
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances for enhanced photodynamic therapy: from new mechanisms to innovative strategies
Xia
Wang
,
Jinlei
Peng
,
Chi
Meng
and
Fude
Feng
 *
*
MOE Key Laboratory of High Performance Polymer Materials and Technology, Department of Polymer Science & Engineering, School of Chemistry & Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: fengfd@nju.edu.cn
First published on 12th July 2024
Abstract
Photodynamic therapy (PDT) has been developed as a potential cancer treatment approach owing to its non-invasiveness, spatiotemporal control and limited side effects. Currently, great efforts have been made to improve the PDT effect in terms of safety and efficiency. In this review, we highlight recent advances in innovative strategies for enhanced PDT, including (1) the development of novel radicals, (2) design of activatable photosensitizers based on the TME and light, and (3) photocatalytic NADH oxidation to damage the mitochondrial electron transport chain. Additionally, the new mechanisms for PDT are also presented as an inspiration for the design of novel PSs. Finally, we discuss the current challenges and future prospects in the clinical practice of these innovative strategies. It is hoped that this review will provide a new angle for understanding the relationship between the intratumoural redox environment and PDT mechanisms, and new ideas for the future development of smart PDT systems.
1. Introduction
Cancer is currently one of the most devastating diseases in the world, presenting a serious threat to human health due to its high morbidity and mortality.1 To date, the classical clinical diagnosis and treatment methods including surgery, chemotherapy and radiotherapy have made great progress. However, some deficiencies still exist in these treatments such as invasive operations, drug resistance, a high recurrence rate, severe systemic toxicity and long-term adverse effects.2–4 Thus, novel therapies with few side effects and excellent antitumour activities are highly needed. Among them, photodynamic therapy (PDT) has drawn increasing interest due to its non-invasiveness, spatiotemporal control, limited side effects and low risk in generating drug resistance.5,6 PDT usually requires three basic components: photosensitizers (PSs), light and oxygen (O2). In the presence of light and O2, the activated PSs can produce toxic reactive oxygen species (ROS) to induce tumour cell death through necrosis, apoptosis, autophagy or ferroptosis.7 As the half-life is usually very short (<40 ns), ROS exert a local effect within a rather short distance (<20 nm).8,9 As a ROS sensitive crucial subcellular organelle, the mitochondrion is an important component of the apoptosis pathway of cancer cells.10–12 Accordingly, the development of mitochondria-targeting PSs has become a promising strategy for efficient PDT.The mechanisms of PDT are generally divided into two categories according to the photophysical and photochemical basis (Fig. 1).13,14 Under light irradiation, the PS at the ground singlet state (S0) is excited to a singlet state (S1), and subsequently populated into a long-lived triplet state (T1) through intersystem crossing (ISC) and internal conversion (IC) steps. For type I PDT, triplet PSs react directly with the intracellular substances through a hydrogen or electron transfer process to produce reactive free radicals, which further react with O2 or other substances to generate ROS such as hydrogen peroxide (H2O2), the hydroxyl radical (˙OH) and superoxide anion (O2˙−).15 The effect of type I PSs is less dependent on the O2 concentration, and therefore type I PSs are preferred for PDT, particularly when O2 supply is insufficient. However, the majority of reported PDT systems are based on type II PSs.16,17 For type II PSs, the triplet excited energy is transferred to O2, generating highly cytotoxic singlet oxygen (1O2).18,19 The effect of O2-dependent type II PSs is limited in the hypoxic tumour microenvironment (TME).
 | ||
| Fig. 1 The photosensitization process and photochemical reaction mechanisms of type I and type II PDT. | ||
Three generations of PSs have been developed for anticancer PDT so far and a variety of PSs (e.g., porphyrin, chlorin, BODIPY) have been commercialized or used in clinical trials.17,20,21 The first generation of PSs is based on a mixture of porphyrin complexes, including hematoporphyrin derivative (HpD), dihematoporphyrin ether (DHE), and Photofrin porfimer sodium. However, low efficiency and poor tissue penetration limit clinical practice in PDT.5 The second generation of PSs in accurate structures (e.g., 5-aminolevulinic acid (5-ALA), phthalocyanines, hypericin, metalloporphyrins) have been developed, but suffer from poor solubility and tumour targeting issues.22 To improve the selectivity to tumours and reduce the damage to normal tissues, the third generation of PSs are equipped with cancer-targeting ligands, including folic acid, peptides, antibody, and aptamer.23 Nevertheless, PDT still faces some challenges that hinder its widespread clinical application: (1) PDT requires light that reaches the target area, so its therapeutic effect is quite restricted to tumours in the skin or areas adjacent to the organs.24,25 (2) The penetration depth of light in tumour tissues strongly depends on its wavelength. However, as most PSs weakly absorb near-infrared (NIR) light, tissue penetration is limited significantly in the use of high-energy short-wavelength excitation (UV, blue, or green light).16,17 (3) Due to the excessive proliferation of cancer cells and insufficient blood supply, the O2 concentration in solid tumours is severely abnormal,26,27 resulting in the significant decrease of PDT efficacy.
In recent years, great efforts have been made to enhance the PDT effect. The strategies mainly focus on the reconstruction of the TME to alleviate tumour hypoxia,28–31 development of novel type I PSs with tumour targeting capability and longer absorption wavelength,32–34 and combination of PDT with other treatments.35–37 Advances in PDT have been introduced comprehensively in previous reviews.23,38–40 Herein, we collectively review the state-of-the-art development of recent PDT strategies that primarily aim at mitochondria and a little bit at other organelles (e.g., lysosome and endoplasmic reticulum) and provide prospects for future high-performance PDT systems that rely on the endogenous redox-environment. The innovative strategies are roughly classified into three categories: (1) the development of novel radicals including the hydrogen radical (H˙) and carbon radical (C˙), (2) activatable PSs based on the TME and light, and (3) photocatalytic oxidation of 1,4-dihydronicotinamide adenine dinucleotide (NADH) (Fig. 2). These methods involve new mechanisms of PDT (e.g., photoredox catalysis) and can be carried out in an O2-independent manner to achieve PDT in the hypoxic TME. However, to promote clinical use of the new mechanisms, developing long-wavelength NIR PSs to increase tissue penetration depth is of great importance. In addition, tumour-targeting therapy needs to be considered to avoid nonspecific damage to surrounding healthy tissues. We hope this review will inspire more significant design of PSs based on new mechanisms for precision cancer therapy.
 | ||
| Fig. 2 The innovative strategies for enhanced PDT. | ||
2. Novel radical-based PDT
Free radicals have attracted increasing attention because they can fight against different types of tumours without causing drug resistance.41 The generation of toxic ROS, which is largely dependent on the concentration of O2 and H2O2, is inadequate on the hypoxic tumour.42 It is crucial to explore more free radicals (e.g., chlorine radicals,43 carbon radicals44 and hydrogen radicals45,46) independent of O2 and H2O2 in PDT. In this section, research on H˙- and C˙-based PDT is discussed in detail.2.1 Hydrogen radical
As a reactive hydrogen species (RHS), H˙ can be generated via ultrasound and ionizing radiation and is reactive to a spectrum of species such as radicals, high valence metals and unsaturated bonds.47,48 Bradley et al. reported that water radiolysis by γ/X-ray irradiation could mediate the activation of a cancer prodrug with the azide group by H˙.49 Studer et al. achieved water activation with a photocatalytic phosphine-mediated radical process under 455 nm light irradiation.50 The generated PR3–H2O radical cation was deprotonated to form a neutral PR3–OH radical intermediate followed by a heterolytic O–H bond cleavage. The PR3–OH radical behaved as a ‘free’ H atom and was used to reduce π systems. H˙ is a two-sided modulator due to its unique redox properties. It is a pro-oxidant due to it scavenging O2 (k = 2.1 × 1010 M−1 s−1) into the hydroperoxyl radical (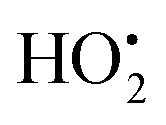 ) or O2˙− under aerobic conditions, and also acts as a powerful reductant in an anaerobic environment.46 To date, H˙-based therapy has been scarcely investigated.45,46,51
) or O2˙− under aerobic conditions, and also acts as a powerful reductant in an anaerobic environment.46 To date, H˙-based therapy has been scarcely investigated.45,46,51
In 2021, we reported a benzotriazole-cored activatable PS (aPS), termed DANO, that was flanked by two triphenylphosphonium (TPP) units to target mitochondria and equipped with an N-nitrosamine moiety as a donor of the nitric oxide radical (˙NO) as well as photocaging functionality. In the co-existence of reduced glutathione (GSH) and visible light, DANO was converted to the photoactive form (DAPS), with concomitant liberation of ˙NO that was initiated by the homolytic N–N bond scission at the N-nitrosamine moiety (Fig. 3).45 Different from the general PDT process, H˙ was detected by the electron spin resonance (ESR) technique and was responsible for the production of O2˙−. Free radical recombination between ˙NO and O2˙− led to immediate formation of peroxynitrite (ONOO−), a reactive nitrogen species (RNS) with greatly increased cytotoxicity relative to ˙NO and O2˙−. The PDT effect on HeLa cells under both normoxia and hypoxia was maximized by the simultaneous production of multiple reactive species that originated from H˙ and ˙NO cascade reactions upon photoactivation of DANO in the GSH-abundant mitochondria. Efficient two-photon excited PDT with DANO was also achieved by using an 800 nm two-photon laser, which showed the potential of DANO in precise cancer phototherapy.
 | ||
Fig. 3 Schematic illustration of the photogeneration of H˙, O2˙−/ , ˙NO, ONOO−, and 1O2 upon coactivation of DANO with GSH and light. Reproduced with permission from ref. 45. Copyright 2021 American Chemical Society. , ˙NO, ONOO−, and 1O2 upon coactivation of DANO with GSH and light. Reproduced with permission from ref. 45. Copyright 2021 American Chemical Society. | ||
To look into the mechanism of H˙ production and its role in the PDT process, our group designed an efficient photochemical H˙ generator based on a donor–acceptor type PS (termed TAF) that consisted of triphenylamine and hexylamine-substituted dibenzothiophene sulfone units (Fig. 4a).46 Mechanically, the triplet excited TAF is reducible by GSH to form TAF˙−, which is protonated and returned to the original state with concomitant release of H˙ (Fig. 4b). The key intermediate radical (TAFH˙) was successfully captured by 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and the adduct product was identified by high resolution mass spectroscopy (HRMS). The role of H˙ in PDT is two-fold. One is the H˙-promoted oxidative effect via scavenging of O2 to produce O2˙− that amplifies the oxidative stress under normoxia, which leads to ferroptosis–apoptosis of tumour cells. The other one is associated with the strong chemical reducing properties of H˙ in causing tumour cell apoptosis and breaking the traditional PDT barrier of extreme hypoxia. In the in vitro study, TAF showed a very high photocytotoxicity index (PI) on HeLa and A549 cells under normoxia and hypoxia. This work establishes a GSH-driven and H˙-based phototherapy and solves the self-limiting hypoxia dilemma of PDT.
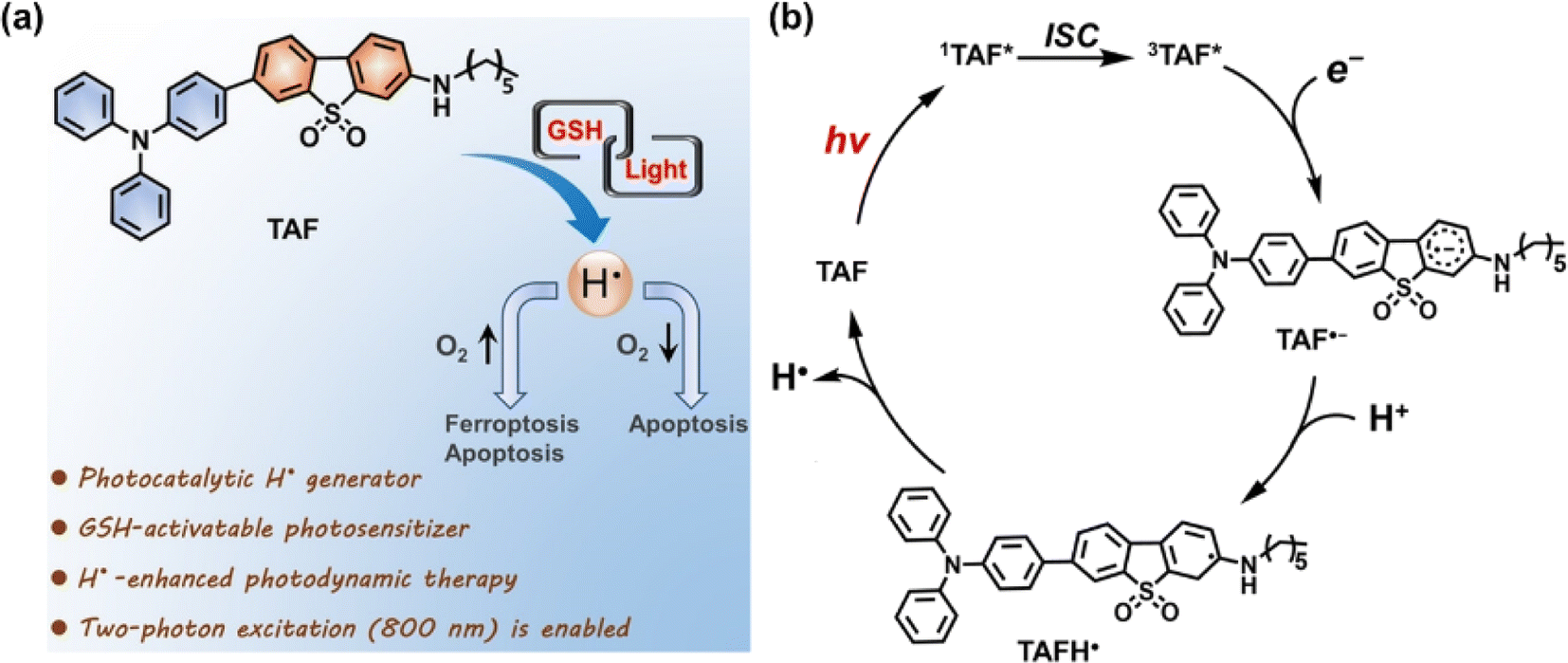 | ||
| Fig. 4 (a) Schematic presentation of GSH-driven H˙ photogeneration for PDT. (b) Proposed mechanism for photocatalytic H˙ generation. Reproduced with permission from ref. 46. Copyright 2022 Wiley-VCH. | ||
2.2 Carbon radical
C˙ can be generated in the absence of O2, presenting significant potential as a candidate cytotoxic species for PDT. Tirapazamine (TPZ), as a hypoxia-activable prodrug (HAP), can be bio-reduced to an alkyl radical (and hydroxyl radical) in the TME, attracting hydrogen from DNA strands and consequently causing DNA cleavage.52 The water-soluble 2,2-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (AIPH) is chemically unstable and is often used as an alkyl radical precursor.53 As an azo compound, AIPH rapidly decomposes to generate cytotoxic alkyl radicals upon exposure to light or heat.54–56Lee et al. constructed a smart nanoformulation system consisting of gold nanorods (GNRs) and AIPH in a poly(lactic-co-glycolic acid) (PLGA) matrix, which could form and release C˙ in response to external NIR light irradiation (Fig. 5a).57 The use of the PLGA polymer matrix allowed for a high loading capacity of AIPH and protection from burst release. In vitro studies demonstrated the nanocomplexes can eradicate CT26 colon cancer cells effectively. In 2019, Yuan et al. prepared Bi2Se3@AIPH through encapsulating AIPH and a phase transition material (lauric acid) in hollow bismuth selenide nanoparticles (Bi2Se3 NPs) (Fig. 5b).58 When circulating in the blood, AIPH was encapsulated without leakage to avoid side effects. In the tumour tissues of mice, Bi2Se3@AIPH presented a strong photothermal (PTT) effect after 808 nm laser irradiation, accompanied by the release of AIPH to produce C˙, leading to GSH depletion and an amplified immune response, showing a tumour growth inhibition rate of 99.6%.
 | ||
| Fig. 5 (a) Synthesis of poly(lactic-co-glycolic acid) (PLGA)/gold nanorod (GNR)/2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (AIPH) nanocomplexes and schematic illustration of the PLGA/GNR/AIPH nanocomplexes allowing near-infrared (NIR) induced heat and free radical generation for efficient dual cancer treatment. Reproduced with permission from ref. 57. Copyright 2018 Elsevier B.V. (b) Theranostic process of Bi2Se3@AIPH for CT and thermal imaging-guided cascaded photothermal and oxygen-independent photodynamic therapy along with immune response. Reproduced with permission from ref. 58. Copyright 2019 Springer Nature. (c) Preparation of P2@IR1061-RGD for combined PTT and TDT in a NIR II window (1000–1700 nm) of the PDXHCC mouse model. Reproduced with permission from ref. 59. Copyright 2021 Wiley-VCH. | ||
Light in the NIR II window (1000–1700 nm) exhibits deep tissue penetration (>1 cm) because of the reduced tissue absorption and scattering, suitable for treatment of deep tumours. Liu et al. reported a degradable nano-system (P2@IR1061-RGD NPs) formed via self-assembly between an amphiphilic thermolabile azo-containing polymer (P2) and a NIR II photothermal dye IR1061 to achieve safe and efficient cancer therapy (Fig. 5c).59 P2@IR1061-RGD NPs were degraded by the photothermal effect of IR1061 with robust C˙ generation once irradiated at 1064 nm, thereby interfering with RNA transcription, as well as inducing cancer cell necrosis or apoptosis even in the hypoxic tumour microenvironment. The nano-system displayed excellent tumour inhibition on a 4T1 breast cancer model as well as a patient-derived xenograft model of hepatocellular carcinoma (PDXHCC), with inhibition rates up to 97% and 100%, respectively, which is promising for clinical therapy on deep tissue hypoxic tumours without serious systemic toxicity.
In 2020, Chao et al. reported an iridium(III) anthraquinone complex (Ir4) as a mitochondria-targeting C˙ initiator (Fig. 6a).60 The anthraquinone moiety was used as a light-activated C˙ initiator as well as a luminescence quencher. In the hypoxic tumour, Ir4 was reduced to anthracene diol (Ir4-red) by the overexpressed reductase, which led to turn-on response of the two-photon excited luminescence. Ir4-red could generate C˙ under two-photon excitation (730 nm) and exhibit dual functions in bioimaging and therapy. In the in vitro (A549 3D multicellular tumour spheroids) and in vivo (male nude mice bearing A549 xenografted tumours) studies, Ir4 presented an outstanding PDT effect for its strong C˙ generating capability. To improve the phototoxic effect and promote clinical application of the metal complex, Chao's group further designed a mitochondria-localized iridium(III) endoperoxide prodrug (2-O–IrAn), which could synergistically release a highly cytotoxic iridium(III) complex (2-IrAn), 1O2, and an alkoxy radical upon two-photon irradiation (750 nm) (Fig. 6b).61 Then 2-O–IrAn was encapsulated into a biotin-functionalized polymer to obtain DSPE-PEG-Biotin@2-O–IrAn NPs in order to provide cancer selectivity and improve the pharmacological properties. Under two-photon irradiation (750 nm, 50 mW, 300 s), the generated nanoparticles nearly fully eradicated the tumour inside A549 tumour-bearing mice within a single treatment via synergistic PDT/photoactivated chemotherapy (PACT).
 | ||
| Fig. 6 (a) Proposed schematic illustration of the PDT mechanism of the Ir4 complex. Reproduced with permission from ref. 60. Copyright 2020 Wiley-VCH. (b) Schematic illustration of the mechanism of the action of the Ir(III) complex by PDT/PACT. Reproduced with permission from ref. 61. Copyright 2022 American Chemical Society. | ||
3. Activatable PSs for PDT
The ability of PSs to generate ROS selectively at the tumour site remains one of the important factors in maximizing the therapeutic effect while minimizing damage to normal cells. However, most PSs are in the “always on” stage, which poses a risk of damage to normal cells during the PDT process.62–64 This issue is alleviated by using activable PSs (aPSs) as PS precursors. The aPSs functionalized with caged groups are originally inactive and their activity can be precisely activated by tumour-specific endogenous stimuli (pH, enzyme, redox, etc.) or exogenous stimuli (ultrasound, temperature, light, etc.),65–67 which contribute to improving treatment efficiency and reducing side effects on healthy tissues. In this section, we will highlight the relevant reports of activatable PDT systems and hope to provide an inspiration for the design of PSs.3.1 TME-activated PSs
TME refers to the local steady-state environment composed of tumour cells, stromal cells, and the extracellular matrix during tumour growth. The TME provides the necessary material basis for tumour occurrence, development, invasion, and metastasis.68,69 In the TME, due to the slow blood flow of oxygen to tumour cells and the rapid cell proliferation, tumours present the hypoxic state.70 In addition, the TME also exhibits the features of lower intercellular pH and higher intracellular GSH and H2O2 concentration.71,72 These unique characteristics of the TME distinguish it from the corresponding normal tissues, thereby helping the design of TME-activated PSs.The real-time monitoring of the drug release induced by hypoxia can obtain feedback on the treatment process in time. In 2022, Kim et al. engineered a compound (DHQ-Cl-Azo) with hypoxia-responsive chemotherapy and PDT functionalities for the cooperative treatment of solid tumours (Fig. 7a).75 DHQ-Cl-Azo was obtained through a covalent connection using a hypoxia-sensitive azo bond (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) bridge between a NIR rhodol-based fluorophore (DHQ-Cl) and the nitrogen mustard drug. In the hypoxic TME, the azo bond of DHQ-Cl-Azo was reduced by azoreductase, leading to the release of the nitrogen mustard for chemotherapy and DHQ-Cl for PDT and diagnosis. DHQ-Cl-Azo helped to eliminate tumour cells in the surface layer during PDT and was beneficial for eradicating hypoxic tumour cells in the tumour center during activation of chemotherapy, thereby maximizing the anti-cancer effect on 4T1 tumour-bearing BALB/c mice.
N–) bridge between a NIR rhodol-based fluorophore (DHQ-Cl) and the nitrogen mustard drug. In the hypoxic TME, the azo bond of DHQ-Cl-Azo was reduced by azoreductase, leading to the release of the nitrogen mustard for chemotherapy and DHQ-Cl for PDT and diagnosis. DHQ-Cl-Azo helped to eliminate tumour cells in the surface layer during PDT and was beneficial for eradicating hypoxic tumour cells in the tumour center during activation of chemotherapy, thereby maximizing the anti-cancer effect on 4T1 tumour-bearing BALB/c mice.
 | ||
| Fig. 7 (a) Principle of DHQ-Cl-Azo for PDT and chemotherapy. Reproduced with permission from ref. 75. Copyright 2022 Elsevier Inc. (b) A reversible hypoxia–normoxia responsive type I photosensitizer for PDT. Reproduced with permission from ref. 76. Copyright 2023 Wiley-VCH. (c) Molecular structures of compounds 1–3. Reproduced with permission from ref. 77. Copyright 2019 American Chemical Society. (d) ICy-N for hypoxia imaging and cancer therapy. Reproduced with permission from ref. 78. Copyright 2019 The Royal Society of Chemistry. | ||
The off–on PSs retain their phototoxic form even after treatment. Thus, it is crucial to alleviate the side effects associated with post-treatment of PDT. Liu et al. reported an on-demand switchable type I PS (TPFN–AzoCF3) that was activated only in the TME (Fig. 7b) and inactivated pre and post PDT.76 TPFN–AzoCF3 was composed of a type I PS (TPFN) and a hypoxia–normoxia cycling responsive group (AzoCF3). The arylazo group served as an off–on–off trigger that reversibly transformed into a hydrazine group depending on hypoxia–normoxia conditions. TPFN–AzoCF3 was converted into TPFN–HZCF3 in hypoxic environments, resulting in PS turn-on and effective ROS generation, but remained non-toxic in normal tissues pre or post PDT, which contributed to minimal side effects. TPFN–AzoCF3 revealed a satisfactory antitumour effect on a xenografted subcutaneous model of HeLa tumours in vivo with safe pre- and post-treatment for PDT.
Overexpressed nitroreductase (NTR) inside hypoxic tumour tissues is another widely studied reductase. Peng et al. developed fluorescein derivatives as smart PSs activated by NTR with thermally activated delayed fluorescence (TADF).77 They designed compound 1 and compound 2 with fluorescence and PDT efficiency in the “off” state. In the presence of NTR, both compounds containing a nitrobenzyl substituent were transformed into compound 3 via intramolecular photoinduced electron transfer (PET) (Fig. 7c). The faster response observed with compound 2 was ascribed to the higher affinity between compound 2 and NTR. Further, efficient PDT on compound 2-treated HeLa cells and HeLa tumour-bearing mice was achieved under 590 nm LED light irradiation. A similar strategy was applied to the iodized hemicyanine dye conjugated with a nitrobenzyl substituent, which could be efficiently used for tumour hypoxia imaging (710 nm) and PDT in 4T1 cells and 4T1 tumour bearing BALB/c mice under 660 nm light irradiation (Fig. 7d).78
In 2022, Yoon et al. constructed an acid-responsive nanoporphyrin (PN3-NP) based on the self-assembly of a water-soluble porphyrin derivative (PN3).81 PN3-NP could transform from nanoparticles into dot-like nanospheres due to the reconstruction of the substructure in the acidic environment. Simultaneously, NIR fluorescence monitoring and synergistic PDT and PTT effects of PN3-NP were also triggered in biofilms, achieving accurate theranostics without damage to normal tissues. Siegwart et al. designed an iodinated, water-soluble NIR BODIPY-based PS that could be activated by leveraging tumour-specific pH for image-guided and efficient PDT against cancer in vivo (Fig. 8a).82 At the physiological pH, the 1O2 release was “turned off” via the PET process, while the quenching process was inhibited and the 1O2 release was “turned on” upon the protonation of the amino groups at acidic pH, inducing cell death via apoptosis in a cancer-selective manner. The PS presented excellent photocytotoxicity on different cancer lines in vitro, and significant anticancer efficacy on athymic nude mice bearing subcutaneous MDA-MB-231 xenograft tumours after treatment with a 600–700 nm laser.
 | ||
| Fig. 8 (a) The pH-activatable BODIPY derivative for 1O2 generation and PDT. Reproduced with permission from ref. 82. Copyright 2018 American Chemical Society. (b) Illustration of the TDPP@PEG–TPZ nanoformulation for phototherapy-enhanced chemotherapy. Reproduced with permission from ref. 83. Copyright 2023 The Royal Society of Chemistry. (c) Reversible switching of ISC and potential application in PDT. Reproduced with permission from ref. 84. Copyright 2019 Wiley-VCH. | ||
Moreover, the acylamide bond was responsive to the acidified TME as well, which is favorable for designing pH-responsive prodrugs. In 2023, Chen et al. utilized acylamide bonds as a linker to synthesize TPZ-functionalized poly(ethylene glycol) (PEG–TPZ), and prepared a multi-responsive nanoplatform for phototherapy-enhanced chemotherapy based on PEG–TPZ encapsulated semiconducting polymer TDPP (Fig. 8b).83 The nanoplatform could release TPZ by the hydrolysis of the acylamide bond. Efficient ROS and photothermal conversion were triggered by laser irradiation, resulting in the consumption of cellular oxygen as well as damage to the blood vessels. The aggravated hypoxia contributed to the activation of TPZ for chemotherapy, which in turn enhanced therapeutic efficacy. TDPP@PEG–TPZ NPs showed a great treatment effect in in vitro (143B cells) and in vivo (143B tumour-bearing mice) experiments.
Reversibly switchable ISC is crucial to deactivate photosensitization when PS is transferred or metabolized away from the pathological region. The availability of conjugated backbone twist (CBT)-enhanced ISC offers a unique opportunity for the reversible on–off switch of PSs. Prasad et al. presented reversible switching of ISC using PE5 through the pH-responsive CBT (Fig. 8c).84 At physiological pH (pH 7.4), PE5 existed mainly in anionic form with no electrostatic interactions to drive the twisted geometry, showing “ISC off”. However, PE5 transformed to a zwitterionic form at pH 6.0 and formation of the CBT conformation occurred, which led to “ISC on” and an efficient PDT effect. In the in vitro study, PE5 exhibited outstanding selectivity for tumour cells.
Compared to traditional PSs with the aggregation-caused quenching (ACQ) effect, aggregation-induced emission (AIE) molecular PSs can restrict intermolecular π–π stacking and intramolecular motions when aggregated, which will be conducive to improving fluorescence imaging and phototherapy.86,87 To develop novel PSs with higher photosensitive properties and specific target response ability, Li et al. synthesized an enzyme-activatable AIE molecular photosensitizer (TPE–TThPy) using the conjugation of tetraphenylethylene (TPE) and tetrahydropyridine (ThPy) with thiophene (T) as the π-bridge (Fig. 9a).88 The TPE–TThPy molecule was responsive to monoamine oxidase (MAOs), a kind of flavinase overexpressed in tumour cells, which converted ThPy to methylpyridine salts (Pys). The cationic TPE–TPys showed mitochondrial targeting properties and ROS photogeneration capability. In vivo studies revealed a strong inhibitory effect of photoexcited TPE–TThPy on nude BALB/c mice bearing a subcutaneous SH-SY5Y xenograft with high MAO-A expression.
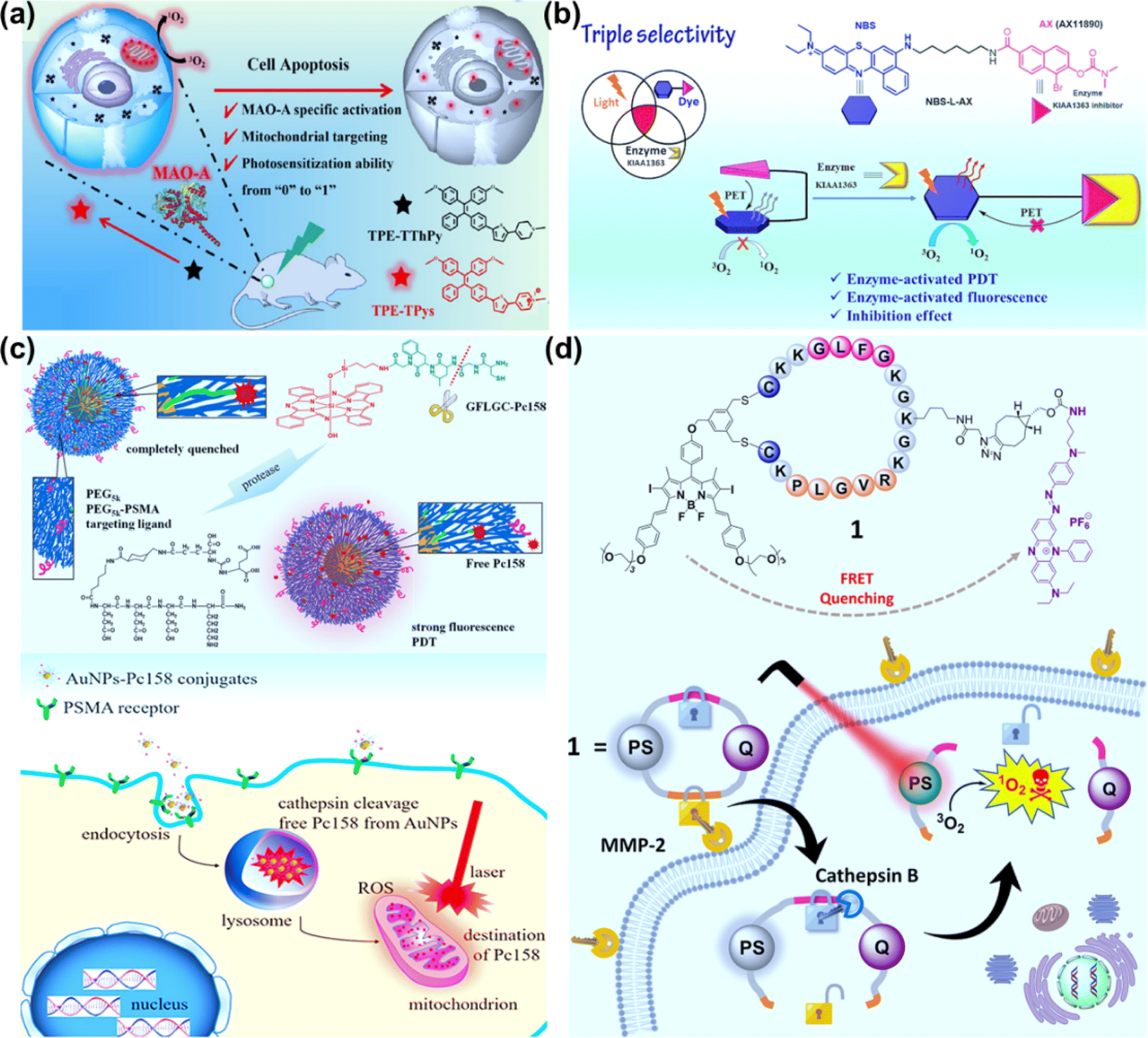 | ||
| Fig. 9 (a) TPE–TThPy as a mitochondria-targeting photosensitizer for MAO-A-activatable PDT. Reproduced with permission from ref. 88. Copyright 2022 Wiley-VCH. (b) Enzyme KIAA1363-activated mechanism for NBS-L-AX based PDT. Reproduced with permission from ref. 90. Copyright 2023 The Royal Society of Chemistry. (c) Cathepsin-activated AuNPs–Pc158 conjugates for selective PDT. Reproduced with permission from ref. 94. Copyright 2020 American Chemical Society. (d) Molecular structure of the double-locked PMB 1 and its dual-enzyme-unlocked mechanism. Reproduced with permission from ref. 95. Copyright 2023 American Chemical Society. | ||
AX11890 has strong binding affinity toward the KIAA1363 enzyme and is an inhibitor of overexpressed KIAA1363 enzyme in breast cancers.89 Foley et al. constructed NBS-L-AX by attaching a benzo[a]phenothiazine moiety with a short linear alkyl chain (NBS-L) to enzyme targeting agent AX11890 (Fig. 9b).90 In normal tissues, NBS-L-AX exhibited a compact sandwiched conformation due to the strong π–π interaction between the NBS-L and AX11890 moieties, which favored a PET process from AX11890 to NBS-L and quenched the fluorescence and PDT activity of NBS-L. In breast cancer cells, when NBS-L-AX was in contact with the KIAA1363, KIAA1363 bound to the AX11890 segment and concomitantly unraveled the sandwiched NBS-L-AX molecule to form a fully extended linear conformation, switching on the fluorescence and PDT activities of the NBS-L segment. In the in vitro experiments, higher photocytotoxicity to KIAA1363-positive MCF-7 cells than KIAA1363-negative HUVEC cells was observed. In the in vivo experiments, NBS-L-AX presented good selectivity on the 4T1 tumour-bearing mice model under 635 nm laser irradiation (100 mW cm−2, 20 min).
The fact that extracellular adenosine triphosphate (ATP) is overexpressed in the tumour tissues has been applied in fabricating nanodrugs for tumour-targeting delivery. Schanze et al. reported metal-free helical nanofibers formed in aqueous solution using the co-assembly of a cationic porphyrin and ATP for PDT.91 The porphyrin–ATP assembly was delivered to the tumour site through whole-body blood circulation and was stabilized via overexpressed extracellular ATP within tumour tissues. The release of porphyrin upon biodegradation of ATP by intracellular phosphatases gave rise to tumour-selective PDT.
Cathepsins are highly abundant within intracellular endolysosomal vesicles in cancer cells. Upregulated cathepsin activity is strongly associated with tumour invasion and metastasis,92,93 which has made cathepsin an excellent tumour-associated biomarker to “turn on” PSs for precise PDT. Basilion et al. designed PSMA-targeting cathepsin activatable AuNPs for treating prostate cancer.94 Silicon phthalocyanine (Pc158) was synthesized and conjugated to gold nanoparticles (AuNPs) via a cathepsin-cleavable linker, GLFGC, to obtain AuNP–Pc158 (Fig. 9c). The AuNP–Pc158 conjugates were inactive and nonfluorescent, while Pc158 released with time upon cathepsin-induced cleavage restored fluorescence and PDT activity. In the in vivo study on bearing PC3pip tumours, AuNP–Pc158 presented tumour specificity via PSMA targeting, and a sustained release of active Pc158 through cathepsin-activation led to multiple sequential PDT treatments.
Dual stimuli-responsive photosensitizing systems can enhance the tumour specificity, functioning as a molecular “AND” logic gate. Lo et al. synthesized a photodynamic molecular beacon (PMB), which could only be activated by the coexistence of matrix metalloproteinase-2 (MMP-2) and cathepsin B (Fig. 9d).95 In the PMB structure, a distyryl boron dipyrromethene (DSBDP)-based PS and a Black Hole Quencher 3 (BHQ-3) moiety were connected with a cyclized peptide containing sequences PLGVR and GFLG. PMB was largely quenched in normal tissues and even in the presence of one of two enzymes due to the highly efficient Förster resonance energy transfer (FRET) process. In the co-existence of MMP-2 and cathepsin B, PMB was cleaved, and the capability of fluorescence emission and ROS generation was recovered. PMB showed a more significant PDT effect on MMP-2-positive A549 and U-87 MG cells than MMP-2-negative HeLa and HEK-293 cells. The PDT effect of PMB was efficient on the A549 tumor-bearing nude mice model using a diode laser at 680 nm (300 mW cm−2, 10 min), causing no damage to the skin of the mice.
Ng et al. reported a thioketal-linked polydopamine (PDA)-based nanosystem (PDA-Dox-Pc-QRH) that could release the immobilised chemotherapeutic and photosensitising drugs (Dox and Pc) upon interactions with the intracellular ROS (Fig. 10a).99 The disaggregation of the Pc molecules resulted in activation upon excitation with NIR light to generate 1O2. The photodynamic reaction accelerated the degradation of the thioketal-linked structure and the release of larger amounts of therapeutic components. The light-triggered on-demand release of Dox induced additional cytotoxicity through a different pathway and reduced the undesired side effects. PDA-Dox-Pc-QRH showed effective in vitro (A549 and A431 cells) and in vivo (A431 tumour-bearing nude mice) anticancer activity via a synergistic treatment manner including chemotherapy and PDT.
 | ||
| Fig. 10 (a) Formulation of PDA-Dox-Pc-QRH for PDT. Reproduced with permission from ref. 99. Copyright 2021 The Royal Society of Chemistry. (b) Construction of H2O2-responsive Ce6@P(EG-a-CPBE) NPs for chemo-PDT synergistic cancer therapy. Reproduced with permission from ref. 100. Copyright 2023 Elsevier B.V. (c) The illustration of TPE–TeV–PPh3-based H2O2-activatable PDT. Reproduced with permission from ref. 101. Copyright 2022 Wiley-VCH. (d) GSH-responsive shape-transformable GdCFS for dual-modal T1/T2 MRI-guided enhanced PDT. Reproduced with permission from ref. 102. Copyright 2024 Wiley-VCH. (e) The activatable photosensitizer (mitoaPS) for GSH and H2O2 mutually responsive PDT. Reproduced with permission from ref. 103. Copyright 2020 Wiley-VCH. (f) Construction of HSeC/IR nanoparticles for cascaded cancer phototherapy. Reproduced with permission from ref. 105. Copyright 2022 Elsevier Ltd. | ||
Generally, the therapeutic effect of PDT is weakened by the upregulated antioxidants (GSH in particular) in the tumour cells. Yan et al. designed H2O2-responsive polymer prodrug nanoparticles (Ce6@P(EG-a-CPBE) NPs) with the GSH-scavenger for chemo-photodynamic synergistic cancer therapy (Fig. 10b).100 Ce6@P(EG-a-CPBE) NPs were constructed by the co-self-assembly of Ce6 and amphiphilic polymer prodrug P(EG-a-CPBE), which was synthesized from a hydrophilic alternating copolymer P(EG-a-PD) by conjugating hydrophobic anticancer drug chlorambucil (CB) via an H2O2-cleavable linker 4-(hydroxymethyl)phenylboronic acid (PBA). In the normal tissues, Ce6@P(EG-a-CPBE) NPs were in the “off” state due to the high stability of the PBA linker, while in the tumour tissues, PBA was cleaved rapidly by endogenous H2O2 to release CB and Ce6 for a synergistic effect. PBA could be transformed into a GSH-scavenger (quinine methide, QM) under intracellular H2O2, thereby preventing the scavenging effect of GSH against 1O2 and inducing stronger oxidative stress. The synergistic chemo-photodynamic therapy of Ce6@P(EG-a-CPBE) NPs was efficient in both in vitro (MCF-7 cells) and in vivo (MCF-7 tumour-bearing mice) studies.
At present, redox-activated PSs with AIE characteristics are drawing increasing interest. In 2022, He et al. reported a type-I PS (TPE–TeV–PPh3) consisting of a tetraphenylethylene group (TPE) as an imaging moiety with AIE properties, triphenylphosphonium (TPP) as a mitochondria-targeting group, and telluroviologen (TeV2+) as a ROS (O2˙−, ˙OH) generating moiety. TPE–TeV–PPh3 was oxidized to luminescent TPE–TeOV–PPh3 by excess H2O2 (Fig. 10c).101 Exposure of TPE–TeV–PPh3 to white light led to killing 7402 cells and MCF-7 cells under normoxia and hypoxia. In the in vivo experiments, the tumour growth of 7402-tumour-bearing nude mice was inhibited by 88% after the PDT procedure (450 nm, 200 mW cm−2, 10 min).
In 2024, Huang et al. developed a GSH-responsive shape-transformable nanotheranostics Ce6 (Gd3+)-FFVLGGGC-SS-PEG (GdCFS) by combining Gd, Ce6 and a peptide (Phe-Phe-Val-Leu-Gly-Gly-Gly-Cys) that was conjugated with polyethylene glycol (PEG) through a disulfide bond, and performed dual-modal T1/T2 magnetic resonance imaging (MRI) specifically at the tumour site (Fig. 10d).102 The spherical GdCFS nanoparticles were responsive to GSH and transformed to nanofibers (NFs), resulting in T1 positive contrast and T2 negative contrast enhancement, accompanied by the generation of ROS. Moreover, the depletion of GSH could potentiate PDT efficacy. In the in vivo studies, GdCFS presented noticeable proliferation inhibition of tumours in C666-1 tumour-bearing BALB/c nude mice.
In view of the availability of GSH and H2O2 in tumours, dual-activatable PDT has advantages in therapeutic controllability. In 2020, our group designed a donor–acceptor type organic PS (mitoaPS) which could be co-activated by GSH and H2O2via domino reactions on the benzothiadiazole (BTD) ring for mitochondria-targeting PDT (Fig. 10e).103 The mitoaPS was a precursor of compound NO2ArSH,104 and double caging modes were responsible for the molecular activation. First, the fluorescence and photosensitizing properties were caged by the electron-withdrawing nitro group. Second, the thiol group, a precursor of the thiyl radical, was caged by a self-immolative boronic ester that was H2O2-specific. The triphenylphosphonium (TPP) ligand-terminated alkyne substituent was attached to the BTD ring to render mitochondria-targeting properties. The mitoaPS was resistant to GSH alone. In the co-existence of GSH and H2O2, the uncaged thiol group on the BTD ring by H2O2 was oxidized into free radical intermediates that caused intramolecular cyclization and nitro disproportionation. GSH participated in the free radical reaction process through hydrogen atom transfer and accelerated the conversion of mitoaPS into an active PS. One of the advantages using the free radical-relevant thiol chemistry lies in the molecular design free of installation of an active site for GSH, which helps reduce the molecular size of the PDT agent and avoid early uncontrolled activation by GSH. The nitro group of mitoaPS was not reducible by enzyme nitroreductase, guaranteeing reaction selectivity. The mutual activation of mitoaPS by GSH and H2O2 in HeLa cells led to “turn on” of both fluorescence and ROS photogeneration, allowing for two-photon excited cellular imaging and hypoxia-tolerant PDT.
In 2022, Qian et al. constructed a dual-activatable PDT nanosystem that could be regulated by external NIR laser irradiation and the tumour-specific redox microenvironment (Fig. 10f).105 The Ce6 was conjugated to hyaluronic acid (HA) via a diselenide linkage to form an amphiphilic polymer (HSeC). The HSeC/IR NPs was assembled from HSeC and IR780 (a PTT agent). During blood circulation, the photoactivity of Ce6 for PDT was “double-locked” by the aggregation-caused quenching (ACQ) effect and FRET from Ce6 to IR780. After selective accumulation into tumours, HSeC/IR NPs were decomposed due to the dissociation of the diselenide bond in the presence of GSH and H2O2. The Ce6 photoactivity was recovered after photothermal degradation of IR780 upon NIR laser irradiation. Consequently, the quenching effect mediated by aggregation and FRET was unlocked by the endogenous redox environment and external NIR light, allowing photoactivation of Ce6 under 660 nm laser irradiation for PDT. HSeC/IR NPs showed low skin 660 nm-photosensitization on the shaved back of healthy mice, while presenting superior tumour suppression via synergetic cascaded PTT–PDT in SCC7 tumour-bearing mice.
3.2 Light-activated PSs
Light-responsive PSs have advantages such as spatiotemporal control of drug activation and reduction of side-effects on healthy tissue.106,107 A variety of light-responsive molecules have been explored for the fabrication of photocages such as derivatives of o-nitrobenzyl, phenacyl, BODIPY, cyanine, and Ru complex structures.32,108–110 In 2021, we established a GSH-light co-responsive PS (DANO) that could be converted to its photoactive form (DAPS), presenting a high performance PDT effect, and a more detailed mechanism is provided in Fig. 3.45In 2022, Tang et al. reported a series of triphenylamine (TPA) derivatives based on 1,4-dihydropyridine (1,4-DHPy) as the light-responsive group and optimized photoactivation efficiency by the substituent effect.111 Amongst these TPA derivatives, TPA–DHPy–Py exhibited the highest photoactivation efficiency (Fig. 11). White light irradiation induced rapid conversion of the non-fluorescent TPA–DHPy–Py into its pyridine counterpart TPA–Py–Py via photo-oxidative dehydrogenation, leading to aggregation-induced near-infrared emission. TPA–DHPy–Py presented type I/II combined photodynamic activity, with remarkable ROS production efficiency, and could precisely discriminate cancer cells from co-cultured cells (cancer cells MDA-MB-231 and normal cells NIH-3T3) without the assistance of extra targeting components because of the larger number of lipid droplets (LDs) in the cancer cells. TPA–DHPy–Py was beneficial for in situ monitoring of LDs and endoplasmic reticulum alteration under ROS-induced stress. Moreover, the tumour growth of MDA-MB-321-bearing nude mice treated with TPA–DHPy–Py was efficiently inhibited after exposure to white light (30 mW cm−2) for 20 min.
 | ||
| Fig. 11 Photoactivatable theranostic probes for simultaneous imaging of LDs and PDT. Reproduced with permission from ref. 111. Copyright 2022 Wiley-VCH. | ||
Sun et al. constructed a Ru-based photocage (PolyTHCRu) with a NIR light-triggered photocleavage reaction (Fig. 12a).112 The commercial anticancer drug tetrahydrocurcumin (THC) was coordinated to the Ru(II) center and formed THCRu with significantly enhanced absorption in the NIR region. The THCRu was coordinated to an amphiphilic polymer bearing pendant benzonitrile groups, forming photocleavable Ru–N coordination bonds. The self-assembled polymeric nanoparticle (PolyTHCRu) was sensitive to 760 nm NIR light which induced the dissociation of the Ru(II) complex from the side chains of polymers in the presence of water molecules. In the in vivo study, upon 760 nm NIR light irradiation, PolyTHCRu inhibited tumour proliferation on 4T1 tumour-bearing mice via combining chemotherapy and PDT. Ding et al. developed CD44-targeting and red-light-activatable Ru–HA@DOX NPs through conjugating hydrophilic biodegradable hyaluronic acid (HA) and hydrophobic photoactivatable ruthenium (Ru) complexes (Fig. 12b).113 Ru–HA@DOX NPs were selectively accumulated at the tumour through an enhanced permeability and retention (EPR) effect and CD44-mediated internalization. Ru–HA@DOX NPs were photolabile at the Ru–N coordination bond under 660 nm laser irradiation, releasing DOX and producing 1O2 for PDT in both in vitro (A549 cells) and in vivo (A549-tumor-bearing mice) studies.
 | ||
| Fig. 12 (a) The photocleavable PolyTHCRu nanoparticles for PDT. Reproduced with permission from ref. 112. Copyright 2023 Wiley-VCH. (b) Formulation of Ru–HA@DOX NPs for chemotherapy–PDT. Reproduced with permission from ref. 113. Copyright 2023 American Chemical Society. | ||
4. Photocatalytic NADH oxidation for PDT
As the electron source of the mitochondrial electron transport chain (mito-ETC), NADH plays an important role in living cells.114 NADH participates in the maintenance of the intracellular redox balance and functions as a coenzyme in over 400 oxidoreductases.71,115 Photoredox catalysis for NADH oxidation to perturb the NADH/NAD+ balance and the electron flow in intracellular mito-ETC is emerging as a novel strategy to induce metabolic dysfunction and cell death.Photocatalysts (PCs) including metal complexes, organic compounds and nano-catalysts catalyze NADH oxidation mainly through two pathways. One is based on a photocatalytic cycle (Fig. 13a).116 The photoexcited PC extracts an electron from NADH, forming the anionic radical PC˙− and NAD˙ radical. In the presence of oxygen, PC is regenerated with concomitant production of O2˙− after electron transfer from PC˙− to oxygen. Then O2˙− extracts an electron from NAD˙ and consumes two protons from the medium to generate H2O2, accompanied by the conversion of NAD˙ into NAD+. Under hypoxia, hemoprotein cytochrome c (Cyt c (Fe3+)) acts as a terminal electron acceptor to enhance the PDT effect via an O2-independent pathway. Another pathway for photocatalytic NADH oxidation is available through photogeneration of ROS which oxidize NADH to NAD+ (Fig. 13b).117 In this section, recent progress on photocatalytic NADH oxidation for PDT via the above two pathways will be discussed.
 | ||
| Fig. 13 (a) Plausible mechanism for PC-mediated photocatalytic NADH oxidation under normoxia and hypoxia. (b) Schematic diagram of photocatalytic NADH oxidation via ROS produced by PS. | ||
4.1 Metal complexes
Transition metal complexes have attractive photochemical and photophysical properties (e.g., excellent photostability, long excited-state lifetime and suitable excited-state reduction potential),6,23 available by coordinating ligands to metal ions in a combinatorial manner.6 The use of transition metal complexes for photocatalytic NADH oxidation is of increasing interest in PDT.118 We will focus on the recent approaches harnessing transition metal complexes such as Ir(III), Ru(II) and Os complexes for PDT via NADH oxidation.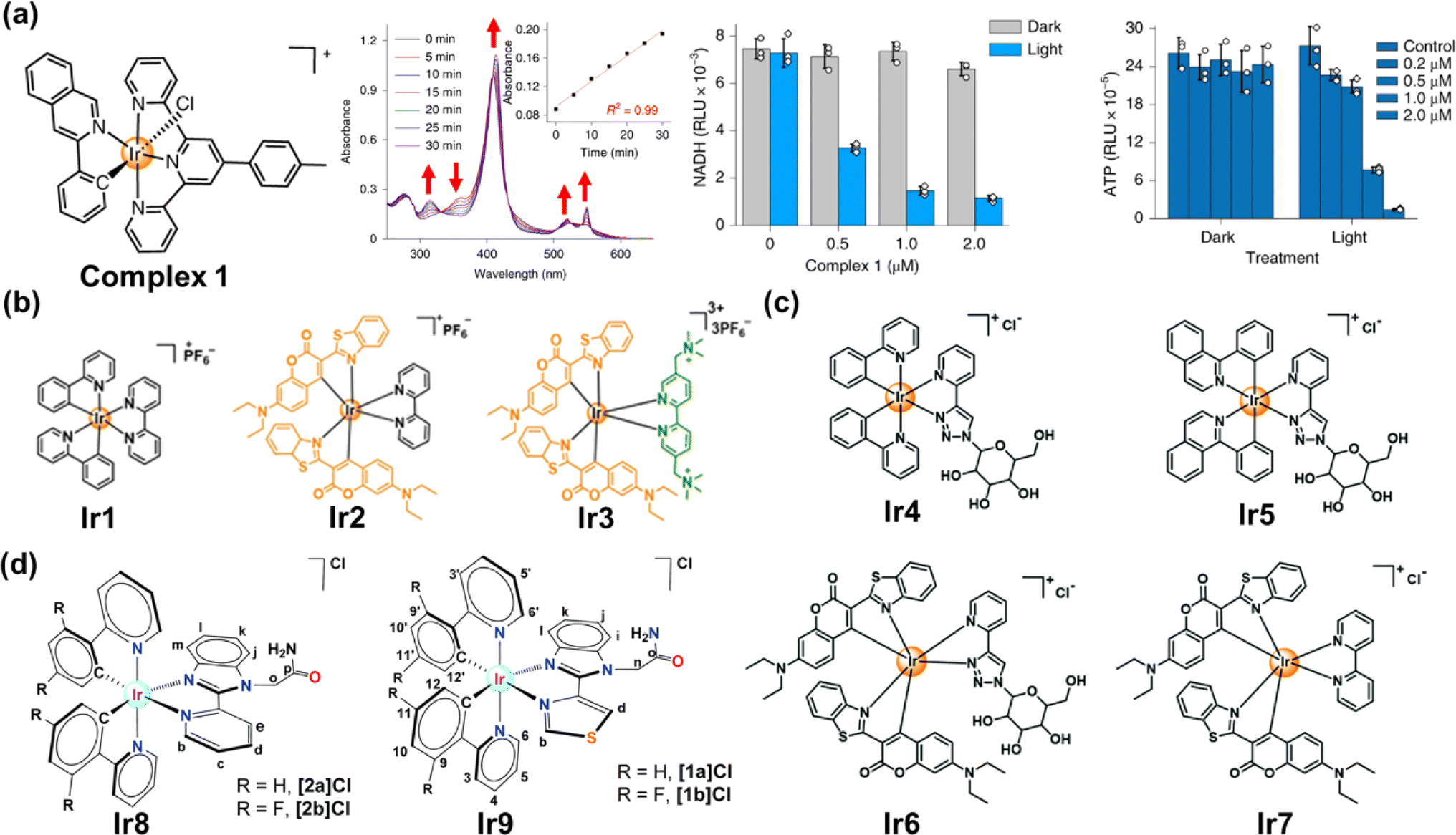 | ||
| Fig. 14 (a) The complex 1 used for photocatalytic NADH oxidation. Reproduced with permission from ref. 116. Copyright 2019 Springer Nature. (b–d) The complexes Ir1–Ir9 used for photocatalytic NADH oxidation. Reproduced with permission from ref. 119. Copyright 2021 Wiley-VCH. Reproduced with permission from ref. 120 and 121. Copyright 2022 The Royal Society of Chemistry. | ||
To date, a variety of Ir(III) complexes have been developed to obtain longer wavelengths and improve biocompatibility. Huang et al. reported a water-soluble coumarin-functionalized Ir(III) photocatalyst (Ir3) that absorbed visible light (Fig. 14b).119 Compared with Ir1 and Ir2, Ir3 presented high TOF (308.0 h−1) on NADH oxidation under normoxia. Ir3 induced remarkable photo-triggered cytotoxicity against various cancer cell lines (1–15 μM), while maintaining low toxicity (>100 μM) on normal cells in the dark. Huang's group further designed three glucose-appended cyclometalated Ir(III) complexes (Ir4–Ir6) containing the N,N-glucose ligand and various N,C-ligands to optimize their photochemical properties and non-glycosylated complex Ir7 (Fig. 14c).120 Ir6 presented the strongest aqueous solubility and the highest photocatalytic NADH oxidation efficiency, exhibiting necro-apoptotic photocytotoxicity on HeLa cells at sub-micromolar concentrations upon light irradiation. Espino et al. prepared four Ir(III) biscyclometallated complexes with N^N′ ancillary ligands containing an amide-substituted benzimidazole fragment and pyridine (Ir8) or thiazolyl (Ir9) rings (Fig. 14d).121 [1a]Cl and [2a]Cl were accumulated primarily in mitochondria, disrupting electron transport via NADH photocatalytic oxidation and presumably inducing mtDNA damage.
Altered cellular metabolism is a hallmark of cancer, where NADH plays an important role in the excess energy required for various anaplerotic reactions.122,123 Maiti et al. reported a quinoline appended Ir(III) complex (QAIC) as a molecular probe to detect endogenous NADH in cancer cells via the “turn-on” luminescence and “turn-off” SERS phenomenon (Fig. 15).124 QAIC could recognize the dynamic fluctuations of NADH and trigger PDT through NADH depletion. The activated N-QAIC in the cellular milieu presented excellent phototoxicity on HepG2 cells by the generation of 1O2 under normoxia and toxic NAD˙ under hypoxia.
 | ||
| Fig. 15 The mechanism of NADH detection with QAIC and laser triggered therapeutic mechanism of N-QAIC. Reproduced with permission from ref. 124. Copyright 2022 Elsevier B.V. | ||
Zhou et al. constructed a PCT system via the use of a Ru(II) complex and Fe(tpy)Cl3 (tpy = 2,2′:6′,2′-terpyridine).128 The Ru(II) complex photocatalyzed NADH oxidation accompanied by the generation of H2O2in situ with a high yield of 73.2%. At a neutral pH, the decomposition of H2O2 was mediated by Fe(tpy)Cl3 and afforded highly cytotoxic ˙OH. To extend the excitation light from blue light into the NIR region, the same group loaded the Ru(II) complex (Ru1) and Fe2+ simultaneously onto the mesoporous silica-encapsulated rare earth-doped upconversion nanoparticles (Fig. 16a).129 Moreover, Zhou's group designed a chloromethyl-modified Ru(II) complex as a mitochondria targeting PS to overcome the limitation of hypoxia in PDT,130 which could be photoexcited to generate a carbon radical in the presence of NADH, accompanied by the oxidation of NADH, thereby giving rise to DNA damage and cancer cell inactivation under hypoxia (3% O2).
 | ||
| Fig. 16 (a) The UCSRF nanoparticle for NIR light-driven photocatalytic NADH oxidation. Reproduced with permission from ref. 129. Copyright 2023 Elsevier Ltd. (b) The prodrug RuAzNM used for chemotherapy and PDT. Reproduced with permission from ref. 131. Copyright 2023 Wiley-VCH. | ||
In 2023, Sadler et al. reported a hypoxia-activated mitochondria-accumulated Ru(II) polypyridyl prodrug, RuAzNM, that was functionalized with an azo group and a nitrogen mustard moiety for multimodal theranostic studies (Fig. 16b).131 In hypoxic tumour microenvironments, the azo group was reduced selectively by reductase and cleaved to generate two primary amine products including the aniline mustard and polypyridyl RuNH2 complex. The aniline mustard triggered oxidative stress and mtDNA crosslinking. Meanwhile, the polypyridyl RuNH2 complex catalyzed NADH oxidation based on its favorable excited-state photoredox potential and superior binding affinity toward NADH. This multimodal therapeutic manner presented an efficient inhibition effect on the HepG2 MCS model.
 | ||
| Fig. 17 (a) The Os(II)-terpyridine complexes used for PDT and photooxidation therapy. Reproduced with permission from ref. 135. Copyright 2020 The Royal Society of Chemistry. (b) The photocatalytic NADH oxidation by light-activating Os2. Reproduced with permission from ref. 117. Copyright 2022 Springer Nature. | ||
4.2 Organic metal-free photocatalysts
For metal complexes, unpredicted metal toxicity, inferior biostability, and limited tissue penetration (UV, blue or green light excitation) arouse concerns in the biomedical field.136,137 Organic photocatalysts alleviate potential metal-associated concerns. At present, the application of organic molecules has emerged as photocatalytic anticancer agents.Huang et al. designed compound bpyPCN with favourable aqueous solubility. The significant PDT performance was ascribed to its excellent ability for 1O2 generation and photocatalytic NADH oxidation.138 To render the absorption of photocatalysts for photoredox catalysis in the NIR spectral range, Kim et al. conjugated a BODIPY moiety to erlotinib (ER, an FDA-approved tyrosine kinase inhibitor) to obtain a photocatalyst (CatER) (Fig. 18a).139 Under normoxia, photoactivation of CatER gave rise to 1O2 production and NADH oxidation with high TOF (49.3 min−1). In vitro studies revealed that CatER exhibited light dose-dependent depletion of NADH and a high level of antiproliferation activity toward A549 cells with NIR irradiation. Additionally, tumour growth was significantly suppressed in BALB/c mice bearing 4T1 tumours.
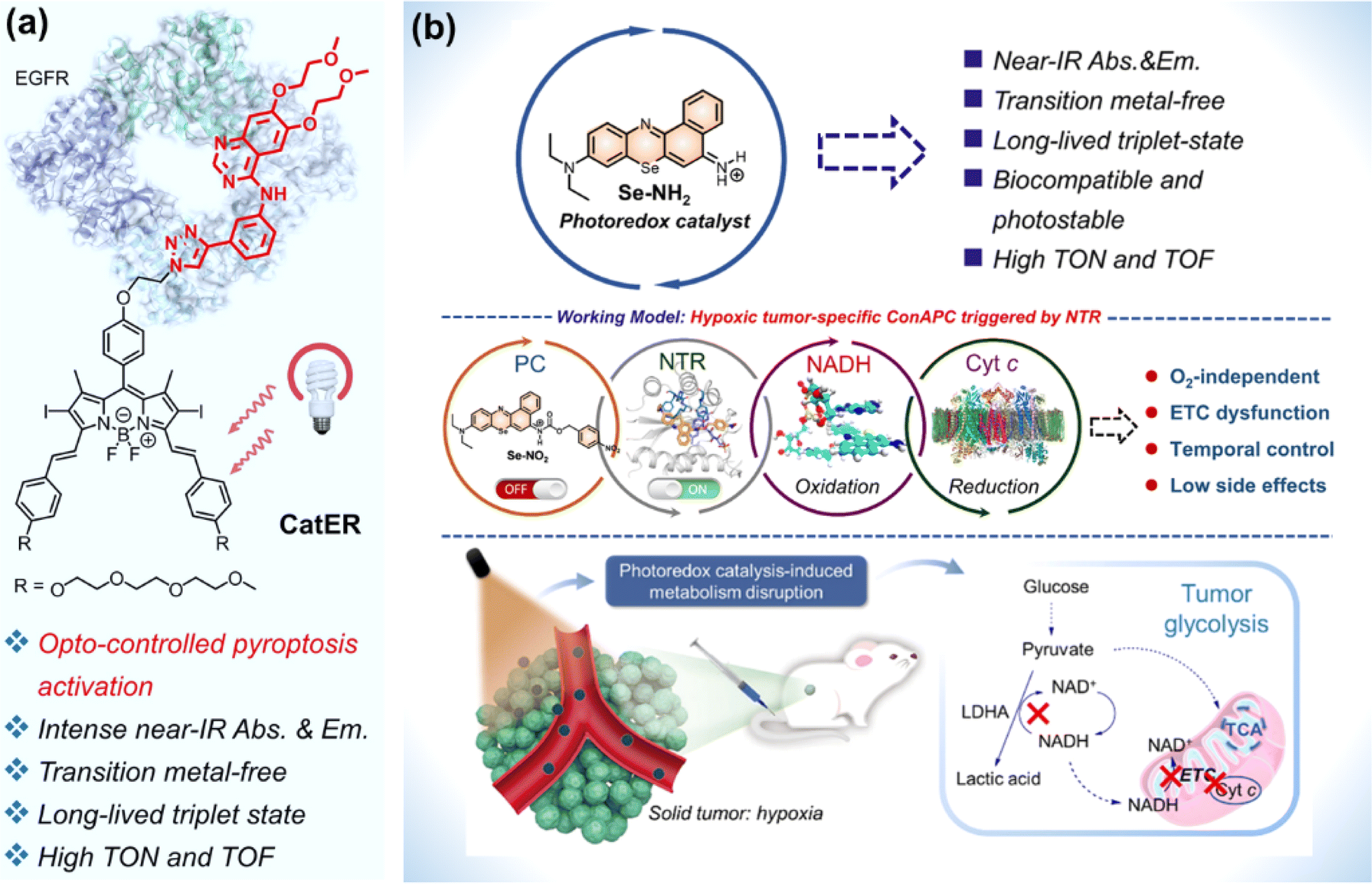 | ||
| Fig. 18 (a) CatER-based photocatalytic NADH oxidation. Reproduced with permission from ref. 139. Copyright 2022 National Academy of Science. (b) Se–NH2 based photoredox catalysis for phototherapy against a hypoxic solid tumour. Reproduced with permission from ref. 140. Copyright 2022 American Chemical Society. | ||
Considering the off-target risk and safety concerns of photocatalysts in living systems, Peng et al. explored a conditionally activatable photoredox catalysis (ConAPC) system and designed ConAPC architecture (Se–NO2) in which the inherent photocatalytic properties were temporarily caged (Fig. 18b).140 Se–NO2 was converted to Se–NH2 by the overexpressed nitroreductase (NTR) in tumours. Se–NH2 was activated by NIR light and catalyzed NADH oxidation with high TOF (18.8 min−1), which triggered the reductive transformation of Cyt c (Fe3+). The hypoxic tumour-specific ConAPC system proceeded independently on O2, and the anti-cancer activity toward hypoxic tumours was achieved through perturbing the mitochondrial respiration ETC and cellular metabolism. In the in vivo PDT experiments on HeLa-tumour-bearing female BALB/c mice, the tumour growth inhibition up to 91% was obtained. This work offers a possibility by leveraging artificial photocatalytic reactions toward the development of facile and intelligent photocatalytic theranostics.
4.3 Nano-catalysts
The EPR effect has been recognized as the central paradigm in tumour-targeted delivery in the past few decades.141 Especially, nano-based drug delivery systems are more beneficial to enhance the EPR effect of the tumour vasculature for in vivo tumour targeting to reduce the systemic toxicity. Recent studies on photocatalytic NADH oxidation using nano-catalysts demonstrate great potential in photoredox catalysis for PDT.142–144Multidrug resistance is a major challenge in chemotherapy and its phenotype is associated with the overexpression of ATP-driven transmembrane efflux pumps in cancer cells.145,146 Therefore, cancer drug resistance can be overcome by inhibiting the production of ATP in cancer cells, which is achievable through oxidizing NADH into NAD+. In 2018, He et al. reported a lipid membrane-coated silica–carbon (LSC) hybrid nanoparticle that targeted mitochondria through pyruvate (Fig. 19a).147 Doxorubicin (DOX) was encapsulated into the LSC NPs to form LSC-D NPs. The size of the LSC-D NPs (∼45 nm) was suitable for the requirement of the EPR effect for in vivo tumour targeting. Upon NIR laser (800 nm) irradiation of the LSC-D NPs, ROS was generated and the oxidation of NADH into NAD+ caused a reduction in the amount of ATP available for the efflux pump. Consequently, multidrug-resistant cancer cells lost their multidrug resistance capability for at least 5 days due to the compromised drug efflux pumps, which created a therapeutic window for chemotherapy (Fig. 19b). The in vivo data showed that the LSC-D NPs in combination with NIR laser treatment effectively inhibited the growth of multidrug-resistant tumours (nude mice with NCI/RES-ADR xenograft) without evident systemic toxicity. This study provides a promising nanotechnology-based strategy for fighting cancer multidrug resistance via photocatalytic NADH oxidation.
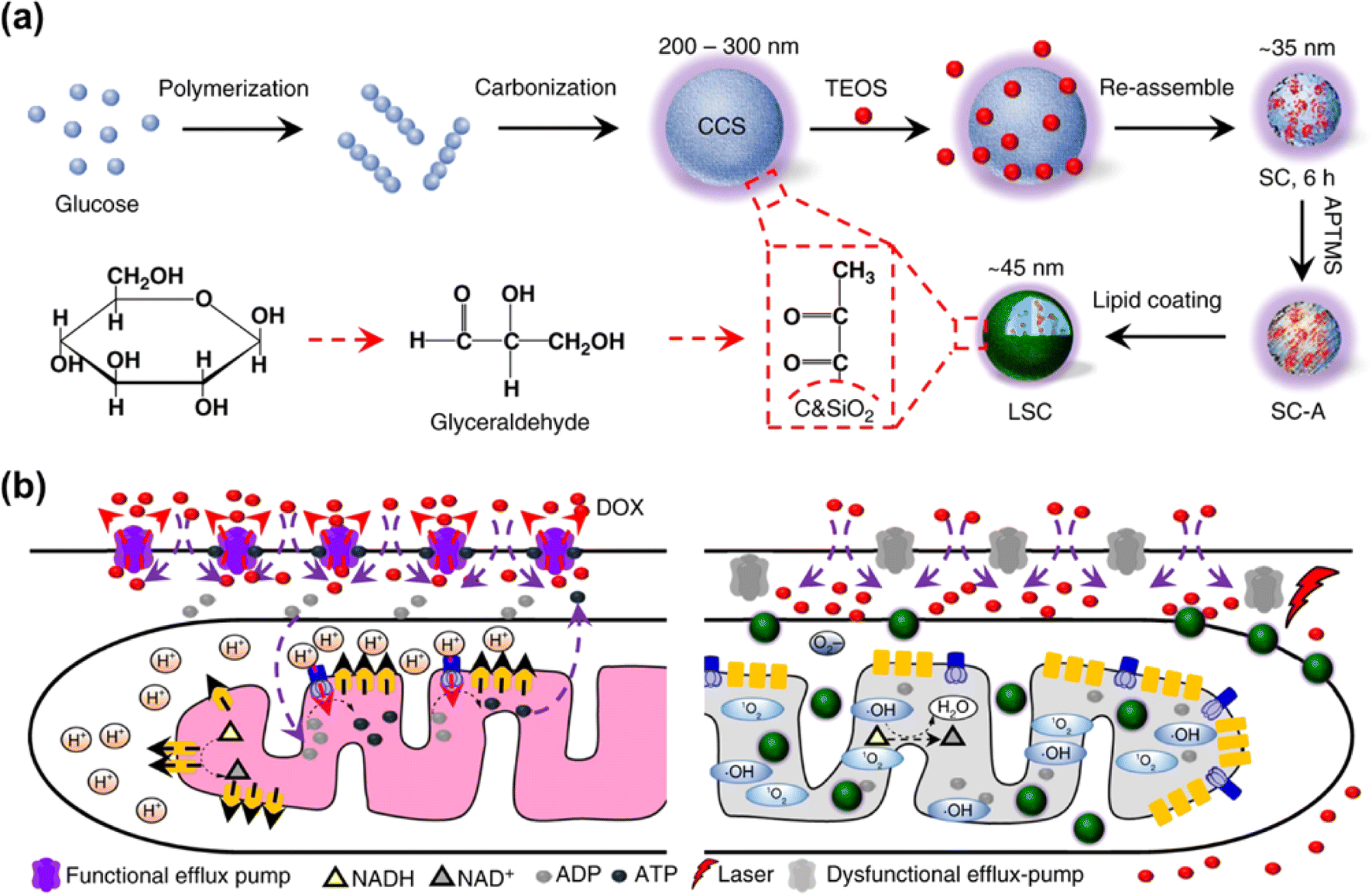 | ||
| Fig. 19 (a) Formulation of LSC NPs. (b) Application of LSC NPs for overcoming the cancer drug resistance. Reproduced with permission from ref. 147. Copyright 2018 Springer Nature. | ||
In 2022, Yang et al. reported a new strategy to boost the type-I PDT reaction by introducing electron acceptors to the existing type-II PS through the supramolecular assembly.148 Three different types of electron acceptors were co-assembled with a type-II PS to form quadruple hydrogen-bonded supramolecular photodynamic agents (DA1, DA2 and DA3) (Fig. 20a). Upon light irradiation of 660 nm, electron transfer took place from the PS to the electron acceptor and then to O2, which led to an efficient production of O2˙−. In addition, this process was accompanied by the generation of a strong oxidizing cationic radical of the PS (PS+˙) that was able to oxidize NADH into NAD+ (Fig. 20b). The supramolecular systems showed excellent activities of photocatalytic NADH oxidation (24.8 min−1 for DA1, 53.7 min−1 for DA2, 24.3 min−1 for DA3). As a result, the supramolecular PS exhibited superior PDT performance in treating tumours at in vitro and in vivo (a subcutaneous tumour model of HeLa in the BALB/c mice) levels. This work will broaden the insight into type-I PDT and inspire researchers to develop effective nano-catalysts for tumour therapy.
 | ||
| Fig. 20 (a) Chemical structures of the photosensitizer (D) and electron acceptors (A1, A2, and A3). (b) Supramolecular photodynamic system for photo-induced oxidation of NADH and generation of O2˙−. Reproduced with permission from ref. 148. Copyright 2022 Springer Nature. | ||
5. Conclusions and future prospects
This review has systematically discussed most recent strategies for PDT based on new mechanisms, including (1) developing novel radicals to reduce the dependence on O2; (2) designing activatable PSs on tumour cells to minimize damage to normal cells; (3) photocatalytic NADH oxidation to perturb the electron flow in intracellular mito-ETC.At present, fortunately, some strategies based on new mechanisms have drawn interest in clinical trials. TLD-1433 is the first Ru-based PS and has completed a phase I clinical trial for the treatment of bladder cancer. The phase II clinical study of TLD-1433 commenced in 2019 in the USA and Canada with a plan to treat 100–125 patients who developed nonmuscle invasive bladder cancer (NMIBC) and are intolerant or unresponsive to bacillus Calmette Guérin (BCG) therapy.149,150 Phototherapy and immunotherapy in combination, namely photoimmunotherapy (PIT), is emerging as a promising therapeutic strategy to both primary and metastatic tumours. PIT utilizes the PDT effect to induce systemic immune responses against tumours,151 maximizing the merits of both therapy modalities, leading to long-term immunity against cancer and increasing the selectivity of cancer therapy. Cetuximab sarotalocan (RM-1929), the first PIT drug in clinical trial, is an antibody–drug conjugate (ADC) carrying cetuximab and organic dye IRdye700DX, which can be activated by NIR irradiation at 690 nm to induce PDT and stimulate the anti-cancer immune response. Currently, cetuximab sarotalocan is being investigated under a world-wide phase III clinical trial and has been conditionally approved in Japan in 2020 for the treatment of unresectable locally advanced or unresectable locoregionally recurrent head and neck carcinoma (HNC).152 Based on the clinical trials of RM-1929 in PIT, in 2023, Mitsunaga et al. discussed abundant cases on how to set up and perform PIT applications in vitro and in vivo from a lot of aspects such as source of antibody, choice of dye and target, and methods used to characterize the labeled antibody, which is useful for targeted elimination of microbes and cells regardless of the target species or drug resistance status of the target.153 Other clinical trials based on innovative PDT are also under process. For example, Roswell Park Cancer Institute Corp put forward a phase I study utilizing PDT to amplify the response to immunotherapy in patients with non-small cell lung cancer (NSCLC) with pleural disease or malignant pleural mesothelioma (NCT04836429). The Mayo Clinic Foundation has proceeded with a phase II study of EUS-guided Verteporfin PDT in solid pancreatic tumours (NCT03033225).
However, most PDT strategies are in the basic research or preclinical phase, and there is a long way to go for clinical applications. One of the major challenges is the development of deep-tissue PDT, which needs to develop PSs with strong absorption in the deep red or NIR regions. Although two-photon excitation has been used to enhance the penetration depth of PDT and improve spatial selectivity, their clinical application is hindered due to the requirement of expensive devices and limitation in the excited domain. Thus, developing chemiluminescence/bioluminescence-based PSs for PDT will be a promising direction. Another important concern is how to improve the tumour-selectivity in PDT theranostics to maximize the therapeutic effect while minimizing damage to normal tissues, which will inspire increasing efforts in harnessing tumour biomarkers (hypoxia, GSH, ROS, NADH, enzyme, pH, etc.) to selectively activate PSs or convert precursors into PSs in tumours. Exogenous 5-ALA is a good case for imaging-guided PDT with good tumour selectivity, as it can be converted into protoporphyrin IX (PpIX), a fluorogenic PS, via a biosynthetic pathway in the tumour cells. In 2022, we reported an example of in-cell synthesis of a trimethine cyanine-type PDT agent via a pseudo-trimerization mechanism using an exogenous iodo-substituted indole derivative as a precursor and intrinsic ROS as the inducer, which provides an opportunity for imaging-guided PDT by taking advantage of the redox environment of tumours.154
Importantly, regarding the efficacy of PDT, the barrier of heterogeneous tumour hypoxia, which affects most clinically used PDT agents, highlights the need to develop new PSs that are active in type I or non-classic photosensitizing mechanisms with minimal oxygen dependence. The exploration of active free radicals for PDT beyond ROS and RNS provides an opportunity to evade the barrier of heterogeneous tumour hypoxia. So far, the intermediates produced in the regular PDT process cover a large scope of reactive oxidative species to treat cancers by photo-oxidative damage. Opposite to the photo-oxidative strategy, reactive reductive species (RRS)-involving PDT is free of O2 dependence and has started to attract attention, which leaves plenty of research space in understanding the biological functions of RRS, designing efficient RRS generators and aiming at hypoxic cancers with satisfactory selectivity and efficacy.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
X. W. and F. F. conceived the idea. X. W., J. P. and C. M. contributed to the writing of the original draft of the manuscript. F. F. reviewed and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22277054, 22077065) for financial support.Notes and references
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- H. J. Burstein, J. Clin. Oncol., 2000, 18, 693 CrossRef CAS PubMed.
- E. Rullier, P. Rouanet, J.-J. Tuech, A. Valverde, B. Lelong, M. Rivoire, J.-L. Faucheron, M. Jafari, G. Portier, B. Meunier, I. Sileznieff, M. Prudhomme, F. Marchal, M. Pocard, D. Pezet, A. Rullier, V. Vendrely, Q. Denost, J. Asselineau and A. Doussau, Lancet, 2017, 390, 469–479 CrossRef PubMed.
- K. Wang and J. E. Tepper, Ca-Cancer J. Clin., 2021, 71, 437–454 CrossRef PubMed.
- J. Tian, B. Huang, M. H. Nawaz and W. Zhang, Coord. Chem. Rev., 2020, 420, 213410 CrossRef CAS.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS.
- S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 11707–11715 CrossRef CAS PubMed.
- X. Li, Y. Zhao, T. Zhang and D. Xing, Adv. Healthcare Mater., 2021, 10, 2001240 CrossRef CAS.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed.
- G. Yang, L. Xu, J. Xu, R. Zhang, G. Song, Y. Chao, L. Feng, F. Han, Z. Dong, B. Li and Z. Liu, Nano Lett., 2018, 18, 2475–2484 CrossRef CAS PubMed.
- X. Guo, N. Yang, W. Ji, H. Zhang, X. Dong, Z. Zhou, L. Li, H.-M. Shen, S. Q. Yao and W. Huang, Adv. Mater., 2021, 33, 2007778 CrossRef CAS.
- C. S. Foote, Photochem. Photobiol., 1991, 54, 659 CrossRef CAS.
- M. S. Baptista, J. Cadet, P. Di Mascio, A. A. Ghogare, A. Greer, M. R. Hamblin, C. Lorente, S. C. Nunez, M. S. Ribeiro, A. H. Thomas, M. Vignoni and T. M. Yoshimura, Photochem. Photobiol., 2017, 93, 912–919 CrossRef CAS PubMed.
- Y.-Y. Wang, Y.-C. Liu, H. Sun and D.-S. Guo, Coord. Chem. Rev., 2019, 395, 46–62 CrossRef CAS.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- M. Dichiara, O. Prezzavento, A. Marrazzo, V. Pittalà, L. Salerno, A. Rescifina and E. Amata, Eur. J. Med. Chem., 2017, 142, 459–485 CrossRef CAS PubMed.
- I. J. Macdonald and T. J. Dougherty, J. Porphyrins Phthalocyanines, 2001, 5, 105–129 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko, E. Kędzierska, K. Knap-Czop, J. Kotlińska, O. Michel, K. Kotowski and J. Kulbacka, Biomed. Pharmacother., 2018, 106, 1098–1107 CrossRef PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- T. Hu, Z. Wang, W. Shen, R. Liang, D. Yan and M. Wei, Theranostics, 2021, 11, 3278–3300 CrossRef CAS PubMed.
- Y. Wu, S. Li, Y. Chen, W. He and Z. Guo, Chem. Sci., 2022, 13, 5085–5106 RSC.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- Z. Yu, P. Zhou, W. Pan, N. Li and B. Tang, Nat. Commun., 2018, 9, 5044 CrossRef PubMed.
- H. Li, X. Fan and J. Houghton, J. Cell. Biochem., 2007, 101, 805–815 CrossRef CAS PubMed.
- D. F. Quail and J. A. Joyce, Nat. Med., 2013, 19, 1423–1437 CrossRef CAS.
- W.-L. Liu, T. Liu, M.-Z. Zou, W.-Y. Yu, C.-X. Li, Z.-Y. He, M.-K. Zhang, M.-D. Liu, Z.-H. Li, J. Feng and X.-Z. Zhang, Adv. Mater., 2018, 30, 1802006 CrossRef.
- L. Deng, D. Sheng, M. Liu, L. Yang, H. Ran, P. Li, X. Cai, Y. Sun and Z. Wang, Biomater. Sci., 2020, 8, 858–870 RSC.
- G. Li, S. Yuan, D. Deng, T. Ou, Y. Li, R. Sun, Q. Lei, X. Wang, W. Shen, Y. Cheng, Z. Liu and S. Wu, Adv. Funct. Mater., 2019, 29, 1901932 CrossRef.
- J. Wan, X. Zhang, D. Tang, T. Liu and H. Xiao, Adv. Mater., 2023, 35, 2209799 CrossRef CAS PubMed.
- C. Wang, Y. Sun, S. Huang, Z. Wei, J. Tan, C. Wu, Q. Chen and X. Zhang, J. Am. Chem. Soc., 2023, 145, 13099–13113 CrossRef CAS PubMed.
- M. Li, T. Xiong, J. Du, R. Tian, M. Xiao, L. Guo, S. Long, J. Fan, W. Sun, K. Shao, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2019, 141, 2695–2702 CrossRef CAS PubMed.
- J. Zhuang, B. Wang, H. Chen, K. Zhang, N. Li, N. Zhao and B. Z. Tang, ACS Nano, 2023, 17, 9110–9125 CrossRef CAS PubMed.
- D. Cen, Q. Zheng, B. Zheng, R. Zhou, X. Xiao, T. Zhang, Z. Huang, T. Yan, J. Yu, X. Li, R. Deng and X. Cai, Adv. Funct. Mater., 2023, 33, 2211402 CrossRef CAS.
- X. Kang, Y. Zhang, J. Song, L. Wang, W. Li, J. Qi and B. Z. Tang, Nat. Commun., 2023, 14, 5216 CrossRef CAS.
- W. Wang, C. Zhu, B. Zhang, Y. Feng, Y. Zhang and J. Li, J. Am. Chem. Soc., 2023, 145, 16642–16649 CrossRef CAS.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- Y. Wan, L.-H. Fu, C. Li, J. Lin and P. Huang, Adv. Mater., 2021, 33, 2103978 CrossRef CAS.
- J. Du, T. Shi, S. Long, P. Chen, W. Sun, J. Fan and X. Peng, Coord. Chem. Rev., 2021, 427, 213604 CrossRef CAS.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS.
- P. Chinna Ayya Swamy, G. Sivaraman, R. N. Priyanka, S. O. Raja, K. Ponnuvel, J. Shanmugpriya and A. Gulyani, Coord. Chem. Rev., 2020, 411, 213233 CrossRef CAS.
- R. Song, H. Wang, M. Zhang, Y. Liu, X. Meng, S. Zhai, C.-c. Wang, T. Gong, Y. Wu, X. Jiang and W. Bu, Angew. Chem., Int. Ed., 2020, 59, 21032–21040 CrossRef CAS PubMed.
- S. Shen, C. Zhu, D. Huo, M. Yang, J. Xue and Y. Xia, Angew. Chem., Int. Ed., 2017, 56, 8801–8804 CrossRef CAS PubMed.
- J. Sun, X. Cai, C. Wang, K. Du, W. Chen, F. Feng and S. Wang, J. Am. Chem. Soc., 2021, 143, 868–878 CrossRef CAS PubMed.
- J. Peng, K. Du, J. Sun, X. Yang, X. Wang, X. Zhang, G. Song and F. Feng, Angew. Chem., Int. Ed., 2023, 62, e202214991 CrossRef CAS PubMed.
- A. Shafferman and G. Stein, Biochim. Biophys. Acta, 1977, 462, 161–170 CrossRef CAS PubMed.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- J. Geng, Y. Zhang, Q. Gao, K. Neumann, H. Dong, H. Porter, M. Potter, H. Ren, D. Argyle and M. Bradley, Nat. Chem., 2021, 13, 805–810 CrossRef CAS PubMed.
- J. Zhang, C. Mück-Lichtenfeld and A. Studer, Nature, 2023, 619, 506–513 CrossRef CAS.
- J. Peng and F. Feng, Chem.–Eur. J., 2024, 30, e202302842 CrossRef CAS PubMed.
- S. B. Reddy and S. K. Williamson, Expert Opin. Invest. Drugs, 2009, 18, 77–87 CrossRef CAS.
- Y. Yoshida, N. Itoh, Y. Saito, M. Hayakawa and E. Niki, Free Radical Res., 2004, 38, 375–384 CrossRef CAS PubMed.
- Y. Wan, G. Lu, J. Zhang, Z. Wang, X. Li, R. Chen, X. Cui, Z. Huang, Y. Xiao, J. Chelora, W. Zhang, Y. Liu, M. Li, H.-Y. Xie and C.-S. Lee, Adv. Funct. Mater., 2019, 29, 1903436 CrossRef.
- Y. Liu, P. Tang, P. Xiao, S. Luo, S. Zhang, H. Zhang, Y. Yang and D. Wu, Adv. Funct. Mater., 2022, 32, 2201666 CrossRef CAS.
- W. Zhang, H. Zhou, D. Gong, H. Wu, X. Huang, Z. Miao, H. Peng and Z. Zha, Adv. Healthcare Mater., 2023, 12, 2202947 CrossRef CAS PubMed.
- G. K. Thirunavukkarasu, G. R. Nirmal, H. Lee, M. Lee, I. Park and J. Y. Lee, J. Ind. Eng. Chem., 2019, 69, 405–413 CrossRef CAS.
- X. Li, Y. Liu, F. Fu, M. Cheng, Y. Liu, L. Yu, W. Wang, Y. Wan and Z. Yuan, Nano-Micro Lett., 2019, 11, 68 CrossRef CAS.
- X. Liu, Y. Yang, M. Ling, R. Sun, M. Zhu, J. Chen, M. Yu, Z. Peng, Z. Yu and X. Liu, Adv. Funct. Mater., 2021, 31, 2101709 CrossRef CAS.
- S. Kuang, L. Sun, X. Zhang, X. Liao, T. W. Rees, L. Zeng, Y. Chen, X. Zhang, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2020, 59, 20697–20703 CrossRef CAS PubMed.
- S. Kuang, F. Wei, J. Karges, L. Ke, K. Xiong, X. Liao, G. Gasser, L. Ji and H. Chao, J. Am. Chem. Soc., 2022, 144, 4091–4101 CrossRef CAS PubMed.
- E. J. Hong, D. G. Choi and M. S. Shim, Acta Pharm. Sin. B, 2016, 6, 297–307 CrossRef PubMed.
- W. Wu, X. Shao, J. Zhao and M. Wu, Adv. Sci., 2017, 4, 1700113 CrossRef PubMed.
- W. Jiang, M. Liang, Q. Lei, G. Li and S. Wu, Cancers, 2023, 15, 585 CrossRef CAS PubMed.
- X. Li, S. Kolemen, J. Yoon and E. U. Akkaya, Adv. Funct. Mater., 2017, 27, 1604053 CrossRef.
- W. Zhai, Y. Zhang, M. Liu, H. Zhang, J. Zhang and C. Li, Angew. Chem., Int. Ed., 2019, 58, 16601–16609 CrossRef CAS PubMed.
- J. Sun, X. Zhang, X. Wang, J. Peng, G. Song, Y. Di, F. Feng and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213765 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- L. Hui and Y. Chen, Cancer Lett., 2015, 368, 7–13 CrossRef CAS PubMed.
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437–447 CrossRef CAS.
- R. A. Cairns, I. S. Harris and T. W. Mak, Nat. Rev. Cancer, 2011, 11, 85–95 CrossRef CAS PubMed.
- H.-W. Liu, X.-X. Hu, K. Li, Y. Liu, Q. Rong, L. Zhu, L. Yuan, F.-L. Qu, X.-B. Zhang and W. Tan, Chem. Sci., 2017, 8, 7689–7695 RSC.
- K. Kiyose, K. Hanaoka, D. Oushiki, T. Nakamura, M. Kajimura, M. Suematsu, H. Nishimatsu, T. Yamane, T. Terai, Y. Hirata and T. Nagano, J. Am. Chem. Soc., 2010, 132, 15846–15848 CrossRef CAS PubMed.
- R. M. Phillips, Cancer Chemother. Pharmacol., 2016, 77, 441–457 CrossRef CAS PubMed.
- J. Xiong, P. Wang, S. Son, C. Zhong, F. Zhang, Z. Mao, Z. Liu and J. S. Kim, Matter, 2022, 5, 1502–1519 CrossRef CAS.
- J. Tian, B. Li, F. Zhang, Z. Yao, W. Song, Y. Tang, Y. Ping and B. Liu, Angew. Chem., Int. Ed., 2023, 62, e202307288 CrossRef CAS PubMed.
- Z. Liu, F. Song, W. Shi, G. Gurzadyan, H. Yin, B. Song, R. Liang and X. Peng, ACS Appl. Mater. Interfaces, 2019, 11, 15426–15435 CrossRef CAS.
- F. Xu, H. Li, Q. Yao, H. Ge, J. Fan, W. Sun, J. Wang and X. Peng, Chem. Sci., 2019, 10, 10586–10594 RSC.
- S. Bhattacharya, B. G. Prajapati and S. Singh, Crit. Rev. Oncol. Hematol., 2023, 185, 103961 CrossRef.
- M. Ashrafizadeh, M. Delfi, A. Zarrabi, A. Bigham, E. Sharifi, N. Rabiee, A. C. Paiva-Santos, A. P. Kumar, S. C. Tan, K. Hushmandi, J. Ren, E. N. Zare and P. Makvandi, J. Controlled Release, 2022, 351, 50–80 CrossRef CAS.
- M. Yang, Z. Özdemir, H. Kim, S. Nah, E. Andris, X. Li, Z. Wimmer and J. Yoon, Adv. Healthcare Mater., 2022, 11, 2200529 CrossRef CAS PubMed.
- H. Xiong, K. Zhou, Y. Yan, J. B. Miller and D. J. Siegwart, ACS Appl. Mater. Interfaces, 2018, 10, 16335–16343 CrossRef CAS.
- J. Zhu, Y. Zhang, Z. Li, X. Bao, Y. Zhou, B. Ma, Y. Xie, P. Yan, Z. Wu, Q. Zhang, J. Zou and X. Chen, Mater. Horiz., 2023, 10, 3014–3023 RSC.
- W. Hu, T. He, H. Zhao, H. Tao, R. Chen, L. Jin, J. Li, Q. Fan, W. Huang, A. Baev and P. N. Prasad, Angew. Chem., Int. Ed., 2019, 58, 11105–11111 CrossRef CAS PubMed.
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2016, 2, 16075 CrossRef.
- P. Cen, J. Huang, C. Jin, J. Wang, Y. Wei, H. Zhang and M. Tian, Aggregate, 2023, 4, e352 CrossRef CAS.
- G. B. Bodedla, X. Zhu and W.-Y. Wong, Aggregate, 2023, 4, e330 CrossRef CAS.
- Y. Hu, S.-Y. Yin, W. Liu, Z. Li, Y. Chen and J. Li, Aggregate, 2023, 4, e256 CrossRef CAS.
- K. R. Shreder, E. C. K. Lin, J. Wu, J. Cajica, C. M. Amantea, Y. Hu, E. Okerberg, H. E. Brown, L. M. Pham, D. M. Chung, A. S. Fraser, E. McGee, J. S. Rosenblum and J. W. Kozarich, Bioorg. Med. Chem. Lett., 2012, 22, 5748–5751 CrossRef CAS PubMed.
- X. Yue, B. Wang, M. Lan, J. Fan, X. Song and J. W. Foley, Chem. Commun., 2023, 59, 4676–4679 RSC.
- Z. Li, S. Li, Y. Guo, C. Yuan, X. Yan and K. S. Schanze, ACS Nano, 2021, 15, 4979–4988 CrossRef CAS.
- D. Tsvirkun, Y. Ben-Nun, E. Merquiol, I. Zlotver, K. Meir, T. Weiss-Sadan, I. Matok, R. Popovtzer and G. Blum, J. Am. Chem. Soc., 2018, 140, 12010–12020 CrossRef CAS PubMed.
- T. Weiss-Sadan, Y. Ben-Nun, D. Maimoun, E. Merquiol, I. Abd-Elrahman, I. Gotsman and G. Blum, Theranostics, 2019, 9, 5731–5738 CrossRef CAS PubMed.
- D. Luo, X. Wang, E. Walker, J. Wang, S. Springer, J. Lou, G. Ramamurthy, C. Burda and J. P. Basilion, ACS Nano, 2020, 14, 15193–15203 CrossRef CAS.
- L. K. B. Tam, J. C. H. Chu, L. He, C. Yang, K.-C. Han, P. C. K. Cheung, D. K. P. Ng and P.-C. Lo, J. Am. Chem. Soc., 2023, 145, 7361–7375 CrossRef CAS PubMed.
- Z. Ruan, P. Yuan, T. Li, Y. Tian, Q. Cheng and L. Yan, Eur. J. Pharm. Biopharm., 2019, 135, 25–35 CrossRef CAS PubMed.
- H. Chen, J. Tian, W. He and Z. Guo, J. Am. Chem. Soc., 2015, 137, 1539–1547 CrossRef CAS.
- H. Chen, W. He and Z. Guo, Chem. Commun., 2014, 50, 9714–9717 RSC.
- G. Dai, J. C. H. Chu, C. K. W. Chan, C. H. J. Choi and D. K. P. Ng, Nanoscale, 2021, 13, 15899–15915 RSC.
- G. Wang, Y. Su, X. Chen, Y. Zhou, P. Huang, W. Huang and D. Yan, Bioact. Mater., 2023, 25, 189–200 CAS.
- Q. Sun, Q. Su, Y. Gao, K. Zhou, W. Song, P. Quan, X. Yang, Z. Ge, Y. Zhang and G. He, Aggregate, 2023, 4, e298 CrossRef CAS.
- Q. Yang, G. Wu, Y. Yang, Y. Zhou, J. Song, H. Gao and W. Huang, Adv. Funct. Mater., 2024, 34, 2402194 CrossRef.
- J. Sun, K. Du, J. Diao, X. Cai, F. Feng and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 12122–12128 CrossRef CAS PubMed.
- J. Sun, X. Li, J. Cao, Q. Sun, Y. Zhang, X. Wang, T. Wu, X. Hu and F. Feng, Chem.–Eur. J., 2019, 25, 9164–9169 CrossRef CAS.
- Y. Li, D. Hu, M. Pan, Y. Qu, B. Chu, J. Liao, X. Zhou, Q. Liu, S. Cheng, Y. Chen, Q. Wei and Z. Qian, Biomaterials, 2022, 288, 121700 CrossRef CAS.
- Y. Zhang, C. Xu, X. Yang and K. Pu, Adv. Mater., 2020, 32, 2002661 CrossRef CAS.
- J. Sun, H. Li, X. Gu and B. Z. Tang, Adv. Healthcare Mater., 2021, 10, 2101177 CrossRef CAS.
- A. P. Gorka, R. R. Nani, J. Zhu, S. Mackem and M. J. Schnermann, J. Am. Chem. Soc., 2014, 136, 14153–14159 CrossRef CAS PubMed.
- J. Liu, H.-J. Butt and S. Wu, Adv. Funct. Mater., 2020, 30, 1907605 CrossRef CAS.
- L. Zhang, P. Wang, X.-Q. Zhou, L. Bretin, X. Zeng, Y. Husiev, E. A. Polanco, G. Zhao, L. S. Wijaya, T. Biver, S. E. Le Dévédec, W. Sun and S. Bonnet, J. Am. Chem. Soc., 2023, 145, 14963–14980 CrossRef CAS.
- X. Chen, N. Niu, D. Li, Z. Zhang, Z. Zhuang, D. Yan, J. Li, Z. Zhao, D. Wang and B. Z. Tang, Adv. Funct. Mater., 2023, 33, 2211571 CrossRef CAS.
- G. He, M. He, R. Wang, X. Li, H. Hu, D. Wang, Z. Wang, Y. Lu, N. Xu, J. Du, J. Fan, X. Peng and W. Sun, Angew. Chem., Int. Ed., 2023, 62, e202218768 CrossRef CAS.
- J.-S. Lan, R.-F. Zeng, Z. Li, Y. Wu, L. Liu, L.-X. Chen, Y. Liu, Y.-T. He, T. Zhang and Y. Ding, ACS Appl. Mater. Interfaces, 2023, 15, 34554–34569 CrossRef CAS.
- H. Wang, J. Chen, Q. Dong, X. Jia, D. Li, J. Wang and E. Wang, Nano Res., 2022, 15, 5256–5261 CrossRef CAS.
- A. Chiarugi, C. Dölle, R. Felici and M. Ziegler, Nat. Rev. Cancer, 2012, 12, 741–752 CrossRef CAS PubMed.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041–1048 CrossRef CAS PubMed.
- N. Lu, Z. Deng, J. Gao, C. Liang, H. Xia and P. Zhang, Nat. Commun., 2022, 13, 2245 CrossRef CAS.
- Z. Fan, J. Huang, H. Huang and S. Banerjee, Chemmedchem, 2021, 16, 2480–2486 CrossRef CAS.
- C. Huang, C. Liang, T. Sadhukhan, S. Banerjee, Z. Fan, T. Li, Z. Zhu, P. Zhang, K. Raghavachari and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 9474–9479 CrossRef CAS PubMed.
- Z. Zhu, L. Wei, Y. Lai, O. W. L. Carter, S. Banerjee, P. J. Sadler and H. Huang, Dalton Trans., 2022, 51, 10875–10879 RSC.
- E. Zafon, I. Echevarría, S. Barrabés, B. R. Manzano, F. A. Jalón, A. M. Rodríguez, A. Massaguer and G. Espino, Dalton Trans., 2022, 51, 111–128 RSC.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- H.-Q. Ju, J.-F. Lin, T. Tian, D. Xie and R.-H. Xu, Signal Transduction Targeted Ther., 2020, 5, 231 CrossRef CAS PubMed.
- S. Shamjith, M. M. Joseph, V. P. Murali, G. S. Remya, J. B. Nair, C. H. Suresh and K. K. Maiti, Biosens. Bioelectron., 2022, 204, 114087 CrossRef CAS PubMed.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- J. Liu, C. Zhang, T. W. Rees, L. Ke, L. Ji and H. Chao, Coord. Chem. Rev., 2018, 363, 17–28 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- X. Da, Z. Wang, Y. Jian, C. Zhang, Y. Hou, Y. Yao, X. Wang and Q. Zhou, Inorg. Chem. Front., 2022, 9, 2544–2556 RSC.
- C. Zhang, J. Huang, X. Guo, X. Da, Z. Dai, M. Hassan, Y. Yu, X. Wang and Q. Zhou, Nano Today, 2023, 50, 101824 CrossRef CAS.
- N. Tian, W. Sun, X. Guo, J. Lu, C. Li, Y. Hou, X. Wang and Q. Zhou, Chem. Commun., 2019, 55, 2676–2679 RSC.
- J. Liu, A. W. Prentice, G. J. Clarkson, J. M. Woolley, V. G. Stavros, M. J. Paterson and P. J. Sadler, Adv. Mater., 2023, 35, 2210363 CrossRef CAS.
- S. A. E. Omar, P. A. Scattergood, L. K. McKenzie, C. Jones, N. J. Patmore, A. J. H. M. Meijer, J. A. Weinstein, C. R. Rice, H. E. Bryant and P. I. P. Elliott, Inorg. Chem., 2018, 57, 13201–13212 CrossRef CAS PubMed.
- P. Zhang and H. Huang, Dalton Trans., 2018, 47, 14841–14854 RSC.
- R. Kushwaha, A. Kumar, S. Saha, S. Bajpai, A. K. Yadav and S. Banerjee, Chem. Commun., 2022, 58, 4825–4836 RSC.
- C. Ge, J. Zhu, A. Ouyang, N. Lu, Y. Wang, Q. Zhang and P. Zhang, Inorg. Chem. Front., 2020, 7, 4020–4027 RSC.
- M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227–6240 RSC.
- Y. M. Wilson, M. Dürrenberger, E. S. Nogueira and T. R. Ward, J. Am. Chem. Soc., 2014, 136, 8928–8932 CrossRef CAS.
- Z. Zhu, L. Wei, A. K. Yadav, Z. Fan, A. Kumar, M. Miao, S. Banerjee and H. Huang, J. Org. Chem., 2023, 88, 626–631 CrossRef CAS PubMed.
- M. Li, Y. Xu, Z. Pu, T. Xiong, H. Huang, S. Long, S. Son, L. Yu, N. Singh, Y. Tong, J. L. Sessler, X. Peng and J. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2210504119 CrossRef CAS PubMed.
- M. Li, K. H. Gebremedhin, D. Ma, Z. Pu, T. Xiong, Y. Xu, J. S. Kim and X. Peng, J. Am. Chem. Soc., 2022, 144, 163–173 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- M. Ikeda-Imafuku, L. L.-W. Wang, D. Rodrigues, S. Shaha, Z. Zhao and S. Mitragotri, J. Controlled Release, 2022, 345, 512–536 CrossRef CAS.
- Y. Takakura and Y. Takahashi, J. Controlled Release, 2022, 350, 486–493 CrossRef CAS PubMed.
- Z. Chen, R. K. Kankala, L. Long, S. Xie, A. Chen and L. Zou, Coord. Chem. Rev., 2023, 481, 215051 CrossRef CAS.
- G. Szakács, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef.
- A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, Adv. Drug Delivery Rev., 2002, 54, 759–779 CrossRef CAS PubMed.
- H. Wang, Z. Gao, X. Liu, P. Agarwal, S. Zhao, D. W. Conroy, G. Ji, J. Yu, C. P. Jaroniec, Z. Liu, X. Lu, X. Li and X. He, Nat. Commun., 2018, 9, 562 CrossRef PubMed.
- K.-X. Teng, L.-Y. Niu, N. Xie and Q.-Z. Yang, Nat. Commun., 2022, 13, 6179 CrossRef CAS PubMed.
- J. Karges, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef CAS.
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Curr. Opin. Chem. Biol., 2020, 56, 23–27 CrossRef CAS PubMed.
- L. Tang, A. Zhang, Z. Zhang, Q. Zhao, J. Li, Y. Mei, Y. Yin and W. Wang, Cancer Commun., 2022, 42, 141–163 CrossRef.
- N. L. Miyazaki, A. Furusawa, P. L. Choyke and H. Kobayashi, Cancers, 2023, 15, 5117 CrossRef CAS PubMed.
- T. Iwase, K. Ito, T. Nishimura, K. Miyakawa, A. Ryo, H. Kobayashi and M. Mitsunaga, Nat. Protoc., 2023, 18, 3390–3412 CrossRef CAS.
- H. Heng, G. Song, X. Cai, J. Sun, K. Du, X. Zhang, X. Wang, F. Feng and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202203444 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |