
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient alkaline water electrolysis with an iron-incorporated yttrium oxide/yttrium phosphide nanorod catalyst on Ni foam: overpotential reduction and electrochemical insights†
Subash
Pandey
 and
Rudy L.
Luck
*
and
Rudy L.
Luck
*
Department of Chemistry, Michigan Technological University, 1400 Townsend Drive, Houghton, MI 49931, USA. E-mail: rluck@mtu.edu
First published on 3rd July 2024
Abstract
The suitability of nonnoble transition metal phosphide-based catalysts for bifunctional water-splitting applications is explored in this study. Here, through a wet impregnation and phosphodization method, a yttrium phosphide catalyst supported by varying ratios of iron-incorporated yttrium oxide nanorod is reported. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio Fe-incorporated Y2O3-supported yttrium phosphide (FeYP11) exhibits efficient bifunctional catalytic activity attributed to heightened charge transfer and facilitated mass transport. The FeYP11 electrocatalyst manifests overpotentials of 301 and 19 mV at 10 mA cm−2 for the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) in an alkaline solution, respectively. Furthermore, electrochemical water splitting is achieved at 10 mA cm−2 and 100 mA cm−2 with cell voltages of 1.53 and 1.56 V, respectively. This investigation provides crucial insights into supported structure catalysts and the cost-effective nature of bifunctional phosphide-based catalysts for water electrolysis.
:1 ratio Fe-incorporated Y2O3-supported yttrium phosphide (FeYP11) exhibits efficient bifunctional catalytic activity attributed to heightened charge transfer and facilitated mass transport. The FeYP11 electrocatalyst manifests overpotentials of 301 and 19 mV at 10 mA cm−2 for the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) in an alkaline solution, respectively. Furthermore, electrochemical water splitting is achieved at 10 mA cm−2 and 100 mA cm−2 with cell voltages of 1.53 and 1.56 V, respectively. This investigation provides crucial insights into supported structure catalysts and the cost-effective nature of bifunctional phosphide-based catalysts for water electrolysis.
Introduction
The utilization of renewable energy sources is widely acknowledged as a solution to address growing environmental issues and worsening energy crises.1–3 A viable approach involves harnessing solar or wind-generated electricity for water electrolysis to produce hydrogen, which then powers fuel cells to generate green energy.4–6 The electrolysis involves two half-reactions: the oxygen evolution reaction (OER) at the anode and the hydrogen evolution reaction (HER) at the cathode.7,8 To overcome the higher activation energy barriers of these reactions, a potential higher than the thermodynamically calculated value of 1.23 V is required.9–11Traditionally, platinum (Pt)-based and iridium/ruthenium (Ir/Ru)-based precious catalysts have been pivotal for HER and OER, respectively.8,12,13 However, their limited supply and high costs hinder widespread application.14–16 As an alternative, non-noble transition metal phosphides (TMPs) are currently under thorough investigation for their electrocatalytic activity in alkaline solutions, offering a cost-effective choice for water-splitting catalysts.17,18 For instance, Wang et al., developed a hierarchical NiCo2O4@Ni2P nanorod array on a Ni rod through a simple hydrothermal method combined with phosphodization. This bifunctional catalyst operated at 10 mV cm−2 current density at a cell voltage of 1.58 V for an overall water-splitting reaction.18 Zhang et al., directly grew inner-porous CoP nanowires on carbon fibers by using a hydrothermal method followed by a phosphodization and an acid-treating process. This functioned with a current density of 10 mA cm−2 at a cell voltage of 1.62 V.19 Wu et al., used a Prussian blue analog synthetic strategy to prepare a bimetallic Fe–Ni phosphide hollow nano frame. This nonnoble catalyst contains abundant active sites and requires an overpotential of 116 and 235 mV for the OER and HER, respectively, in an alkaline solution to deliver a current density of 10 mA cm−2.11 Despite these advancements, there is still a need to enhance the electrochemical performance of non-noble, phosphide-based catalysts as viable alternatives to precious materials, especially for industrial-scale optimization.
Y2O3 is known for its high chemical and thermal stability, along with its basic nature. It serves as an excellent substrate for dispersed metal nanoparticles and helps immobilize the active surface area, making it widely used in catalysis.20 Y2O3 is also particularly suitable for oxygen activation.21 Various morphologies of yttrium oxide nanoparticles, such as spherical, rod, tube, prism, and sheets,22 can be synthesized using techniques such as the solvothermal, hydrothermal, microwave-assisted, and precipitation methods.22–25 Ruthenium nanoparticles on Y2O3via a precipitation method were synthesized and evaluated by Feng et al. as a decomposition catalysis for NH3 to produce COx-free H2.26 Okura et al., studied H2 generation using Ni/Y2O3 prepared by a wet impregnation method.27 Zhang et al., prepared Y2O3 support using hydrothermal methods and incorporated Ni through a wet impregnation method also for H2 production from NH3.28 We chose the hydrothermal method to produce Y2O3 nanorods and to accomplish Fe incorporation due to its simplicity, cost-effectiveness, and control morphology.29 Additionally, nanorods offer a large surface area,28 and to our knowledge, Fe-impregnated Y2O3 has not been previously reported.
The performance of an electrocatalyst can be improved by modulating the electronic structure of TMPs through modulation of their chemical composition.30–32 In this context, the incorporation of foreign ions, such as Fe3+, has a tuning effect on the local atomic structure, enhancing water-splitting activity.15,30,33 This study focuses on the influence of Fe3+ incorporation into yttrium oxide (Y2O3) and the formation of yttrium phosphide (YP5) on the nanorod surface as a cost-effective catalyst. This Y2O3-supported system on a Ni foam substrate exhibits remarkable bifunctional performance in a 1 M KOH solution. Specifically, the 50 wt% Fe3+ incorporated FeYP (FeYP11) catalyst demonstrates excellent performance for HER with catalytic performance (19 mV at a current density of 10 mA cm−2) comparable to a 5% Pt–C (24 mV at a current density of 10 mA cm−2) catalyst. Moreover, it exhibits an optimized overpotential of 301 mV at 10 mA cm−2 for OER, similar to reported robust OER catalysts.34,35 By pairing the bi-functional FeYP11 catalyst, the electrolyzer achieves outstanding performance, reaching current densities of 10 mA cm−2, 50 mA cm−2, and 100 mA cm−2 at relatively low cell voltages of 1.53, 1.55, and 1.56 mV, respectively. This work highlights a new supported system, a nonnoble metal phosphide catalyst, specifically a Fe-impregnated Y2O3-supported yttrium phosphide material, as a novel methodology for structure modulation and enhanced catalytic performance. The synthesis, a straightforward procedure, is both scalable and cost-effective. One resulting compound, FeYP11, tested as a catalyst for the OER and HER, demonstrated notable advantages such as reduced overpotential and increased activity compared to existing catalysts. This work emphasizes FeYP11 as an efficient electrode material for generating green hydrogen, while also suggesting that the Y2O3 substrate could support catalysts for future developments in water-splitting applications.
Methods
Preparation of Y2O3 nanoparticles
A solution consisting of 12.6 mmol of YCl3·6H2O and 33.3 mmol of urea dissolved in 50 mL of nano-pure water was prepared. 50 mL of 0.179 mol of NaOH(aq.) was added dropwise into this solution under vigorous stirring for 30 minutes. The resulting solution, containing a white precipitate, was transferred to a Teflon-lined autoclave and maintained at 170 °C for 24 hours. After cooling to room temperature, the precipitate was obtained by filtration and washed several times with ethanol and water until the solution reached a neutral pH. The precipitate was placed in a crucible and calcined at 600 °C in the air for 3 hours.Fe3+ incorporation
A wet impregnation method was employed to obtain the different weight percentages of Fe3+ in Y2O3 nanorod (FeYO). Various amounts of Fe3+ (0.10, 0.33, 0.50, and 0.66 g) and 3 g of polyvinylpyrrolidone (PVP) were dissolved in 50 mL of ethanol and stirred for 15 minutes. Subsequently, 0.9, 0.66, 0.50, or 0.33 g of Y2O3 powder was added, respectively, to achieve a total mass of 1 g and the solution was further stirred for 10 hours. The resulting precipitate was then filtered, washed with ethanol and water six times, dried at 100 °C for 1 hour, and calcined in air at 600 °C for 3 hours. The obtained dry precipitates were labeled as FeYO19, FeYO12, FeYO11, and FeYO21 for 0.10, 0.33, 0.50, and 0.66 g of Fe3+ incorporated Y2O3, respectively.Phosphodization
The phosphodization was conducted in a tube furnace at 800 °C under a flow of nitrogen gas. For each composition of FeYO, a crucible containing 0.5 g of the different products was placed in the center of the tube furnace. Another crucible with 0.25 g of red phosphorus powder was positioned near the FeYO crucible. A partial phosphodization of Y2O3 is performed to create the number of heterointerface regions. Nitrogen gas was passed into the tube furnace from one end with the other end connected to a mercury bubbler. The tube furnace was then heated gradually to 800 °C and maintained at that temperature for 2 hours and then allowed to cool to room temperature. The obtained products were labeled as FeYP19, FeYP12, FeYP11, and FeYP21 for iron-incorporated yttrium oxide/yttrium phosphide catalyst, corresponding to the use of 0.1, 0.33, 0.5, and 0.66 g Fe3+ in the wet impregnating preparations, respectively.Characterizations
Powder X-ray patterns of the as-prepared FeYP11 were recorded for phase identification and crystalline size measurements. The diffraction patterns of FeYP11 were collected using a Scintag, Inc. (Division of Thermal ARL. Dearborn MI) XDS-2000 θ/θ powder diffractometer with CuKα radiation (voltage = 40 kV, current = 35 mA, X-ray wavelength = 0.15405929 nm, beam slits = 1 to 2 mm, receiving slit = 0.3 to 0.5 mm, scan range = 15.0 to 65.0° at 0.02° at scan rate of 0.5). MDI Jade® V8.7 @10/18/22 software was employed for profile fitting and the JCPDS ICDD PDF-4+ database was used for indexing. For the characterization of morphology and topography, an energy dispersive X-ray spectrum (EDX), a JEOL JSM-6400 field-emission scanning electron microscope (FE-SEM) located in Michigan Tech's ACMAL laboratory was utilized. The EDX spectra of powder were collected using adhesive carbon tape. To minimize the charging effect, FE-SEM images were collected using transmission electron microscope (TEM) cells. The samples were dissolved in a mixture of isopropyl alcohol and nano pure water and a drop of each sample was poured into a TEM cell. This prepared sample in a TEM cell was also used for STEM characterization. Further morphological and elemental characterization, as well as particle size measurement, were conducted using a transmission electron microscope (FEI 200 kV Titan Themis STEM) in the ACMAL Laboratory.Electrochemical measurements
To fabricate the working electrode, 9 mg of each catalyst was dissolved in a mixture of 20 μL of 5% Nafion solution, 600 μL of ethanol, and 380 μL of nano-pure water. The solution was ultrasonicated for 30 minutes to form a homogenous ink. Subsequently, 500 μL of the sonicated ink was dropped on Ni foam within a 1 × 1 cm area at the rate of 10 μL min−1 resulting in a catalyst loading of 0.45 mg cm−2. The electrode was air-dried overnight at room temperature and then at 80 °C for 1 hour. All electrochemical measurements were performed using a CHI420 EQCM instrument. A three-electrode system with a KOH electrolyte was used for OER and HER measurement. N2 gas was bubbled through the electrolyte solution for 30 min before the measurements, and this inert environment was maintained throughout the experiment. The catalyst-loaded Ni foam served as the working electrode, Pt-wire functioned as the counter electrode, and an Ag/AgCl filled with 3 M KCl was used as the reference electrode. The performance of the catalyst-loaded Ni foam electrode includes the contribution from the Ni foam substrate. All the electrode potentials were converted to the reversible hydrogen electrode (i.e., vs. RHE) using the equation shown in the ESI.† Electrochemical measurements were conducted using a Pt counter electrode and measurements using a graphite rod counter electrode were also examined. This consisted of obtaining an LSV plot after subjecting the system to fifty CV cycles at a scan rate of 100 mV s−1 from 0.9 to 2 V vs. RHE in a 1 M KOH solution. Similar results were obtained with the Pt and graphite rod counter electrodes under these conditions.Results and discussion
Synthesis and characterization of catalyst
Iron-incorporated yttrium oxide/yttrium phosphide (FeYP) nanorods were synthesized using an impregnation method followed by phosphodization, as illustrated in Fig. 1. Initially, Y2O3 nanorods were prepared hydrothermally under high pH conditions. This results in nanorods with a higher surface area to volume ratio which can serve as support for ion impregnations. Different concentrations of Fe3+ impregnating solutions were used to saturate the Y2O3 nanopowders. After removing the excess impregnated solutions and subjecting them to heating at 600 °C under the atmosphere, Fe-incorporated Y2O3 (FeYO) powders were obtained. The phosphodization of the FeYO sample was accomplished at high temperatures under an inert atmosphere and resulted in the Y2O3-supported FeYP catalyst material. The final compounds, with Fe3+ and Y2O3 ratios of 1:9, 1:2, 1:1, and 2:1, supporting yttrium phosphide (FeYP) were labelled as FeYP19, FeYP12, FeYP11, and FeYP21, respectively. Our objective here was to precisely incorporate Fe3+ to observe the effects of saturating Y2O3 nanorods with different weight percentages of impregnating solutions. This was followed by partial phosphodization on the surface to create electroactive multiple heterointerfaces, aimed at enhancing bifunctional water-splitting performance.
 | ||
| Fig. 1 Schematic illustration of the preparation process of the Fe-incorporated yttrium phosphide, and the VESTA-generated polyhedral crystalline structure of FeYP11. | ||
The field-emission scanning electron microscopy (FE-SEM) image in Fig. S1(a) (ESI†) reveals the nanorod-like structure of yttrium oxide, characterized by an average thickness of 818 nm and an average length of 7.37 μm, with a nano-fiber-like morphology at the end. This nanorod structure is known to possess a high specific surface area, facilitating maximum electrolyte penetration.36 Y2O3 nanorods serve as a foundation for Fe incorporation and are known to aid in the deprotonation of water forming *OOH, enhancing active sites, and promoting electrocatalytic activity in water-splitting reactions.36,37 Following Fe incorporation in Y2O3 forming nanorod (FeYO11), the average thickness decreases to 691 nm, and the average length becomes 7.16 μm (Fig. S1(b), ESI†). The doped Y2O3 diameter is reduced due to the incorporation of Fe3+ ions, analogous to findings reported by Rathore et al. in their study on Fe-doped NiCo2Se4.36 The FeYP11 formed after phosphodization shows an average diameter of 806 nm and an average length of 7.58 μm (Fig. 2(c)). An increase in width is observed after phosphodization. The FE-SEM images of FeYP19, FeYP12, FeYP11, and FeYP21 (Fig. 2(a)–(d)) depict the preservation of the nanorod-like morphology after phosphodization. In FeYP21, two phases of nanorod yttrium phosphide and amorphous FeO are observed. The energy dispersive X-ray (EDX) spectra of FeYP19, FeYP12, FeYP11, and FeYP21 (Fig. S1(c)–(f), ESI†) confirm the presence of yttrium, iron, oxygen, and phosphorous, providing robust evidence that Fe-incorporated yttrium phosphide supported by yttrium oxide was successfully prepared. The EDX spectra of FeYP12, and FeYP11 show that ∼3.5–3.9% of Fe is present in the saturated Fe–Y2O3 nanorods. Using Fe3+ in greater quantities than Y2O3 under identical conditions leads to the formation of separate Fe phases alongside Y2O3, as indicated by FE-SEM images (Fig. 2(d)). As a result, the EDX analysis of FeYP21 reveals a significant presence of Fe (∼67%).
 | ||
| Fig. 2 FE-SEM images of (a) FeYP19, (b) FeYP12, (c) FeYP11, and (d) Fe-YP21. | ||
X-ray diffraction pattern (XRD) of FeYP11 (Fig. 3(a)) matches well with the cubic(I) phase of Y2O3 (JCPDS no. 00-041-1105) and the tetragonal phase of YPO4 (JCPDS no. 01-084-0335), showing that Y2O3 supported yttrium phosphide is successfully synthesized. We believe that the prepared yttrium phosphide is oxidized to yttrium phosphate after some time. A peak at 45.3° attributed to the delta phase of Fe is observed. The absence of iron phosphide phases suggests that iron is embedded within the Y2O3 nanorods, and phosphodization exclusively took place on the surface of the Y2O3 nanorod. The XRD spectrum of FeYP21 (Fig. S4, ESI†) shows that an additional phase of hematite (Fe2O3, JCPSD no. 01-071-5088) is formed along with yttrium phosphide (YP5, JCPDS no. 04-010-2346). The increase in Fe3+ incorporation to a 2:1 ratio with Y2O3 under similar experimental conditions led to the formation of a new Fe2O3 phase along with the Y2O3 phase. The TEM image of FeYP11 in Fig. 3(b) and (c) reveals the inner porous structure of the nanorod morphology. A similar kind of nanorod-like structure is observed in FeYP19 (Fig. S2(a), ESI†), and FeYP12 (Fig. S3(a), ESI†). In FeYP21 (Fig. S4(b), ESI†) spherical shaped Fe2O3 nanoparticles are polydisperse on the nanorod surface. The XRD pattern in Fig. 3 did not correspond to FeP and FePO4 phases (Fig. S5, ESI†). The HR-TEM image (Fig. 3(d)) illustrates that the (200) and (101) interplanar spacing of tetragonal Y(PO4) is 0.347 and 0.457 nm respectively, and the interplanar spacing of cubic Y2O3 is 0.276 and 0.427 nm, respectively. The different lattice fringes of yttrium phosphide on yttrium oxide from the HR-TEM images support the XRD results of FeYP11. The partial phosphodization of Fe-incorporated yttrium oxide shows the presence of both crystalline and amorphous regions, which give rise to abundant interface regions for catalytically active sites. The HR-TEM image of FeYP19 (Fig. S2(c), ESI†) shows the lattice fringes with a spacing of 0.308 and 0.375 nm. Similarly, the HR-TEM image of FeYP12 (Fig. S3(c), ESI†) shows the lattice fringes with a spacing of 0.352 and 0.469 nm, corresponding to the (200) planes of Y(PO4) and the (211) planes of Y2O3, respectively. By carefully matching the lattice fringes of spherical Fe2O3 and nanorod YP5 in FeYP21 (Fig. S4(d), ESI†), the interplanar spacing of 0.27 nm can be matched with a (104) plane of Fe2O3 and the fringe spacing of 0.335 nm is matched with a (120) plane of YP5. The corresponding selected area diffraction (SAED) pattern verifies the composition of yttrium phosphide supported by yttrium oxide with bright spots indexed to (200), (220), (202), (321), and (400) planes of Y(PO4), and (222), (400), and (431) planes of Y2O3. In Fig. 3(f) the high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) elemental mapping of a FeYP11 nanorod demonstrates homogeneous distribution of all elements. Due to the similar X-ray energies of yttrium and phosphorous, distinguishing between them using EDS mapping is not possible.
 | ||
| Fig. 3 (a) XRD pattern (Fe* refers to the delta phase for iron), (b) and (c) TEM images, (d) HR-TEM images with corresponding lattice fringe pattern, (e) SAED pattern, and (f) elemental mapping analysis of FeYP11. | ||
Electrochemical characterization
The OER activity of different weight percentages of Fe in YP was assessed using Ni foam as the substrate. The catalyst's effectiveness relied on the contribution of both Ni foam and FeYP. For electrode activation, 50 cycles of cyclic voltammetric scans were performed from 0.9 to 2 V vs. RHE in 1 M KOH solution at a scan rate of 100 mV s−1. In Fig. 4(a), the linear sweep voltammograms (LSV) illustrate the OER activity of different weight percentages of Fe in YP with FeYP11 showing higher activity than other samples. The activity of commercially available RuO2 and Ni foam substrate was also determined for comparison with the synthesized material's performance.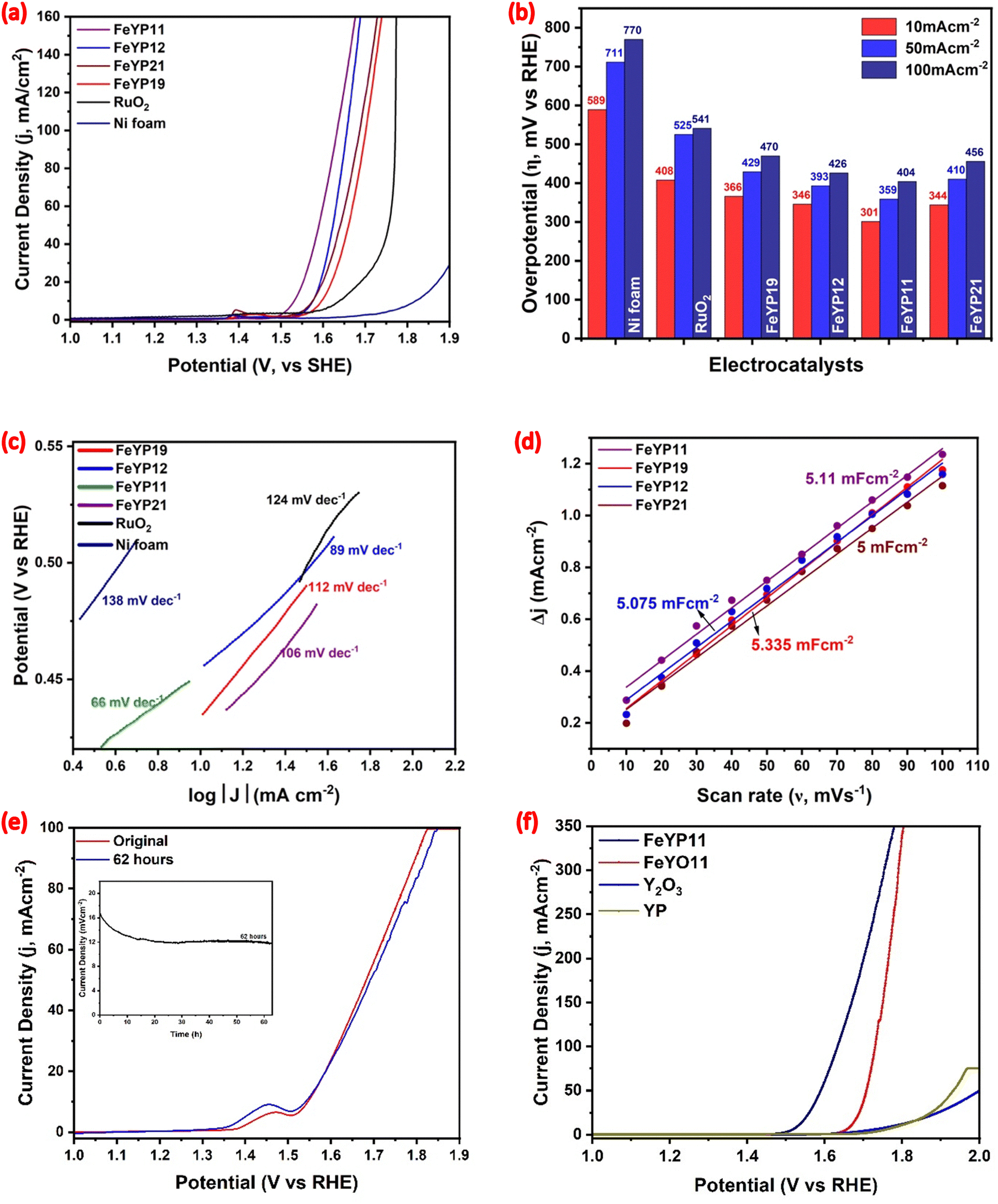 | ||
| Fig. 4 OER performance of the prepared samples. (a) Polarization curves in a 1 M KOH solution (with 100% iR correction)38 at a potential window of 0.9 V to 2 V with a scan rate of 10 mV s−1, (b) comparative bar diagrams of overpotential at different current densities, (c) Tafel plots, (d) double-layer capacitance measurements, (e) the chronopotentiometry curve of Fe-YP11 in 1 M KOH, and (f) polarization curves of FeYP11, FeYO11, Y2O3 and YP. | ||
The overpotential required for each active material and Ni foam to attain current densities of 10, 50, and 100 mA cm−2 is displayed in Fig. 4(b). Among them, FeYP11 on Ni foam emerges as the most active electroactive catalyst. Specifically, it requires overpotentials of 301, 343, and 347 mV to generate 10, 50, and 100 mA cm−2, respectively. FeYP19, FeYP12, and FeYP21 on Ni foam closely align with FeYP11 in terms of overpotential, Fig. 4(b). In comparison, the commercial RuO2 on Ni foam exhibits an overpotential of 408 mV at a current density of 10 mA cm−2. Notably, all of the prepared catalyst materials display higher activity than commercial RuO2. The Ni foam substrate, with a 589 mV overpotential, shows less activity and confirms that the higher OER activity achieved for the FeYP11 electrode on the Ni substrate is mainly from the contribution of the prepared electrode material rather than the substrate. Additionally, iR uncompensated activity was collected and compared with 100% iR-compensated results, Fig. S6 (ESI†).38 After 100% iR compensation, an improvement in the overpotential becomes apparent. Initially, the FeYP11 catalyst necessitates an overpotential of 332 mV to achieve a current density of 10 mA cm−2 without iR compensation, Fig. S7 (ESI†). However, after correcting for solution resistance, the overpotential decreases by 31 mV to deliver the same current density of 10 mA cm−2.
The Tafel slopes were determined for Ni foam, commercial RuO2, and FeYP19, FeYP12, FeYP11, and FeYP21 all on Ni foam using polarization scans collected at 5 mV s−1 (shown in Fig. 4(c)). FeYP11 produced a Tafel slope of 66 mV dec−1, which is lower than those for FeYP12 (89), FeYP21 (106), FeYP19 (112), RuO2 (124) and Ni foam (138 mV dec−1). The smaller Tafel slope of FeYP11 suggests that a small increase in potential results in a notable change in the current density, suggesting high electrocatalytic activity. Tafel slope values help to determine the OER pathway and rate-determining step for a catalyst. Typically, the OER pathway in an alkaline solution involves four steps, see Section S1.2 in ESI.† The Tafel slope for FeYP11, approximately 60 mV dec−1, implies that the second elementary step (OHads + OH− ↔ Oads + H2O + e−) is the rate-determining step, having a higher activation energy barrier (ΔG2).12 The electrochemical active surface area (ECASA) sheds light on the factors influencing the material's electrocatalytic performance. The double-layer capacitance (Cdl) value which is proportional to the ECASA, signifies the catalyst's activity for electrochemical performance. Cdl is calculated from the cyclic voltammogram of the non-faradic region (Fig. S8, ESI†) and illustrated in Fig. 4(d). The calculated Cdl values for FeYP11 (5.11), FeYP19 (5.335), FeYP12 (5.075), and FeYP21 (5 mF cm−2) reveal that the varying Fe weight percentages do not significantly change the active surface area. However, despite similar ECASA values, the superior electrochemical OER performance of FeYP11 suggests that other factors contribute to its catalytic activity. The LSV plot for OER after normalization using ECASA (Fig. S9(a), ESI†) shows that FeYP11 exhibits a lower overpotential than other electrode materials, emphasizing the intrinsic catalytic activity of FeYP11 active material.
FeYP11 exhibits the best OER performance compared to the other catalysts requiring only 331 mV (without iR compensation) overpotential at a geometric current density of 10 mA cm−2. The stability test for water electrolysis with FeYP11 conducted without iR compensation reveals a 30% change in current density after 62 hours of continuous electrolysis at 1.573 V vs. RHE in 1 M KOH. Interestingly, polarization curves show a negligible change in current density at 1.573 V, indicating robust stability even after 62 hours of continuous operation.
Additionally, the electrocatalytic OER performance was measured for Y2O3, YP-NF (NF = nickel foam), and 50% Fe incorporated yttrium oxide in 1 M KOH. Fig. 4(f) displays the iR-corrected LSV curves for FeYP11, Y2O3, YP-NF (YP) and 50% FeYO-NF (FeYO11), revealing significantly superior catalytic activity for FeYP11. Specifically, FeYO11 requires an overpotential of 440 mV, YP requires an overpotential of 560 mV and Y2O3 requires an ultrahigh potential of 552 mV to achieve a current density of 10 mA cm−2, both much higher than that for FeYP11. Furthermore, the polarization curves of FeYO11 and yttrium oxide (Fig. 4(f)) display a higher overpotential value for the OER reaction, suggesting that Fe3+ incorporation and then phosphodization generates more active sites, significantly improving the OER performance of the FeYP11 catalyst.
The electrocatalytic performance of the aforementioned varying Fe weight percentages incorporated into YP catalysts for HER was determined, with commercial Ni foam and 5 wt% Pt–C included for comparison. The 100% iR-compensated polarization curve is displayed in Fig. 5(a) while the corresponding overpotential values at 10, 50, and 100 mA cm−2 current densities are presented in Fig. 5(b). All four Fe-incorporated YP samples exhibit significantly lower overpotential values compared to commercial Ni foam, with FeYP11 standing out as the most active catalyst. To generate 10 mA cm−2, FeYP11 requires only 17 mV overpotential using a graphite rod as counter electrode, Fig. 5a, similar to the 19 mV overpotential using a Pt counter electrode which is lower than 5% Pt–C by 5 mV. The redox chemistry of Pt is similar under both alkaline and acidic conditions and reduction of oxidized Pt occurs “below 0 V vs. Ag/AgCl in 1 M KOH.”39 It is likely that the oxidation and reduction of Pt occurs with our systems but, under the initial measurements, the observed differences between the different electrodes is probably due to the nature of the electrodes and not from Pt deposited onto them (Fig. 5).40 APt wire was used recently as a counter electrode in HER experiments in alkaline solutions.41,42
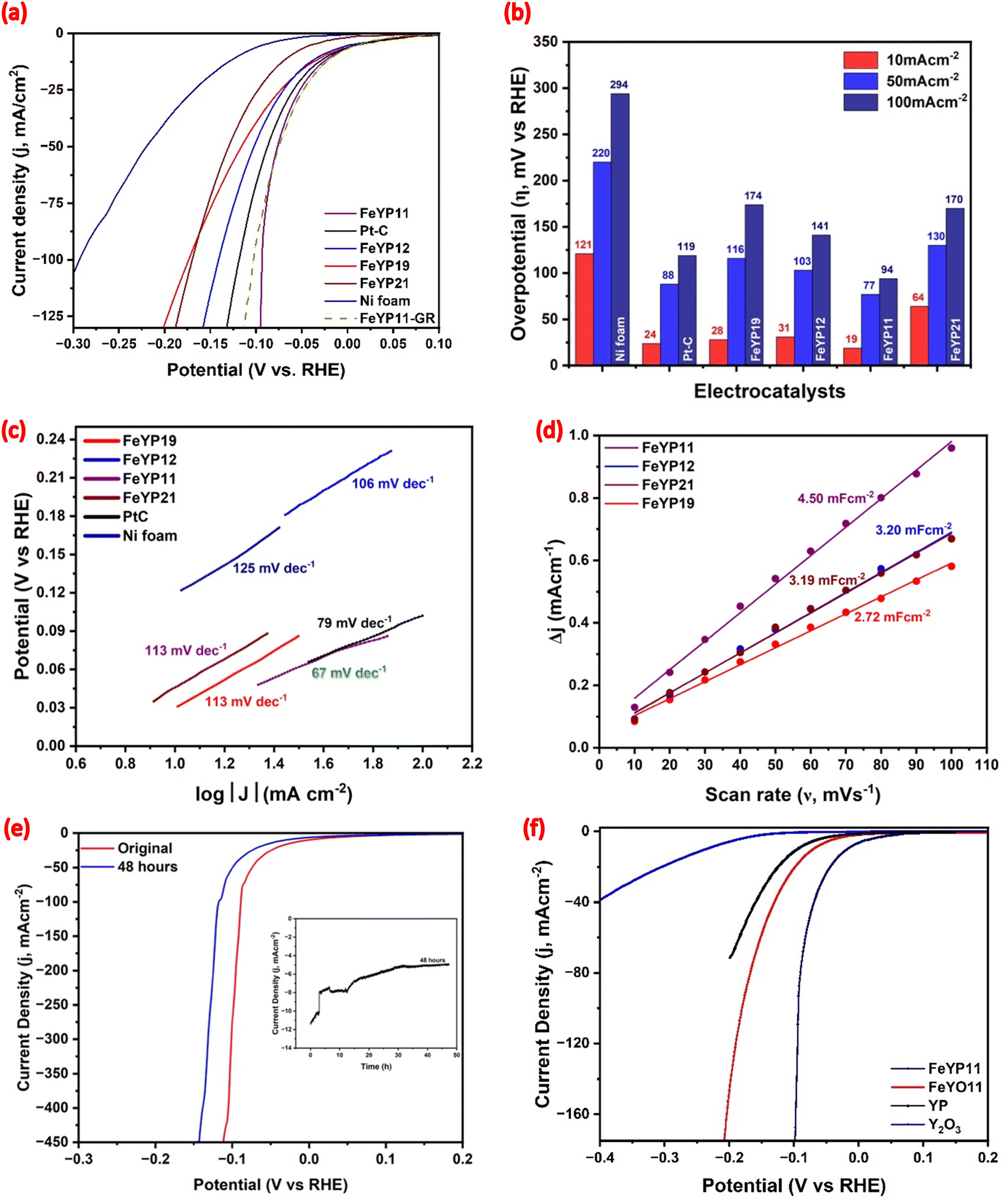 | ||
| Fig. 5 HER performance of the as-prepared samples. (a) Polarization curves at a 10 mV s−1 scan rate, (b) comparative bar diagrams of overpotential at different current densities, (c) Tafel plots, (d) double-layer capacitance measurements, (e) the chronopotentiometry curve of FeYP11, and (f) polarization curves of FeYP11, FeYO11, YP and Y2O3 in 1 M KOH solution. | ||
The other three materials, FeYP19, FeYP12, and FeYP21 require 28, 31, and 64 mV at a current density of 10 mA cm−2. The HER performance shows enhancement with a rise in the Fe:Y weight ratio to 1:1, suggesting improvement in the charge transfer mechanism within the nanorod structure and the generation of additional active sites. Conversely, a decline in activity is observed as the Fe:Y weight ratio increases to 2:1, attributed to the separation of two-phase particles. Additionally, the overpotential without iR compensation is measured and compared with 100% iR-compensated data (Fig. S10(a), ESI†). FeYP11 requires a 40 mV overpotential to achieve a 10 mA cm−2 current density. This experiment was conducted three times and resulted in almost identical LSV plots upon completion (Fig. S11, ESI†).
The Tafel slopes exhibit distinct values for various FeYP compositions: 113 for FeYP19, 106 for FeYP12, 67 for FeYP11, 113 for FeYP21, and 79 mV dec−1 for 5% Pt–C. The relatively small Tafel slope of 67 mV dec−1 for FeYP11 further confirms the superior catalytic efficacy in driving hydrogen production. The alkaline HER pathway involves three steps, as detailed in the ESI.† Tafel slope values around 30, 40, or 120 mV dec−1 indicate that the HER mechanism is controlled by the Tafel, Heyrovsky, or Volmer steps, respectively.43 While the Tafel slopes of FeYP19, FeYP12, and FeYP21 are nearer those expected for the Volmer step (near 120), the Tafel slope of FeYP11 at 67 mV dec−1 presents challenges in explaining the mechanism solely based on this value. The Cdl of the FeYP series of catalysts is calculated by scanning the HER-activated electrode in a non-faradic region (1.223 to 1.323 V vs. RHE, Fig. S12, ESI†) to estimate the ECASA for the origins of their HER activities. The resulting Cdl values for FeYP19, FeYP12, FeYP11, and FeYP21 are 2.72, 3.20, 4.50, and 3.19 mF cm−2, respectively (Fig. 6(d)). The higher Cdl values suggest more exposed sites for FeYP11. Also, the ECASA-normalized current density (Fig. S13(a), ESI†) for FeYP11 is larger than other materials under the same overpotential, indicating better intrinsic HER activities.
 | ||
| Fig. 6 (a) Half-cell HER and OER polarization curves at a scan rate of 10 mV s−1, (b) polarization curves of FeYP11 (+)//FeYP11 (−) in a two-electrode system, (c) two-electrode measurements for overall water-splitting performance for 60 hours, and (d) amount of H2 and O2 gas evolved at a constant overpotential of 406 mV. | ||
Fe is known to enhance the activity for OER and HER reactions with a variety of catalysts referred to as “The Fe Effect”.44 The LSV plot in Fig. 4(a) indicates that the current value for OER increased from FeYP19 to FeYP11. However, from FeYP11 to FeYP21, the current value decreased. Similar trends were also observed with the HER study (Fig. 5(a)). The calculated Cdl values reveal that the varying Fe weight percentages do not significantly change the active surface area. Incorporating Fe into the composites can result in interfacial charge transfer, modifications of the electronic structure, enhanced charge transport, and increases in the current.45 However, in FeYP21, the Fe phase separates, (Fig. 2(d)), and forms hematite, and thus a decrease in activity was observed.
Assessing the long-term stability is another important metric for HER performance evaluations. The durability of FeYP11 is tested using a time-based coulometric method without iR compensation at the constant overpotential value of 45 mV vs. RHE and LSV methods with 100% iR compensation. During the LSV measurement, the current density fluctuated from 13.3 to 8.2 mA cm−2 at an overpotential of 19 mV after 48 hours. Coulometric measurement for 48 hours shows an activity loss of ∼56% for FeYP11.
To gain insight into the effect of Fe incorporation and phosphodization, LSV measurements were conducted for FeYO11, YP, and Y2O3 at 10 mV s−1 with 100% iR compensation in 1 M KOH, as shown in Fig. 5(f). The results show that FeYO11 displayed an overpotential of 70 mV, YP requires an overpotential of 95 mV while Y2O3 exhibited an overpotential of 236 mV vs. RHE at a current density of 10 mA cm−2. This observation suggests a significant enhancement in activity following Fe incorporation, with a subsequent increase through phosphodization.
FeYP11 stands out as the most active catalyst, evident in its superior performance in both HER and OER. To assess the overall water-splitting capability, a 1 M KOH electrolyzer was constructed using FeYP11 as both the anode and cathode. In Fig. S14(a) (ESI†), a digital photograph of the fabricated electrolyzer cell capturing a water-splitting reaction is presented. The combination of the corresponding half-cell HER and OER polarization curves of FeYP11 and Pt–C (cathode) with RuO2 (anode) are shown in Fig. 6(a) with marked potential ranges for 10 mA cm−2. The overpotential at 10 mA cm−2 from the combination of the corresponding half-cell of FeYP11 is 320 mV, significantly outperforming Pt–C with RuO2 by 110 mV. The iR-compensated LSV of the as-fabricated FeYP11//FeYP11 electrolyzer displays low working potentials of 1.53, 1.55, and 1.56 V to deliver 10, 50, and 100 mA cm−2, respectively. This underscores the exceptional efficiency of FeYP11 in driving water-splitting reactions. An illustration of the iR compensated and uncompensated overpotential for the as-fabricated FeYP11//FeYP11 system is presented in Fig. S15 (ESI†).
Moreover, a stability assessment of FeYP11//FeYP11 was conducted through a time-based coulometric method (Fig. 6(c)) at 1.59 V over 63 hours. The LSV plot, before and after 63 hours without iR compensation, indicates an increase in overpotential from 312 to 395 mV required to deliver a current density of 10 mA cm−2 for FeYP11//FeYP11. The current density versus time plot (inset Fig. 6(c)) reveals a decrease of approximately 36% after 63 hours. For faradic efficiency calculations, a two-electrode configuration was established using two measuring cylinders of 10 mL. The gas displacement method in the 2-electrode setup is depicted in Fig. S14(b) (ESI†) (along with the associated video). The collected H2 and O2 gases at the cathode and anode, respectively, were consistent with the amounts calculated using Faraday's law based on the current–time plot. The faradic efficiency for HER is approximately 98.5% and for OER it is around 107%. The rate of formation of H2 and O2 is equal to 0.00427 and 0.0021 μmol cm−2 s−2, respectively. A listing of related Fe and Ni catalysts deposited on Ni foam reveals that the cell voltage for FeYP11 at 1.53 V is within the reported range from 1.42–1.63 V, (Table S1, ESI†). Considering these comprehensive performance metrics, FeYP11 is a good option for bifunctional water-splitting applications, showcasing the potential of low-cost, non-noble electrocatalysts.
Conclusion
In summary, we introduced catalytically active iron (Fe) into a nearly catalytically inactive Y2O3 nanorod, followed by phosphodization. The resulting FeYP11 catalyst, when applied to Ni foam, demonstrated outstanding HER activity with a minimal overpotential of 19 mV, and it also exhibited robust OER activity, necessitating an overpotential of 301 mV to achieve current densities of 10 mA cm−2. The observed performance in both OER and HER activities is influenced by the quantity of Fe3+ employed during the impregnation process, further enhanced through the phosphodization of the Fe-incorporated Y2O3 oxide catalyst. However, using a greater quantity of Fe3+ compared to Y2O3 under equivalent experimental circumstances leads to the formation of a distinct phase and results in diminished electrochemical performance. This study describes a cost-effective method for preparing high-performing and bifunctional metal phosphide catalysts in a supported system. Further studies on these materials implementing strategies regarding the use of Pt as a counter electrode are envisaged.46Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial assistance of the Chemistry Department at Michigan Technological University in paying for this publication.References
- M. Amini, Y. Mousazade, Z. Zand, M. Bagherzadeh and M. M. Najafpour, Ultra-Small and Highly Dispersive Iron Oxide Hydroxide as an Efficient Catalyst for Oxidation Reactions: A Swiss-Army-Knife Catalyst, Sci. Rep., 2021, 11(1), 6642, DOI:10.1038/s41598-021-85672-x.
- Z. He, J. Zhang, Z. Gong, H. Lei, D. Zhou, N. Zhang, W. Mai, S. Zhao and Y. Chen, Activating Lattice Oxygen in NiFe-Based (Oxy)Hydroxide for Water Electrolysis, Nat. Commun., 2022, 13(1), 2191, DOI:10.1038/s41467-022-29875-4.
- Z. Gao, F. Liu, L. Wang and F. Luo, Hierarchical Ni2P@NiFeAlOx Nanosheet Arrays as Bifunctional Catalysts for Superior Overall Water Splitting, Inorg. Chem., 2019, 58(5), 3247–3255, DOI:10.1021/acs.inorgchem.8b03327.
- B. Winther-Jensen and D. R. MacFarlane, New Generation, Metal-Free Electrocatalysts for Fuel Cells, Solar Cells and Water Splitting, Energy Environ. Sci., 2011, 4(8), 2790–2798, 10.1039/C0EE00652A.
- K. Wu, K. Sun, S. Liu, W.-C. Cheong, Z. Chen, C. Zhang, Y. Pan, Y. Cheng, Z. Zhuang, X. Wei, Y. Wang, L. Zheng, Q. Zhang, D. Wang, Q. Peng, C. Chen and Y. Li, Atomically Dispersed Ni–Ru–P Interface Sites for High-Efficiency pH-Universal Electrocatalysis of Hydrogen Evolution, Nano Energy, 2021, 80, 105467, DOI:10.1016/j.nanoen.2020.105467.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Recommended Practices and Benchmark Activity for Hydrogen and Oxygen Electrocatalysis in Water Splitting and Fuel Cells, Adv. Mater., 2019, 31(31), 1806296, DOI:10.1002/adma.201806296.
- C. Hu, K. Yue, J. Han, X. Liu, L. Liu, Q. Liu, Q. Kong, C.-W. Pao, Z. Hu, K. Suenaga, D. Su, Q. Zhang, X. Wang, Y. Tan and X. Huang, Misoriented High-Entropy Iridium Ruthenium Oxide for Acidic Water Splitting, Sci. Adv., 2023, 9(37), eadf9144, DOI:10.1126/sciadv.adf9144.
- A. Kumar, S. K. Purkayastha, A. K. Guha, M. R. Das and S. Deka, Designing Nanoarchitecture of NiCu Dealloyed Nanoparticles on Hierarchical Co Nanosheets for Alkaline Overall Water Splitting at Low Cell Voltage, ACS Catal., 2023, 13(16), 10615–10626, DOI:10.1021/acscatal.3c02096.
- H. Liu, M. Jin, D. Zhan, J. Wang, X. Cai, Y. Qiu and L. Lai, Stacking Faults Triggered Strain Engineering of ZIF-67 Derived Ni-Co Bimetal Phosphide for Enhanced Overall Water Splitting, Appl. Catal., B, 2020, 272, 118951, DOI:10.1016/j.apcatb.2020.118951.
- A. G. Rajan and E. A. Carter, Microkinetic Model for pH- and Potential-Dependent Oxygen Evolution during Water Splitting on Fe-Doped β-NiOOH, Energy Environ. Sci., 2020, 13(12), 4962–4976, 10.1039/D0EE02292F.
- Y. Wu, Y. Yi, Z. Sun, H. Sun, T. Guo, M. Zhang, L. Cui, K. Jiang, Y. Peng and J. Sun, Bimetallic Fe-Ni Phosphide Carved Nanoframes toward Efficient Overall Water Splitting and Potassium-Ion Storage, Chem. Eng. J., 2020, 390, 124515, DOI:10.1016/j.cej.2020.124515.
- X. Xie, L. Du, L. Yan, S. Park, Y. Qiu, J. Sokolowski, W. Wang and Y. Shao, Oxygen Evolution Reaction in Alkaline Environment: Material Challenges and Solutions, Adv. Funct. Mater., 2022, 32(21), 2110036, DOI:10.1002/adfm.202110036.
- B. You, N. Jiang, M. Sheng, M. W. Bhushan and Y. Sun, Hierarchically Porous Urchin-Like Ni2P Superstructures Supported on Nickel Foam as Efficient Bifunctional Electrocatalysts for Overall Water Splitting, ACS Catal., 2016, 6(2), 714–721, DOI:10.1021/acscatal.5b02193.
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. Goddard, S. Chen and Z. Ren, High-Performance Bifunctional Porous Non-Noble Metal Phosphide Catalyst for Overall Water Splitting, Nat. Commun., 2018, 9(1), 2551, DOI:10.1038/s41467-018-04746-z.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, Identification of Highly Active Fe Sites in (Ni,Fe)OOH for Electrocatalytic Water Splitting, J. Am. Chem. Soc., 2015, 137(3), 1305–1313, DOI:10.1021/ja511559d.
- H. Wen, L.-Y. Gan, H.-B. Dai, X.-P. Wen, L.-S. Wu, H. Wu and P. Wang, In Situ Grown Ni Phosphide Nanowire Array on Ni Foam as a High-Performance Catalyst for Hydrazine Electrooxidation, Appl. Catal., B, 2019, 241, 292–298, DOI:10.1016/j.apcatb.2018.09.043.
- B. H. R. Suryanto, Y. Wang, R. K. Hocking, W. Adamson and C. Zhao, Overall Electrochemical Splitting of Water at the Heterogeneous Interface of Nickel and Iron Oxide, Nat. Commun., 2019, 10(1), 5599, DOI:10.1038/s41467-019-13415-8.
- Q. Wang, H. Wang, X. Cheng, M. Fritz, D. Wang, H. Li, A. Bund, G. Chen and P. Schaaf, NiCo2O4@Ni2P Nanorods Grown on Nickel Nanorod Arrays as a Bifunctional Catalyst for Efficient Overall Water Splitting, Mater. Today Energy, 2020, 17, 100490, DOI:10.1016/j.mtener.2020.100490.
- S. Zhang, T. Xiong, X. Tang, Q. Ma, F. Hu and Y. Mi, Engineering Inner-Porous Cobalt Phosphide Nanowire Based on Controllable Phosphating for Efficient Hydrogen Evolution in Both Acidic and Alkaline Conditions, Appl. Surf. Sci., 2019, 481, 1524–1531, DOI:10.1016/j.apsusc.2019.03.250.
- K. Narasimharao and A. Alshehri, Gold Supported Yttrium Oxide Nanorods for Catalytic Oxidative Cracking of n-Propane to Light Olefins, Fuel, 2020, 278, 118375, DOI:10.1016/j.fuel.2020.118375.
- G. J. Hutchings and S. H. Taylor, Designing Oxidation Catalysts, Catal. Today, 1999, 49(1), 105–113, DOI:10.1016/S0920-5861(98)00414-3.
- M. Hajizadeh-Oghaz, R. S. Razavi, M. Barekat, M. Naderi, S. Malekzadeh and M. Rezazadeh, Synthesis and Characterization of Y2O3 Nanoparticles by Sol–Gel Process for Transparent Ceramics Applications, J. Sol-Gel Sci. Technol., 2016, 78(3), 682–691, DOI:10.1007/s10971-016-3986-3.
- L. Wen, X. Sun, Q. Lu, G. Xu and X. Hu, Synthesis of Yttria Nanopowders for Transparent Yttria Ceramics, Opt. Mater., 2006, 29(2), 239–245, DOI:10.1016/j.optmat.2005.09.003.
- A. Dupont, C. Parent, B. Le Garrec and J. M. Heintz, Size and Morphology Control of Y2O3 Nanopowders via a Sol–Gel Route, J. Solid State Chem., 2003, 171(1), 152–160, DOI:10.1016/S0022-4596(02)00202-5.
- R. V. Mangalaraja, K. V. S. Ramam, J. Ravi and C. P. Camurri, Synthesis of Nanocrystalline Yttria by Microwave-Assisted Citrate-Gel Decomposition Technique, J. Mater. Process. Technol., 2008, 197(1), 292–295, DOI:10.1016/j.jmatprotec.2007.06.041.
- J. Feng, L. Liu, X. Ju, J. Wang, X. Zhang, T. He and P. Chen, Highly Dispersed Ruthenium Nanoparticles on Y2O3 as Superior Catalyst for Ammonia Decomposition, ChemCatChem, 2021, 13(6), 1552–1558, DOI:10.1002/cctc.202001930.
- K. Okura, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, Ammonia Decomposition over Nickel Catalysts Supported on Rare-Earth Oxides for the On-Site Generation of Hydrogen, ChemCatChem, 2016, 8(18), 2988–2995, DOI:10.1002/cctc.201600610.
- R.-B. Zhang, Z.-A. Tu, S. Meng, G. Feng, Z.-H. Lu, Y.-Z. Yu, T. R. Reina, F.-Y. Hu, X.-H. Chen and R.-P. Ye, Engineering Morphologies of Yttrium Oxide Supported Nickel Catalysts for Hydrogen Production, Rare Met., 2023, 42(1), 176–188, DOI:10.1007/s12598-022-02136-5.
- G. De, W. Qin, J. Zhang, J. Zhang, Y. Wang, C. Cao and Y. Cui, Effect of OH− on the Upconversion Luminescent Efficiency of Y2O3:Yb3+, Er3+ Nanostructures, Solid State Commun., 2006, 137(9), 483–487, DOI:10.1016/j.ssc.2005.12.034.
- X. Meng, J. Han, L. Lu, G. Qiu, Z. L. Wang and C. Sun, Fe2+-Doped Layered Double (Ni, Fe) Hydroxides as Efficient Electrocatalysts for Water Splitting and Self-Powered Electrochemical Systems, Small, 2019, 15(41), 1902551, DOI:10.1002/smll.201902551.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Tuning Electronic Structure of NiFe Layered Double Hydroxides with Vanadium Doping toward High Efficient Electrocatalytic Water Oxidation, Adv. Energy Mater., 2018, 8(15), 1703341, DOI:10.1002/aenm.201703341.
- T. Zhao, X. Shen, Y. Wang, R. K. Hocking, Y. Li, C. Rong, K. Dastafkan, Z. Su and C. Zhao, In Situ Reconstruction of V-Doped Ni2P Pre-Catalysts with Tunable Electronic Structures for Water Oxidation, Adv. Funct. Mater., 2021, 31(25), 2100614, DOI:10.1002/adfm.202100614.
- Y. Sasaki, A. Iwase, H. Kato and A. Kudo, The Effect of Co-Catalyst for Z-Scheme Photocatalysis Systems with an Fe3+/Fe2+ Electron Mediator on Overall Water Splitting under Visible Light Irradiation, J. Catal., 2008, 259(1), 133–137, DOI:10.1016/j.jcat.2008.07.017.
- W. Li, Y. Yin, K. Xu, F. Li, K. Maliutina, Q. Wu, C. Li, B. Zhu and L. Fan, Enhancement of Oxygen Evolution Activity of Perovskite (La0.8Sr0.2)0.95MnO3-δ Electrode by Co Phase Surface Modification, Catal. Today, 2021, 364, 148–156, DOI:10.1016/j.cattod.2020.02.015.
- F. Li, N. Mushtaq, T. Su, Y. Cui, J. Huang, M. Sun, M. Singh, X. Zhao, K. Maliutina, Y. Zhang, C. He, M. Yang, B. Zhu and L. Fan, NCNT Grafted Perovskite Oxide as an Active Bifunctional Electrocatalyst for Rechargeable Zinc-Air Battery, Mater. Today Nano, 2023, 21, 100287, DOI:10.1016/j.mtnano.2022.100287.
- D. Rathore, S. Ghosh, J. Chowdhury and S. Pande, Fe-Doped NiCo2Se4 Nanorod Arrays as Electrocatalysts for Overall Electrochemical Water Splitting, ACS Appl. Nano Mater., 2023, 6(4), 3095–3110, DOI:10.1021/acsanm.3c00265.
- Y. Xue, J. Fang, X. Wang, Z. Xu, Y. Zhang, Q. Lv, M. Liu, W. Zhu and Z. Zhuang, Sulfate-Functionalized RuFeOx as Highly Efficient Oxygen Evolution Reaction Electrocatalyst in Acid, Adv. Funct. Mater., 2021, 31(32), 2101405, DOI:10.1002/adfm.202101405.
- W. Zheng, iR Compensation for Electrocatalysis Studies: Considerations and Recommendations, ACS Energy Lett., 2023, 8(4), 1952–1958, DOI:10.1021/acsenergylett.3c00366.
- R. Chen, C. Yang, W. Cai, H.-Y. Wang, J. Miao, L. Zhang, S. Chen and B. Liu, Use of Platinum as the Counter Electrode to Study the Activity of Nonprecious Metal Catalysts for the Hydrogen Evolution Reaction, ACS Energy Lett., 2017, 2(5), 1070–1075, DOI:10.1021/acsenergylett.7b00219.
- M. A. Bird, S. E. Goodwin and D. A. Walsh, Best Practice for Evaluating Electrocatalysts for Hydrogen Economy, ACS Appl. Mater. Interfaces, 2020, 12(18), 20500–20506, DOI:10.1021/acsami.0c03307.
- S. Yadav, V. Dao, W. Wang, K. Chen, C. Kim, G.-C. Kim and I.-H. Lee, Promising Ce Single-Atom-Dispersed Nitrogen-Doped Graphene Catalysts for the Hydrogen Evolution Reaction, Mater. Adv., 2023, 4(24), 6498–6506, 10.1039/D3MA00536D.
- A. Ganguly, R. J. McGlynn, A. Boies, P. Maguire, D. Mariotti and S. Chakrabarti, Flexible Bifunctional Electrode for Alkaline Water Splitting with Long-Term Stability, ACS Appl. Mater. Interfaces, 2024, 16(10), 12339–12352, DOI:10.1021/acsami.3c12944.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Insight on Tafel Slopes from a Microkinetic Analysis of Aqueous Electrocatalysis for Energy Conversion, Sci. Rep., 2015, 5(1), 13801, DOI:10.1038/srep13801.
- S. Anantharaj, S. Kundu and S. Noda, “The Fe Effect”: A Review Unveiling the Critical Roles of Fe in Enhancing OER Activity of Ni and Co Based Catalysts, Nano Energy, 2021, 80, 105514, DOI:10.1016/j.nanoen.2020.105514.
- M. A. Khan, S. I. Woo and O.-B. Yang, Hydrothermally Stabilized Fe(III) Doped Titania Active under Visible Light for Water Splitting Reaction, Int. J. Hydrogen Energy, 2008, 33(20), 5345–5351, DOI:10.1016/j.ijhydene.2008.07.119.
- G. Jerkiewicz, Applicability of Platinum as a Counter-Electrode Material in Electrocatalysis Research, ACS Catal., 2022, 12(4), 2661–2670, DOI:10.1021/acscatal.1c06040.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00362d |
| This journal is © The Royal Society of Chemistry 2024 |