Polythiophene synthesis via halogen dance†‡
Keisuke
Shono
,
Yugo
Sumino
,
Shota
Tanaka
,
Shunsuke
Tamba
and
Atsunori
Mori
*
Department of Chemical Science and Engineering, Kobe University, 1-1 Rokkodai, Nada, Kobe 657-8501, Japan. E-mail: amori@kobe-u.ac.jp
First published on 16th May 2014
Abstract
Polymerization of 2,4-dibromo-3-hexyl-5-lithiated thiophene that is formed by deprotonation of 2,5-dibromo-3-hexylthiophene with LDA and following halogen dance takes place in the presence of a nickel(II) or palladium(II) catalyst to afford poly(3-bromo-4-hexylthiophen-2,5-diyl).
Introduction
Polythiophenes have recently attracted much attention as materials for a variety of electronic devices.1 Development of a facile method for the preparation of polythiophenes is of particular interest in organic and organometallic chemistry as well as polymer synthesis. Cross coupling of metalated thiophenes with thienyl halides in the presence of a transition metal catalyst is one of the most practical preparative methods to form the thiophene–thiophene bond leading to polythiophenes, and various types of polythiophenes are indeed synthesized by the cross coupling methodology.2 In particular, the reaction of 2-halo-5-metalated thiophene, which is in situ formed by the reaction of the corresponding 2,5-dihalothiophene developed by Rieke, McCullough, and Yokozawa, is extensively studied to afford poly(3-substituted thiophen-2,5-diyl)s with remarkably excellent head-to-tail regioregularity in controllable manners of molecular weight and molecular weight distribution based on the amount of catalyst loading.3On the other hand, we have been engaged in the development of transition metal-catalyzed coupling reactions, in which coupling at the C–H bond of heteroaromatic compounds has been of our particular concern.4 We have recently developed several polymerizations of thiophene derivatives that occur by the condensation at the C–H and C–X bonds leading to polythiophenes, where deprotonative metalation of halothiophene has played an important role for the successful polymerization.5 Accordingly, development of a new class of metalation protocols for heteroaromatic compounds is highly important in the synthesis of π-conjugated polymers. Concerning the polythiophene synthesis, organomagnesium (Grignard reagents), zinc, boron, and tin reagents have been mainly employed as metallic species.2–5 In addition to the above metals, we have shown that organolithium species of thiophene derivatives, indeed, can be available for nickel(II)-catalyzed polythiophene synthesis, and lithiation was performed by deprotonation at the C–H bond or by lithium–halogen exchange at the carbon–halogen bond.6 Among lithiation of (hetero)aromatic compounds, an alternative choice also for the formation of organolithium species is halogen dance, which occurs in intra- or intermolecular halogen–lithium exchange.7 In our preliminary communication on Murahashi coupling polymerization of thiophene derivatives with organolithium species, we have partially described that organolithium of thiophene formed by halogen dance can also serve as an organometallic monomer for polythiophene synthesis;6 however, few studies on the detail have been discussed. Herein we report halogen dance of thiophene derivatives toward the formation of lithiated monomer, polymerization of the thus formed metalated species, and transformation of the polythiophene formed through halogen dance.
Results and discussion
First, we studied the optimum conditions for halogen dance with 2,5-dibromo-3-hexylthiophene (1) as shown in Scheme 1. Deprotonation of 1 at the 4-position was carried out with LDA (lithium diisopropylamide: LiNiPr2) at −78 °C. After stirring for 5 min, progress of halogen dance was confirmed by treatment with H2O to obtain 35% of 2,4-dibromo-3-hexylthiophene (2). Since the progress of deprotonation under similar conditions was also found to be ca. 45%, which was confirmed by treatment of a deprotonated mixture with iodine to observe unreacted ca. 55% of 1, these results suggested that the lithiated species at the 4-position was mostly rearranged into 5-lithio-4-brominated thiophene at −78 °C through halogen dance.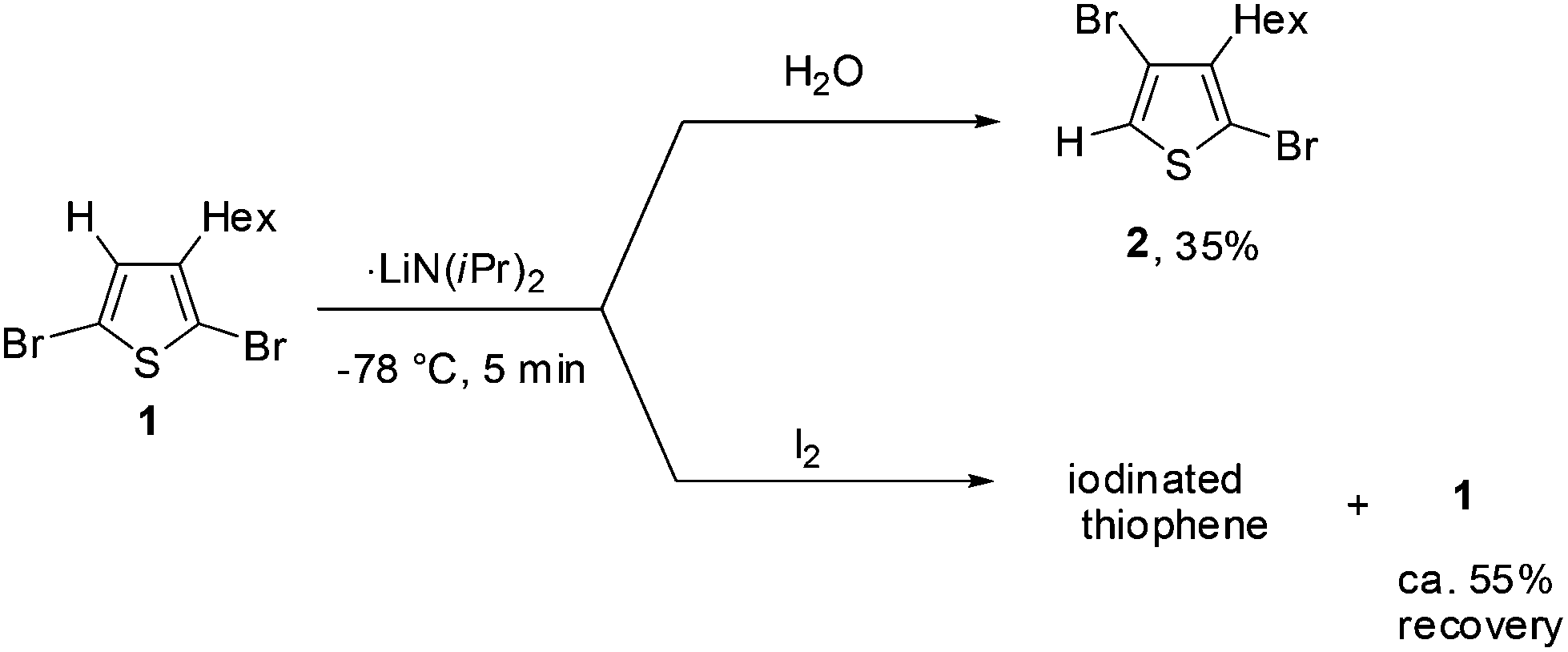 | ||
| Scheme 1 Deprotonation of 1 with LDA. | ||
Halogen dance under several conditions was then examined as summarized in Table 1. When 1 was treated with 1.1 equivalents of LDA at 0 °C and room temperature for 5 min, 90%, respectively, of 2 was obtained by treatment with water. On the other hand, deprotonation with LTMP (lithium 2,2,6,6-tetramethylpiperidide) was found slower to observe conversion to 2 in only 75% after stirring at room temperature for 30 min whereas further reaction to 4 h improved the reaction to 81%. The reaction of 1 with Cy2NLi (lithium dicyclohexylamide) underwent halogen dance smoothly to observe, after treatment with water, conversion to 2 in 85% (30 min) and 96% (5 h), respectively. It was also found that the reaction of 1 with lithium diethylamide (Et2NLi) at room temperature for 30 min resulted in 85% conversion. By contrast, LHMDS (lithium hexamethyldisilazide) was found to hardly deprotonate 1 at room temperature for 30 min. Conditions at 60 °C for 24 h were required to afford 2 in 77% conversion.
| Lithium amide | Temp. (°C) | Time | Conversion to 2b (%) |
|---|---|---|---|
| a The reaction was carried out with 1 (0.5 mmol) and 1.1 equiv. of lithium amide in THF. b Confirmed by 1H NMR after quenching the crude reaction mixture with water. c Lithium diisopropylamide. d Lithium 2,2,6,6-tetramethylpiperidide. e Lithium dicyclohexylamide. f Lithium hexamethyldisilazide. | |||
| LDAc | −78 | 5 min | 35 |
| 0 | 5 min | 90 | |
| r.t. | 5 min | 90 | |
| LTMPd | r.t. | 30 min | 75 |
| r.t. | 4 h | 81 | |
| Cy2NLie | r.t. | 30 min | 85 |
| r.t. | 5 h | 96 | |
| Et2NLi | r.t. | 30 min | 85 |
| LHMDSf | r.t. | 30 min | 0 |
| 60 | 24 h | 77 | |
Halogen dance with magnesium amide was also studied. When the reaction of 1 was examined with the Knochel–Hauser base8 TMPMgCl·LiCl (chloromagnesium-2,2,6,6-tetramethylpiperidide lithium chloride salt), halogen dance hardly took place at room temperature for 3 h. However, the reaction with iPr2NMgCl·LiCl (1.2 equiv.) resulted in ca. 60% halogen dance, which was confirmed by quenching with water to afford 2 (Scheme 2). Since treatment with iodine suggested that the amount of deprotonation was 68%, most of the metalated thiophene magnesium species induced halogen dance. Although the reaction of iPr2NMgCl·LiCl at elevated temperature (60 °C) was also examined, little improvement of the reaction was observed.
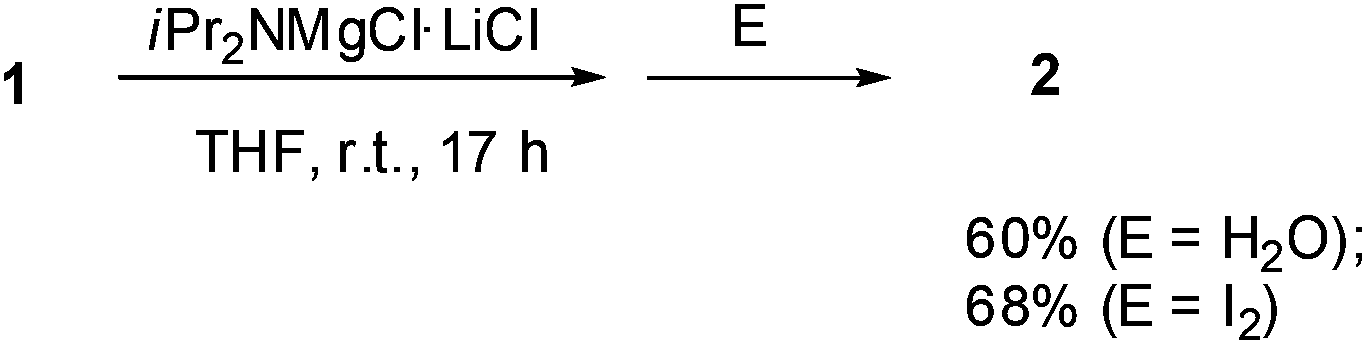 | ||
| Scheme 2 Attempted halogen dance of 1 with iPr2NMgCl·LiCl. | ||
With the studied reaction conditions of halogen dance, polymerization of the thus obtained lithiated thiophene species was carried out. After 2,5-dibromothiophene 1 was added to a THF solution of LDA at −78 °C, the reaction mixture was gradually raised to 0 °C over 30 min to induce halogen dance. Then, 2.0 mol% of the nickel catalyst bearing the N-heterocyclic carbene (NHC) ligand NiCl2(PPh3)IPr9 was added to the resulting mixture and stirring was continued at 60 °C for 1 h to afford polymer 3 in 77% isolated yield. SEC analysis was revealed to show Mn = 7900 (Mw/Mn = 1.28) (Scheme 3).
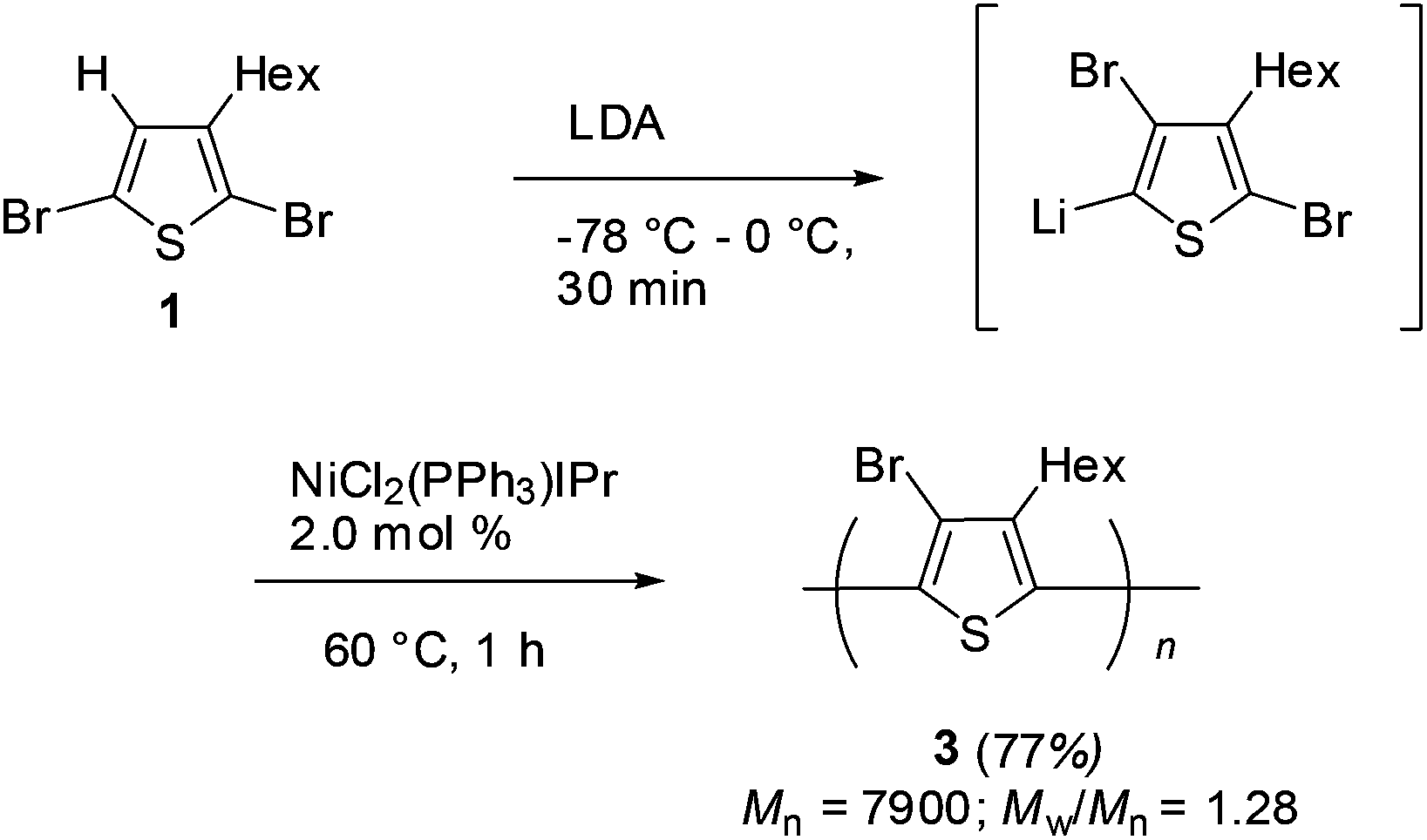 | ||
| Scheme 3 Halogen dance polymerization of 1 with LDA. | ||
Fig. 1 shows the 1H NMR spectrum of the obtained polymer indicating that the proton signal observed at 6.8 ppm disappeared. Broadening of alkyl signals corresponding to the substituent at the 3-position also supports polymer formation. In addition, elemental analysis of the obtained polythiophene indicated bromine contents of 32.83%, showing good correspondence to Br(C10H13BrS)32H (33.27%). The result suggests that the monomer unit of 3 contains bromine atoms. In contrast with poly(3-hexylthiophen-2,5-diyl), the polymer bearing a bromine atom at the 4-position 3 was found to be highly soluble in organic solvents such as THF, chloroform, and hexane. The results suggest that π-conjugation between the thiophene rings in the polymer chain is less extended.
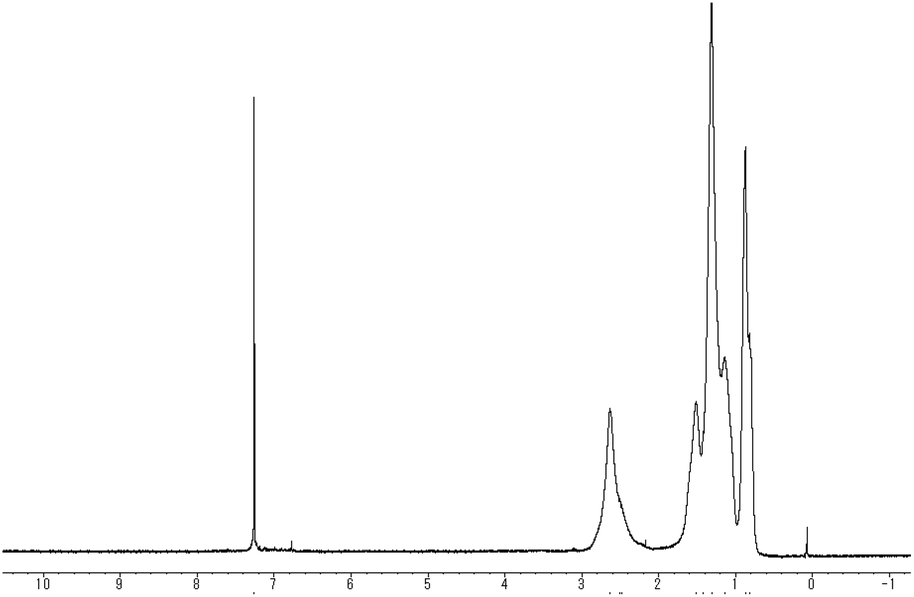 | ||
| Fig. 1 1H NMR spectrum (CDCl3) of 3. | ||
Table 2 represents halogen dance polymerization of 1 under several conditions. Compared with the polymerization conditions shown in Scheme 3 (entry 1), it was found that polymerization with NiCl2(PPh3)IPr also proceeded at room temperature in 78% yield (Mn = 7900; Mw/Mn = 1.28) (entry 2). On the other hand, other nickel catalysts bearing bidentate phosphine ligands dppp and dppf were less effective to afford oligomers with inferior yields (entries 3 and 4). Considering that nickel catalysts bearing bidentate phosphines effectively undergo polymerization of 3-hexylthiophene, polymerization to afford 5 would be less reactive because of the steric effect of the bromine substituent. A palladium catalyst was also found to undergo polymerization when NHC is employed as a ligand.10 The use of palladium catalysts bearing NHC ligands (Umicore CX31 and CX32)11 induced polymerization to afford 3 of slightly higher molecular weight although the molecular weight distribution was much broader (entries 5 and 6). However, polymerization with PEPPSI12-SIPr at room temperature afforded 3 of higher molecular weight (entry 7). The lithiated monomer via halogen dance with Cy2NLi and LTMP was also found to afford polymer 3 smoothly (entries 8–11).
| Entry | Halogen dance (temp., time) | Catalyst (mol%) | Polymerization temp. (°C), time (h) | Yield (%) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
M
w/Mnb |
|---|---|---|---|---|---|---|
| a The reaction was carried out with 1 (0.5 mmol) and 1.1 equiv. of lithium amide in THF for the metalation and 1.0–5.0 mol% of catalyst was employed for the polycondensation. b M n and Mw/Mn values were estimated by SEC analysis using CHCl3 as an eluent. c IPr: 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene. d [1,3-Bis(2,6-diisopropylphenyl)imidazol-2-ylidene]chloro[3-phenylallyl]palladium(II). e [1,3-Bis(2,6-diisopropylphenyl)imidazolin-2-ylidene]chloro[3-phenylallyl]palladium(II). f (1,3-Bis(2,6-diisopropylphenyl)imidazolidene)(3-chloropyridyl)palladium(II) dichloride. | ||||||
| 1 | LDA (0, 30 min) | NiCl2(PPh3)IPrc (2.0) | 60, 1 | 77 | 7900 | 1.28 |
| 2 | LDA (0, 30 min) | NiCl2(PPh3)IPrc (1.3) | r.t., 1 | 78 | 5100 | 1.45 |
| 3 | LDA (r.t., 2 h) | NiCl2dppp (2.0) | 60, 24 | 44 | 1200 | |
| 4 | LDA (r.t., 3.5 h) | NiCl2dppf (2.0) | 60, 24 | 62 | 1100 | |
| 5 | LDA (r.t., 2 h) | Umicore CX31d (5.0) | 60, 24 | 43 | 4400 | 3.36 |
| 6 | LDA (r.t., 2 h) | Umicore CX32e (5.0) | 60, 24 | 56 | 6000 | 3.48 |
| 7 | LDA (0, 30 min) | PEPPSI-SIPrf (1.3) | r.t., 72 | 67 | 7700 | 1.66 |
| 8 | Cy2NLi (r.t., 5 h) | NiCl2(PPh3)IPr (1.0) | 60, 24 | 72 | 10100 |
1.61 |
| 9 | Cy2NLi (r.t., 5 h) | NiCl2(PPh3)IPr (1.0) | r.t., 24 | 41 | 6700 | 1.56 |
| 10 | Cy2NLi (r.t., 5 h) | PEPPSI-SIPrf (1.0) | r.t., 24 | 53 | 6100 | 1.57 |
| 11 | LTMP (r.t., 8 h) | PEPPSI-SIPrf (1.0) | r.t., 24 | 72 | 5600 | 1.54 |
Table 3 summarizes the results on the relationship of molecular weight with catalyst loading in the halogen dance polymerization. It was found that the molecular weight of 3 increased as the amount of catalyst loading decreased. However, the molecular weight was found much inferior to the theoretical line based on the ideal living polymerization.
| 1/Ni cat. | Yield (%) | M (theor.) | M n | M w/Mnc |
|---|---|---|---|---|
| a The reaction was carried out with 1 and 1.1 equiv. of Cy2NLi in THF for halogen dance (−78 °C–r.t. over 5 h) and 1.0–3.0 mol% of NiCl2(PPh3)IPr was employed for the polymerization. b Theoretical molecular weight based on the ratio of 1/nickel catalyst at 100% conversion. c M n and Mw/Mn values were estimated by SEC analysis using CHCl3 as an eluent. | ||||
| 33 | 65 | 8200 | 2800 | 1.22 |
| 50 | 61 | 12300 |
3300 | 1.28 |
| 67 | 78 | 16300 |
4400 | 1.42 |
| 100 | 72 | 24500 |
10100 |
1.61 |
A thiophene derivative bearing a methyl substituent at the 3-position was also subjected to halogen dance polymerization. The reaction of 2,5-dibromo-3-methylthiophene 4 was treated with lithium dicyclohexylamide at −78 °C and then gradually raised to room temperature over 4.5 h. Addition of the palladium catalyst PEPPSI-SIPr (1.0 mol%) at room temperature induced polymerization in a similar manner to the case of 1 to afford the corresponding polymer 5 in 85% yield after stirring for 24 h (Mn = 6100, Mw/Mn = 1.71). The reaction of chlorothiophene was also performed with 5-bromo-2-chloro-3-hexylthiophene (6). Treatment of 6 with LDA and subsequent addition of NiCl2(PPh3)IPr (1.0 mol%) resulted in polymerization at room temperature for 24 h to afford the corresponding polymer 3 in a quantitative yield (Mn = 4900, Mw/Mn = 1.58). These results are depicted in Scheme 4.
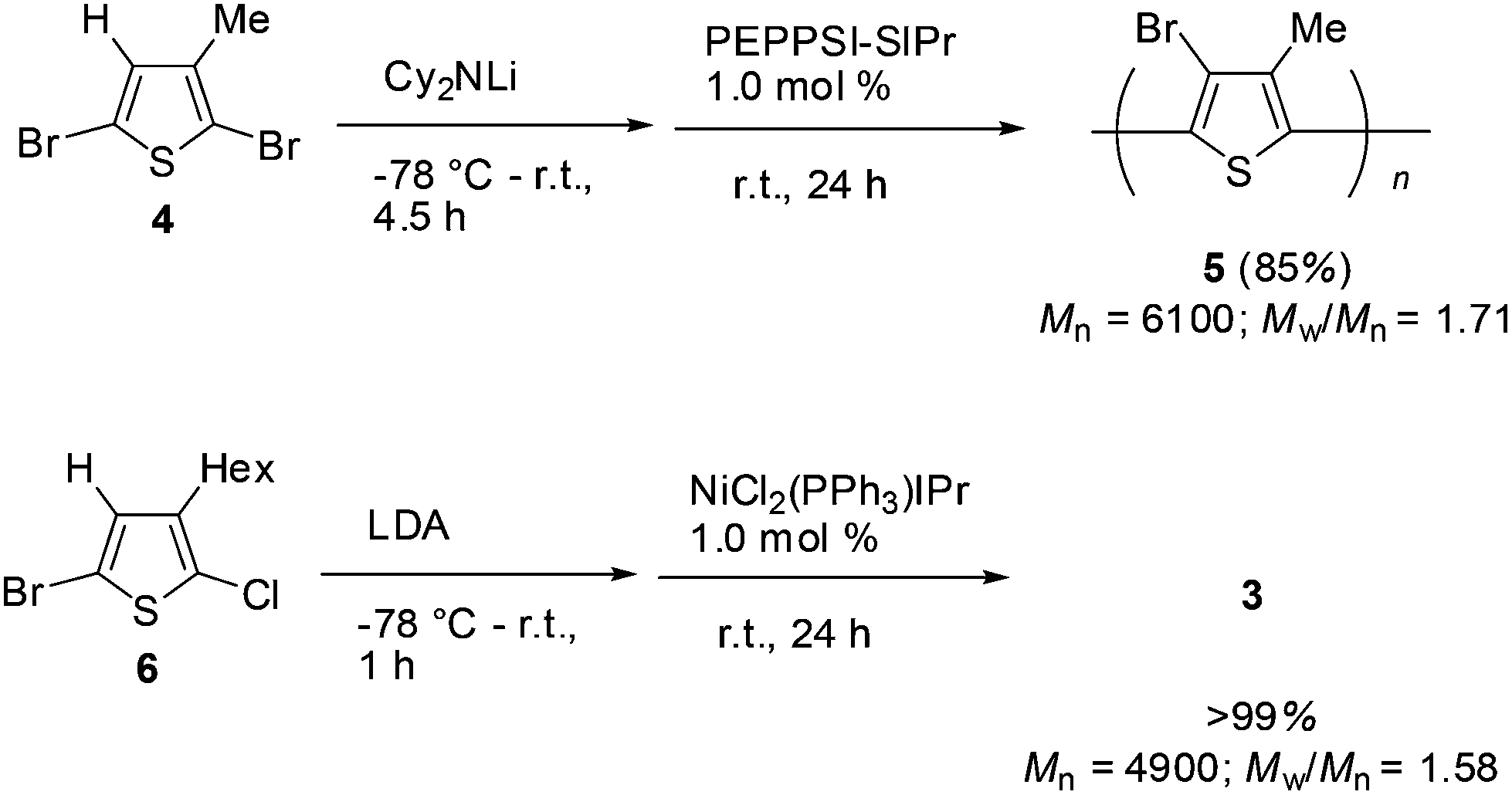 | ||
| Scheme 4 Halogen dance polymerization of 4 and 6. | ||
Polymer 3 obtained by halogen dance polymerization was subjected to transformation. It is interesting that polymerization of 2,5-dihalo-3-alkylthiophene gives polymers of different structures by switching the metalation protocol. The usual GRIM (Grignard metathesis)3 metalation and the following transition-metal-catalyzed polycondensation gives poly(3-alkylthiophen-2,5-diyl) whereas halogen dance is shown to lead to poly(3-alkyl-4-bromo-thiophen-2,5-diyl). We envisaged that the obtained polymer by halogen dance would be transformed by reduction of the bromine atom toward poly(3-alkylthiophen-2,5-diyl). Accordingly, the polymer obtained by halogen dance (Mn = 7100; Mw/Mn = 1.55) was treated with tBuMgCl (2.0 equiv.) in the presence of NiCl2dppp at room temperature for 24 h. Measurement of the 1H NMR spectrum revealed that a new signal at ca. 7 ppm, which is identical with the methine proton of the thiophene ring of P3HT, appeared. A comparison of the integral value with that of the CH2 signal of the hexyl group adjacent to the thiophene ring suggested that ca. 60% of bromine atoms were reduced to hydrogen (Scheme 5). The color of the polymer was changed from yellow to dark purple. Measurements of UV-vis spectra of 3 and the transformed polymer (λmax shifted to 357 nm to 376 nm) also suggested that extended π-conjugation increased close to that of P3HT (λmax = ca. 450 nm).
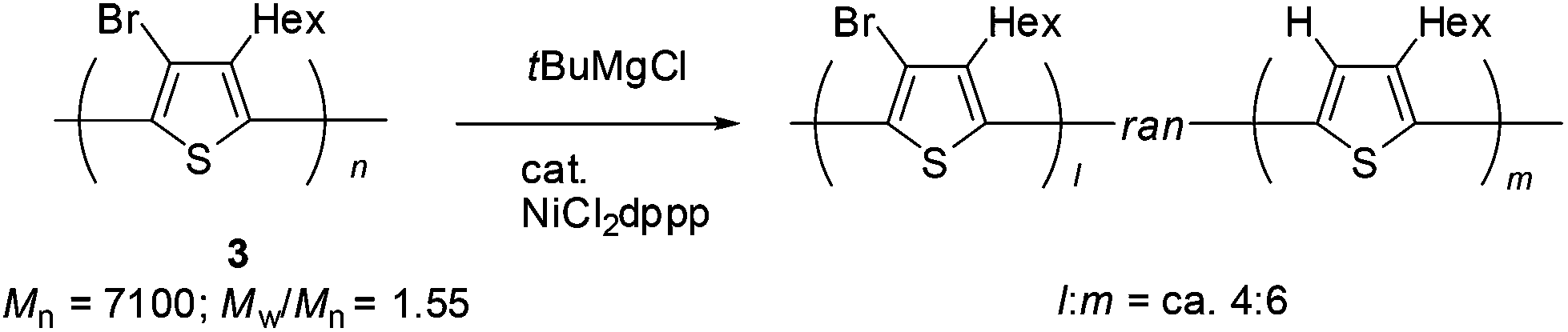 | ||
| Scheme 5 Transformation of 3. | ||
Conclusion
In conclusion, we have shown a new class of polymerizations with organolithium species of a thiophene derivative, which was generated by halogen dance of 2,5-dihalo-3-alkylthiophene by treatment with lithium amide. The reaction afforded polythiophene bearing a bromine atom as a substituent of the thiophene ring. The reaction was found to proceed in the presence of the nickel(II) or palladium(II) catalyst via cross coupling polycondensation of organolithium. Debromination of the obtained polymer increased the contents of extended π-conjugation. If a suitable halogen dance is designed in not only a thiophene ring but also another class of aromatic rings, this strategy enhances a variety of halogen-containing polymer materials, which allow a wide range of organic transformation reactions.Acknowledgements
This work was partially supported by a Grant-in-Aid (B) for Scientific Research from the Japan Society for the Promotion of Science (JSPS), Japan (no. 22350042 and 25288049). S.T. and S.T. thank JSPS for a Research Fellowship for Young Scientists. The authors thank Umicore, Japan KK for the kind donation of palladium catalysts CX31 and CX32.Notes and references
- (a) P. M. Beaujunge and J. M. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009 CrossRef PubMed; (b) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 197, 899 Search PubMed.
- (a) F. Diederich and P. J. Stang, Metal-Catalyzed Cross-Coupling Reaction, Wiley-VCH, Weinheim, Germany, 1998 Search PubMed; (b) A. Mori and M. S. Mohamed-Ahmed, in Cross-coupling Polymerization, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2007, vol. 11, pp. 653–690 Search PubMed; (c) T. Yamamoto, Bull. Chem. Soc. Jpn., 2010, 83, 431 CrossRef CAS; (d) B. Carsten, F. He, H.-J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493 CrossRef CAS PubMed.
- (a) T. A. Chen and R. D. Rieke, J. Am. Chem. Soc., 1992, 114, 10087 CrossRef CAS; (b) T. A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233 CrossRef CAS; (c) R. S. Loewe, S. M. KIhersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250 CrossRef CAS; (d) E. E. Sheina, J. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526 CrossRef CAS; (e) M. C. Iovu, E. E. Sheina, R. R. Gil and R. D. McCullough, Macromolecules, 2005, 38, 8649 CrossRef CAS; (f) A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169 CrossRef CAS; (g) R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542 CrossRef CAS PubMed; (h) R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 753 CrossRef CAS; (i) E. L. Lanni and A. J. McNeil, J. Am. Chem. Soc., 2009, 131, 16573 CrossRef CAS PubMed; (j) R. Tkachov, V. Senkovskyy, H. Komber, J.-U. Sommer and A. Kiriy, J. Am. Chem. Soc., 2010, 132, 7803 CrossRef CAS PubMed; (k) H. A. Bronstein and C. K. Luscombe, J. Am. Chem. Soc., 2009, 131, 12894 CrossRef CAS PubMed; (l) M. Wong, J. Hollinger, L. M. Kozycz, T. M. ScCormick, Y. Lu, D. C. Burns and D. S. Seferos, ACS Macro Lett., 2012, 1, 1266 CrossRef CAS; (m) Z. J. Bryan, M. L. Smith and A. J. McNeil, Macromol. Rapid Commun., 2012, 33, 842 CrossRef CAS PubMed. For reviews: (n) T. Yokozawa and A. Yokoyama, Chem. Rev., 2009, 109, 5595 CrossRef CAS PubMed; (o) R. D. McCullough, Adv. Mater., 1998, 10, 93 CrossRef CAS; (p) I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202 CrossRef CAS PubMed; (q) K. Okamoto, J. Zhang, J. B. Housekeeper, S. R. Marder and C. K. Luscombe, Macromolecules, 2013, 46, 8059 CrossRef CAS; (r) K. Okamoto and C. K. Luscombe, Polym. Chem., 2011, 2, 2424 RSC; (s) M. C. Stefan, M. P. Bhatt, P. Sista and H. D. Magurudeniya, Polym. Chem., 2012, 3, 1693 RSC.
- (a) A. Mori, A. Sekiguchi, K. Masui, T. Shimada, M. Horie, K. Osakada, M. Kawamoto and T. Ikeda, J. Am. Chem. Soc., 2003, 125, 1700 CrossRef CAS PubMed; (b) K. Masui, H. Ikegami and A. Mori, J. Am. Chem. Soc., 2004, 126, 5074 CrossRef CAS PubMed; (c) M. Takahashi, K. Masui, H. Sekiguchi, N. Kobayashi, A. Mori, M. Funahashi and N. Tamaoki, J. Am. Chem. Soc., 2006, 128, 10930 CrossRef CAS PubMed; (d) K. Kobayashi, A. Sugie, M. Takahashi, K. Masui and A. Mori, Org. Lett., 2005, 7, 5083 CrossRef CAS PubMed; (e) D. Monguchi, T. Fujiwara, H. Furukawa and A. Mori, Org. Lett., 2009, 11, 1607 CrossRef CAS PubMed; (f) D. Monguchi, A. Yamamura, T. Fujiwara, T. Somete and A. Mori, Tetrahedron Lett., 2010, 51, 850 CrossRef CAS PubMed; (g) S. Mitsuda, T. Fujiwara, K. Kimigafukuro, D. Monguchi and A. Mori, Tetrahedron, 2012, 68, 3585 CrossRef CAS PubMed; (h) S. Tamba, Y. Okubo, S. Tanaka, D. Monguchi and A. Mori, J. Org. Chem., 2010, 75, 6998 CrossRef CAS PubMed. For a review: (i) A. Mori and A. Sugie, Bull. Chem. Soc. Jpn., 2008, 81, 548 CrossRef CAS.
- (a) S. Tamba, S. Tanaka, Y. Okubo, S. Okamoto, H. Meguro and A. Mori, Chem. Lett., 2011, 40, 398 CrossRef CAS; (b) S. Tamba, K. Shono, A. Sugie and A. Mori, J. Am. Chem. Soc., 2011, 133, 9700 CrossRef CAS PubMed; (c) S. Tamba, S. Mitsuda, F. Tanaka, A. Sugie and A. Mori, Organometallics, 2012, 31, 2263 CrossRef CAS; (d) S. Tamba, Y. Okubo, A. Sugie and A. Mori, Polym. J., 2012, 44, 1209 CrossRef CAS; (e) S. Tamba, K. Fuji, K. Meguro, S. Okamoto, T. Tendo, R. Komobuchi, A. Sugie, T. Nishino and A. Mori, Chem. Lett., 2013, 42, 281 CrossRef CAS; (f) A. Mori, K. Ide, S. Tamba, S. Tsuji, Y. Toyomori and T. Yasuda, Chem. Lett., 2014, 41, 640 CrossRef. For a review, see: (g) A. Mori, J. Synth. Org. Chem., Jpn., 2011, 69, 1202 CrossRef CAS. See also: (h) R. D. McCullough and R. D. Lowe, J. Chem. Sco., Chem. Commun., 1992, 70 RSC; (i) Q. Wang, R. Takita, T. Kikuzaki and F. Ozawa, J. Am. Chem. Soc., 2010, 132, 11420 CrossRef CAS PubMed; (j) J. Hassan, E. Schulz, C. Gozzi and M. Lemaire, J. Mol. Catal. A: Chem., 2003, 195, 125 CrossRef CAS; (k) B. Bonillo and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 18916 CrossRef CAS PubMed; (l) Y. Fujinami, J. Kuwabara, W. Lu, H. Hayashi and T. Kanbara, ACS Macro Lett., 2012, 1, 67 CrossRef CAS; (m) M. Wakioka, N. Ichihara and Y. Kitano, Macromolecules 201447626 Search PubMed.
- K. Fuji, S. Tamba, K. Shono, A. Sugie and A. Mori, J. Am. Chem. Soc., 2013, 135, 12208 CrossRef CAS PubMed.
- (a) M. Schunürch, M. Spina, A. F. Kahn, M. D. Mihovilovic and P. Stanetty, Chem. Soc. Rev., 2007, 36, 1046 RSC; (b) S. Kano, Y. Yuasa, T. Yokomatsu and S. Shibuya, Heterocycles, 1983, 20, 2035 CrossRef CAS.
- (a) C. R. Hauser and H. G. Walker, J. Am. Chem. Soc., 1947, 69, 295 CrossRef CAS; (b) A. Krasovskiy, V. Krasovskaya and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 2958 CrossRef CAS PubMed.
- IPr: 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene. See: W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS.
- Attempted reaction with Pd(tBu3P)2, which was shown to induce several polymerizations of (hetero)arylenes,3,5,6 resulted in giving little polymerization.
- [1,3-Bis(2,6-diisopropylphenyl)imidazol-2-ylidene]chloro[3-phenylallyl]palladium(II): (CX31) and [1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene]chloro[3-phenylallyl]palladium(II) (CX32), respectively. Y. Horiguchi and S. P. Nolan, J. Synth. Org. Chem., Jpn., 2009, 67, 653 CrossRef CAS.
- PEPPSI: pyridine-enhanced precatalyst preparation stabilization and initiation, (1,3-bis(2,6-diisopropylphenyl)imidazolidene)(3-chloropyridyl)palladium(II) dichloride. (a) C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem. – Eur. J., 2006, 12, 4743 CrossRef CAS PubMed; (b) M. G. Organ, S. Calimisiz, M. Sayah, K. H. Hoi and A. J. Lough, Angew. Chem., Int. Ed., 2009, 48, 2383 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Professor Max Malacria upon cerebration of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and spectroscopic data. See DOI: 10.1039/c4qo00109e |
| This journal is © the Partner Organisations 2014 |