSO solution, 0.03 mmol/2.4 mL, 10 mol%, 2.4 mL), CH3CN (2.4 mL) and DI water (1.2 mL). The vial was capped and the reaction mixture was stirred at 70 °C for 5 min, and then cooled down to room temperature and stirred for 1 h. The reaction was quenched by the addition of 3 mL of water and the resulting mixture was extracted with EtOAc (5 mL × 3). The combined organic phase was dried over Na2SO4, and then concentrated under vacuum. The desired product was obtained after purification by flash chromatography column on silica gel (gradient eluent of EtOAc in hexanes: 0–30%, v/v).Time-yield curve in formation of benzophenone (Fig. 1, red markers)
Parallel experiments were carried out with the procedure described below. An 8 mL vial was charged with magnetic stir bar, ArBF3K (1a, 0.3 mmol), 2-oxo-2-phenylacetic acid (4, 0.6 mmol, 2.0 equiv.), K2S2O8 (0.6 mmol, 2.0 equiv.), followed by Pd(OAc)2 (DMSO solution, 0.0075 mmol/1.2 mL, 2.5 mol%, 1.2 mL) and DI water (1.8 mL, DMSO:DI water = 1/1.5, v/v, 3 ml in total). The vial was capped and then the reaction mixture was stirred at room temperature. At each interval, NaOH (1 N, 3 mL) was added and the reaction mixture was extracted with EtOAc (5 mL × 3). The combined organic phase was concentrated under vacuum. The yields of benzophenone were determined by 1H NMR using CH2Br2 as an internal standard.
Time-yield curve in formation of N,N-diethylbenzamide at room temperature (Fig. 1, purple markers)
Parallel experiments were carried out with the procedure described below. An 8 mL vial was charged with magnetic stir bar, PhBF3K (1a, 0.3 mmol), 2-(diethylamino)-2-oxoacetic acid (5, 0.6 mmol, 2.0 equiv.), K2S2O8 (0.9 mmol, 3.0 equiv.), followed by Pd(OAc)2 (DMSO solution, 0.015 mmol/2.4 mL, 5 mol%, 2.4 mL), CH3CN and DI water (DMSO:CH3CN:DI water = 4/4/2, v/v/v, 6 ml in total). The vial was capped and then the reaction mixture was stirred at room temperature. At each interval, NaOH (1 N, 3 mL) was added and the reaction mixture was extracted with EtOAc (5 mL × 3). The combined organic phase was concentrated under vacuum. The yields of N,N-diethylbenzamide were determined by 1H NMR using CH2Br2 as an internal standard.
Time-yield curve in formation of N,N-diethylbenzamide, heated (Fig. 1, orange and blue markers)
Parallel experiments were carried out with the procedure described below. An 8 mL vial was charged with magnetic stir bar, PhBF3K (1a, 0.3 mmol), 2-(diethylamino)-2-oxoacetic acid (5, 0.6 mmol, 2.0 equiv.), K2S2O8 (0.9 mmol, 3.0 equiv.), followed by Pd(OAc)2 (DMSO solution, 0.015 mmol/2.4 mL, 5 mol%, 2.4 mL), CH3CN and DI water (DMSO:CH3CN:DI water = 4/4/2, v/v/v, 6 ml in total). The vial was capped and the reaction mixture was stirred at 70 °C for 10 min, and then stirred at room temperature. At each interval, the vial is cooled with water bath immediately, and then 3 mL water was added and the reaction mixture was extracted with EtOAc (5 mL × 3). The combined organic phase was concentrated under vacuum. The yields of N,N-diethylbenzamide were determined by 1H NMR using CH2Br2 as an internal standard.
Time-yield curve in formation of ethyl benzoate (Fig. 1, pink and green markers)
Parallel experiments were carried out with the procedure described below. An 8 mL vial was charged with magnetic stir bar, PhBF3K (1a, 0.3 mmol potassium 2-ethoxy-2-oxoacetate (2a, 0.6 mmol, 2.0 equiv.), K2S2O8 (0.9 mmol, 3.0 equiv.), followed by Pd(OAc)2 (DMSO solution, 0.03 mmol/2.4 mL, 10 mol%, 2.4 mL), CH3CN and DI water (DMSO:CH3CN:DI water = 4/4/2, v/v/v, 6 ml in total). The vial was capped and the reaction mixture was stirred at 70 °C for 5 min, and then stirred at room temperature. At each interval, the vial is cooled with water bath immediately, and then 3 mL of water was added and the reaction mixture was extracted with EtOAc (5 mL × 3). The combined organic phase was concentrated under vacuum. The yields of ethyl benzoate were determined by 1H NMR using CH2Br2 as an internal standard.
Control experiments with TEMPO in the coupling reactions
The reactions were performed with standard procedures described above or in our previous reports14 except that TEMPO was added into the vial before Pd(OAc)2 and the solvents. Product yields were determined by 1H NMR using CH2Br2 as an internal standard.Acknowledgements
We are grateful for the financial support from Indiana University Purdue University Indianapolis. We thank Mingzhong Li (Indina University Purdue University Indianapolis) and Cong Wang (Indina University Purdue University Indianapolis) for their preliminary work. The Bruker 500 MHz NMR was purchased using funds from an NSF-MRI award (CHE-0619254).Notes and references
- For recent reviews, see: (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651 CrossRef CAS PubMed; (c) L.-X. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS; (d) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS; (e) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed; (f) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS; (g) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS; (h) M. Catellani, E. Motti and N. Della Ca’, Acc. Chem. Res., 2008, 41, 1512 CrossRef CAS PubMed; (i) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS; (j) R. Giri, B.-F. Shi, K. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (k) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (l) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (m) S. De Ornellas, T. E. Storr, T. J. Williams, C. G. Baumann and I. J. S. Fairlamb, Curr. Org. Chem., 2011, 8, 79 CAS; (n) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (o) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (p) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (q) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910 RSC; (r) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS; (s) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (t) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (u) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248 CrossRef CAS PubMed; (v) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (w) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed.
- For recent reviews, see: (a) O. Baudoin, Angew. Chem., Int. Ed., 2007, 46, 1373 CrossRef CAS PubMed; (b) L. J. Goossen, K. Goossen, N. Rodriguez, M. Blanchot, C. Linder and B. Zimmermann, Pure Appl. Chem., 2008, 80, 1725 CrossRef CAS; (c) L. J. Goossen, N. Rodriguez and K. Goossen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef CAS; (d) L. J. Goossen, F. Collet and K. Goossen, Isr. J. Chem., 2010, 50, 617 CrossRef CAS; (e) S. M. Bonesi and M. Fagnoni, Chem. – Eur. J., 2010, 16, 13572 CrossRef CAS; (f) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (g) R. Shang and L. Liu, Sci. China, Ser. B, 2011, 54, 1670 CrossRef CAS; (h) J. Cornella and I. Larrosa, Synthesis, 2012, 653 CAS.
- M. Nilsson, Acta Chem. Scand., 1966, 20, 423 CrossRef CAS.
- A. G. Myers, D. Tanaka and R. J. Mannion, Am. Chem. Soc., 2002, 124, 11250 CrossRef CAS.
- (a) L. J. Goossen, G. J. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef CAS PubMed; (b) L. J. Goossen, N. Rodriguez, B. Melzer, C. Linder, G. J. Deng and L. M. Levy, J. Am. Chem. Soc., 2007, 129, 4824 CrossRef CAS.
- (a) L. J. Goossen, F. Rudolphi, C. Oppel and N. Rodríguez, Angew. Chem., Int. Ed., 2008, 47, 3043 CrossRef CAS; (b) L. J. Goossen, B. Zimmermann and T. Knauber, Angew. Chem., Int. Ed., 2008, 47, 7103 CrossRef CAS; (c) Y. Zheng, G. Yu, J. Wu and W.-M. Dai, Synlett, 2010, 1075–1080 Search PubMed.
- R. Shang, Y. Fu, J.-B. Li, S.-L. Zhang, Q.-X. Guo and L. Liu, J. Am. Chem. Soc., 2009, 131, 5738 CrossRef CAS.
- (a) F. Minisci, A. Citterio and C. Giordano, Acc. Chem. Res., 1983, 16, 27 CrossRef CAS; (b) F. Minisci, E. Vismara and F. Fontana, Heterocycles, 1989, 28, 489 CrossRef CAS; (c) F. Fontana, F. Minisci, M. C. N. Barbosa and E. Vismara, J. Org. Chem., 1991, 56, 2866 CrossRef CAS; (d) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef; (e) C. Punta and F. Minisci, Trends Heterocycl. Chem., 2008, 13, 1 CAS.
- P. Fang, M.-Z. Li and H.-B. Ge, J. Am. Chem. Soc., 2010, 132, 11898 CrossRef CAS.
- Z.-Y. Li and G.-W. Wang, Org. Lett., 2015, 17, 4866 CrossRef CAS.
- For recent reviews, see: (a) F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 2419 CAS; (b) O. Baudoin, Eur. J. Org. Chem., 2005, 4223 CrossRef CAS; (c) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS; (d) N. Miyaura, Bull. Chem. Soc. Jpn., 2008, 81, 1535 CrossRef CAS; (e) M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2009, 48, 3565 CrossRef CAS; (f) G. A. Molander and B. Canturk, Angew. Chem., Int. Ed., 2009, 48, 9240 CrossRef CAS; (g) N. Miyaura, Synlett, 2009, 2039 CAS; (h) C. E. I. Knappke and A. J. V. Wangelin, Angew. Chem., Int. Ed., 2010, 49, 3568 CrossRef CAS PubMed; (i) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS; (j) M. M. Heravi and E. Hashemi, Tetrahedron, 2012, 68, 9145 CrossRef CAS; (k) M. Blangetti, H. Rosso, C. Prandi, A. Deagostino and P. Venturello, Molecules, 2013, 18, 1188 CrossRef CAS.
- Selected recent examples of CO insertion: (a) M.-J. Dai, B. Liang, C.-H. Wang, Z.-J. You, J. Xiang, G.-B. Dong, J.-H. Chen and Z. Yang, Adv. Synth. Catal., 2004, 346, 1669 CrossRef CAS; (b) P. J. Tambade, Y. P. Patil, A. G. Panda and B. M. Bhanage, Eur. J. Org. Chem., 2009, 3022 CrossRef CAS; (c) M. V. Khedkar, P. J. Tambade, Z. S. Qureshi and B. M. Bhanage, Eur. J. Org. Chem., 2010, 6981 CrossRef CAS; (d) F. Jafarpour, P. Rashidi-Ranjbar and A. O. Kashani, Eur. J. Org. Chem., 2011, 2128 CrossRef CAS; (e) H.-L. Li, M. Yang, Y.-X. Qi and J.-J. Xue, Eur. J. Org. Chem., 2011, 2662 CrossRef.
- (a) L. J. Goossen and K. Ghosh, Angew. Chem., Int. Ed., 2001, 40, 3458 CrossRef CAS; (b) L. J. Goossen, Chem. Commun., 2001, 2084 CAS; (c) L. J. Goossen, L. Winkel, A. Dohring, K. Ghosh and J. Paetzold, Synlett, 2002, 1237 CrossRef; (d) R. Kakino, S. Yasumi, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2002, 75, 137 CrossRef CAS; (e) R. Kakino, H. Narahashi, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2002, 75, 1333 CrossRef CAS.
- (a) M.-Z. Li, C. Wang and H.-B. Ge, Org. Lett., 2011, 13, 2062 CrossRef CAS PubMed; (b) M.-Z. Li, C. Wang, P. Fang and H.-B. Ge, Chem. Commun., 2011, 6587 RSC.
- (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed; (b) D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS PubMed; (c) A. R. Dick, J. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 12790 CrossRef CAS; (d) G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275 CrossRef CAS; (e) X. Tong, M. Beller and M. K. Tse, J. Am. Chem. Soc., 2007, 129, 4906 CrossRef CAS PubMed; (f) L. L. Welbes, T. W. Lyons, K. A. Cychosz and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 5836 CrossRef CAS; (g) W.-Y. Yu, W. N. Sit, K.-M. Lai, Z.-Y. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2008, 130, 3304 CrossRef CAS; (h) P. A. Sibbald, C. F. Rosewall, R. D. Swartz and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 15945 CrossRef CAS; (i) G.-W. Wang and T.-T. Yuan, J. Org. Chem., 2010, 75, 476 CrossRef CAS PubMed; (j) T. W. Lysons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef; (k) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS.
- (a) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 CrossRef CAS; (b) D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef CAS PubMed.
- G. A. Molander, W. Febo-Ayala and L. Jean-Gerard, Org. Lett., 2009, 11, 3830 CrossRef CAS PubMed.
- R. Peters, M. Althaus and A.-L. Nagy, Org. Biomol. Chem., 2006, 4, 498 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Data table of the time-yield curve and copies of 1H and 13C NMR spectra of new compounds. See DOI: 10.1039/c5qo00349k |
| This journal is © the Partner Organisations 2016 |