
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A step towards hydroformylation under sustainable conditions: platinum-catalysed enantioselective hydroformylation of styrene in gamma-valerolactone†
Péter
Pongrácz
c,
László
Kollár
bc and
László T.
Mika
*a
aDepartment of Chemical and Environmental Process Engineering, Budapest University of Technology and Economics, Budapest, Hungary. E-mail: laszlo.t.mika@mail.bme.hu; Fax: +36 1 463 3197; Tel: +36 1 463 1263
bDepartment of Inorganic Chemistry, University of Pécs and Szentágothai Research Center, Ifjúság u. 6., Pécs, H-7624, Hungary
cMTA-PTE Research Group for Selective Chemical Syntheses, Ifjúság u. 6., Pécs, H-7624, Hungary
First published on 8th September 2015
Abstract
Platinum-catalysed enantioselective hydroformylation of styrene was performed in γ-valerolactone (GVL) as a proposed environmentally benign reaction medium. Optically active bidentate ligands, possessing various types of chirality elements e.g. central (BDPP), axial (BINAP, SEGPHOS, DM-SEGPHOS, DTBM-SEGPHOS) and planar/central (JOSIPHOS) elements, were applied in in situ generated Pt-diphosphine-tin(II)chloride catalyst systems. In general, slightly higher activities and regioselectivities towards a branched aldehyde (2-phenylpropanal) were obtained in toluene as a reference conventional solvent. However, higher chemoselectivities towards aldehydes (up to 98%) in GVL were obtained at lower temperatures. The application of GVL proved to be also advantageous regarding enantioselectivity: although moderate enantioselectivities were obtained in both solvents, in most cases higher ee values were detected in GVL. From the mechanistic point of view, the formation of different catalytic intermediates and/or different kinetics can be envisaged from the different temperature dependences of ee in GVL and toluene. The 31P-NMR characterization of catalyst species in GVL was also provided.
Introduction
Solvents are intrinsic parts of many chemical reactions and the “solvent friendly chemical thinking” has evolved due to many advantages in both industrial and laboratory operations.1 Thus, the industrial activities involving solvents result in the release of 10–15 million tons of solvents into the atmosphere annually, some of which are leading to serious environmental concerns and economic issues. Consequently, the replacement of conventional organic solvents with green alternatives having low vapour pressure even at high temperature, low or no toxicity, low flammability and limited negative impacts on the environment is a crucial part in the development of greener and cleaner chemical technologies.2 Although, “solvent free” transformation could offer environmentally friendly solutions, many thousands if not millions of reactions can only be operated in the presence of solvents. According to FDA guidelines3 the use of several common organic solvents has to be avoided such as benzene and chlorinated hydrocarbons or limited including toluene, hexane etc., just to name a few. However, if a solvent is crucial to perform the target transformation, an alternative that has no or limited impact on the environment and health has to be selected. Several alternative solvents such as water,4 ‘fluorous’ solvents5 and ionic liquids,6 supercritical carbon-dioxide7 have been proposed for better solutions and some of them have industrial importance, e.g. Ruhrchemie/Rhone-Poulenc aqueous biphasic hydroformylation process.8 Moreover, the application on a non-toxic reaction medium is fundamentally important in the pharmaceutical industry, where the residual solvent traces could result in serious health issues.The intensive research activity on biomass conversion has led to the identification of several platform molecules such as γ-valerolactone (GVL)9 which could replace the currently used fossil-based chemicals including solvents. Due to the environmentally friendly chemical and physical properties, GVL has been considered as a sustainable liquid.9,10 It is renewable and can efficiently be produced by hydrogenation of levulinic acid (LA).11 GVL has already been used for the production of alkanes,11e transportation fuels,12 ionic liquids,13 1,4-pentanediol,11d,e adipic acid,14 polymers,15 illuminating liquids, lighter fluids16etc. Since it was firstly suggested by Horváth, only a few studies have been published concerning the utilization of GVL as a solvent. It was shown that GVL could be used as a reaction medium for acid catalysed dehydration of various carbohydrates and wastes.17 We demonstrated that the selective hydrogenation of LA could be performed in GVL and/or in a GVL–water mixture.11a–c Very recently, heterogeneous Sonogashira,18 Heck19 and Hiyama20 cross-coupling reactions have been performed in GVL as well.
The very low toxicity of GVL9 makes it a particularly attractive reaction medium in the synthesis of pharmaceuticals. Since chiral aldehydes are potential precursors of non-steroidal anti-inflammatory drugs (NSAI), such as ibuprofen, naproxen or suprofen,21,22 the enantioselective hydroformylation of vinyl aromatics to 2-arylpropanals in the presence of rhodium and platinum catalysts is of utmost importance. Soon after the recognition of the hydroformylation activity of in situ generated platinum–monophosphine–tin(II)halide systems,23 the corresponding ‘preformed’ PtCl(SnCl3)(chiral diphosphine) catalysts and PtCl2(chiral diphosphine) + tin(II) halide systems were tested in enantioselective oxo-synthesis.24,24a In addition, to the most widely used vinyl aromatics, 1,1-disubstituted olefins were also used as model substrates in highly enantioselective hydroformylation.25 During the past three decades, various types of chiral mono- and bidentate phosphorus ligands have been tested in platinum-based hydroformylation with the aim of increasing the activity, chemo-, regio- and enantioselectivities.26 Since the industrial hydroformylation was carried out in alternative solvents to facilitate catalyst recycling,27 even the enantioselective hydroformylation was performed in water,28 in ionic liquids,29 as well as in fluorous/scCO2 media.30
We report here the investigation of asymmetric hydroformylation of styrene (Scheme 1) in the presence of platinum-chiral diphosphine-tin(II)chloride in situ generated catalyst systems comparing their activity and selectivities in toluene as a conventional solvent and in γ-valerolactone as a non-toxic and environmentally benign solvent. It has to be also emphasized that to the best of our knowledge, some of the diphosphines (the SEGPHOS family) were not tested in hydroformylation at all.
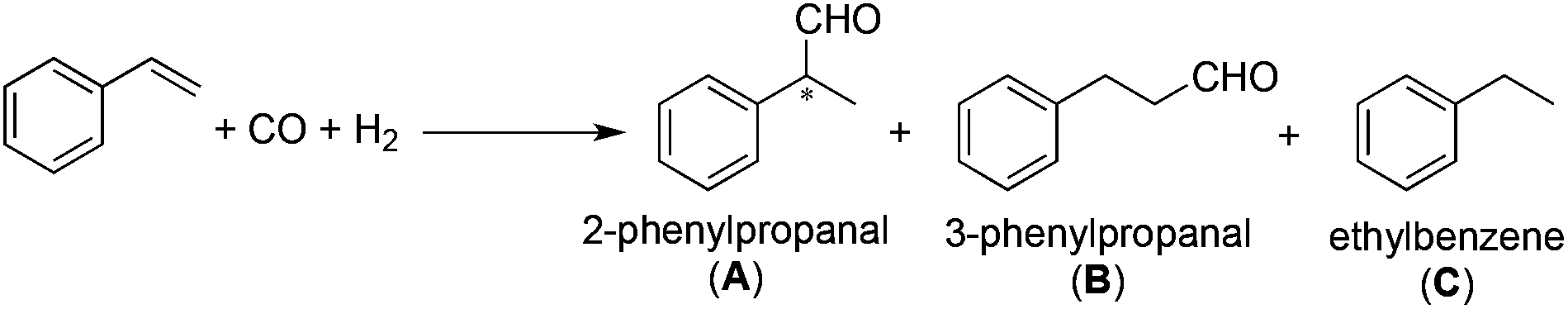 | ||
| Scheme 1 Hydroformylation of styrene. | ||
Results and discussion
Initially, the formation of the corresponding platinum complexes of the applied ligands in gamma-valerolactone was investigated. The 31P-NMR measurement clearly established the in situ formation of PtCl2(diphosphine) complexes from PtCl2(PhCN)2 (0.005 mmol) and 0.005 mmol ligands (S,S)-BDPP, (R)-SEGPHOS, (R),(S)-JOSIPHOS and (R)-BINAP (Fig. 1) representing a square-planar geometry in GVL (0.6 mL). The 1J (31P,195Pt) coupling constants of the diagnostic value show that phosphorus donor atoms are coordinated trans to the chloro ligands (Table 1). Similarly, the excellent solubility of the PtCl2(diphosphine) complexes and SnCl2 in GVL allows us to study the structure of the PtCl(SnCl3)(diphosphine) complexes formed in the ‘carbene-like’ insertion of tin(II)-chloride into the Pt–Cl bond. The formation of the trichlorostannato ligand was undoubtedly proved by increasing the chemical shift and decreasing the 1J (31P,195Pt) coupling constants of the phosphorus trans to SnCl3. The 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two isomers (I and II) was formed when JOSIPHOS possessing two chemically different phosphorus atoms was used. It is worth mentioning that the NMR characteristics were found to be almost identical to those obtained in CDCl3.25c It should be emphasized that the characterization of bidentate phosphine modified Pt- and even Pt/SnCl3-complexes in GVL has not yet been described. Accordingly, it can be proposed that GVL could also be a good solvent for characterization of transition metal complexes, as well.
:1 mixture of two isomers (I and II) was formed when JOSIPHOS possessing two chemically different phosphorus atoms was used. It is worth mentioning that the NMR characteristics were found to be almost identical to those obtained in CDCl3.25c It should be emphasized that the characterization of bidentate phosphine modified Pt- and even Pt/SnCl3-complexes in GVL has not yet been described. Accordingly, it can be proposed that GVL could also be a good solvent for characterization of transition metal complexes, as well.
 | ||
| Fig. 1 Ligands used in Pt-catalysed hydroformylation. | ||
|
|
||||
|---|---|---|---|---|
| Complex | δ (PA) (ppm) | 1 J (Pt, PA) (Hz) | δ (PB) (ppm) | 1 J (Pt, PB) (Hz) |
| a PtCl2(PhCN)2/SnCl2 = 1/2, PA: trans to Cl; PB: trans to SnCl3. b P A trans to SnCl3, PBtrans to Cl. c Chemically different phosphorus atoms; solvent: GVL; for spectra, see ESI Fig. S1–S9. | ||||
| PtCl2(BDPP) | 6.8 | 3394 | — | — |
| PtCl2(SEGPHOS) | 7.5 | 3662 | — | — |
| PtCl2(JOSIPHOS)c | −0.7 | 3570 | 44.4 | 3537 |
| PtCl2(BINAP) | 9.7 | 3665 | — | — |
| PtCl(SnCl3)(BDPP) | 7.8 | 3302 | 13.1 | 2780 |
| PtCl(SnCl3)(SEGPHOS) | 6.8 | 3512 | 17.2 | 3110 |
| PtCl(SnCl3)(JOSIPHOS) (I) | 1.3 | 3450 | 59.5 | 3072 |
| PtCl(SnCl3)(JOSIPHOS) (II)b | 11.7 | 3048 | 58.9 | 3496 |
| PtCl(SnCl3)(BINAP) | 8.4 | 3515 | 18.8 | 3070 |

Since the in situ formation of precursors of catalytically active species from PtCl2(PhCN)2, optically active bidentate phosphine, and tin(II)chloride in GVL was established, they were applied for the hydroformylation of styrene. Various types of chiral ligands possessing stereogenic centres (BDPP, entries 1–4), perpendicular dissymmetric planes due to restricted rotation (BINAP, entries 5–18; SEGPHOS, entries 19 and 20; DM-SEGPHOS, entries 21–26; DTBM-SEGPHOS, entries 27–31) and both stereogenic centres and planar elements of chirality (JOSIPHOS, entries 32–36) were used (Fig. 1 and Table 1). As a major aim of this paper, the applicability of GVL in hydroformylation was tested and the most important issues such as activity, chemo-, regio- and enantioselectivities were investigated. As a comparison, toluene as a generally used solvent in platinum-catalysed hydroformylation was also investigated in all cases. It has to be added that the SEGPHOS family was not tested in hydroformylation, even while using GVL as a solvent.
All catalysts show remarkable catalytic activity in GVL under standard ‘oxo-conditions’ (p(CO) = p(H2) = 40 bar, 60–100 °C). As generally observed in the hydroformylation of styrene (Scheme 1), in addition to the branched and linear formyl regioisomers (2-phenylpropanal (A) and 3-phenylpropanal (B), respectively) the hydrogenation by-product ethylbenzene (C) was also formed.
The catalytic activities of the above systems in GVL were comparable to those observed in toluene. However, in general lower activities were obtained in GVL when experiments were carried out under identical conditions (compare for instance entries 1 and 3 (BDPP), entries 6 and 12 (BINAP, entries 19 and 20 (SEGPHOS), entries 22 and 25 (DM-SEGPHOS), and entries 32 and 34 (JOSIPHOS). The only exception is DTBM-SEGPHOS which formed more active catalysts in GVL (entries 27 and 29). It has to be added that the activities obtained with the Pt-DTBM-SEGPHOS catalyst in both solvents fall far behind those generally observed in platinum-catalysed hydroformylation of styrene.23–25 It should be noted that the hydroformylation carried out above 120 °C resulted in the formation of some alcohols as the side-products, i.e., the hydrogenation products of the aldehydes (A and B) (entries 5 and 11).
As for the chemoselectivity towards hydroformylation, in general the GVL-based systems provided higher aldehyde selectivities. When chemoselectivities at higher reaction temperatures (100 °C) are compared, the values fall in the range of 80–88%, with similar chemoselectivities in GVL and toluene. For instance, 81 and 85% (BDPP), 88 and 86% (BINAP), 83 and 86% (DM-SEGPHOS), and 82 and 80% (JOSIPHOS) were obtained in toluene and GVL, respectively. However, in some cases more pronounced differences in chemoselectivities obtained in GVL and toluene at a lower temperature (80 °C) were observed. For instance, 86 and 93% for BDPP were obtained.
When the partial pressure of hydrogen was increased from 40 to 80 bar (while carbon monoxide pressure was kept at 40 bar) a decrease in aldehyde selectivity was detected, expectedly (entries 12 and 15). A similar effect was observed when hydrogen partial pressure was increased from 20 to 60 bar (while carbon monoxide partial pressure was kept at 20 bar) (entries 13 and 14). As in the case of activity, the DTBM-SEGPHOS-containing systems behave in a different way (entries 29 and 30). As for the regioselectivity, the linear aldehyde (B) predominated over the branched one (A) in all the cases as observed generally when platinum–diphosphine catalysts were used in the hydroformylation of styrene.25 It can be stated that the application of GVL instead of toluene resulted in a drop of ca. 10%. While regioselectivities in GVL are in general below 30%, the corresponding values in toluene fall in the range of 36–48% (BINAP), 36–46% (DM-SEGPHOS). No pronounced effect of the change of the partial pressures on regioselectivities was observed.
Although moderate enantioselectivities were obtained using the above systems, some important phenomena regarding enantioselectivity were observed. The application of GVL instead of toluene resulted in two important features. First, the ee values obtained in GVL were higher in the cases of all six ligands. Second, the strong dependence of the enantioselectivity on the reaction temperature was not observed neither in the case of BDPP nor that of BINAP. As published before, the formation of (S)-2-phenylpropanal was found to be favoured at low temperatures while that of the (R)-enantiomer at higher temperatures when (S,S)-BDPP was used in toluene.25c,d The results were rationalized on the basis of a kinetic phenomenon.31 A similar change of the absolute configuration in the temperature range of 40–100 °C was observed with the Pt-BINAP system.26f
The GVL-based system differs substantially from the toluene-based one in the cases of both ligands: neither the application of the Pt-BDPP system nor that of the Pt-BINAP system resulted in the reversal of the enantioselectivity as a function of temperature. The formation of the (S)-2-phenylpropanal as the predominating enantiomer was observed both with (S,S)-BDPP (entries 3 and 4) and (R)-BINAP (entries 11–18) in the temperature range investigated.
To investigate the effect of the hydrogen partial pressure, a rather complicated picture was obtained. While the increase of the hydrogen pressure from 20 to 60 bar (entries 13 and 14), as well as from 40 to 80 bar (entries 12 and 15) has no effect on the ee-s in the case of BINAP, some increase was observed with DTBM-SEGPHOS (entries 29 and 30) and a substantial decrease with JOSIPHOS (entries 35 and 36).
The reproducibility of the catalytic results was confirmed by repeating the experiments performed in the presence of BDPP in GVL (Table 2, entry 4) at 80 °C. The parallel experiments resulted practically in the same chemo-, regio- and enantioselectivities. That is, the selectivity values differ by less than 1.5%.
| Entry | Ligand (L) | Solvent | T (°C) | Time (h) | Conv. (%) |
R
cb (%) |
R
brc (%) |
eed (%) |
|---|---|---|---|---|---|---|---|---|
| A: 2-phenylpropanal, B: 3-phenylpropanal, C: ethylbenzene.a Reaction conditions: Pt/styrene = 1/100, Pt/SnCl2 = 1/2; p(CO) = p(H2) = 40 bar, 1 mmol of styrene, solvent: 5 mL of toluene (or GVL).b Chemoselectivity towards aldehydes (A, B) [(moles of A + moles of B)/(moles of A + moles of B + moles of C) × 100].c Regioselectivity towards the branched aldehyde (A) [moles of A/(moles of A + moles of B) × 100].d Enantioselectivities were determined by chiral GC.e ca. 8% alcohols as side-products.f p(CO) = p(H2) = 20 bar.g ca. 5% alcohols as side-products.h p(CO) = 20 bar, p(H2) = 60 bar.i p(CO) = 40 bar, p(H2) = 80 bar. | ||||||||
| 1 | (S,S)-BDPP | Toluene | 100 | 3 | 99 | 81 | 33 | 7 (R) |
| 2 | (S,S)-BDPP | Toluene | 80 | 20 | 99 | 86 | 36 | 14 (R) |
| 3 | (S,S)-BDPP | GVL | 100 | 20 | 91 | 85 | 23 | 2 (S) |
| 4 | (S,S)-BDPP | GVL | 80 | 96 | 99 | 93 | 26 | 18 (S) |
| 5e | (R)-BINAP | Toluene | 120 | 20 | 99 | 78 | 48 | 16 (S) |
| 6 | (R)-BINAP | Toluene | 100 | 20 | 99 | 88 | 44 | 28 (S) |
| 7f | (R)-BINAP | Toluene | 100 | 20 | 96 | 82 | 48 | 30 (S) |
| 8 | (R)-BINAP | Toluene | 80 | 70 | 99 | 92 | 45 | 2 (S) |
| 9 | (R)-BINAP | Toluene | 60 | 144 | 99 | 94 | 36 | 19 (R) |
| 10 | (R)-BINAP | Toluene | 40 | 120 | 16 | 91 | 31 | 32 (R) |
| 11g | (R)-BINAP | GVL | 130 | 20 | 99 | 59 | 20 | 5 (S) |
| 12 | (R)-BINAP | GVL | 100 | 20 | 83 | 86 | 19 | 26 (S) |
| 13f | (R)-BINAP | GVL | 100 | 20 | 58 | 84 | 22 | 16 (S) |
| 14h | (R)-BINAP | GVL | 100 | 20 | 94 | 84 | 21 | 15 (S) |
| 15i | (R)-BINAP | GVL | 100 | 20 | 98 | 72 | 21 | 25 (S) |
| 16 | (R)-BINAP | GVL | 80 | 72 | 99 | 94 | 21 | 10 (S) |
| 17 | (R)-BINAP | GVL | 60 | 240 | 70 | 90 | 26 | 28 (S) |
| 18 | (R)-BINAP | GVL | 40 | 336 | 46 | 95 | 23 | 49 (S) |
| 19 | (R)-SEGPHOS | Toluene | 100 | 24 | 99 | 82 | 48 | 21 (S) |
| 20 | (R)-SEGPHOS | GVL | 100 | 20 | 14 | 80 | 23 | 45 (S) |
| 21 | (S)-DM-SEGPHOS | Toluene | 120 | 5 | 83 | 77 | 36 | 1 (S) |
| 22 | (S)-DM-SEGPHOS | Toluene | 100 | 20 | 99 | 83 | 39 | 10 (S) |
| 23 | (S)-DM-SEGPHOS | Toluene | 80 | 72 | 99 | 90 | 46 | 21 (S) |
| 24 | (S)-DM-SEGPHOS | GVL | 120 | 20 | 30 | 55 | 25 | 24 (S) |
| 25 | (S)-DM-SEGPHOS | GVL | 100 | 20 | 45 | 86 | 24 | 14 (S) |
| 26 | (S)-DM-SEGPHOS | GVL | 80 | 71 | 10 | 92 | 32 | 36 (S) |
| 27 | (S)-DTBM-SEGPHOS | Toluene | 100 | 24 | 2 | 73 | 23 | n.d. |
| 28 | (S)-DTBM-SEGPHOS | Toluene | 100 | 48 | 11 | 74 | 42 | 40 (S) |
| 29 | (S)-DTBM-SEGPHOS | GVL | 100 | 24 | 17 | 82 | 16 | 46 (S) |
| 30i | (S)-DTBM-SEGPHOS | GVL | 100 | 24 | 20 | 93 | 24 | 56 (S) |
| 31 | (S)-DTBM-SEGPHOS | GVL | 80 | 48 | 20 | 98 | 12 | 57 (S) |
| 32 | (R)-(S)-JOSIPHOS | Toluene | 100 | 24 | 99 | 82 | 28 | 13 (S) |
| 33 | (R)-(S)-JOSIPHOS | Toluene | 60 | 48 | 99 | 88 | 34 | 6 (S) |
| 34 | (R)-(S)-JOSIPHOS | GVL | 100 | 24 | 52 | 80 | 22 | 31 (S) |
| 35 | (R)-(S)-JOSIPHOS | GVL | 60 | 72 | 6 | 97 | 30 | 70 (S) |
| 36i | (R)-(S)-JOSIPHOS | GVL | 60 | 240 | 13 | 94 | 28 | 36 (S) |
Additionally, efforts were made to separate and re-use the catalyst. Using PtCl2(PhCN)2 together with (S,S)-BDPP or (R)-BINAP ligands, we were able to separate the products as GVL solution from the catalyst by vacuum (10 mmHg) distillation at 80–82 °C. The glue-like residue containing the catalyst, which remained at the bottom of the distillation unit, was re-dissolved in GVL and reused in the next catalytic run. The activity and also the regioselectivity of the reaction decreased in those reactions carried out at 100 °C, reflecting the partial degradation of the catalyst. When recirculation experiments with the Pt-BINAP-tin(II)chloride system were carried out at a lower temperature (85 °C) the decrease in the catalytic activity was accompanied by the slight decrease in regioselectivity. Surprisingly, some increase in chemo- and enantioselectivities was observed (Table 3).
| Cycle | Conv. (%) | R c (%) | R br (%) | ee (%) |
|---|---|---|---|---|
| a Reaction conditions: Pt/styrene = 1/100, Pt/SnCl2 = 1/2; p(CO) = p(H2) = 40 bar, 1 mmol of styrene, solvent: 5 mL of GVL, reaction time 24 h. For Rc, Rbr and ee, see footnotes in Table 2. | ||||
| 1. | 76 | 94 | 16 | 22 (S) |
| 2. | 57 | 96 | 14 | 20 (S) |
| 3. | 33 | 97 | 12 | 26 (S) |
| 4. | 25 | 97 | 12 | 25 (S) |
The reused catalysts were investigated by 31P-NMR after removal of volatile compounds from the reaction mixture. Surprisingly, the characteristic 1/4/1 pattern of the PtCl2(diphosphine) complex could be seen but no presence of the corresponding trichlorostannato complex (Table 1) was detected. In addition, neither the signals of the uncoordinated diphosphine and its oxide nor that of the hemioxide was present even after the catalyst was re-circulated three times (ESI Fig. S10 and S11†). It can be assumed that the complete dissociation of tin(II)chloride in the presence of the rest of the reaction products led to the decreased activity of the catalyst. Presumably, the partial degradation of the catalyst cannot be related to the (partial) oxidation of the diphosphine ligand.
Conclusions
We have demonstrated that γ-valerolactone can be an excellent solvent in the enantioselective hydroformylation of styrene in the presence of platinum–diphosphine–tin(II)chloride ‘in situ’ generated catalysts. Various chiral diphosphines with central, axial and planar elements of chirality were tested. Although more active catalysts were formed in widely used toluene, the higher chemo- and enantioselectivities, as well as the green properties of GVL would deserve further investigations.Experimental
The platinum(II)chloride–dibenzonitrile complex was prepared as described before.32 The chiral ligands were purchased from Sigma-Aldrich and were used without further purification. Gamma-valerolactone was purchased from Sigma-Aldrich Kft., Budapest, Hungary and was purified by vacuum distillation (2 Torr, 80–82 °C). The purified GVL was stored under nitrogen.In a typical hydroformylation experiment, a solution of PtCl2(PhCN)2 (2.4 mg, 0.005 mmol), chiral diphosphine (0.005 mmol), and tin(II) chloride (1.9 mg, 0.01 mmol) in 5 mL of GVL containing 0.115 mL (1.0 mmol) of styrene was transferred under argon into a 100 mL stainless steel autoclave. The reaction vessel was pressurized to 80 bar total pressure (CO/H2 = 1/1) and placed in an oil bath of appropriate temperature and the mixture was stirred with a magnetic stirrer. Samples were taken from the mixture and the pressure was monitored throughout the reaction. After cooling and venting of the autoclave, the pale yellow solution was removed and immediately analysed by GC and chiral GC (on a capillary Cyclodex-column, (S)-2-phenylpropanal was eluted before the (R) enantiomer). For appropriate determination of enantiomeric excess, 10 mL of hexane was added to a sample of the reaction mixture (2 mL) and washed with water (twice 10 mL). The hexane phase was dried over Na2SO4, filtered and concentrated to a colorless oil. The CH2Cl2 solution of this GVL free sample was applied for chiral GC.
Acknowledgements
This work was supported by the Hungarian Scientific Research Fund (Grant No. OTKA K113177), “Environmental industry related innovative trans- and interdisciplinary research team development in the University of Pécs knowledge base” (SROP-4.2.2.D-15/1/KONV-2015–0015), “Synthesis of supramolecular systems, examination of their physicochemical properties and their utilization for separation and sensor chemistry” project (SROP-4.2.2.A-11/1/KONV-2012-0065) and National Research, Development and Innovation Office – NKFIH (PD 116559). L. T. Mika is grateful to the support of János Bolyai Research Scholarship of the Hungarian Academy of Sciences. The present scientific contribution is dedicated to the 650th anniversary of the foundation of the University of Pécs, Hungary.Notes and references
- C. Reichardt, Org. Process Res. Dev., 2007, 11, 105 CrossRef CAS.
- (a) F. M. Kerton, Alternative Solvents for Green Chemistry, RSC, Cambridge, 2009 Search PubMed; (b) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, UK, 1998 Search PubMed.
- http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/ManualofPoliciesProcedures/ucm124407.pdf .
- (a) F. Joó, in Aqueous Organometallic Catalysis, Catalysis by Metal complexes, ed. B. R. James and P. W. N. M. van Leeuwen, Kluwer Academic Pub., Dordrecht/Boston/London, 2001, vol. 23, p. 152 Search PubMed; (b) B. Cornils, W. A. Herrmann and W. E. Eckl, J. Mol. Catal. A: Chem., 1997, 116, 27 CrossRef CAS; (c) E. G. Kuntz, CHEMTECH, 1987, 570 CAS; (d) F. Joó and Á. Kathó, J. Mol. Catal. A: Chem., 1997, 116, 3 CrossRef; (e) Aqueous-Phase Organometallic Catalysis, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed; (f) B. Cornils, J. Mol. Catal. A: Chem., 1999, 143, 1 CrossRef CAS.
- (a) I. T. Horváth, Acc. Chem. Res., 1998, 31, 641 CrossRef; (b) I. T. Horváth and J. Rábai, Science, 1994, 266, 72 Search PubMed.
- (a) T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS PubMed; (b) J. P. Hallet and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef PubMed; (c) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS PubMed.
- W. Leitner, Acc. Chem. Res., 2002, 35, 746 CrossRef CAS PubMed.
- (a) E. G. Kuntz, CHEMTECH, 1987, 17, 570 CAS; (b) B. Cornils and E. Wiebus, CHEMTECH, 1995, 25, 33 CAS.
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238 RSC.
- V. Fábos, G. Koczó, H. Mehdi, L. Boda and I. T. Horváth, Energy Environ. Sci., 2009, 2, 767 Search PubMed.
- (a) J. M. Tukacs, M. Novák, G. Dibó and L. T. Mika, Catal. Sci. Technol., 2014, 4, 2908 RSC; (b) J. M. Tukacs, D. Király, A. Strádi, G. Novodárszki, Z. Eke, G. Dibó, T. Kégl and L. T. Mika, Green Chem., 2012, 14, 2057 RSC; (c) V. Fábos, L. T. Mika and I. T. Horváth, Organometallics, 2014, 33, 181 CrossRef; (d) F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510 CrossRef CAS PubMed; (e) H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49 CrossRef CAS; (f) J. M. Tukacs, B. Fridrich, G. Dibó, E. Székely and L. T. Mika, Green Chem., 2015 10.1039/C5GC01099C.
- (a) J. Q. Bond, D. M. Alonso, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110 CrossRef CAS PubMed.
- (a) D. Fegyverneki, L. Orha, L. Láng and I. T. Horváth, Tetrahedron, 2010, 66, 1078 CrossRef CAS; (b) A. Strádi, M. Molnár, M. Óvári, G. Dibó, F. U. Richter and L. T. Mika, Green Chem., 2013, 15, 1857 RSC; (c) A. Strádi, M. Molnár, P. Szakál, D. Gáspár, G. Dibó and L. T. Mika, RSC Adv., 2015, 5, 72529 RSC.
- P. K. Wong, C. Li, L. Stubbs, M. van Menurs, D. G. A. Kumband, C. Y. Lim and E. Drent, PTC/SG2012/000107, 2012.
- M. Chalid, H. J. Heeres and A. A. Broekhuis, J. Appl. Polym. Sci., 2012, 123, 3556 CrossRef CAS.
- V. Fábos, M. Y. Lui, Y. F. Mui, Y. Y. Wong, L. T. Mika, L. Qi, E. Cséfalvay, V. Kovács, T. Szűcs and I. T. Horváth, ACS Sustainable Chem. Eng., 2015, 3, 1899 CrossRef.
- (a) L. Qui and I. T. Horváth, ACS Catal., 2012, 2, 2247 CrossRef; (b) L. Qi, Y. F. Mui, S. W. Lo, M. Y. Lui, G. R. Akien and I. T. Horváth, ACS Catal., 2014, 4, 1470 CrossRef CAS.
- G. Strappaveccia, L. Luciani, E. Bartollini, A. Marrocchi, F. Pizzo and L. Vaccaro, Green Chem., 2015, 17, 1071 RSC.
- G. Strappaveccia, E. Ismalaja, C. Petrucci, D. Lanari, A. Marrocchi, M. Drees, A. Facchetti and L. Vaccaro, Green Chem., 2015, 17, 365 RSC.
- E. Ismalaj, G. Strappaveccia, E. Ballerini, F. Elisei, O. Piermatti, D. Gelman and L. Vaccaro, ACS Sustainable Chem. Eng., 2014, 2, 2461 CrossRef CAS.
- (a) C. Botteghi, S. Paganelli, A. Schionato and M. Marchetti, Chirality, 1991, 3, 355 CrossRef CAS PubMed; (b) S. Gladiali, J. C. Bayón and C. Claver, Tetrahedron: Asymmetry, 1995, 6, 1453 CrossRef CAS.
- (a) M. Jouffroy, D. Semeril, D. Armspach and D. Matt, Eur. J. Org. Chem., 2013, 6069 CrossRef CAS; (b) H. Fernandez-Perez, J. Benet-Buchholz and A. Vidal-Ferran, Org. Lett., 2013, 15, 3634 CrossRef CAS PubMed; (c) T. Adint, G. Wong and C. Landis, J. Org. Chem., 2013, 78, 4231 CrossRef CAS PubMed; (d) X. Wang and S. Buchwald, J. Org. Chem., 2013, 78, 3429 CrossRef CAS PubMed; (e) X. Zheng, B. Cao, T. Liu and X. Zhang, Adv. Synth. Catal., 2013, 355, 679 CrossRef CAS; (f) I. Mon, J. Amilan and A. Vidal-Ferran, Chem. – Eur. J., 2013, 19, 2720 CrossRef CAS PubMed; (g) R. Bellini and J. Reek, Chem. – Eur. J., 2012, 18, 13510 CrossRef CAS PubMed; (h) S. Chikkali, R. Bellini, B. de Bruin, J. I. van der Vlugt and J. N. H. Reek, J. Am. Chem. Soc., 2012, 134, 6607 CrossRef CAS PubMed; (i) T. Gadzikwa, R. Bellini, H. Dekker and J. N. H. Reek, J. Am. Chem. Soc., 2012, 134, 2860 CrossRef CAS PubMed; (j) G. Noonan, J. Fuentes, C. Cobley and M. Clarke, Angew. Chem., Int. Ed., 2012, 51, 2477 CAS; (k) X. Wan and S. Buchwald, J. Am. Chem. Soc., 2011, 133, 19080 CrossRef PubMed; (l) A. Watkins and C. R. Landis, Org. Lett., 2011, 13, 164 CrossRef CAS PubMed.
- (a) C. Y. Hsu and M. Orchin, J. Am. Chem. Soc., 1975, 97, 3553 CrossRef CAS; (b) I. Schwager and J. F. Knifton, J. Catal., 1976, 45, 256 CrossRef CAS.
- (a) G. Consiglio and P. Pino, Helv. Chim. Acta, 1976, 59, 642 CrossRef CAS; (b) Y. Kawabata, T. Hayashi and I. Ogata, J. Chem. Soc., Chem. Commun., 1979, 462 RSC; (c) T. Hayashi, Y. Kawabata, T. Isoyama and I. Ogata, Bull. Chem. Soc. Jpn., 1981, 54, 3438 CrossRef CAS.
- (a) G. Consiglio, P. Pino, L. I. Flowers and C. U. Pittmann Jr., J. Chem. Soc., Chem. Commun., 1983, 612 RSC; (b) P. Haelg, G. Consiglio and P. Pino, J. Organomet. Chem., 1985, 296, 281 CrossRef CAS; (c) G. Consiglio, F. Morandini, M. Scalone and P. Pino, J. Organomet. Chem., 1985, 279, 193 CrossRef CAS; (d) L. Kollár, G. Consiglio and P. Pino, J. Organomet. Chem., 1987, 330, 305 CrossRef.
- (a) G. Parrinello and J. K. Stille, J. Am. Chem. Soc., 1987, 109, 7122 CrossRef CAS; (b) S. Mutez, A. Mortreux and F. Petit, Tetrahedron Lett., 1988, 29, 1911 CrossRef CAS; (c) L. Kollár, J. Bakos, I. Tóth and B. Heil, J. Organomet. Chem., 1988, 350, 277 CrossRef; (d) L. Kollár, J. Bakos, I. Tóth and B. Heil, J. Organomet. Chem., 1989, 370, 257 CrossRef; (e) L. Kollár, J. Bakos, B. Heil, P. Sándor and G. Szalontai, J. Organomet. Chem., 1990, 385, 147 CrossRef; (f) L. Kollár, P. Sándor and G. Szalontai, J. Mol. Catal., 1991, 67, 191 CrossRef; (g) L. Kollár, T. Kégl and J. Bakos, J. Organomet. Chem., 1993, 453, 155 CrossRef; (h) S. Gladiali, D. Fabbri and L. Kollár, J. Organomet. Chem., 1995, 491, 91 CrossRef CAS; (i) B. Jedlicka, W. Weissensteiner, T. Kégl and L. Kollár, J. Organomet. Chem., 1998, 563, 37 CrossRef CAS; (j) C. G. Arena, F. Faraone, C. Graiff and A. Tiripicchio, Eur. J. Inorg. Chem., 2002, 711 CrossRef CAS; (k) C. Hegedűs, J. Madarász, H. Gulyás, A. Szőllősy and J. Bakos, Tetrahedron Asymmetry, 2001, 12, 2867 CrossRef.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702 CrossRef CAS PubMed.
- I. Tóth, I. Guo and B. E. Hanson, J. Mol. Catal., 1997, 116, 217 CrossRef.
- Y. Chauvin, L. Mussmann and H. Olivier, Angew. Chem., Int. Ed. Engl., 1995, 34, 2698 CrossRef CAS.
- G. Francio and W. Leitner, Chem. Commun., 1999, 1663 RSC.
- (a) C. P. Casey, S. C. Martins and M. A. Fagan, J. Am. Chem. Soc., 2004, 126, 5585 CrossRef CAS PubMed; (b) P. Pongrácz, T. Papp, L. Kollár and T. Kégl, Organometallics, 2014, 33, 1389 CrossRef.
- F. R. Hartley, Organomet. Chem. Rev., Sect. A, 1970, 6, 119 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01778e |
| This journal is © The Royal Society of Chemistry 2016 |