Glutaraldehyde in bio-catalysts design: a useful crosslinker and a versatile tool in enzyme immobilization
Oveimar
Barbosa
a,
Claudia
Ortiz
b,
Ángel
Berenguer-Murcia
c,
Rodrigo
Torres
a,
Rafael C.
Rodrigues
*d and
Roberto
Fernandez-Lafuente
*e
aEscuela de Química, Grupo de investigación en Bioquímica y Microbiología (GIBIM), Universidad Industrial de Santander, Edificio Camilo Torres 210, Bucaramanga, Colombia
bEscuela de Bacteriología y Laboratorio Clínico, Universidad Industrial de Santander, Bucaramanga, Colombia
cInstituto Universitario de Materiales, Departamento de Química Inorgánica, Universidad de Alicante, Campus de San Vicente del Raspeig, Ap. 99, 03080 Alicante, Spain
dBiocatalysis and Enzyme Technology Lab, Institute of Food Science and Technology, Federal University of Rio Grande do Sul, ICTA-UFRGS, Av. Bento Gonçalves, 9500, P.O. Box 15090, ZC 91501-970, Porto Alegre, RS, Brazil. E-mail: rafaelcrodrigues@ufrgs.br
eDepartamento de Biocatalisis, ICP-CSIC, Campus UAM-CSIC, C/Marie Curie 2 Cantoblanco, 28049 Madrid, Spain. E-mail: rfl@icp.csic.es
First published on 28th October 2013
Abstract
Glutaraldehyde is one of the most widely used reagents in the design of biocatalysts. It is a powerful crosslinker, able to react with itself, with the advantages that this may bring forth. In this review, we intend to give a general vision of its potential and the precautions that must be taken when using this effective reagent. First, the chemistry of the glutaraldehyde/amino reaction will be commented upon. This reaction is still not fully clarified, but it seems to be based on the formation of 6-membered heterocycles formed by 5 C and one O. Then, we will discuss the production of intra- and inter-molecular enzyme crosslinks (increasing enzyme rigidity or preventing subunit dissociation in multimeric enzymes). Special emphasis will be placed on the preparation of cross-linked enzyme aggregates (CLEAs), mainly in enzymes that have low density of surface reactive groups and, therefore, may be problematic to obtain a final solid catalyst. Next, we will comment on the uses of glutaraldehyde in enzymes previously immobilized on supports. First, the treatment of enzymes immobilized on supports that cannot react with glutaraldehyde (only inter and intramolecular cross-linkings will be possible) to prevent enzyme leakage and obtain some enzyme stabilization via cross-linking. Second, the cross-linking of enzymes adsorbed on aminated supports, where together with other reactions enzyme/support crosslinking is also possible; the enzyme is incorporated into the support. Finally, we will present the use of aminated supports preactivated with glutaraldehyde. Optimal glutaraldehyde modifications will be discussed in each specific case (one or two glutaraldehyde molecules for amino group in the support and/or the protein). Using preactivated supports, the heterofunctional nature of the supports will be highlighted, with the drawbacks and advantages that the heterofunctionality may have. Particular attention will be paid to the control of the first event that causes the immobilization depending on the experimental conditions to alter the enzyme orientation regarding the support surface. Thus, glutaraldehyde, an apparently old fashioned reactive, remains the most widely used and with broadest application possibilities among the compounds used for the design of biocatalyst.
1. Introduction
Enzymes are biocatalysts which catalyze most metabolic reactions in living beings. In vivo, they are highly specific (modifying just one substrate among a collection of similar ones), chemo/enantio/regio-selective (yielding just one substrate among several possible) and very active under very mild environmental conditions (atmospheric pressure, room temperature, aqueous medium). Thus, enzymes have been considered the ideal catalyst from an environmental point of view, in reactions involving complex or labile compounds.1–5However, enzymes perform their function inside living beings under complex regulations and stress. This causes enzymes to become inhibited by many compounds. Thus, their in vivo stability can hardly be measured in days, and their exceptional properties may not be found when they are utilized for modifying other substrates (different to the physiological ones) or even performing other reactions (e.g., using hydrolases as transferases in kinetically controlled synthesis). Moreover, the soluble nature of the enzymes prevents their extended use (and subsequent re-use) in industry. These properties are far from the requirements of an industrial catalyst.6
The solution to these limitations is the main subject of enzyme technology. The researcher may use a handful of different tools, such as microbiology,2 genetic approaches,7–10 immobilization,11–17 chemical modification,18,19 and medium and reactor engineering to shortcut these enzyme limitations. Many of these tools may be used in an integrated way.20–23
Physicochemical tools, such as chemical modification or enzyme immobilization, have special interest in this context. Due to the requirements of producing a heterogeneous catalyst, enzyme immobilization becomes a necessity from this perspective.14 This strategy has been investigated by many authors as a way to improve enzyme properties. It has been shown that enzyme stability may be significantly improved if an intense multipoint covalent attachment (MCA) between an enzyme molecule and a rigid support, via a short spacer arm, is achieved.17
This phenomenon causes all groups involved in the immobilization to keep their relative positions when the enzyme is submitted to any conformational change. The area of the enzyme involved in the immobilization may be also critical to maximize the stabilizing effect of the MCA.22 Immobilization may also tune some other enzyme properties, such as activity, resistance to inhibition, selectivity or specificity.17 Chemical modification of enzymes may be used to further improve enzyme stability (e.g., via chemical crosslinking),24 and may be also used as another tool to alter enzyme catalytic features.18 Moreover, as it has been revised, chemical modification or immobilization of enzymes may be designed to simplify or improve one another.21
Glutaraldehyde is the reagent that the present review is devoted to. It has been used in many instances as protein cross-linker, as an activator of supports, and as crosslinker of enzymes and supports.25,26 In this review, we intend to give a wide vision of the prospects of this very interesting and versatile molecule in the design of biocatalysts.
2. Chemistry of glutaraldehyde
Glutaraldehyde is a bi-functional reagent with the capacity to polymerize.27,28 Glutaraldehyde may react with different enzyme moieties, mainly involving primary amino groups of proteins, although it may eventually react with other groups (thiols, phenols, and imidazoles).27–30 However, the exact structure of the main structures related to protein crosslinking or enzyme immobilization is still not fully clarified. Fig. 1 shows some of these proposed structures. It is clear that the structure of the glutaraldehyde relevant for the modification of enzymes and supports is not a linear one, but some kind of fairly stable cycles (activated support may be washed with an excess of distilled water without losing glutaraldehyde moieties).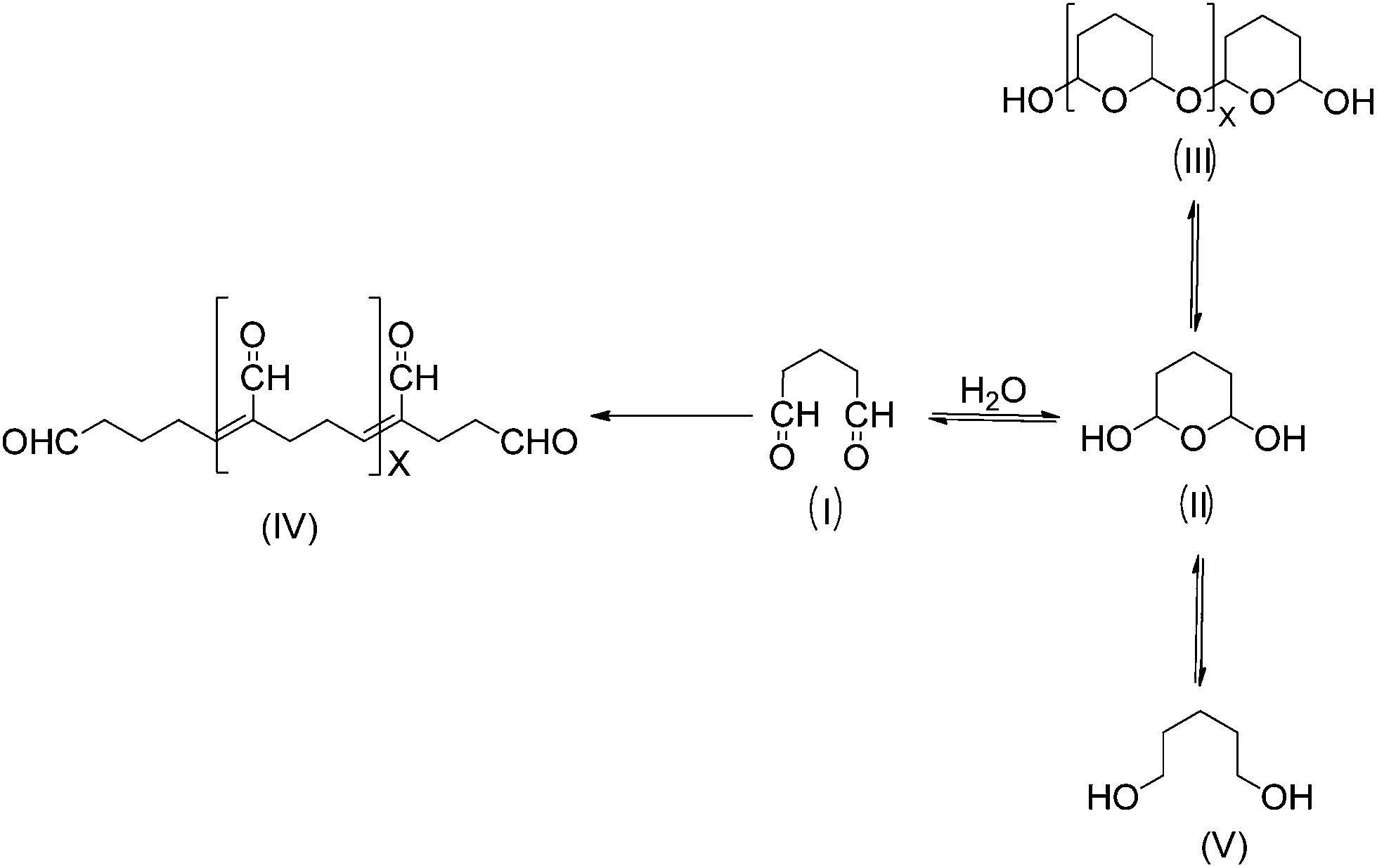 | ||
| Fig. 1 Possible structures of glutaraldehyde in aqueous solution. | ||
The reaction mechanism of glutaraldehyde with proteins implies that it is not limited to just one mechanism. This is because the main reactive species of glutaraldehyde are found in equilibrium between their monomeric and polymeric conformations.27,28,31,32 Moreover, every structure can react in a different way with the protein. For instance, under both acidic and neutral conditions, aldehyde groups from glutaraldehyde can react with proteins by formation of Schiff bases. In this case, a nucleophilic attack takes place by the ε-amino group from lysine to glutaraldehyde (see Fig. 2a). However, Schiff bases are unstable at acidic conditions and are broken up, regenerating both the aldehyde and the amine groups. For this reason, several procedures have recommended reduction by NaBH4 or NaBCNH3 as a final step in order to stabilize the Schiff base into a secondary amine. Nevertheless, some studies suggest that protein preparations treated with a reducing agents does not cause an striking increase in the enzyme stability.33 Additionally, lysine residues of protein treated with glutaraldehyde without further chemical reduction are not regenerated by incubation in HCl 6 M at 110 °C by 24 h.34 Consequently, it is possible that a mechanism of formation of Schiff bases between proteins and glutaraldehyde could not be carried out. In this sense, an additional reduction step would not be necessary in order to stabilize the reaction product.27,28,35 Therefore, the mechanism of glutaraldehyde at neutral and acidic conditions would be mediated by hemiacetal cyclic conformations from both the monomer and polymer of glutaraldehyde. This cyclic hemiacetal of glutaraldehyde reacts by nucleophilic substitution of the amino groups from lysines with the hydroxy group of glutaraldehyde according to the Fig. 2.
 | ||
| Fig. 2 Reactions of glutaraldehyde with proteins under acidic or neutral conditions. | ||
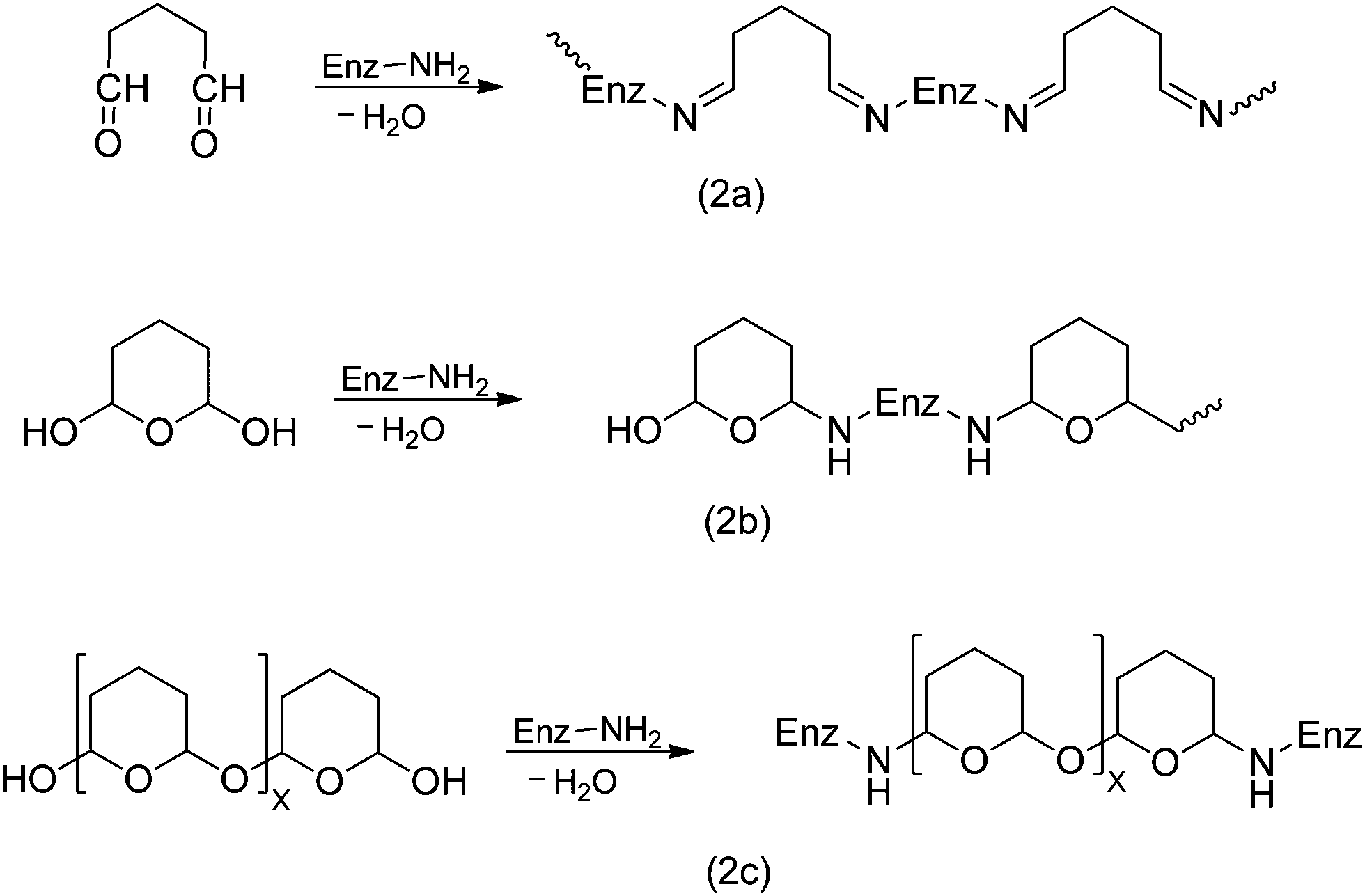
On the other hand, under basic conditions it has been proposed that glutaraldehyde quickly suffers intramolecular aldolic condensations, producing a polymeric form of an a α,β-unsaturated aldehyde, which may react with amino groups from proteins through two mechanisms: Firstly by formation of Schiff bases between internal aldehyde groups from the polymeric form of glutaraldehyde and primary amino groups from the protein (Fig. 3, reaction 1). In this case, the obtained product could be stabilized by a resonance effect of conjugated double C–C bonds.32 The second mechanism involves a Michael addition to the double C–C linkage.27 However, this reaction results in a less stable product due to loss of resonance effect of the conjugated double C–C bonds, which would be labile under acidic conditions (Fig. 3).
 | ||
| Fig. 3 Schiff base (1) and Michael-type (2) reactions of glutaraldehyde with proteins under basic conditions. | ||
Prof. Monsan,36,37 has shown that it is relatively simple to only have one or two molecules of glutaraldehyde per amino group in a support. This result, coupled to some more recent ones that confirm this, suggested a different reactivity of the amino/glutaraldehyde moiety with free glutaraldehyde compared to the capacity of free glutaraldehyde molecule to polymerize, as under the described conditions to get this amino/glutaraldehyde/glutaraldehyde moiety there is not a massive precipitation of glutaraldehyde polymers.38 Amino/glutaraldehyde reacted with free glutaraldehyde much more rapidly that free glutaraldehyde with free glutaraldehyde molecules. It also showed that the two glutaraldehyde molecules/amino moiety groups in the support possessed the structure that exhibited the highest reactivity versus amino groups in a protein. If just one glutaraldehyde molecule was attached to the amino group, this moiety exhibited a low reactivity versus amino groups. However, this group, together with the already commented reactivity versus free glutaraldehyde molecules, exhibited a very high reactivity versus other similar amino/glutaraldehyde moieties, being this activation of a protein the preferred way to get crosslinkings as we will discuss later. This different reactivity of the amino/glutaraldehyde moiety compared to the glutaraldehyde or the amino/glutaraldehyde/glutaraldehyde moiety should be found in the presence of an amino group in the heterocycle of the glutaraldehyde ring.39,40 Amino/glutaraldehyde/glutaraldehyde has a similar reactivity when compared to free glutaraldehyde, as the second glutaraldehyde group will be very similar to free glutaraldehyde. If the conditions were forced (e.g., increasing the pH value or the glutaraldehyde concentration) to get a higher degree of polymerization on the amino group, the free glutaraldehyde may also react; giving large glutaraldehyde aggregates that may precipitate and the suspension will turn white.
3. Glutaraldehyde as protein crosslinker
The first use of glutaraldehyde was to preserve and fix tissues41,42 in some instances combined with formaldehyde.25 This is achieved through the formation of intermolecular crosslinking. Nowadays, glutaraldehyde remains as one of the most potent crosslinker reagents, even with clinical applications.43–45 In part, these very good crosslinker features are a consequence of the capacity of glutaraldehyde to react with itself or with protein groups already modified with a glutaraldehyde molecule.28The protein crosslinkings may be among groups placed on different protein molecules (intermolecular crosslinking) or between groups placed in the same molecule (intramolecular crosslinkings).24,46 Both kinds of crosslinkings have interest in specific cases.
3.1. Glutaraldehyde as intermolecular crosslinker of proteins
As previously commented, this was the first use of glutaraldehyde, using its crosslinker potential to fix tissues.41 However, in this review we will focus on the use of glutaraldehyde in the design of biocatalysts.The addition of glutaraldehyde to a protein solution may produce the chemical aggregation of the enzyme, causing protein molecules to react among themselves, and can directly yield a “solid biocatalyst” (Fig. 4). Although this immobilization strategy has not been widely used, it is possible to find diverse examples in literature. For example, insoluble trypsin was prepared by the use of glutaraldehyde to produce intermolecular crosslinks and the insoluble trypsin thus prepared exhibited enzymatic activity toward casein.47 Later, glutaraldehyde was reacted in aqueous solutions with papain to form a water-insoluble product with enzymatic activity after activation by reducing agents. A rapid reaction of glutaraldehyde with the essential sulfhydryl of papain was not involved in the reaction since after activation the insoluble enzyme retained esterolytic and proteolytic activity.48
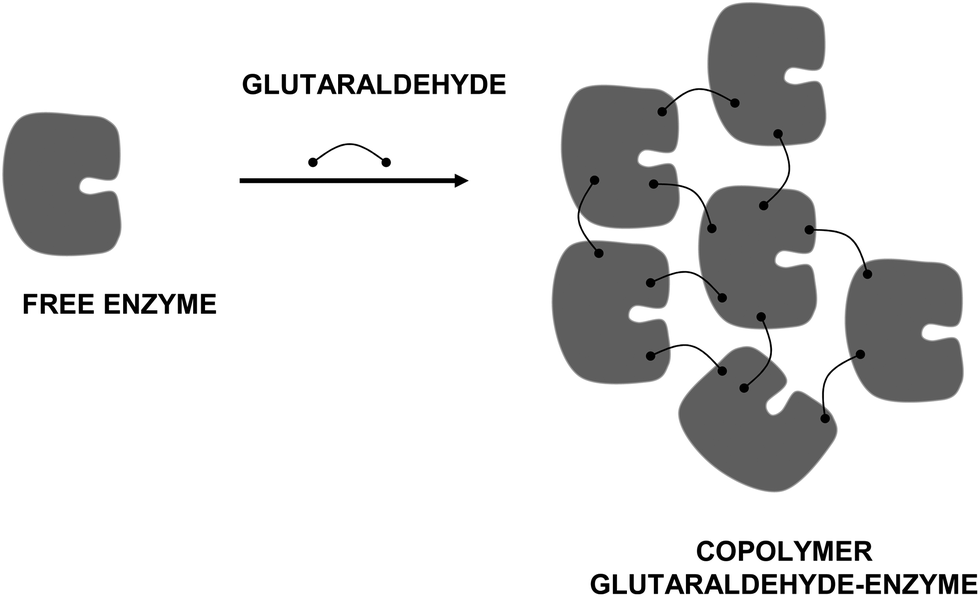 | ||
| Fig. 4 Immobilization of enzyme via copolymerization with glutaraldehyde. | ||
In another example, a purified preparation of extracellular alkaline proteinase of Trichoderma koningii was insolubilized by intermolecular crosslinking with glutaraldehyde.49 The optimum operational temperature of the insolubilized enzyme increased by 20 °C. The immobilized enzyme was also relatively more stable and activity was less depended on the presence of ions or detergents than the soluble enzyme. More surprisingly, an enhanced affinity to casein, hemoglobin and bovine serum albumin was found, although with a lower Vmax values.
Intermolecular cross-linking of the protease stem bromelain with 0.25 and 1.25% glutaraldehyde results in the formation of a large molecular mass, multimeric and soluble aggregate having comparable activity to the unmodified bromelain. Both 0.25 and 1.25% glutaraldehyde cross-linked bromelain preparations were more stable against urea, guanidine hydrochloride and temperature-induced inactivation, and exhibited slightly better storage stability compared to the unmodified protease. Such a high molecular weight, soluble, active and stable preparation may be useful in industry, i.e. in the textile industry for improving the properties of a fabric without loss of fabric strength and shape.50
On the other hand, glutaraldehyde is the intermolecular crosslinker of some of the recent carrier-free immobilization protocols, such as crosslinked enzyme crystals (CLECs)51,52 (Fig. 5) or aggregates (CLEAs) (Fig. 6).53–56 While using a conventional homo-bi-functional reagent the ideal strategy seems to modify around 50% of the reactive groups of the protein to maximize the possibility of crosslinking, the chemistry of glutaraldehyde causes this option not to be the optimal one, as amino/glutaraldehyde moieties reacts better with other amino/glutaraldehyde groups than one amino group.33 Moreover, amino/glutaraldehyde/glutaraldehyde did not react easily with other amino/(glutaraldehyde)2 groups, making it inconvenient to excessively modify the proteins. Thus, to reach protein crosslinking using glutaraldehyde, it seems adequate to use moderate concentrations of glutaraldehyde, high enough to ensure the activation of most amino groups with one molecule of glutaraldehyde, but not too high so as not to reach activations with two molecules of glutaraldehyde per amino group (e.g., 0.1–1% (v/v) glutaraldehyde at pH 7 for 1 hour).36 Milder activation of the amino groups of the enzyme may produce a milder crosslinking. However, in some instances it may be necessary to use suboptimal crosslinking conditions if the protein is especially sensible to glutaraldehyde modifications. In certain cases, as if the enzyme has a Lys group in its catalytic site (e.g., some aldolases), glutaraldehyde must be discarded and other crosslinking reagent should be used, such as aldehyde dextran.57
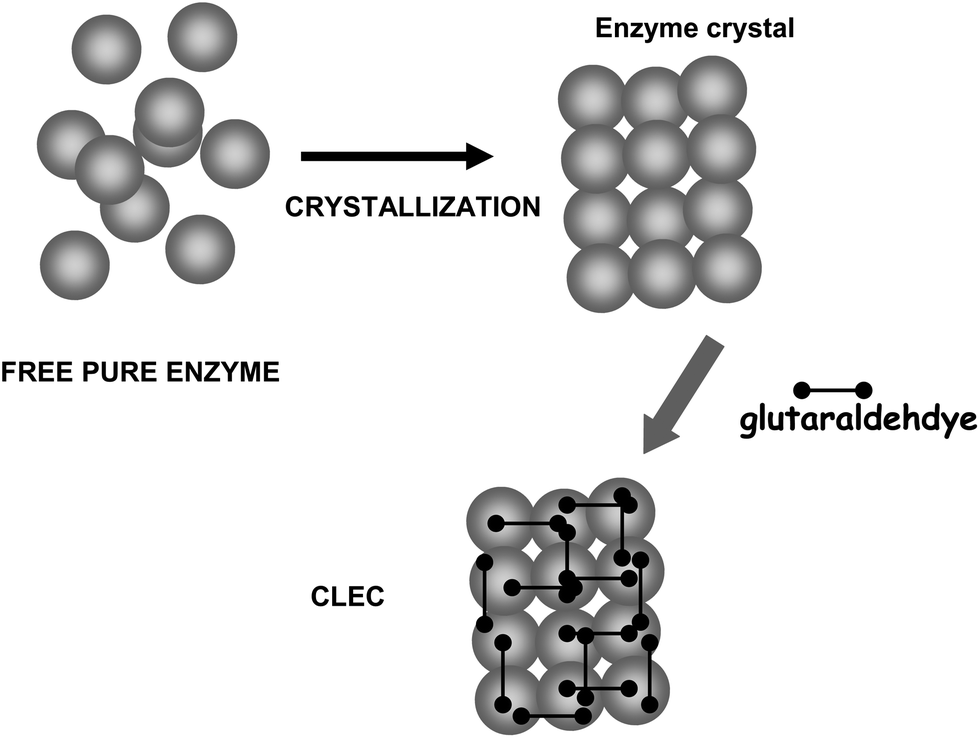 | ||
| Fig. 5 Preparation of crosslinked enzyme crystals (CLECs). | ||
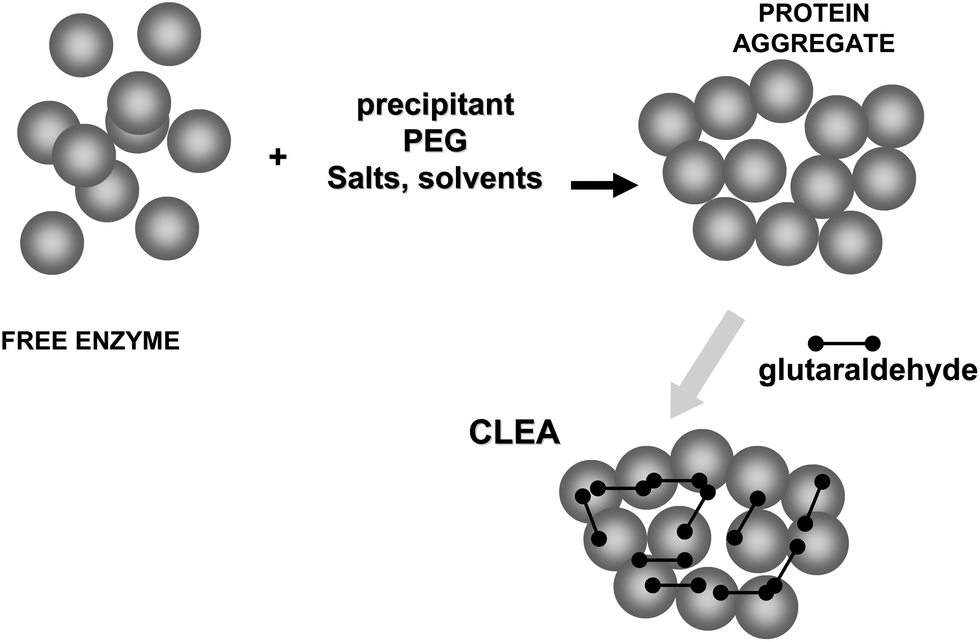 | ||
| Fig. 6 Preparation of crosslinked enzyme aggregates (CLEAs). | ||
To achieve a proper crosslinking in this kind of immobilization strategies may be a problem, mainly if the protein has not many Lys on its surface (Fig. 7). This may make the formation of large aggregates difficult and enable the release of enzyme molecules (individual molecules or small aggregates) from the solid. In some cases, it is even not possible to get a solid in aqueous medium. Using CLEAs some solutions have been offered to overcome this situation. Taking advantage of the fact that the enzyme did not need to be pure to prepare CLEAs, the co-precipitation of the target enzyme with inert proteins having a high density of superficial Lys groups (e.g. bovine serum albumin)58–63 or with polymers having many amino groups (e.g., polyethylenimine)64–67 has been proposed (Fig. 8). Both strategies permit the formation of physically stable CLEAs, but they also produce a decrease in the volumetric loading of the target enzyme, as part of the volume will be occupied by the inert protein or the polymer. As an alternative, a strategy based on the use of chemically aminated enzymes with their protein surface enriched on reactive groups has been proposed (Fig. 9).68 This way, it is not necessary to mix the target protein with any other molecule or polymer and the volumetric loading of the CLEA is similar to that of proteins that may be directly used to prepare CLEAs.
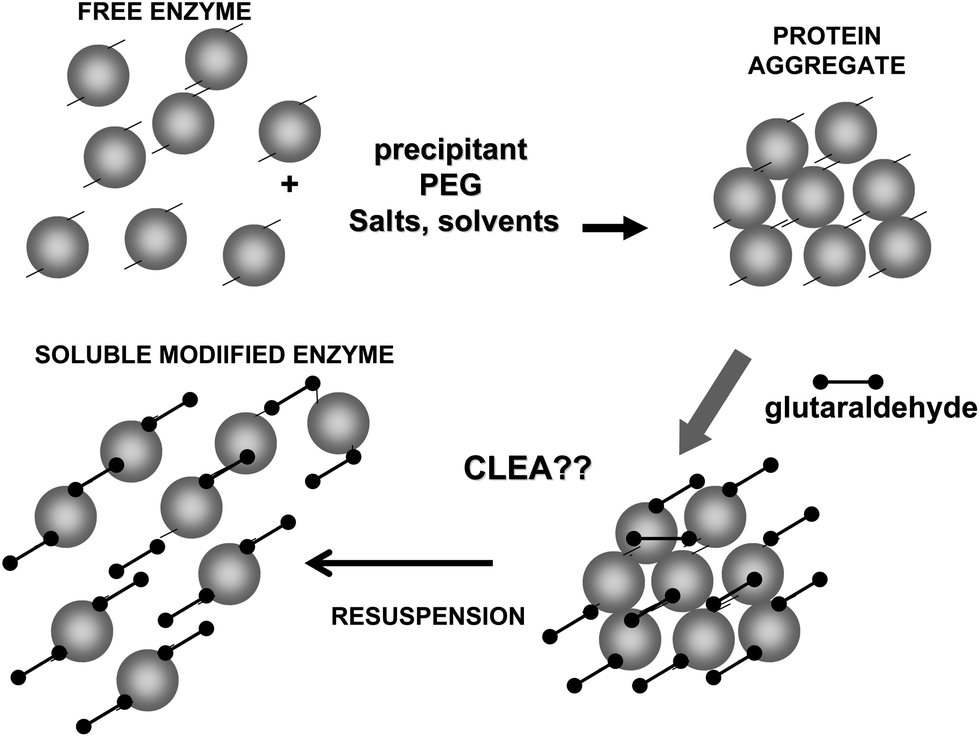 | ||
| Fig. 7 Problems in the crosslinking step during CLEAs preparation using enzymes poor in Lys residues. | ||
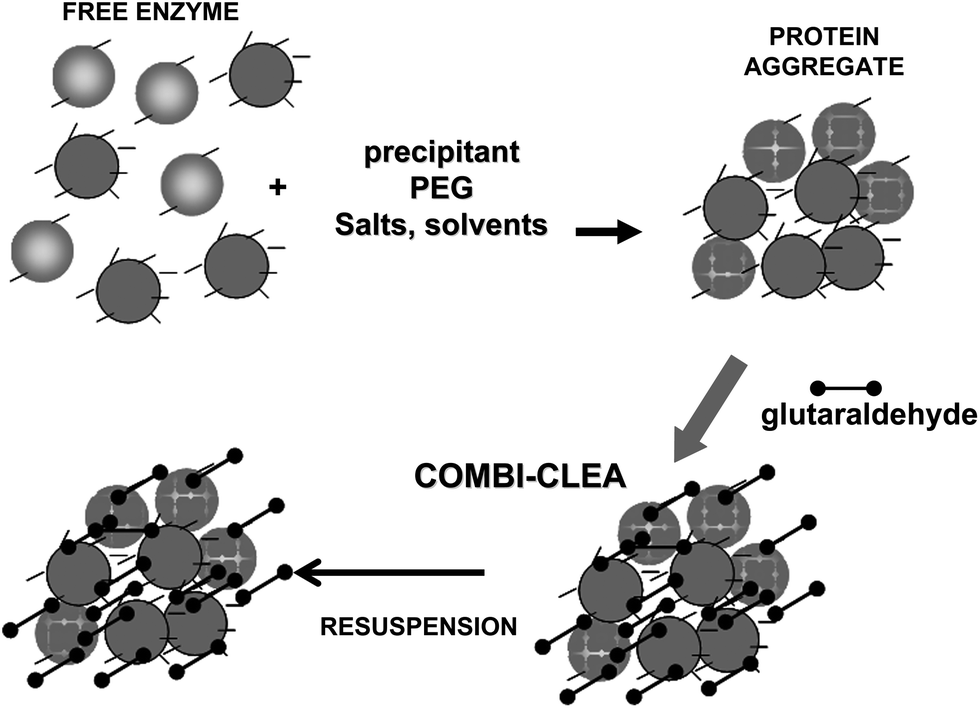 | ||
| Fig. 8 Preparation of combi-CLEAs using proteins having many Lys residues to facilitate the crosslinking step. | ||
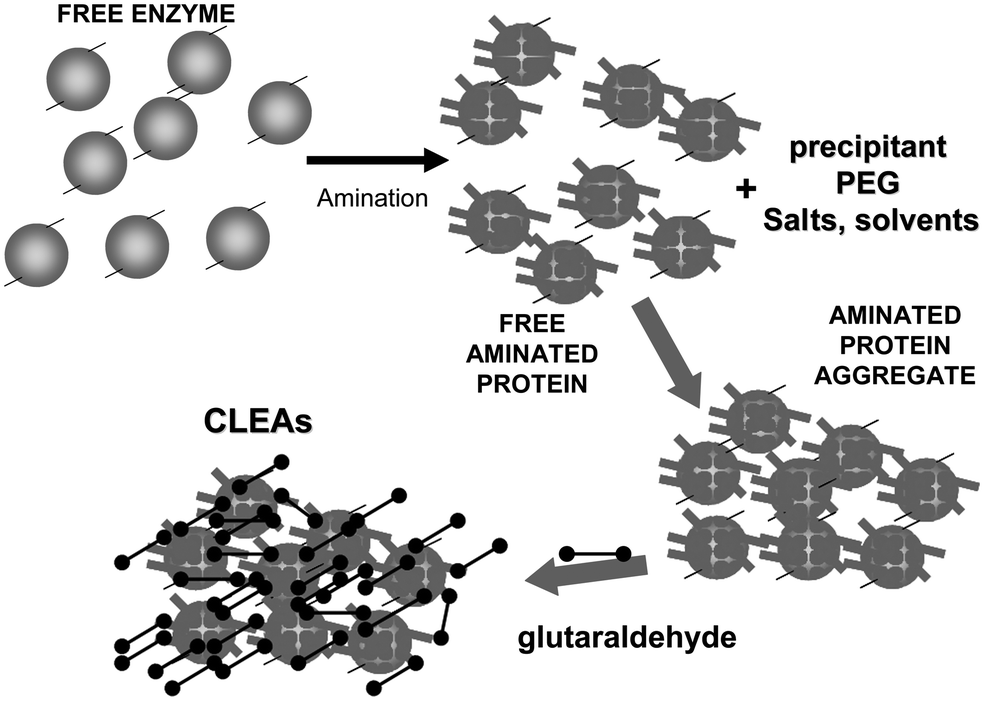 | ||
| Fig. 9 Amination of enzymes that are poor in Lys residues to facilitate the crosslinking step in CLEA preparation. | ||
3.2. Glutaraldehyde as intramolecular crosslinker of proteins
The introduction of intramolecular crosslinkings in an enzyme structure is one of the most widely used techniques to increase enzyme stability, using genetic or chemical routes.24,69,70 If the crosslinking agent is short enough, the relative mobility of one group relative to the other crosslinked group should be minimized.71 Furthermore, if the researcher is able to introduce several crosslinkings on the enzyme surface, the final result should be a global rigidification of the overall structure of the enzyme (Fig. 10).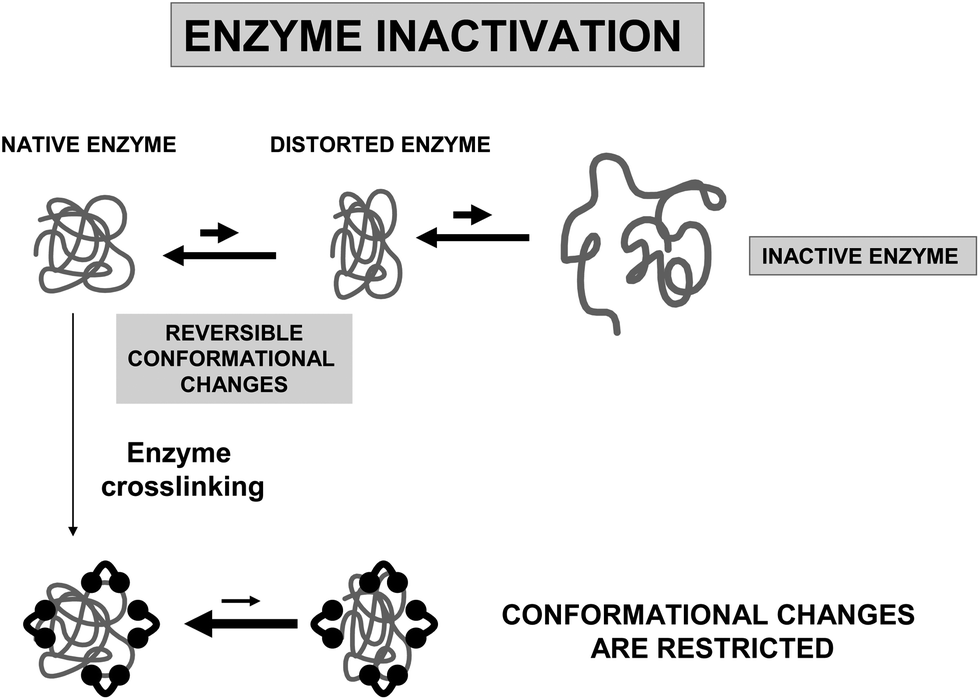 | ||
| Fig. 10 Effect of intramolecular crosslinking on the stability of enzymes. | ||
However, to introduce several intramolecular crosslinkings in a protein molecule and to get a significant stabilization effect is not a simple task, as many problems may rise. We will detail some of the most relevant ones.
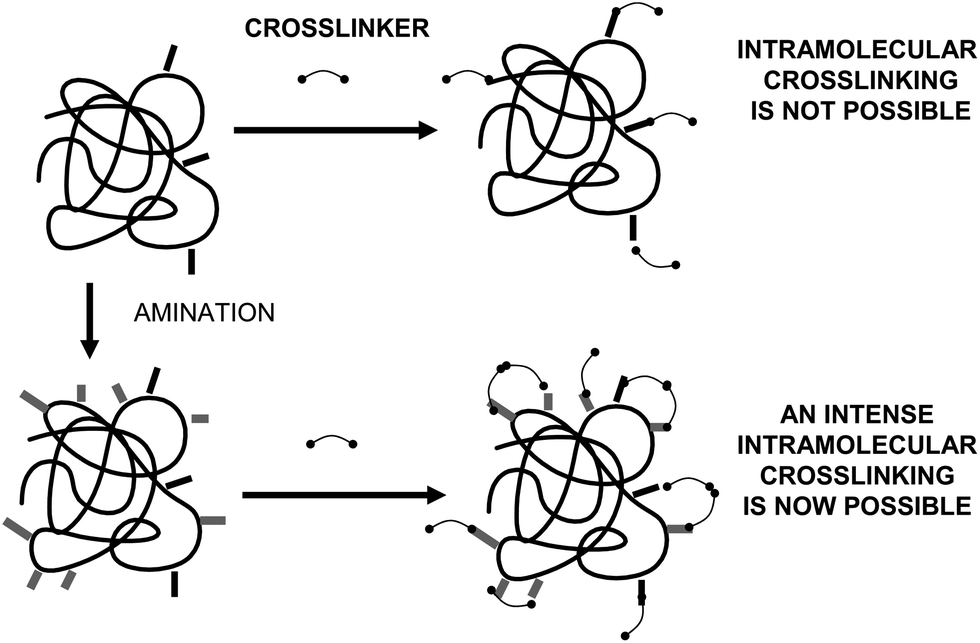 | ||
| Fig. 11 Amination of enzyme surfaces to facilitate the promotion of an intense intramolecular crosslinking. | ||
Even when using immobilized enzymes, to fully prevent enzyme intermolecular crosslinking may be hard. Using porous supports, if the immobilization rate is much higher than the diffusion rate, the enzymes may be packed so near each other that the enzyme molecules may be crosslinked with each other even using short reagents like glutaraldehyde (Fig. 12). If the immobilization rate is slow enough, the distance between enzyme molecules may be enough to almost fully prevent enzyme intermolecular crosslinking.83 Recently, by changing the immobilization conditions, a lipase has been immobilized on octyl agarose and treated with glutaraldehyde.84 Under one immobilization condition, immobilization was quite slow (in presence of ethanol) and intermolecular crosslinking was almost negligible and it was possible to analyze the effect of intramolecular modifications. Under another condition, immobilization rate was very high and the enzyme molecules were so near that both modifications could be achieved. The comparison between both derivatives (one having only intramolecular modifications, the other having both intramolecular and intermolecular modifications) permitted to determine the positive effects of the intermolecular crosslinking in the enzyme stability.
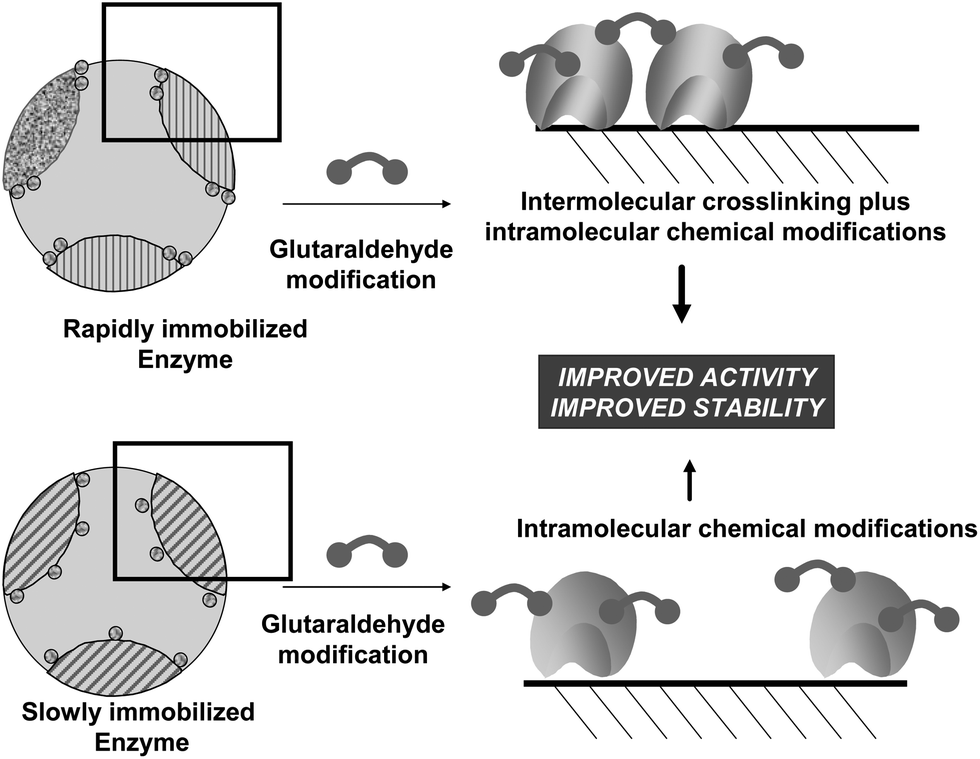 | ||
| Fig. 12 Effect of the immobilization rate on the possibilities of intermolecular crosslinking between immobilized enzyme molecules. | ||
3.3. Crosslinking of multimeric enzymes
Many enzymes are formed by several subunits, which are in association/dissociation equilibrium, usually being the associated form the most active and stable.85–87 In other cases, some cascade reactions may require the joint action of several different enzymes that require their being associated.88–90 If the researcher is able to crosslink these structures, the system will work in a more adequate fashion. However, the difficulties commented for the intramolecular crosslinking remain valid here, even with one additional key point: the reactive groups at the crosslinking distance need to be placed in different enzyme subunits (Fig. 13).23 This is why crosslinking of these structures may be achieved in a better way using polymers, as they should not confer an intense structural rigidity, but may be useful to keep together the different enzyme subunits or maintain the multi enzymatic complex assembled (e.g., dextran aldehyde,91 polyehtylenimine92,93). Nevertheless, glutaraldehyde has been used in certain cases to stabilize, even though only in a small percentage of the total enzyme structures, the subunit–subunit interactions. For example, the crosslinking of the hetero-oligomeric glucose dehydrogenase from a moderate thermophilic bacterium, SM4, using glutaraldehyde as crosslinking reagent has been reported.94 The treatment permitted glucose dehydrogenase to gain high thermal stability without loss of its catalytic activity and thus increase the activity of the enzyme at high temperature. The authors concluded that by chemical cross-linking of the subunits, the dissociation of alpha and beta subunits was prevented. Consequently, its quaternary structure was stabilized, and thus the thermal stability of the glucose dehydrogenase was enhanced. Also, D-lactate dehydrogenase from Limulus polyphemus is a homodimer which is composed of identical subunits that has been crosslinked with glutaraldehyde to show a relation of reactivation with reassociation of the dimer.95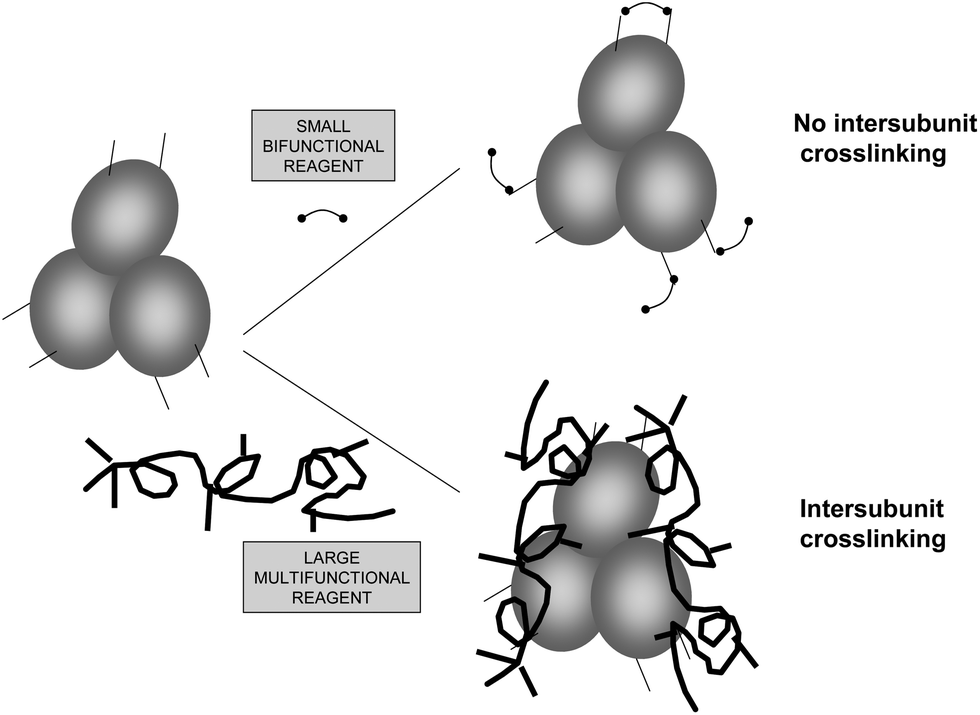 | ||
| Fig. 13 Chemical crosslinking of subunits in multimeric enzymes. | ||
In another paper, primase/helicase produced by bacteriophage T7 that can form both hexamers and heptamers was studied. These oligomers were stabilized via cross-linking with glutaraldehyde and purified.96 The authors detected how the percentage of each oligomeric form could be altered by the presence of either dTTP or β,γ-methylene. Heptamers are unable to efficiently bind either single-stranded DNA or double-stranded DNA, thus the authors postulated that a switch between heptamer to hexamer may provide a ring-opening mechanism for the single-stranded DNA binding pathway.
NTPDase1 and NTPDase2 enzymes from rats were expressed in Xenopus laevis oocytes and their quaternary structure was analyzed.97 The treatment with glutaraldehyde permitted to detect that native NTPD-ase1 and NTPD-ase2 occur in oligomeric form. Dynamic alterations in oligomeric state may induce changes in substrate preference and thus influence the pattern of in situ extracellular nucleotide degradation.
In some instances, glutaraldehyde may have an indirect use to stabilize a multimeric enzyme, by crosslinking a polymer to the enzyme surface avoiding the polymer release. The coating of the surface of multimeric enzymes with a poly-ionic polymer that may simultaneously interact with several enzyme subunits, preventing enzyme dissociation has proved to prevent enzyme subunit dissociation if the polymer can become adsorbed on the enzyme (Fig. 14).93 This strategy has been recently utilized to stabilize the enzyme glutamate dehydrogenase from Thermus thermophilus and formate dehydrogenase from Pseudomonas sp., using polyethyleneimine as the “crosslinking” polymer. Both enzymes were inactivated by dissociation at acidic pH value, and the coating of their surface with polyethyleneimine prevented this (Fig. 14). However, the reversible nature of the polymer adsorption permitted the polymer to become desorbed under certain conditions (e.g., high ionic strength, drastic pH value), and the protective effect of the polymer coating was lost. This problem was solved by a further treatment with glutaraldehyde, crosslinking the enzyme and the polymer: the new composite was fully stable under enzyme dissociation conditions. This composite could be used in soluble form, or ionically exchanged on a cation exchanger.93 When a similar strategy was applied to the hexameric glutamate dehydrogenase from E. coli, the polymer protection was successful, but the enzyme was rapidly inactivated by incubation with glutaraldehyde.92 Thus, this strategy not only requires that the polyethyleneimine may stabilize the multimeric structure of the enzyme without inactivating it, but also requires that the enzyme was resistant to the treatment with glutaraldehyde.
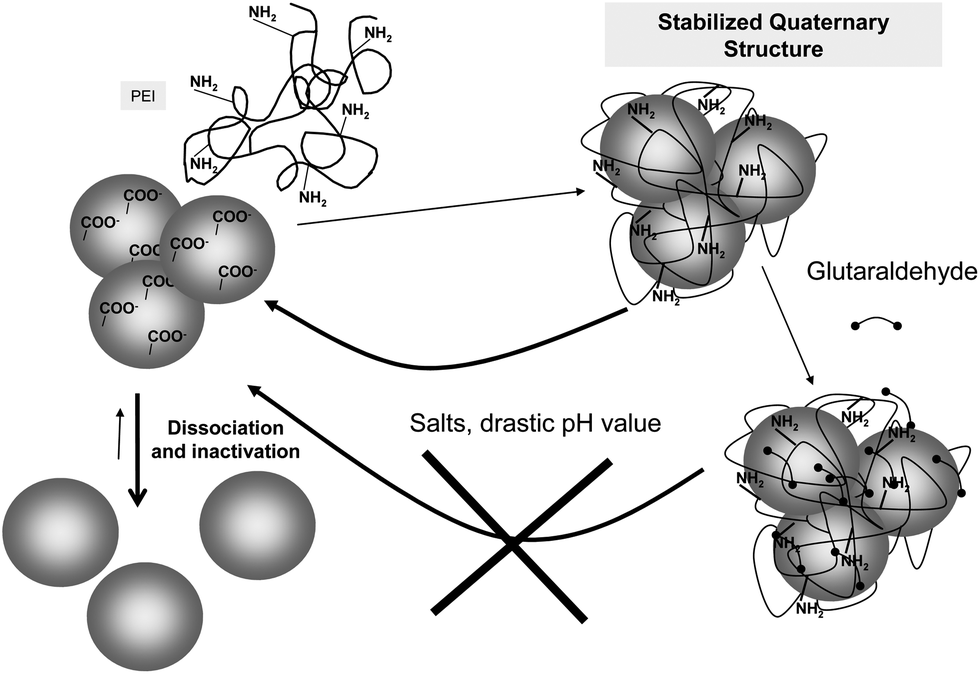 | ||
| Fig. 14 Stabilization of polyethyleneimine/enzyme composites via glutaraldehyde crosslinking to prevent subunit dissociation. | ||
4. Intermolecular crosslinking of immobilized proteins via reversible immobilization using supports unreactive towards glutaraldehyde
Enzyme immobilization by physical adsorption (using ion exchange, immobilized chelates or hydrophobic adsorption) is the simplest protocol to immobilize enzymes, does not require enzyme nor support treatment, immobilization is rapid, and the support does not require special storage conditions.14 However, this enzyme immobilization strategy has two disadvantages. The first one, the enzyme may be released from the support under certain experimental conditions.98,99 Using ion exchangers, this may occur when changing the pH value or increasing the ionic strength. Using hydrophobic adsorption, if the reaction requires the presence of co-solvents or detergents the enzyme may be released to the medium. The second problem is that physical adsorption may have no positive effects on enzyme stability, even as the support retains its capacity to interact with the enzyme, the immobilization may produce a certain destabilization of the enzyme.14Thus, to prevent enzyme desorption and improve enzyme stability, it is relatively common to find reports where the researcher, after enzyme reversible immobilization, treats the immobilized enzyme with glutaraldehyde. In most cases, a certain enzyme stabilization is observed and enzyme desorption is at least partially avoided under conditions where previously all the immobilized enzyme became desorbed (Fig. 12). Furthermore, this occurs using supports with which the glutaraldehyde can not react, thus the enzyme cannot become immobilized on the support. This is the case of supports having aliphatic acyl moieties or quaternary amine groups as active groups on the support; glutaraldehyde hardly can react with these groups in the support.
To explain these results, we should go back to point 3.2 of this review. Immobilization via physical adsorption is usually much faster than enzyme diffusion inside the support pores and in most cases the treatments of the biocatalyst involving glutaraldehyde described in the literature are performed using fully protein loaded biocatalysts. Under these conditions, the enzymes are packed together, so close to each other that the distance is in the range of glutaraldehyde crosslinking. Furthermore, if two reactive groups are conveniently confronted, intermolecular crosslinking will occur (Fig. 12).84 Moreover, intramolecular modifications (one point or crosslinkings) will also take place, reinforcing in some cases the stabilization effects. Regarding enzyme release, it now becomes necessary to simultaneously release all adsorbed enzymes, otherwise the aggregate will remain adsorbed. This makes that the “strength” of the adsorption increases exponentially with the size of the aggregate, and under conditions where individual molecules were fully desorbed, now the enzyme remains fully adsorbed on the support.84
As in certain cases the reactivity of the support with the glutaraldehyde cannot be fully discarded due to its unknown nature, the lack of glutaraldehyde reaction with the support may be verified via different strategies. For example, it is possible to check if we can “preactivate” the support, to achieve a covalent attachment between the enzyme molecules and the support after washing the support with an excess of water to eliminate all free glutaraldehyde molecules and later offering the enzyme. The use of Schiff reactive may also help to verify if some reactive glutaraldehyde remain bound to the support after the washings. Second, slowing down the immobilization rate of the enzyme to increase the distance between enzyme molecules, and checking if the glutaraldehyde treatment still avoids desorption of a significant percentage of the enzyme molecules.
One drawback of this strategy is that desorption of very large chemical aggregates may be fully impossible even under the most drastic conditions, transforming a reversible immobilization method in an irreversible one. The researcher should carefully evaluate the convenience of this treatment, considering the stability requirements of the enzyme and the real risks of enzyme desorption during the use of the biocatalyst versus the possibility of having to discard the support after enzyme inactivation.14
This strategy has been used in several examples to prepare crosslinked enzyme aggregates in an immobilized form, that some authors call CLEAs (see above), but which really are an altogether different thing. Hierarchically ordered mesocellular mesoporous carbon was used as a host for enzyme immobilization.100 To improve the retention of enzymes, the adsorbed enzymes were cross-linked using glutaraldehyde. The resulting preparation showed a significant stabilization with high enzyme loadings. For example, 0.5 g chymotrypsin could be loaded in 1 g of silica with no activity decrease observed with rigorous shaking over one month. In contrast, adsorbed chymotrypsin without any crosslinking treatment resulted in a lower loading, which further decreased due to continuous leaching of adsorbed chymotrypsin under shaking. The activity of crosslinked enzyme aggregates was 10 times higher than that of the adsorbed chymotrypsin.
In two papers, both α-chymotrypsin and a lipase were immobilized in SBA-15 mesoporous silica by crosslinking adsorbed enzymes to give the so-called one-dimensional crosslinked enzyme aggregates.101,102 This simple approach resulted in one-dimensional crosslinked enzyme aggregates in the linear pore channels of SBA-15, which was very effective in preventing the enzyme leaching and consequently improving the enzyme stability.
In another research effort, meso-structured onion-like silica was produced with a 200–300 nm sized primary meso-structured onion building unit, with each onion unit having highly curved mesopores of 10 nm diameter in a multishell structure.103 Nanoscale enzyme reactors in these supports were prepared via a two-step process of enzyme adsorption and subsequent enzyme cross-linking with glutaraldehyde, which effectively prevents the leaching of cross-linked enzyme aggregates from highly curved mesopores. This improved the enzyme stability as well as the enzyme loading.
In another example, β-glucosidase was immobilized onto mesocellular silica foams and later crosslinked with glutaraldehyde.104 This resulted in the formation of crosslinked enzyme aggregates of nanometer scale. The final catalyst was more stable and presented a lower Km than that of its free counterpart.
Other authors really produced CLEAs inside the pores of supports, taking advantage of the increase in size of the enzyme when an aggregate was formed. However, they are neither real CLEAs, as the enzyme is not previously precipitated (Fig. 15). This is the case of the examples described by Prof Hartman's group. Using chloroperoxidase, they showed that the formation of cross-linked chloroperoxidase aggregates in the pores of mesocellular foam materials results in active biocatalysts that are more resistant to leaching than the conventional catalyst prepared by physisorption of chloroperoxidase. Small-angle neutron scattering experiments clearly confirm that the chloroperoxidase-CLEAs are located in the pores of the mesocellular foams.105 Later, they extended the studies to glucose oxidase. The formation of cross-linked enzyme aggregates of glucose oxidase in the pores of mesocellular foams was obtained. The enzymes can enter the ultra-large cavities connected through the smaller windows, where their agglomeration and cross-linking with glutaraldehyde will take place. After cross-linking, the diameter of the aggregates is larger than the diameter of the pore entrance and, thus, the enzymes are trapped in the pores of the support.106 Finally, glutaraldehyde cross-linked enzyme aggregates of chloroperoxidase and glucose oxidase were grown in large-pore mesocellular foams, improving operational stability in the oxidation of indol.107
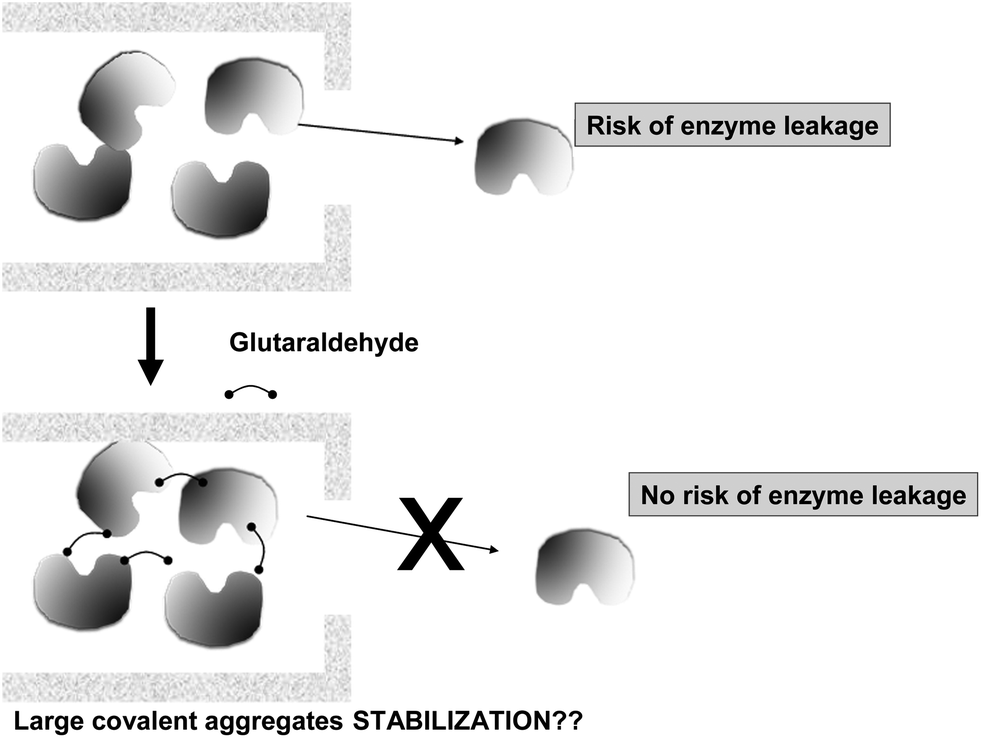 | ||
| Fig. 15 Production of enzyme chemical aggregates to prevent enzyme leakage on mildly adsorbed enzymes. | ||
5. Crosslinking of supports bearing primary amino groups and ionically exchanged proteins
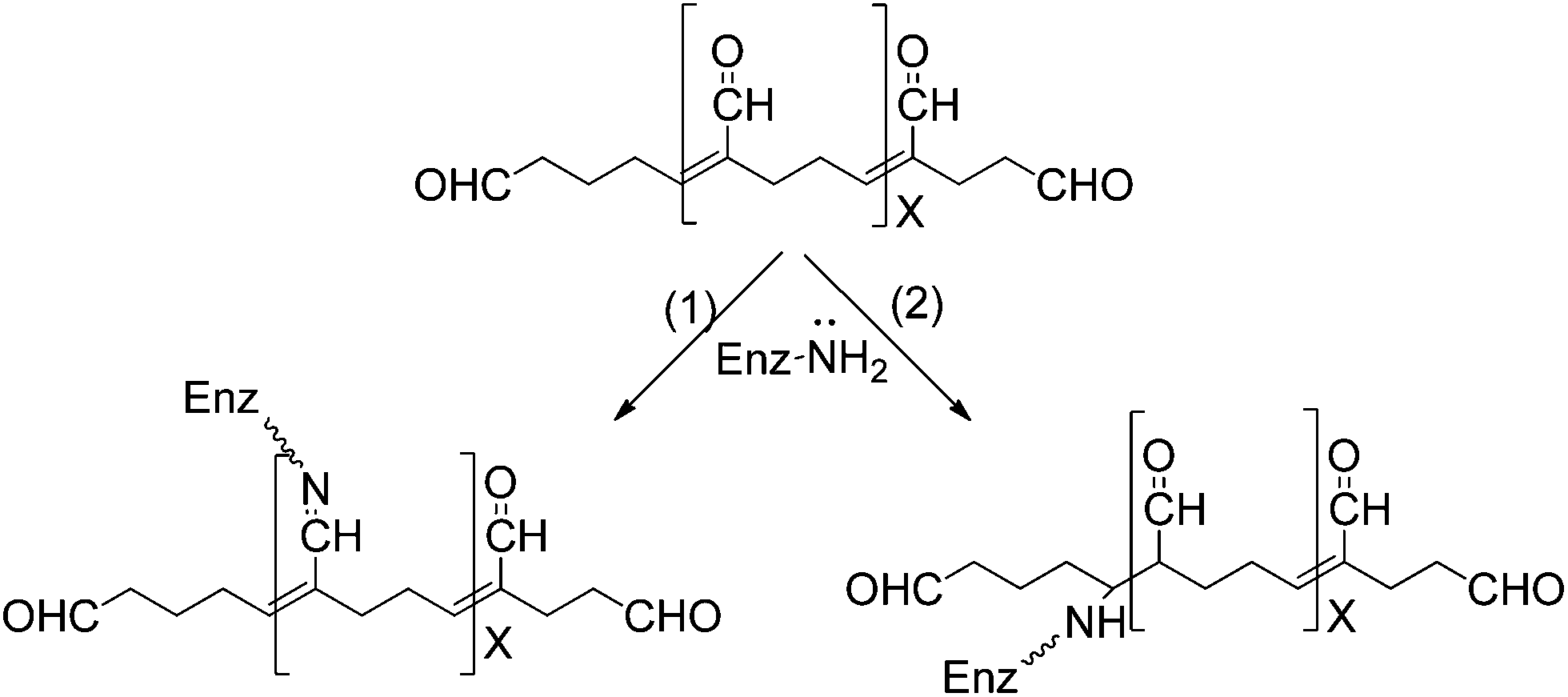
In this new example of use of glutaraldehyde to immobilize enzymes in preexisting supports, as in the case explained above, the treatment with glutaraldehyde is performed after the enzyme is adsorbed in the support, in this case via ion exchange. However, this is a fully different case from the one described above. Now, the researcher knows that the support has primary amino groups, and that, therefore, it is likely to modify the support with glutaraldehyde, and finally obtain enzyme-support covalent bonds.108 Thus, in this case, the enzyme may experience three different kinds of modifications caused by glutaraldehyde (Fig. 16):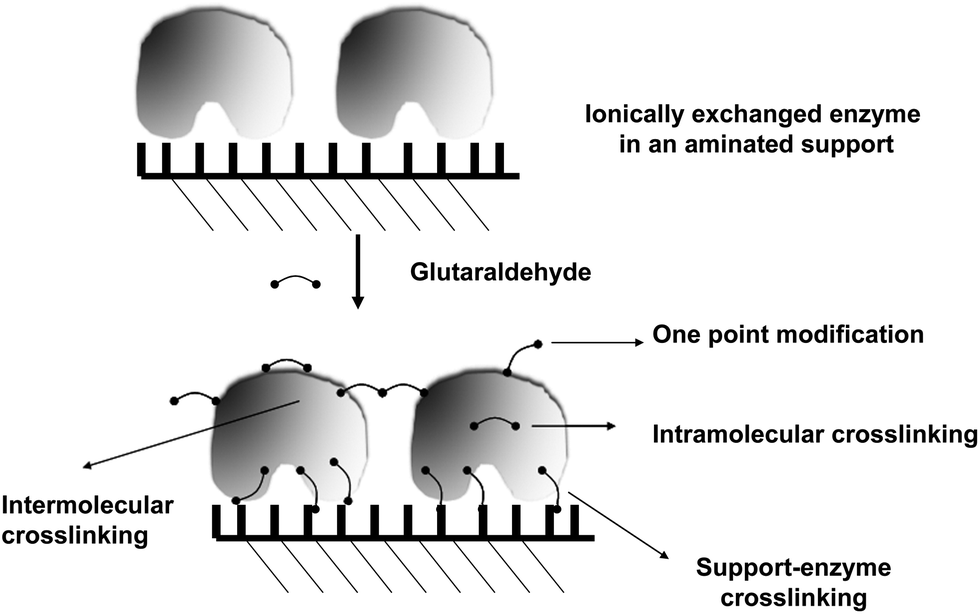 | ||
| Fig. 16 Crosslinking of enzymes and aminated supports after enzyme ionic adsorption. | ||
(1) Intramolecular modifications (one point modifications or crosslinking, see above).
(2) Interprotein crosslinking. If the ion exchange immobilization has been rapid enough, the enzymes will be very near each other and it is likely to have enzyme–enzyme crosslinking (see above).
(3) Support-enzyme reaction. The support and the enzyme molecules may react with each other. In fact, if the treatment with glutaraldehyde is performed in a way that enables the modification of each reactive group in the support and the enzyme with one glutaraldehyde molecule, we will have the situation where the highest prospects of enzyme support reaction may occur. And this reaction will take place in a relative wide range of pH value, as the reaction will occur between amino/glutaraldehyde moieties; although considering the stability of the groups and its reactivity, some reports suggest that pH values around 8.5 may be the most adequate.33 That way, the support will behave as a large multi-crosslinking reagent, fixing the positions of all the protein groups involved in the reaction with the support, whose relative position must remain unaltered under any distorting condition, promoting an increase of the overall enzyme structure, which becomes more intense when a more intense multipoint covalent attachment is achieved.17,109–111
Thus, this strategy has some positive points, as the good reactivity of the amino glutaraldehyde moieties with similar groups in a relatively wide range of pHs, that include neutral pH, the fact that the glutaraldehyde treatment is performed on a previously immobilized enzyme, and the possibilities of having some positive inter or intra-molecular modifications. This last point is also the drawback of the strategy, as the enzyme should be fully modified, not only in the groups involved in the immobilization, and in some cases these modifications may be negative for enzyme stability or activity. However, in many instances when there is a comparison between immobilization on preactivated supports (see below) or treatment with glutaraldehyde after enzyme ion exchange, stabilization improves using this strategy.108,112,113
In other many cases, a comparison was not performed but results using the treatment of previously adsorbed enzymes were very positive regarding stabilization and prevention of enzyme leakage.114–119
The treatment of lipases adsorbed on anion exchanger supports has been in some cases used to modulate enzyme properties. Thus, the open form of some lipases was fixed by glutaraldehyde treatment of adsorbed lipases in the presence of detergents.120 In other cases, the random chemical modification was enabled to modulate the lipase selectivity or specificity.121
This strategy may be only used when the support is able to immobilize the protein via ionic exchange. That means that the support should present a high enough number of ionic groups to permit the adsorption of the enzyme on the support. Moreover, the enzyme should have the capacity to become adsorbed to anion exchangers. Considering that this is a multipoint process (several ion bridges need to be produced to fix the enzyme to the support),122–124 the strategy presents the problem that the support should never be physically inert. Furthermore, the inertness of the support surface may be in many instances a desired feature of the support, to prevent uncontrolled support-enzyme interactions that may affect enzyme stability (sometimes in a positive sense, but in another may have a negative impact in the enzyme stability).14,125 Another negative point to be considered is the possibility of reactivating the enzyme after inactivation, the folding–unfolding strategy may not work if the unfolded enzyme may interact with the support.14
Moreover, the strategy does not offer a large versatility. It may be possible that the immobilization via ionic exchange under different conditions of pH and ionic strength may involve different protein areas, but that is the only way of altering the enzyme orientation regarding the support.126–128 The immobilization of an enzyme via different areas may be interesting to reach optimum stabilization (involving the most labile area of the protein in the immobilization)22,129–132 or to tune the enzyme catalytic properties (selectivity, specificity, activity).17
6. Activation of supports with glutaraldehyde
In this last case, the support is pre-activated with glutaraldehyde. Following the results from Monsan, the optimal activation regarding the chemical reactivity of the support should be two molecules of glutaraldehyde per amino group in the support (obtained, e.g., by incubating the support in 1 M glutaraldehyde at pH 7 by 12–16 h).37 It is very likely the most widespread form of using glutaraldehyde to immobilize enzymes (see for example133–141) and one of the first papers concerning the use of glutaraldehyde to prepare immobilize enzymes may be found in ref. 142.Compared to the strategy described above, this method has some advantages and drawbacks. As a drawback, we can consider the higher difficulty on having an intense multipoint covalent attachment, as now the reaction is between amino/glutaraldehyde/glutaraldehyde moieties and the ε amino groups of the Lys residues (and the terminal amino group). The Lys groups have a pK of 10.7, and the stability of the glutaraldehyde groups in the support is not good at alkaline pH value while at neutral pH value the reactivity of the amino groups is relatively low. At neutral pH value, the most reactive amino group is the terminal one, with a pK value between 7 and 8, being much more reactive that the addition of the reactivities of all Lys external groups.143 However, it should be considered that after the first immobilization, if some nucleophiles of the protein are in the area exposed to the support, the high apparent concentration of the different groups may permit the establishment of some new covalent enzyme-support bonds.144 Moreover, the preactivated support may not be stored for a long time under wet conditions due to the relatively low stability of the groups.
The first advantage compared to the case described in Section 5, is that now only the groups involved in the covalent immobilization are modified by glutaraldehyde. Moreover, the support may immobilize enzymes even if they are very poorly activated, because the enzyme is covalently fixed to the support just with one point as the glutaraldehyde-protein bonds are stable. Thus, using very lowly activated amino supports (e.g., 1 reactive group under each projected area of the protein) immobilization using glutaraldehyde activated supports will be directly performed by a covalent reaction by the most reactive amino group on the enzyme (very likely the terminal amino group of the enzyme if immobilization is performed at neutral pH value).143 However, immobilization will be very slow due to the low activation of the support, and will offer no chance of reaching an intense multipoint covalent attachment.14
On the other hand, using highly activated supports; the features of the spacer arm (amino/glutaraldehyde/glutaraldehyde) convert the supports in a heterofunctional support. The term “heterofunctional support” may be applied to supports that present several functionalities on its surface that are able to interact with different groups of an enzyme, and these interactions may be different under different conditions (Fig. 17).145,146
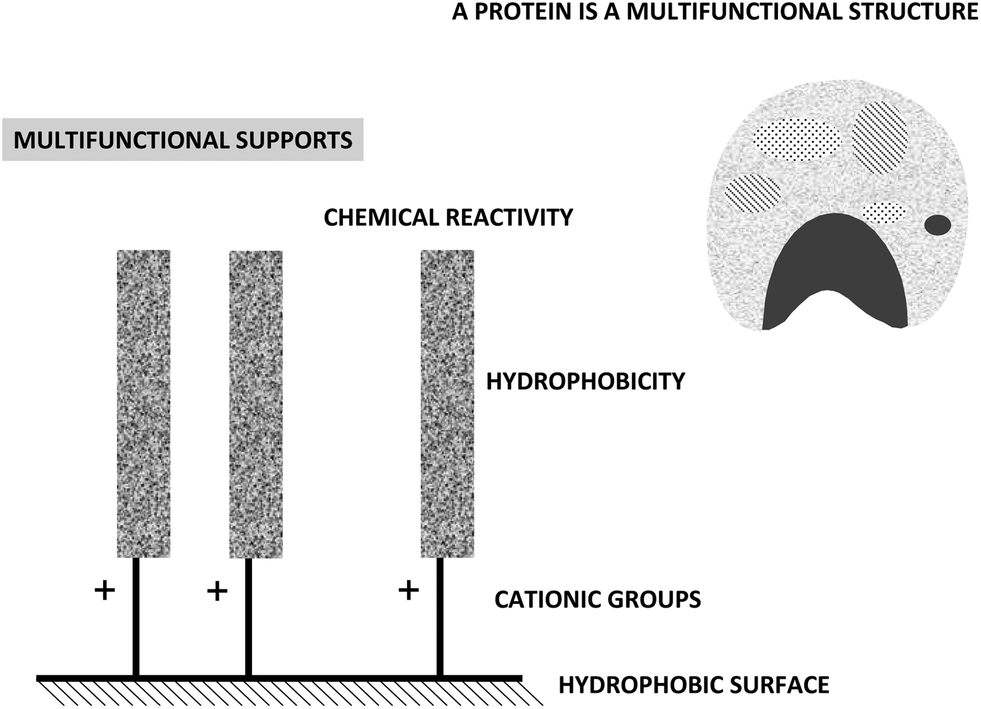 | ||
| Fig. 17 Multifunctional supports. | ||
Immobilization of enzymes on a heterofunctional support may be more versatile. If the researcher is able to benefit from this fact it may be an advantage, but if this fact is not considered, the understanding of the results may be really complex. The use of heterofunctional supports to immobilize proteins has been recently discussed in the literature.147 In this review we will focus on glutaraldehyde activated supports.
The multifunctionality of the supports having a high surface density of amino groups and activated with glutaraldehyde is a direct consequence of the way they are prepared. Their preparation requires the modification of supports bearing primary amino groups with two glutaraldehyde molecules, if the optimal protocol is followed.28,36,37 The final result is a support having spacer arms bearing one or two amino groups (cationic groups that may function as anion exchangers), a fairly hydrophobic moiety formed by the glutaraldehyde dimer and the covalent reactive group.
In this way, a support highly activated with glutaraldehyde may give three different kinds of interactions with a protein: hydrophobic, anionic exchange and covalent (Fig. 18).84 Biomacromolecules are only immobilized on supports via ionic exchange or hydrophobic interactions when several enzyme-support interactions may be established, being the one point interaction insufficient to immobilize a protein.128 Thus, these interactions will only have a real impact on enzyme immobilization when using supports bearing several amino-(glutaraldehyde)n moieties under each enzyme molecule (Fig. 18).148–153 Thus, using highly activated glutaraldehyde supports, the three immobilization causes may be able to immobilize a protein molecule. If the researcher is aware of this fact, it is possible to permit that one or the other immobilization cause may be the dominant one, by playing with the experimental conditions.38 This permits to immobilize enzymes following different orientations regarding the support surface. If there is a very small amount of groups on the support, (e.g. just one spacer arm per projected area of the enzyme), this multi-interaction will no longer be possible or will be so slow, that the only relevant cause of immobilization will be the one point covalent immobilization, as explained above.143,148–153
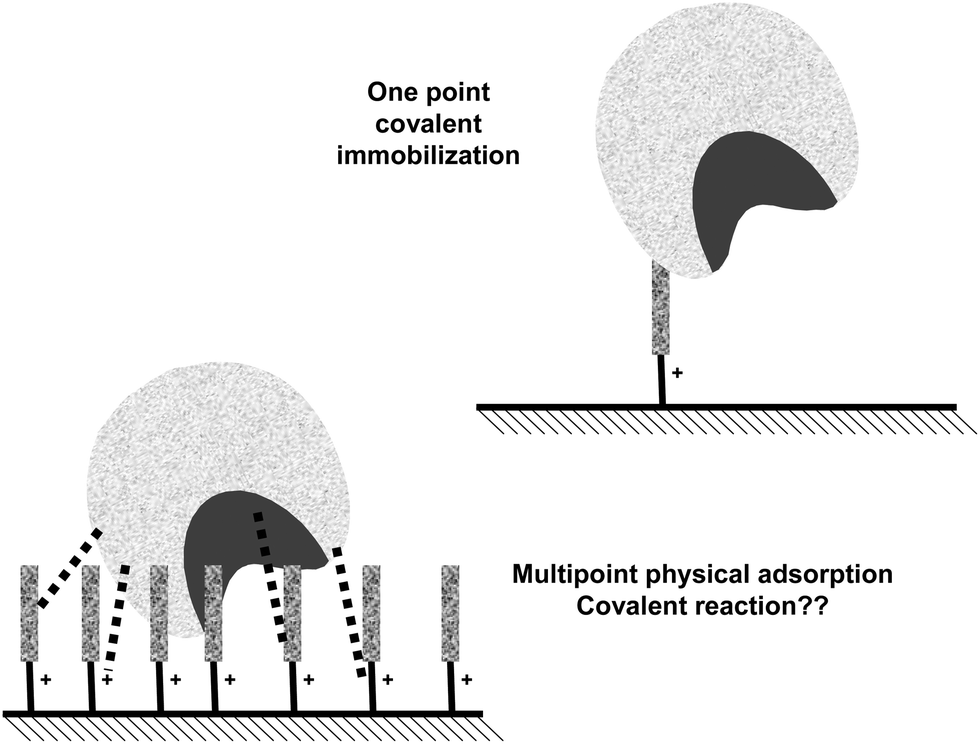 | ||
| Fig. 18 Effect of the superficial density of glutaraldehyde groups on the possibilities of physical adsorption of the enzyme on the support surface. | ||
This means that even if all enzyme molecules are immobilized (incorporated to the support) in a very rapid fashion using a highly activated glutaraldehyde support, there is no guarantee that covalent immobilization of the enzyme on the support has taken place. In fact, the immobilization may be very strong, and the enzyme may remain immobilized under conditions where the enzyme may be fully released from the mother amino support, and still the enzyme molecules may be just adsorbed, as now the adsorption may be mixed, ionic/hydrophobic.84 To ensure that covalent immobilization has taken place, the enzyme should remain immobilized under conditions where both ionic and hydrophobic interactions may be broken (e.g., using cationic detergents, using guanidine, increasing the ionic strength in the presence of non-ionic detergents).84
Using highly activated supports, it has been shown that in most cases an ionic exchange with the amino groups in the support is the first step in the immobilization of most enzymes.22,38,84,154–156
One effect of this first ionic adsorption is that, even though glutaraldehyde is able to immobilize enzymes via just one attachment due to the stability of the bond formed, the activation degree of the support has an exponential effect on the immobilization reaction rate of proteins, not the expected linear effect using reactive groups in the support able to immobilize the proteins via just one point.143 This is because the researcher is measuring the rate of ionic exchange of the enzyme on the support, which requires the establishment of several enzyme-support interactions, and thus it is exponentially dependent on the surface density of amino groups on the support although the covalent reaction should be of order 1.38,148–153 This immobilization mechanism is much faster that the direct covalent attachment via glutaraldehyde–enzyme covalent reaction, being this way the first cause of immobilization for most enzyme molecules.
This multifunctionality of the glutaraldehyde supports is an advantage in certain cases. The rapid ionic exchange of the enzyme on the support keeps the enzyme from being in soluble form before being covalently immobilized (thus avoiding some inactivation causes, such as interaction with interfaces or autolysis).16,17 If enzyme immobilization via ion exchange has a positive effect on enzyme stability, enzyme inactivation by distortion will also be slowed down.
However, the main advantage of the multifunctionality of glutaraldehyde is that we can alter the enzyme orientation on the support by changing the immobilization conditions, favoring one mechanism or another as the first immobilization cause (Fig. 18).
If the researcher wishes to have a first hydrophobic adsorption, this can be achieved using a high enough ionic strength. The glutaraldehyde dimer is not very hydrophobic, but may be hydrophobic enough if the surface density of the groups in the support is high enough. Using very high ionic strength, the ionic exchange will be rolled out, and the areas of the protein with high concentration of external hydrophobic groups may be involved in the first enzyme adsorption and delimit the area where the reactive groups of the enzyme which will react with the support should be located. After enzyme hydrophobic adsorption, the reactive groups of the enzyme near the support surface may produce some covalent reactions. However, this will be produced after enzyme immobilization, and there is no guarantee that the enzyme will finally have any covalent attachment with the support (e.g., if there are no reactive groups on the enzyme surface in that area).
The second possibility is to permit ionic exchange of the enzyme prior to covalent immobilization. Using most enzymes, the use of low ionic strength is enough to reach this situation (an ionic strength that permits ionic exchange of the proteins on the non-activated glutaraldehyde amino support).38,84 In this case, the enzyme will be first immobilized on the support by ionic exchange and this area will be the one where nucleophiles capable of reacting with the glutaraldehyde moieties should be located. Ionic exchange may also involve different enzyme regions depending on the experimental conditions and activation degree of the support. Ionic exchange at different pH values may in certain enzymes change the area where the highest concentration of available anionic charged groups may be found. Furthermore, the ionic strength may determine the area involved because the higher the ionic strength, the more restrictive the immobilization becomes (requiring more enzyme support-interactions).126,127
Finally, it is possible to immobilize the enzyme via a direct first covalent attachment, involving the most reactive exposed group of the enzyme (usually the terminal amino group). Using most enzymes, the moderate ionic strength used to prevent ionic exchange (100–250 mM of NaCl) is not enough to force the hydrophobic adsorption of the protein on the support (that is not very hydrophobic) and a direct covalent immobilization may be the first cause for the enzyme immobilization.38,84
Lipases are a special case of enzymes that permit a new alternative to the immobilization on glutaraldehyde activated supports. This has special interest as they are perhaps the most used enzymes in industry and academic studies due to their broad range of substrates and reactions, accompanied of a high enantio- and regio-selectivity or specificity, together to good stability in different reaction medium and wide availability.157–159
Lipases are complex enzymes that usually have two conformations, a closed one where the active center is secluded from the medium by a polypeptide chain (lid or flap), and an open form, where the lid is displaced and the active center of the lipase is exposed to the medium (Fig. 19).160 This open form presents a very large hydrophobic area exposed to the medium, formed by the hydrophobic residues around the active center of the lipase and the hydrophobic groups in the internal face of the lid.161 The exposition to a hydrophilic medium (e.g., an aqueous buffer) of this large hydrophobic area is unfavorable, thus the enzyme in aqueous homogeneous media will be mainly in the closed form. However, this open form is readily adsorbed on the hydrophobic surface of the oil drops even at very low ionic strength (interfacial activation) (Fig. 19).162,163
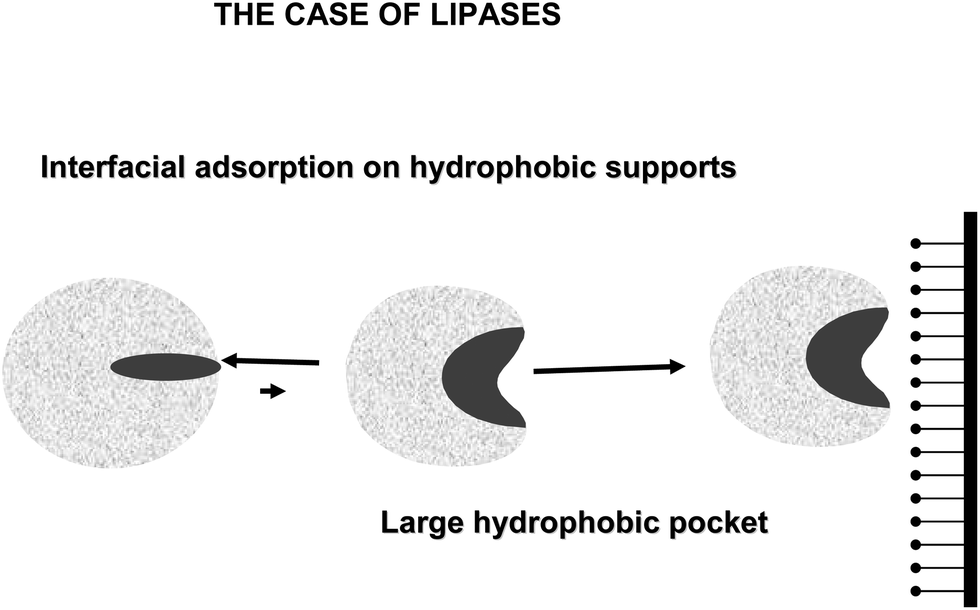 | ||
| Fig. 19 Interfacial activation of lipases. | ||
It has been shown that lipases may become adsorbed via a similar mechanism on any other hydrophobic surface (hydrophobic supports, hydrophobic proteins, other “open” lipases).164–166 Thus, the use of hydrophobic supports to immobilize lipases at low ionic strength has been suggested as a simple way to obtain the open and stabilized form of the immobilized lipases.167 Although this interfacial activation is the specific feature of lipases, the lid may be quite different from one lipase to another, e.g., lipase B from Candida antarctica has a very small lid that can not fully seclude the active center from the medium,168 while the lipase from Bacillus thermocatenulatus has a double lid and a very complex movement governing its activation.169 This immobilization is based on the fact that the hydrophobic support mimics the natural substrate of lipases. And highly activated glutaraldehyde activated supports offers a fairly hydrophobic surface to this interfacial activation of lipases.
In fact, it has been shown that using lipases, it is at least possible to immobilize the enzyme via 4 different mechanisms on highly activated glutaraldehyde supports.84
Thus, the control of the immobilization on these supports using lipases is more complex, but the support offers a new immobilization orientation.
Due to the tendency of the open form of the lipases to become adsorbed versus hydrophobic interfaces,164,170,171 if the immobilization is just performed at low ionic strength, the enzyme will be immobilized by both immobilization mechanisms: interfacial activation and ionic exchange (Fig. 20).84 Depending on the enzyme, the support and the immobilization conditions, one or the other immobilization cause may be predominant, but most likely the other cause will always be present in a major or minor percentage. This mixture of lipase orientations is not the ideal situation if the researcher wishes to tune lipase properties via immobilization and may also make difficult to understand the results. In fact, this is the usual situation that we may find in the literature.
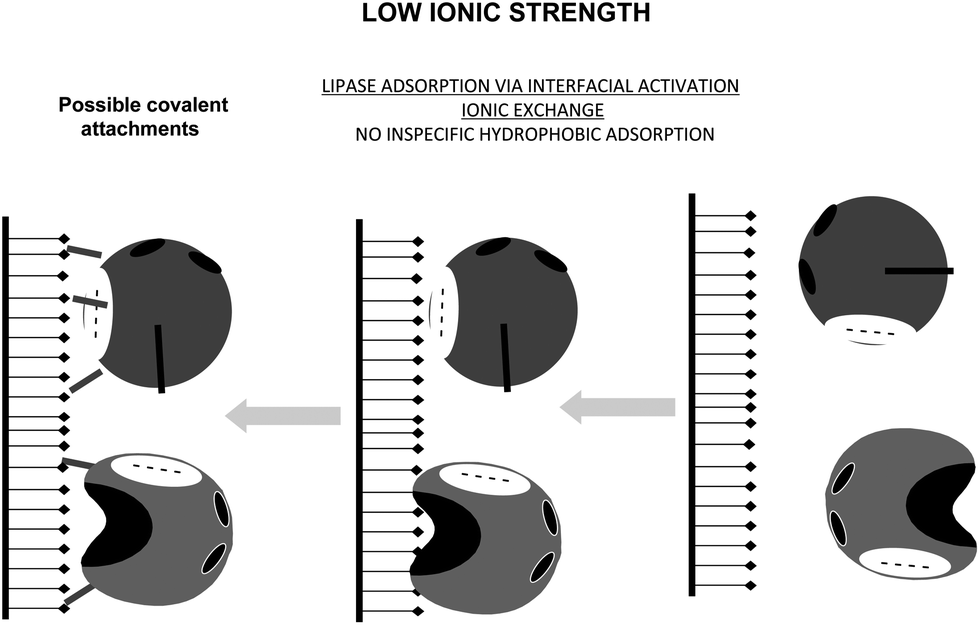 | ||
| Fig. 20 Immobilization of lipases at low ionic strength on glutaraldehyde activated supports. | ||
This situation may be avoided by using non-ionic detergents, which prevent the interfacial activation of the lipase versus a hydrophobic support (Fig. 21).164 Performing the immobilization in the presence of Triton X-100 at low ionic strength, lipases are mainly immobilized on the glutaraldehyde supports via a first ionic exchange.84 Using moderate ionic strength, the enzyme immobilization will mainly proceed via interfacial activation (Fig. 22).
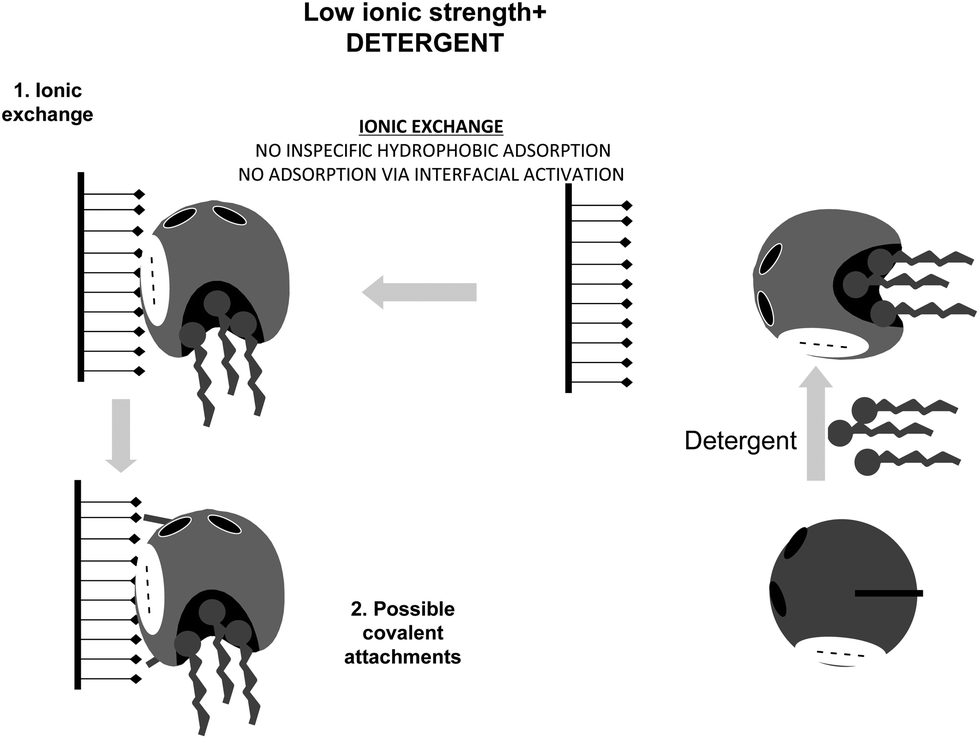 | ||
| Fig. 21 Immobilization of lipases at low ionic strength in the presence of a detergent on glutaraldehyde activated supports. | ||
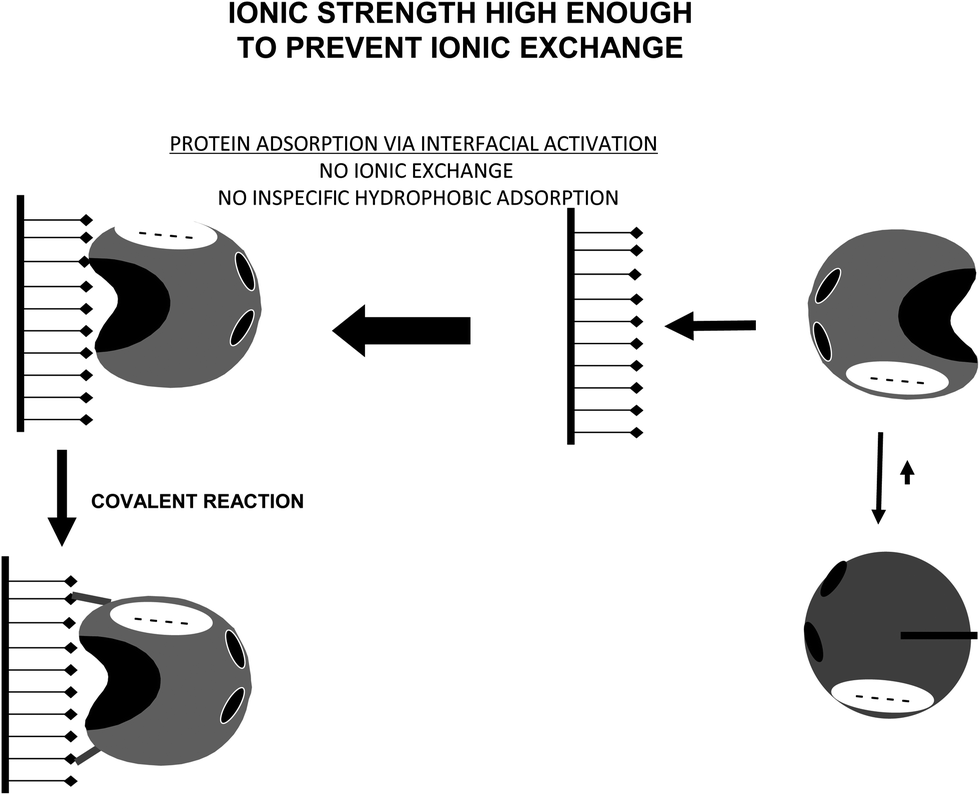 | ||
| Fig. 22 Immobilization of lipases at high enough ionic strength to prevent ionic exchange on glutaraldehyde activated supports. | ||
The use of very high ionic strength will cause the lipase to adopt mainly the closed form, permitting as a first immobilization step the conventional hydrophobic adsorption via hydrophobic groups located on the lipase surface (Fig. 23). It has been reported that interfacial activation of lipases on hydrophobic supports is slowed at high ionic strength.164 That way, using an ionic strength just high enough able to prevent the ionic exchange of the lipase, lipases will become immobilized on the support first via a rapid interfacial activation on the support, which is much faster than the direct covalent attachment.84
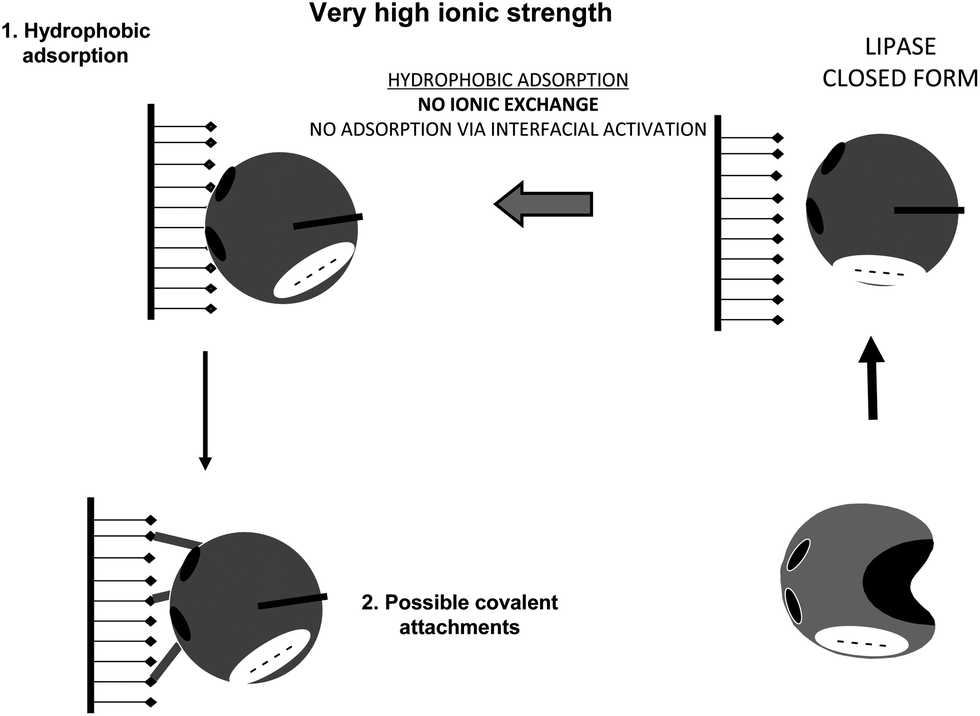 | ||
| Fig. 23 Immobilization of lipases at very high ionic strength (more than 1 M) on glutaraldehyde activated supports. | ||
The only way to ensure that the first step in the immobilization of a lipase in these supports is a covalent attachment is to simultaneously prevent ionic exchange and interfacial activation. This may be achieved using simultaneously moderately high ionic strength and detergents, or ionic detergents (Fig. 24).84
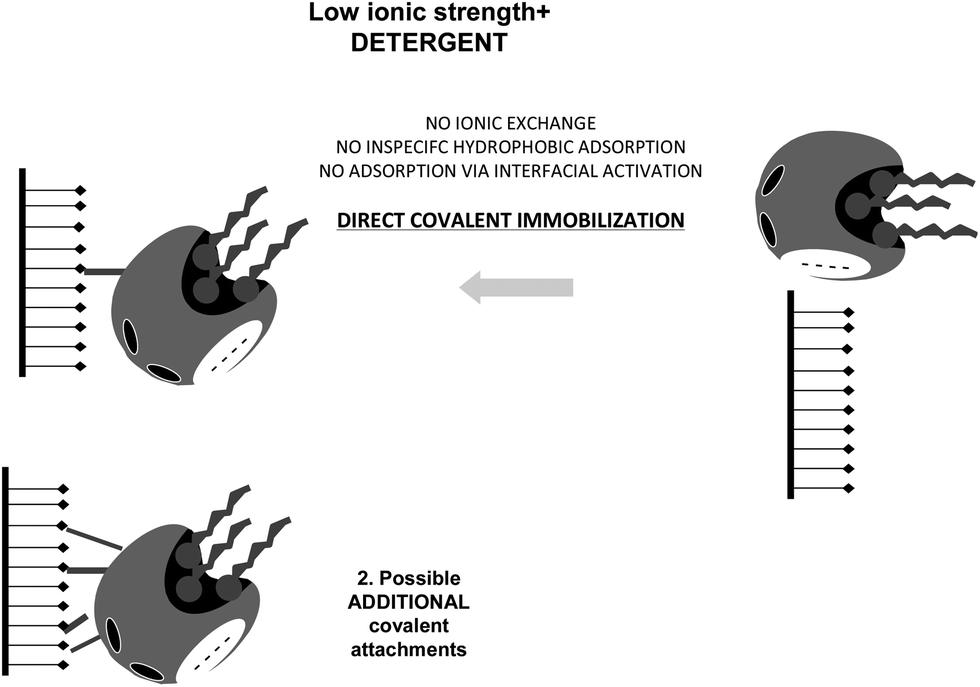 | ||
| Fig. 24 Immobilization of lipases at moderate ionic strength and in the presence of detergents on glutaraldehyde activated supports. | ||
Whatever the enzyme used, if we have a situation where the first phenomenon is covalent immobilization, the surface density of groups in the support will have a first order effect on the rate of enzyme immobilization.143 If this is not the case (mainly at the highest support activation degree), another cause may be the responsible for the first enzyme immobilization.
Thus, it is possible to immobilize enzymes on glutaraldehyde supports via different first events. This may lead to different orientations of the enzyme on the support, offering different stabilities.38 In the case of lipases, they also exhibited different catalytic behavior (e.g., specificity was altered).84 This way, it is possible to have, using the same immobilization support, enzymes immobilized by different areas, with different numbers of enzyme molecule-support covalent bonds and different enzyme-support unspecific interactions.147 We should bear in mind that, due to the proximity between the groups of the support and of the enzyme, interactions between immobilized enzyme and support will be produced over time even though they may not be enough to be the only cause for immobilization.144
In another, more difficult to classify case, a lipase was immobilized on electrospun and ethanol-dispersed polystyrene–poly(styrene-co-maleic anhydride) nanofibers in the form of enzyme precipitate coatings.172 Lipase precipitate coatings were prepared in a three-step process, consisting of lipase covalent attachment, lipase precipitation, and crosslinking of precipitated lipases onto the covalently attached lipases via glutaraldehyde treatment.
A similar approach was used to immobilize β-glucosidase. The enzyme was immobilized on polymer nanofibers. Then, additional enzyme molecules were crosslinked onto the covalently attached enzyme molecules via glutaraldehyde treatment.173
7. Conclusions
Glutaraldehyde, an apparently old fashioned reagent in use for a long time in the design of biocatalysts, remains one of the most interesting tools in enzyme crosslinking and immobilization (Fig. 25). However, to achieve optimal results, it is necessary to understand the different reactivities of the amino/glutaraldehyde and amino/glutaraldehyde/glutaraldehyde, together with the fact that, in immobilization of enzymes on glutaraldehyde pre-activated supports, it is a heterofunctional support that permits altering the enzyme orientation on the support surface. Thus, glutaraldehyde remains one of the most potent and versatile tools in enzyme technology, and the new knowledge on its reactivity and possibilities opens new opportunities for the future. | ||
| Fig. 25 Glutaraldehyde, a versatile reagent in enzyme biocatalyst design. | ||
Acknowledgements
This work has been supported by grant CTQ2009-07568 from Spanish Ministerio de Ciencia e Innovacion, Grant no.1102-489-25428 from COLCIENCIAS and Universidad Industrial de Santander (VIE-UIS Research Program) and CNPq and FAPERGS (Brazil). Á. Berenguer-Murcia thanks the Spanish Ministerio de Ciencia e Innovacion for a Ramon y Cajal fellowship (RyC-2009–03813). The authors would like to thank Mr Ramiro Martinez (Novozymes, Spain S.A) for his kind support to our research.References
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- S. G. Burton, D. A. Cowan and J. M. Woodley, Nat. Biotechnol., 2002, 20, 37–45 CrossRef CAS PubMed.
- J. M. Woodley, Trends Biotechnol., 2008, 26, 321–327 CrossRef CAS PubMed.
- A. Liljeblad and L. T. Kanerva, Tetrahedron, 2006, 62, 5831–5854 CrossRef CAS PubMed.
- S. Panke, M. Held and M. Wubbolts, Curr. Opin. Biotechnol., 2004, 15, 272–279 CrossRef CAS PubMed.
- H. E. Schoemaker, D. Mink and M. G. WubboLts, Science, 2003, 299, 1694–1697 CrossRef CAS PubMed.
- F. H. Arnold, Nature, 2001, 409, 253–257 CrossRef CAS PubMed.
- F. H. Arnold, Chem. Eng. Sci., 1996, 51, 5091–5102 CrossRef CAS.
- J. M. Woodley, Curr. Opin. Chem. Biol., 2013, 17, 310–316 CrossRef CAS PubMed.
- A. R. Fersht and L. Serrano, Curr. Opin. Struct. Biol., 1993, 3, 75–83 CrossRef CAS.
- R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernandez-Lafuente, Chem. Soc. Rev., 2013, 45, 6290–6307 RSC.
- M. L. Verma, C. J. Barrow and M. Puri, Appl. Microbiol. Biotechnol., 2013, 97, 23–39 CrossRef CAS PubMed.
- E. T. Hwang and M. B. Gu, Eng. Life Sci., 2013, 13, 49–61 CrossRef CAS.
- C. Garcia-Galan, A. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- D. Brady and J. Jordaan, Biotechnol. Lett., 2009, 31, 1639–1650 CrossRef CAS PubMed.
- P. V. Iyer and L. Ananthanarayan, Process Biochem., 2008, 43, 1019–1032 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS PubMed.
- A. Díaz-Rodríguez and B. G. Davis, Curr. Opin. Chem. Biol., 2011, 15, 211–219 CrossRef PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem.–Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- D. A. Cowan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2011, 49, 326–346 CrossRef CAS PubMed.
- R. C. Rodrigues, Á. Berenguer-Murcia and R. Fernandez-Lafuente, Adv. Synth. Catal., 2011, 353, 2216–2238 CrossRef CAS.
- K. Hernandez and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2011, 48, 107–122 CrossRef CAS PubMed.
- R. Fernandez-Lafuente, Enzyme Microb. Technol., 2009, 45, 405–418 CrossRef CAS PubMed.
- S. S. Wong and L. J. C. Wong, Enzyme Microb. Technol., 1992, 14, 866–874 CrossRef CAS.
- E. A. Woodroof, J. Bioeng., 1978, 2, 1–9 CAS.
- A. D. Russell, Infection Control and Hospital Epidemiology, 1994, 15, 724–733 CAS.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, BioTechniques, 2004, 37, 790–802 CAS.
- D. R. Walt and V. I. Agayn, TrAC, Trends Anal. Chem., 1994, 13, 425–430 CrossRef CAS.
- A. F. S. A. Habeeb and R. Hiramoto, Arch. Biochem. Biophys., 1968, 126, 16–26 CrossRef CAS.
- Y. Wine, N. Cohen-Hadar, A. Freeman and F. Frolow, Biotechnol. Bioeng., 2007, 98, 711–718 CrossRef CAS PubMed.
- C. Aso and Y. Aito, Die Makromolekulare Chemie, 1962, 58, 195–203 CrossRef CAS.
- F. M. Richards and J. R. Knowles, J. Mol. Biol., 1968, 37, 231–233 CrossRef CAS.
- R. Fernandez-Lafuente, C. M. Rosell, V. Rodriguez and J. M. Guisan, Enzyme Microb. Technol., 1995, 17, 517–523 CrossRef CAS.
- A. H. Korn, S. H. Feairheller and E. M. Filachoine, J. Mol. Biol., 1972, 65, 525–529 CrossRef CAS.
- T. J. Johnson, Eur. J. Cell Biol., 1978, 45, 160–169 Search PubMed.
- P. Monsan, G. Puzo and H. Mazarguil, Biochimie, 1975, 57, 1281–1292 CrossRef CAS.
- P. Monsan, J. Mol. Catal., 1978, 3, 371–384 CrossRef CAS.
- L. Betancor, F. López-Gallego, A. Hidalgo, N. Alonso-Morales, G. Dellamora-Ortiz, C. Mateo, R. Fernández-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2006, 39, 877–882 CrossRef CAS PubMed.
- K. Ziegler, I. Schits and H. Zahn, in Protein Crosslinking: Biochemical and Biological Aspects, ed. M. Friedman, Plenum, New York, 1977, vol. 86a, pp. 345–354 Search PubMed.
- T. Imoto and H. Yamada, in Protein Function: A Practical Approach, ed. T. E. Creighton, IRL Press, Oxford, UK, 1989, pp. 247–278 Search PubMed.
- D. T. Cheung, N. Perelman, E. C. Ko and M. E. Nimni, Connect. Tissue Res., 1985, 13, 109–115 CrossRef CAS.
- E. A. Talman and D. R. Boughner, Ann. Thorac. Surg., 1995, 60, S369–S373 CrossRef CAS.
- S. Mathapati, D. K. Bishi, S. Guhathakurta, K. M. Cherian, J. R. Venugopal, S. Ramakrishna and R. S. Verma, Mater. Sci. Eng., C, 2013, 33, 1561–1572 CrossRef CAS PubMed.
- Y. Liu, L. Ma and C. Gao, Mater. Sci. Eng., C, 2012, 32, 2361–2366 CrossRef CAS PubMed.
- P. Sinha, D. Zurakowski, T. K. Susheel Kumar, D. He, C. Rossi and R. A. Jonas, J. Thorac. Cardiovasc. Surg., 2012, 143, 224–227 CrossRef CAS PubMed.
- C. P. Govardhan, Curr. Opin. Biotechnol., 1999, 10, 331–335 CrossRef CAS.
- A. F. S. A. Habeeb, Arch. Biochem. Biophys., 1967, 119, 264–268 CrossRef CAS.
- E. F. Jansen and A. C. Olson, Arch. Biochem. Biophys., 1969, 129, 221–227 CrossRef CAS.
- H. K. Manonmani and R. Joseph, Process Biochem., 1993, 28, 325–329 CrossRef CAS.
- T. Anwar, B. Ahmad and H. Younus, Biocatal. Biotransform., 2007, 25, 453–458 CrossRef CAS.
- F. A. Quiocho and F. M. Richards, Proc. Natl. Acad. Sci. U. S. A., 1964, 52, 833–839 CrossRef CAS.
- N. L. St. Clair and M. A. Navia, J. Am. Chem. Soc., 1992, 114, 7314–7316 CrossRef CAS.
- L. Cao, F. Van Rantwijk and R. A. Sheldon, Org. Lett., 2000, 2, 1361–1364 CrossRef CAS PubMed.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- R. A. Sheldon, R. Schoevaart and L. M. Van Langen, Biocatal. Biotransform., 2005, 23, 141–147 CrossRef CAS.
- R. A. Sheldon, Appl. Microbiol. Biotechnol., 2011, 92, 467–477 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, L. M. Van Langen, F. Van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2004, 86, 273–276 CrossRef CAS PubMed.
- S. Shah, A. Sharma and M. N. Gupta, Anal. Biochem., 2006, 351, 207–213 CrossRef CAS PubMed.
- I. Matijošyte, I. W. C. E. Arends, S. de Vries and R. A. Sheldon, J. Mol. Catal. B: Enzym., 2010, 62, 142–148 CrossRef PubMed.
- F. Šulek, D. P. Fernández, Z. Knez, M. Habulin and R. A. Sheldon, Process Biochem., 2011, 46, 765–769 CrossRef PubMed.
- T. Dong, L. Zhao, Y. Huang and X. Tan, Bioresour. Technol., 2010, 101, 6569–6571 CrossRef CAS PubMed.
- J. J. Karimpil, J. S. Melo and S. F. D'Souza, J. Mol. Catal. B: Enzym., 2011, 71, 113–118 CrossRef CAS PubMed.
- J. Cruz, O. Barbosa, R. C. Rodrigues, R. Fernandez-Lafuente, R. Torres and C. Ortiz, J. Mol. Catal. B: Enzym., 2012, 80, 7–14 CrossRef CAS PubMed.
- J. Pan, X. D. Kong, C. X. Li, Q. Ye, J. H. Xu and T. Imanaka, J. Mol. Catal. B: Enzym., 2011, 68, 256–261 CrossRef CAS PubMed.
- L. Wilson, G. Fernández-Lorente, R. Fernández-Lafuente, A. Illanes, J. M. Guisán and J. M. Palomo, Enzyme Microb. Technol., 2006, 39, 750–755 CrossRef CAS PubMed.
- F. López-Gallego, L. Betancor, A. Hidalgo, N. Alonso, R. Fernández-Lafuente and J. M. Guisán, Biomacromolecules, 2005, 6, 1839–1842 CrossRef PubMed.
- J. Yan, X. Gui, G. Wang and Y. Yan, Appl. Biochem. Biotechnol., 2012, 166, 925–932 CrossRef CAS PubMed.
- M. Galvis, O. Barbosa, M. Ruiz, J. Cruz, C. Ortiz, R. Torres and R. Fernandez-Lafuente, Process Biochem., 2012, 47, 2373–2378 CrossRef CAS PubMed.
- V. P. Torchilin and K. Martinek, Enzyme Microb. Technol., 1979, 1, 74–82 CrossRef CAS.
- A. E. Habibi, K. Khajeh, H. Naderi-Manesh, B. Ranjbar and M. Nemat-Gorgani, J. Biotechnol., 2006, 123, 434–442 CrossRef CAS PubMed.
- K. Martinek and V. P. Torchilin, Methods Enzymol., 1988, 137, 615–626 CAS.
- V. P. Torchilin, A. V. Maksimenko, V. N. Smirnov, I. V. Berezin, A. M. Klibanov and K. Martinek, Biochim. Biophys. Acta, 1978, 522, 277–283 CrossRef CAS.
- V. P. Torchilin, A. V. Maksimenko, V. N. Smirnov, I. V. Berezin and K. Martinek, Biochim. Biophys. Acta, Enzymol., 1979, 568, 1–10 CrossRef CAS.
- N. M. Young, C. R. MacKenzie, S. A. Narang, R. P. Oomen and J. E. Baenziger, FEBS Lett., 1995, 377, 135–139 CrossRef CAS.
- U. Arnold and M. Schöpfel, FEBS J., 2012, 279, 2508–2519 CrossRef CAS PubMed.
- V. G. H. Eijsink, A. Bjork, S. Gaseidnes, R. Sirevag, B. Synstad, B. V. D. Burg and G. Vriend, J. Biotechnol., 2004, 113, 105–120 CrossRef CAS PubMed.
- V. Grazu, F. López-Gallego and J. M. Guisán, Process Biochem., 2012, 47, 2538–2541 CrossRef CAS PubMed.
- O. Abian, V. Grazú, J. Hermoso, R. González, J. L. García, R. Fernández-Lafuente and J. M. Guisán, Appl. Environ. Microbiol., 2004, 70, 1249–1251 CrossRef CAS.
- K. L. Carraway and D. E. Koshland Jr, Methods Enzymol., 1972, 25, 616–623 CAS.
- K. L. Carraway, P. Spoerl and D. E. Koshland Jr, J. Mol. Biol., 1969, 42, 133–137 CrossRef CAS.
- V. P. Torchilin, A. V. Maksimenko, V. N. Smirnov, I. V. Berezin, A. M. Klibanov and K. Martinek, Biochim. Biophys. Acta, Enzymol., 1979, 567, 1–11 CrossRef CAS.
- R. Tyagi and M. N. Gupta, Biochemistry, 1998, 63, 334–344 CAS.
- J. M. Bolivar, A. Hidalgo, L. Sánchez-Ruiloba, J. Berenguer, J. M. Guisán and F. López-Gallego, J. Biotechnol., 2011, 155, 412–420 CrossRef CAS PubMed.
- O. Barbosa, R. Torres, C. Ortiz and R. Fernandez-Lafuente, Process Biochem., 2012, 47, 1220–1227 CrossRef CAS PubMed.
- O. M. Poltorak, E. S. Chukhray and I. Y. Torshin, Biochemistry, 1998, 63, 303–311 CAS.
- R. W. Lencki, J. Arul and R. J. Neufeld, Biotechnol. Bioeng., 1992, 40, 1427–1434 CrossRef CAS PubMed.
- O. M. Poltorak, E. S. Chukhrai and I. Y. Torshin, Russ. J. Phys. Chem. A, 2000, 74, S400–S410 Search PubMed.
- A. J. D. Silva, D. P. Gómez-Mendoza, M. Junqueira, G. B. Domont, E. Ximenes Ferreira Filho, M. V. de Sousa and C. A. O. Ricart, Proteomics, 2012, 12, 2729–2738 CrossRef PubMed.
- T. Ohtsuki, Suyanto, S. Yazaki, S. Ui and A. Mimura, Lett. Appl. Microbiol., 2005, 40, 111–116 CrossRef CAS PubMed.
- H. Schagger, W. A. Cramer and G. Von Jagow, Anal. Biochem., 1994, 217, 220–230 CrossRef CAS.
- M. Fuentes, R. L. Segura, O. Abian, L. Betancor, A. Hidalgo, C. Mateo, R. Fernandez-Lafuente and J. M. Guisan, Proteomics, 2004, 4, 2602–2607 CrossRef CAS PubMed.
- C. Garcia-Galan, O. Barbosa and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2013, 52, 211–217 CrossRef CAS PubMed.
- J. M. Bolivar, J. Rocha-Martin, C. Mateo, F. Cava, J. Berenguer, R. Fernandez-Lafuente and J. M. Guisan, Biomacromolecules, 2009, 10, 742–747 CrossRef CAS PubMed.
- T. Yamazaki, W. Tsugawa and K. Sode, Biotechnol. Lett., 1999, 21, 199–202 CrossRef CAS.
- M. Gerl, R. Rudolph and R. Jaenicke, Biol. Chem. Hoppe-Seyler, 1985, 366, 447–454 CrossRef CAS.
- D. J. Crampton, M. Ohi, U. Qimron, T. Walz and C. C. Richardson, J. Mol. Biol., 2006, 360, 667–677 CrossRef CAS PubMed.
- B. U. Failer, A. Aschrafi, G. Schmalzing and H. Zimmermann, Eur. J. Biochem., 2003, 270, 1802–1809 CrossRef CAS.
- R. A. Sheldon and S. Van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- P. Jochems, Y. Satyawali, L. Diels and W. Dejonghe, Green Chem., 2011, 13, 1609–1623 RSC.
- J. Lee, J. Kim, H. Jia, M. I. Kim, J. H. Kwak, S. Jin, A. Dohnalkova, H. C. Park, H. N. Chang, P. Wang, J. W. Grate and T. Hyeon, Small, 2005, 1, 744–753 CrossRef CAS PubMed.
- M. I. Kim, J. Kim, J. Lee, S. Shin, H. B. Na, T. Hyeon, H. G. Park and H. N. Chang, Microporous Mesoporous Mater., 2008, 111, 18–23 CrossRef CAS PubMed.
- I. K. Moon, J. Kim, J. Lee, H. Jia, B. N. Hyon, K. Y. Jong, H. K. Ja, A. Dohnalkova, J. W. Grate, P. Wang, T. Hyeon, G. P. Hyun and N. C. Ho, Biotechnol. Bioeng., 2007, 96, 210–218 CrossRef PubMed.
- S. H. Jun, J. Lee, B. C. Kim, J. E. Lee, J. Joo, H. Park, J. H. Lee, S. M. Lee, D. Lee, S. Kim, Y. M. Koo, C. H. Shin, S. W. Kim, T. Hyeon and J. Kim, Chem. Mater., 2012, 24, 924–929 CrossRef CAS.
- R. Reshmi and S. Sugunan, J. Mol. Catal. B: Enzym., 2013, 85–86, 111–118 CAS.
- D. Jung, M. Paradiso, D. Wallacher, A. Brandt and M. Hartmann, ChemSusChem, 2009, 2, 161–164 CrossRef CAS PubMed.
- D. Jung, M. Paradiso and M. Hartmann, J. Mater. Sci., 2009, 44, 6747–6753 CrossRef CAS.
- D. Jung and M. Hartmann, Catal. Today, 2010, 157, 378–383 CrossRef CAS PubMed.
- F. López-Gallego, L. Betancor, C. Mateo, A. Hidalgo, N. Alonso-Morales, G. Dellamora-Ortiz, J. M. Guisán and R. Fernández-Lafuente, J. Biotechnol., 2005, 119, 70–75 CrossRef PubMed.
- A. S. Bommarius and M. F. Paye, Chem. Soc. Rev., 2013, 42, 6534–6565 RSC.
- U. Hanefeld, L. Cao and E. Magner, Chem. Soc. Rev., 2013, 42, 6211–6212 RSC.
- S. Cantone, V. Ferrario, L. Corici, C. Ebert, D. Fattor, P. Spizzo and L. Gardossi, Chem. Soc. Rev., 2013, 42, 6262–6276 RSC.
- L. Betancor, F. López-Gallego, A. Hidalgo, N. Alonso-Morales, G. Dellamora-Ortiz, J. M. Guisán and R. Fernández-Lafuente, J. Biotechnol., 2006, 121, 284–289 CrossRef CAS PubMed.
- M. Filho, B. C. Pessela, C. Mateo, A. V. Carrascosa, R. Fernandez-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2008, 42, 265–271 CrossRef CAS PubMed.
- D. Radva, J. Spanyol and J. Kosáry, Food Technol. Biotechnol., 2011, 49, 257–262 CAS.
- C. Z. Guidini, J. Fischer, L. N. S. Santana, V. L. Cardoso and E. J. Ribeiro, Biochem. Eng. J., 2010, 52, 137–143 CrossRef CAS PubMed.
- C. Pan, B. Hu, W. Li, Y. Sun, H. Ye and X. Zeng, J. Mol. Catal. B: Enzym., 2009, 61, 208–215 CrossRef CAS PubMed.
- C. Sisak, Z. Csanádi, E. Rónay and B. Szajáni, Enzyme Microb. Technol., 2006, 39, 1002–1007 CrossRef CAS PubMed.
- A. Kumar and S. S. Kanwar, Enzyme Res., 2011, 2011, 718949 CrossRef PubMed.
- A. Kumar and S. S. Kanwar, Bioresour. Technol., 2011, 102, 2162–2167 CrossRef CAS PubMed.
- G. Fernández-Lorente, J. M. Palomo, C. Mateo, R. Munilla, C. Ortiz, Z. Cabrera, J. M. Guisán and R. Fernández-Lafuente, Biomacromolecules, 2006, 7, 2610–2615 CrossRef PubMed.
- J. M. Palomo, R. L. Segura, G. Fernandez-Lorente, R. Fernandez-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2007, 40, 704–707 CrossRef CAS PubMed.
- M. Filho, B. C. Pessela, C. Mateo, A. V. Carrascosa, R. Fernandez-Lafuente and J. M. Guisán, Process Biochem., 2008, 43, 1142–1146 CrossRef CAS PubMed.
- M. Fuentes, P. Batalla, V. Grazu, B. C. C. Pessela, C. Mateo, T. Montes, J. A. Hermoso, J. M. Guisan and R. Fernandez-Lafuente, Biomacromolecules, 2007, 8, 703–707 CrossRef CAS PubMed.
- B. C. C. Pessela, R. Munilla, L. Betancor, M. Fuentes, A. V. Carrascosa, A. Vian, R. Fernandez-Lafuente and J. M. Guisán, J. Chromatogr., A, 2004, 1034, 155–159 CrossRef CAS PubMed.
- C. Mateo, O. Abian, G. Fernández-Lorente, J. Pedroche, R. Fernández-Lafuente, J. M. Guisan, A. Tam and M. Daminati, Biotechnol. Prog., 2002, 18, 629–634 CrossRef CAS PubMed.
- B. C. C. Pessela, M. Fuentes, C. Mateo, R. Munilla, A. V. Carrascosa, R. Fernandez-Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2006, 39, 909–915 CrossRef CAS PubMed.
- B. C. C. Pessela, L. Betancor, F. Lopez-Gallego, R. Torres, G. M. Dellamora-Ortiz, N. Alonso-Morales, M. Fuentes, R. Fernández-Lafuente, J. M. Guisán and C. Mateo, Enzyme Microb. Technol., 2005, 37, 295–299 CrossRef CAS PubMed.
- M. Fuentes, C. Mateo, B. C. C. Pessela, P. Batalla, R. Fernandez-Lafuente and J. M. Guisán, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 849, 243–250 CrossRef CAS PubMed.
- A. Schellenberger and R. Ulbrich, Biomed. Biochim. Acta, 1989, 48, 63–67 CAS.
- J. Mansfeld, G. Vriendl, O. R. Veltman, B. Van Den Burg, G. Venema, V. G. H. Eijsink and R. Utbrich-Hofmann, FASEB J., 1997, 11 Search PubMed.
- R. Ulbrich-Hofmann, U. Arnold and J. Mansfeld, J. Mol. Catal. B: Enzym., 1999, 7, 125–131 CrossRef CAS.
- J. Mansfeld, G. Vriend, B. Van Den Burg, V. G. H. Eijsink and R. Ulbrich-Hofmann, Biochemistry, 1999, 38, 8240–8245 CrossRef CAS PubMed.
- S. Pahujani, S. S. Kanwar, G. Chauhan and R. Gupta, Bioresour. Technol., 2008, 99, 2566–2570 CrossRef CAS PubMed.
- A. Pal and F. Khanum, Process Biochem., 2011, 46, 1315–1322 CrossRef CAS PubMed.
- S. Nagar, A. Mittal, D. Kumar, L. Kumar and V. K. Gupta, Process Biochem., 2012, 47, 1402–1410 CrossRef CAS PubMed.
- T. N. Nwagu, B. N. Okolo and H. Aoyagi, J. Microbiol. Biotechnol., 2012, 22, 628–636 CrossRef CAS.
- G. Yang, J. Wu, G. Xu and L. Yang, Colloids Surf., B, 2010, 78, 351–356 CrossRef CAS PubMed.
- M. Yiitolu and Z. Temoçin, J. Mol. Catal. B: Enzym., 2010, 66, 130–135 CrossRef PubMed.
- M. P. Klein, L. P. Fallavena, J. D. N. Schöfer, M. A. Z. Ayub, R. C. Rodrigues, J. L. Ninow and P. F. Hertz, Carbohydr. Polym., 2013, 95, 465–470 CrossRef CAS PubMed.
- M. P. Klein, M. R. Nunes, R. C. Rodrigues, E. V. Benvenutti, T. M. H. Costa, P. F. Hertz and J. L. Ninow, Biomacromolecules, 2012, 13, 2456–2464 CrossRef CAS PubMed.
- S. G. Valerio, J. S. Alves, M. P. Klein, R. C. Rodrigues and P. F. Hertz, Carbohydr. Polym., 2013, 92, 462–468 CrossRef CAS PubMed.
- W. L. Stanley, G. G. Watters, S. H. Kelly, B. G. Chan, J. A. Garibaldi and J. E. Schade, Biotechnol. Bioeng., 1976, 18, 439–443 CrossRef CAS.
- C. Mateo, O. Abian, M. Bernedo, E. Cuenca, M. Fuentes, G. Fernandez-Lorente, J. M. Palomo, V. Grazu, B. C. C. Pessela, C. Giacomini, G. Irazoqui, A. Villarino, K. Ovsejevi, F. Batista-Viera, R. Fernandez-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2005, 37, 456–462 CrossRef CAS PubMed.
- J. M. Bolivar, C. Mateo, C. Godoy, B. C. C. Pessela, D. S. Rodrigues, R. L. C. Giordano, R. Fernandez-Lafuente and J. M. Guisan, Process Biochem., 2009, 44, 757–763 CrossRef CAS PubMed.
- C. Mateo, G. Fernández-Lorente, O. Abian, R. Fernández-Lafuente and J. M. Guisán, Biomacromolecules, 2000, 1, 739–745 CrossRef CAS.
- C. Mateo, V. Grazu, J. M. Palomo, F. Lopez-Gallego, R. Fernandez-Lafuente and J. M. Guisan, Nat. Protoc., 2007, 2, 1022–1033 CrossRef CAS PubMed.
- O. Barbosa, R. Torres, C. Ortiz, Á. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Biomacromolecules, 2013, 14, 2433–2462 CrossRef CAS PubMed.
- A. Kumar, I. Y. Galaev and B. Mattiasson, J. Chromatogr., B: Biomed. Sci. Appl., 2000, 741, 103–113 CrossRef CAS.
- B. C. C. Pessela, R. Torres, M. Fuentes, C. Mateo, R. Munilla, A. Vian, A. V. Carrascosa, J. L. Garcia, J. M. Guisán and R. Fernandez-Lafuente, J. Chromatogr., A, 2004, 1055, 93–98 CrossRef CAS PubMed.
- F. H. Arnold, Biotechnology, 1991, 9, 151–156 CrossRef CAS PubMed.
- E. Hochuli, H. Döbeli and A. Schacher, J. Chromatogr., A, 1987, 411, 177–184 CrossRef CAS.
- V. Gaberc-Porekar and V. Menart, J. Biochem. Biophys. Methods, 2001, 49, 335–360 CrossRef CAS.
- M. Fuentes, C. Mateo, R. Fernández-Lafuente and J. M. Guisán, Biomacromolecules, 2006, 7, 540–544 CrossRef CAS PubMed.
- W. S. Adriano, E. H. C. Filho, J. A. Silva, R. L. C. Giordano and L. R. B. Gonçalves, Braz. J. Chem. Eng., 2005, 22, 529–538 CrossRef CAS PubMed.
- W. S. Adriano, E. H. C. Filho, J. A. Silva and L. R. B. Gonçalves, Biotechnol. Appl. Biochem., 2005, 41, 201–207 CrossRef CAS PubMed.
- W. S. Adriano, D. B. Mendonça, D. S. Rodrigues, E. J. Mammarella and R. L. C. Giordano, Biomacromolecules, 2008, 9, 2170–2179 CrossRef CAS PubMed.
- K. E. Jaeger and T. Eggert, Curr. Opin. Biotechnol., 2002, 13, 390–397 CrossRef CAS.
- K. E. Jaeger and M. T. Reetz, Trends Biotechnol., 1998, 16, 396–403 CrossRef CAS.
- R. D. Schmid and R. Verger, Angew. Chem., Int. Ed., 1998, 37, 1609–1633 CrossRef CAS.
- R. Verger, Trends Biotechnol., 1997, 15, 32–38 CrossRef CAS.
- Z. S. Derewenda, U. Derewenda and G. G. Dodson, J. Mol. Biol., 1992, 227, 818–839 CrossRef CAS.
- N. Miled, F. Beisson, J. De Caro, A. De Caro, V. Arondel and R. Verger, J. Mol. Catal. B: Enzym., 2001, 11, 165–171 CrossRef CAS.
- P. Reis, K. Holmberg, H. Watzke, M. E. Leser and R. Miller, Adv. Colloid Interface Sci., 2009, 147–148, 237–250 CrossRef CAS PubMed.
- R. Fernandez-Lafuente, P. Armisén, P. Sabuquillo, G. Fernández-Lorente and J. M. Guisán, Chem. Phys. Lipids, 1998, 93, 185–197 CrossRef CAS.
- J. M. Palomo, C. Ortiz, M. Fuentes, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, J. Chromatogr., A, 2004, 1038, 267–273 CrossRef CAS PubMed.
- J. M. Palomo, M. M. Peñas, G. Fernández-Lorente, C. Mateo, A. G. Pisabarro, R. Fernández-Lafuente, L. Ramírez and J. M. Guisán, Biomacromolecules, 2003, 4, 204–210 CrossRef CAS PubMed.
- J. M. Palomo, G. Muoz, G. Fernández-Lorente, C. Mateo, R. Fernández-Lafuente and J. M. Guisán, J. Mol. Catal. B: Enzym., 2002, 19-20, 279–286 CrossRef CAS.
- J. Uppenberg, S. Patkar, T. Bergfors and T. A. Jones, J. Mol. Biol., 1994, 235, 790–792 CrossRef CAS PubMed.
- C. Carrasco-López, C. Godoy, B. de las Rivas, G. Fernández-Lorente, J. M. Palomo, J. M. Guisán, R. Fernández-Lafuente, M. Martínez-Ripoll and J. A. Hermoso, J. Biol. Chem., 2009, 284, 4365–4372 CrossRef PubMed.
- J. M. Palomo, M. Fuentes, G. Fernández-Lorente, C. Mateo, J. M. Guisan and R. Fernández-Lafuente, Biomacromolecules, 2003, 4, 1–6 CrossRef CAS PubMed.
- G. Fernández-Lorente, J. M. Palomo, M. Fuentes, C. Mateo, J. M. Guisán and R. Fernández-Lafuente, Biotechnol. Bioeng., 2003, 82, 232–237 CrossRef PubMed.
- H. J. An, H. J. Lee, S. H. Jun, S. Y. Hwang, B. C. Kim, K. Kim, K. M. Lee, M. K. Oh and J. Kim, Bioprocess Biosyst. Eng., 2011, 34, 841–847 CrossRef CAS PubMed.
- S. M. Lee, L. H. Jin, J. H. Kim, S. O. Han, H. B. Na, T. Hyeon, Y. M. Koo, J. Kim and J. H. Lee, Bioprocess Biosyst. Eng., 2010, 33, 141–147 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |