
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature-dependent structural dynamics in covalent organic frameworks observed by cryogenic infrared spectroscopy†
Silas O.
Frimpong
a,
Nathan
McLane
 b,
Matthew
Dietrich
a,
Garrison A.
Bauer
a,
Michael R.
Baptiste
a,
Leah G.
Dodson
*a and
Mercedes K.
Taylor
*a
b,
Matthew
Dietrich
a,
Garrison A.
Bauer
a,
Michael R.
Baptiste
a,
Leah G.
Dodson
*a and
Mercedes K.
Taylor
*a
aDepartment of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, USA. E-mail: mkt@umd.edu; ldodson@umd.edu
bInstitute for Physical Science and Technology, University of Maryland, College Park, MD 20742, USA
First published on 12th August 2024
Abstract
Understanding the structural dynamics of covalent organic frameworks (COFs) in response to external temperature change is necessary for these materials' application at cryogenic temperatures. Herein, we report reversible structural dynamics observed in covalent organic frameworks as the temperature varies from 298 K to 30 K. A series of frameworks (COF-300, COF-300-amine, and COF-V) was studied in situ using a cryogenic infrared spectroscopy system. We observed peak shifts in the Fourier-transform infrared (FTIR) spectrum of COFs as temperature cooled to 30 K, and these peak shifts were reversed as temperature returned to 298 K. Comparison of these materials showed different degrees of temperature-dependent change, through the quantitative degree of the peak shift and a qualitative description of which peaks shifted. A general IR peak shift towards a higher frequency as temperature decreased was observed, with COF-300 exhibiting quantitatively larger blue shifts in key vibrational modes as compared with the other frameworks. The nature of the conformational changes giving rise to the IR shifts was studied using quantum-chemistry calculations on model systems. The results of the calculations indicate that key peak shifts arise from a pedal motion experienced by the frameworks during cooling. This understanding of temperature-dependent framework dynamics will enhance the development, selection, and application of covalent organic frameworks at extreme temperatures.
1. Introduction
Covalent organic frameworks (COFs) are porous materials constructed from organic building blocks, which covalently link in an orderly manner to form a crystalline framework.1 These frameworks are characterized by their crystallinity,1 tunability,2 high surface area,3 and stability,3 a combination of properties that opens the door to diverse applications. Consequently, covalent organic frameworks have attracted research attention in the areas of catalysis,4 energy storage,5 sensors,6,7 gas separation and storage,8 metal capture,9,10 and water purification.11,12The emphasis on application has led covalent organic frameworks to be relentlessly functionalized and optimized, in a quest to design well-defined materials with industrial relevance.13 In particular, increasing effort has been devoted to the design of structurally dynamic materials that respond reversibly to external stimuli.14,15 The structural flexibility and enhanced stability of covalent organic frameworks allow them to undergo reversible transformations without decomposition, thereby maintaining the framework's integrity.16 Conformational interconversions, including breathing, swelling, and linker rotation, are common features in covalent organic frameworks.16–19
External stimuli have also been shown to influence bond vibrations in these materials.19–21 When temperature change is the external stimulus, frameworks have been found to undergo changes in lattice vibrations as well as changes in structure.17,18 Other materials properties have been studied as a function of temperature using various spectroscopies, particularly using Raman spectroscopy.22–25 However, few variable-temperature studies probe the framework behavior at cryogenic conditions, particularly at temperatures below 77 K.
This knowledge gap is important to address because of the promising cryogenic applications of covalent organic frameworks. In one recent example, Oh et al. explored the cryogenic flexibility of covalent organic frameworks for quantum sieving.26 They were able to separate isotopes of hydrogen at cryogenic conditions due to a general contraction of the framework, which gave rise to selectivity for hydrogen over deuterium. Hence, studying conformational changes under cryogenic conditions is crucial for the development of future applications, especially in adsorptive separations.
Fourier-transform infrared spectroscopy (FTIR) has recently been explored as a powerful technique to study framework behavior in response to external stimuli. Fang et al. conducted an in situ FTIR analysis to study the behavior of covalent organic frameworks in response to external pressure.20 Upon applying pressure from 0 to 12 GPa, they observed a blue shift in most of the modes in the framework, with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N functional group experiencing the greatest effect. Because of the wealth of chemical information provided by FTIR spectroscopy, we used this technique to understand framework behavior at cryogenic temperatures.
N functional group experiencing the greatest effect. Because of the wealth of chemical information provided by FTIR spectroscopy, we used this technique to understand framework behavior at cryogenic temperatures.
We selected three well-known frameworks as representatives of three distinct structure types within the family of covalent organic frameworks (Fig. 1). The first, COF-300, is a framework with three-dimensional connectivity (3D COF) based on imine linkages.27,28 The second framework, which we term COF-300-amine, is the amine-linked analog of COF-300.29,30 The third framework, in contrast, is a two-dimensional material in which imine linkages connect monomers in two-dimensional planes; these 2D sheets are stacked through noncovalent interactions to yield a crystalline framework named COF-V.31
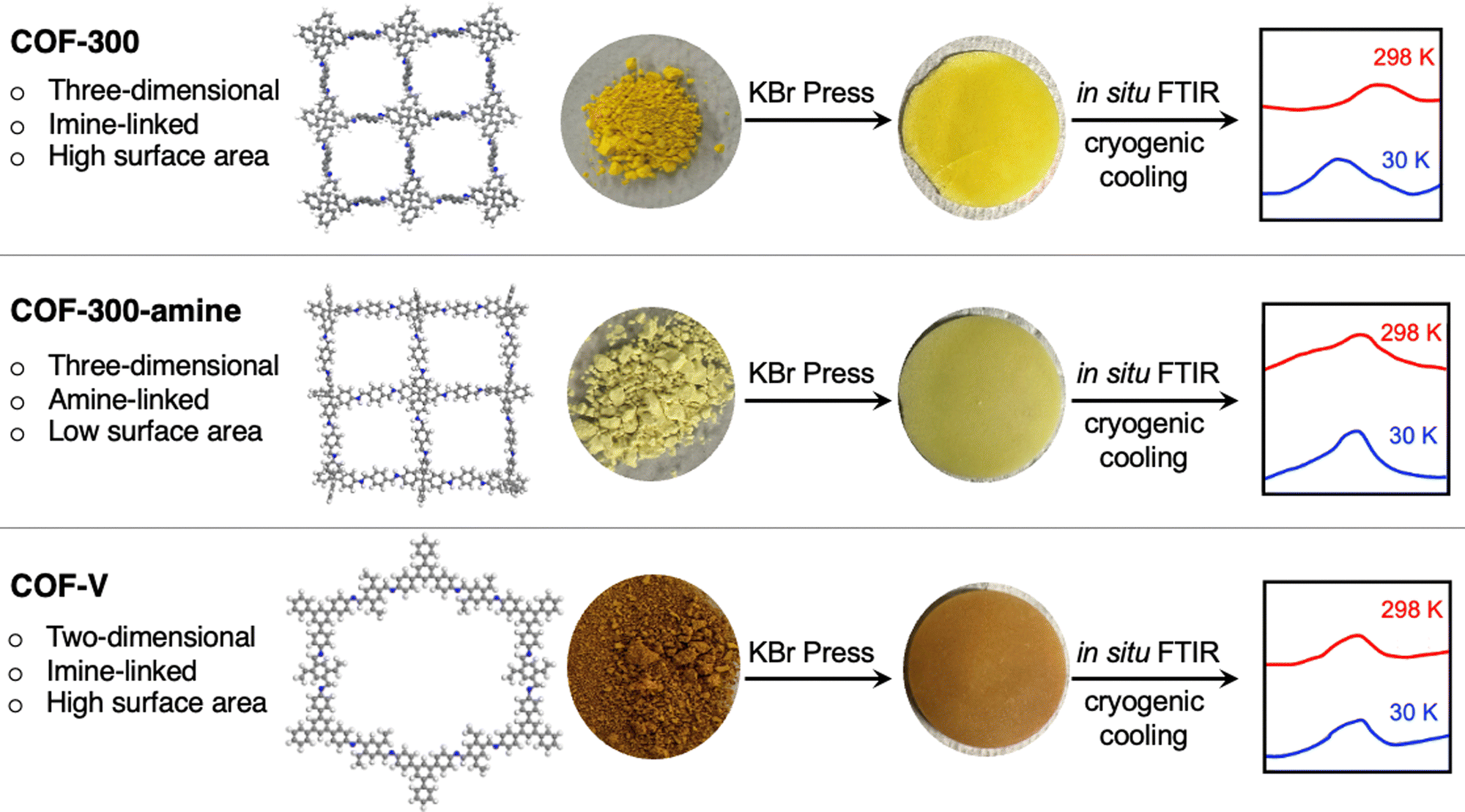 | ||
| Fig. 1 Schematic depiction of the in situ FTIR analysis performed on three covalent organic frameworks. The image of the COF-300 structure was created from crystallographic data published in ref. 28; the images of the COF-300-amine and COF-V structures are simply illustrations created using the program Materials Studio. | ||
Using a novel FTIR spectroscopy system, we herein show that extremely low temperatures induce structural changes in these frameworks, encoded in changes in the IR spectra. Our cryogenic FTIR system was used to carry out an in situ structural study of these COFs at temperatures down to 30 K. Further comparison of these COFs revealed different degrees of change through the quantitative degree of the peak shift and a qualitative assignment of shifted peaks. Shifting peak positions in the IR spectra were identified using quantum-chemistry calculations to reveal the functional groups most impacted by cryogenic cooling. This is the first in situ study of the structural dynamics of covalent organic frameworks at cryogenic conditions as low as 30 K.
2. Experimental methods
2.1. Synthesis and characterization of covalent organic frameworks
We synthesized three known covalent organic frameworks for this study. Adapted from a previously reported solvothermal method,27 COF-300 was synthesized by reacting tetrakis(4-aminophenyl)methane (TAPM; 1 eq.) and terephthaldehyde (2 eq.) in a solution of 1,4-dioxane and 6 M aqueous acetic acid. The mixture was degassed, sealed, and subjected to an elevated temperature of 120 °C for 72 hours. The synthesis of crystalline COF-300 was confirmed using powder X-ray diffraction (PXRD; see Fig. S4, ESI†).Synthesis of COF-300-amine was carried out by the reduction of COF-300.29 Treating imine-linked COF-300 with NaBH4 (38 eq.) in anhydrous methanol at room temperature for 24 hours in the presence of 1,4-benzenedicarboxylic acid (1 eq.) yielded amine-linked COF-300-amine. This was confirmed using solid-state 13C cross-polarization magic-angle spinning NMR spectroscopy, which shows the disappearance of an imine carbon at 158 ppm and the appearance of amine carbon at 52 ppm in COF-300-amine (Fig. S1 and S2, ESI†).
The third framework, COF-V, was synthesized by combining 1,3,5-tris(4-aminophenyl)benzene (TPB) and 2,5-divinylterephthalaldehyde (DVA) in a solution of acetonitrile and 12 M acetic acid at room temperature for 72 h.31 This afforded a brownish-yellow powder that was characterized by PXRD (Fig. S6, ESI†) and solid-state NMR (Fig. S3, ESI†). All covalent organic frameworks underwent extensive solvent exchanges and were vacuum-dried at 80 °C overnight before subsequent analysis.
2.2. Cryogenic FTIR experiment
Cluster models (Fig. S17, ESI†) were used to approximate the vibrational spectra of the extended frameworks and to assign observed peaks to specific functional groups. Geometries of the cluster models were optimized at the B3LYP level of theory including empirical dispersion (DFT-D3), using the cc-pVTZ basis.33,34 Minimum structures were confirmed by the absence of imaginary frequencies, and the calculated harmonic vibrational frequencies were scaled by 0.968 to account for anharmonicities35,36
The effect of structural changes on vibrational spectra was simulated by carrying out relaxed geometry scans over the main dihedral angle in small molecules (N-benzylideneaniline and N-benzylaniline) that represent molecular fragments of COF-300 and COF-300-amine (Fig. S18, ESI†). The C–CN–C dihedral angle was scanned in N-benzylideneaniline, and the C–C–N–C dihedral angle was scanned in N-benzylaniline. Each dihedral angle was scanned in +10° increments from the global minimum structure (defined as 0°) to a maximum of 20°. Geometries of the molecular fragments were reoptimized and harmonic frequencies were calculated at each point using B3LYP/6-311G++(d,p), including empirical dispersion (DFT-D3).
3. Results and discussion
A high-resolution cryogenic FTIR spectroscopic system was used to acquire low-temperature condensed-phase IR spectra of the covalent organic frameworks (Fig. 1). Samples were initially compressed at a pressure of 12 tons per inch2 with KBr to form 1-inch diameter pellets with thicknesses ranging from 0.025–0.075 inches. To ensure that the preparation process did not cause a deformation of the frameworks, the samples were observed by IR and PXRD after compression at the same pressure, without KBr (Fig. S4–S6 and S19–S21, ESI†). The IR spectra and PXRD patterns are unchanged before and after compression, indicating that the preparation method did not cause any structural change to the frameworks.The FTIR spectra of the covalent organic frameworks were then collected in situ at various temperatures from room temperature (measured to be 298 K) to as low as 30 K. As temperature decreased, we observed a gradual peak shift to higher frequency (shorter wavelengths) in most of the modes. This peak shift was shown to be reversible; when samples were then heated back to room temperature, the peaks returned to their original positions. Samples could be cycled repeatedly between 298 K and 30 K with no loss or change in peak shifting.
Of the three frameworks studied, COF-300 showed the greatest extent of peak shifting. While almost all peaks in the IR spectra (∼30 peaks from 700 to 1700 cm−1) were observed to shift by some amount in the COF-300 sample, we focus on assigning and describing five modes that are observed to shift by the greatest extent in COF-300 (without duplicating similar modes, i.e., nearly-degenerate CN stretches). Our peak assignments are given in Fig. 2, and an illustration of the regions of each mode is given in Fig. 3. Peak assignments came from the literature wherever possible, but peaks were assigned and confirmed based on our quantum chemistry calculations if no literature assignments were available. Peak shifts were quantified in two ways: (1) by evaluating the overall peak shift from room temperature to the lowest temperature (ν30K–ν298K), where a positive value indicates that a peak blue shifted when the sample became cold; and (2) by performing a linear fit of the peak centers observed as a function of temperature for all six temperatures used in these experiments, where slopes (cm−1 K−1) that are negative indicate that the peak blue shifts as a function of decreasing temperature (Fig. 4).
 | ||
| Fig. 2 FTIR spectra at varying temperatures for COF-300. Peaks marked with an asterisk are from H2O and CO2 contaminants in the KBr pellets and not from the samples under investigation. Spectra from 4000–700 cm−1 are given in Fig. S22 (ESI†). | ||
 | ||
| Fig. 3 Depiction of the functional groups containing the vibrational modes analyzed herein for COF-300 (a; R = H), COF-V (a; R = CHCH2), and COF-300-amine (b). | ||
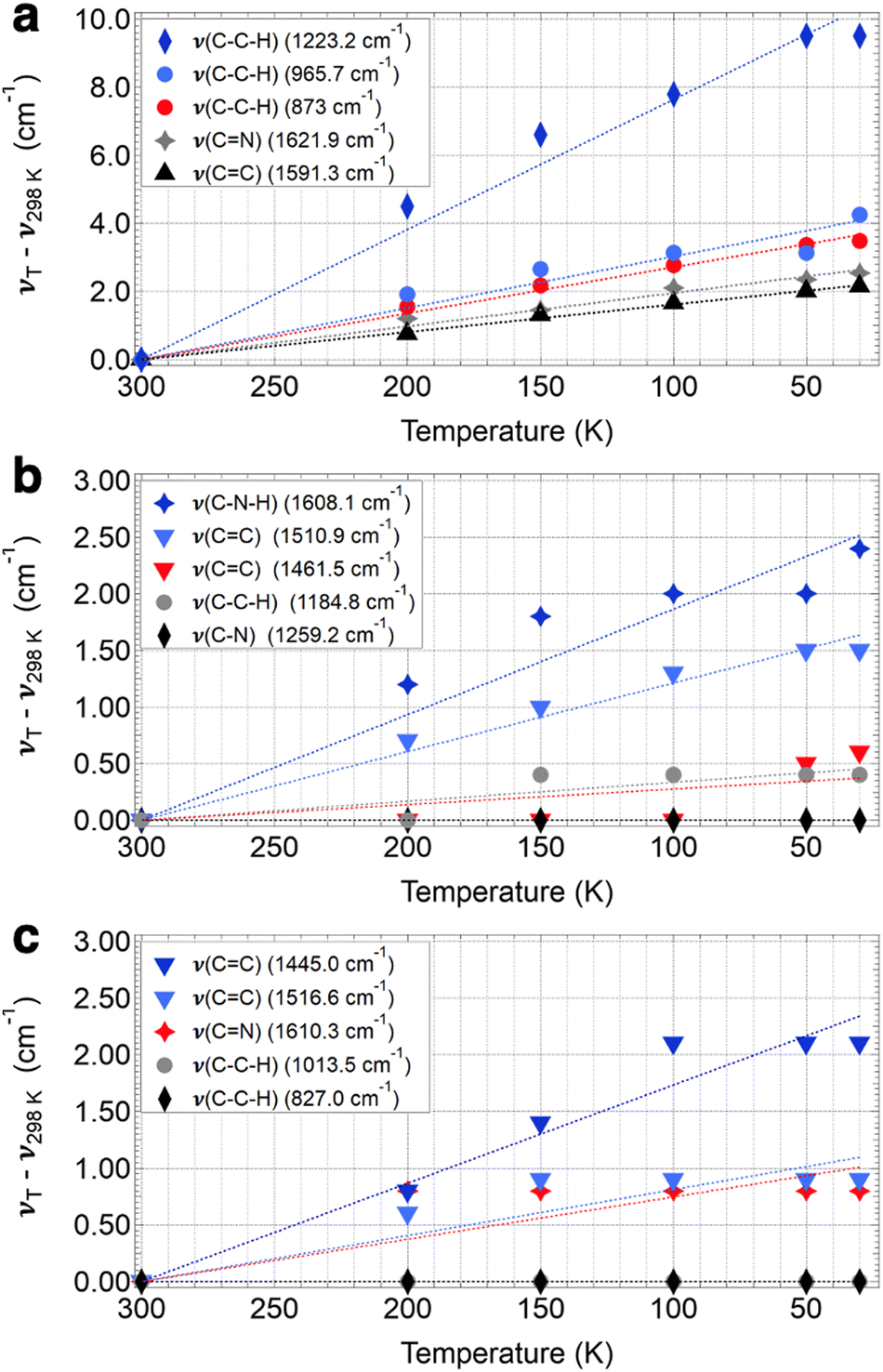 | ||
| Fig. 4 Peak shifts as a function of temperature for COF-300 (a), COF-300-amine (b), and COF-V (c). Symbols are the peak shift for each temperature T and dotted lines are the linear fit for the temperature dependence of the shift for each mode, which was constrained to intersect 0 cm−1 at room temperature. | ||
For COF-300, the peak observed to undergo the greatest shift as a function of temperature is the C–C–H imine/aromatic stretch, centered at 1223.2 cm−1 at room temperature and shifting by 9.5 cm−1 to 1232.7 cm−1 at 30 K, with a slope of −0.038 cm−1 K−1. The C–C–H bend (965.7 cm−1 at 298 K) shifts by about half as much, at a rate of −0.015 cm−1 K−1, resulting in an overall ν30K–ν298K peak shift of 4.3 cm−1. Another C–C–H bend appears at 873.0 cm−1 at 298 K and shifts at a slightly slower rate of −0.013 cm−1 K−1, resulting in an overall ν30K–ν298K peak shift of 3.5 cm−1. Two higher-energy peaks are observed to shift significantly in the COF-300 spectra. The first appears at 1621.9 cm−1 at room temperature and is assigned as the CN imine stretch.18,37,38 This peak shifts by 2.5 cm−1 at 30 K, at a rate of −0.0098 cm−1 K−1. Finally, the peak assigned to the CC aromatic stretch at 1591.3 cm−1 shifts by 2.2 cm−1 at 30 K, at a rate of −0.0081 cm−1 K−1.
Similar modes in the COF-300-amine structure shift by smaller amounts—if at all (Fig. 5a). The peak that shifts by the largest amount in COF-300-amine is the C–N–H bend (1608.1 cm−1 at 298 K),18,37,39 which blue shifts by 2.4 cm−1 at 30 K (−0.0093 cm−1 K−1). Both CC aromatic stretches shift by less than 2 cm−1; at room temperature, the peaks appear at 1510.9 and 1461.5 cm−1, and at 30 K they blue shift by 1.5 and 0.6 cm−1, respectively (−0.0061 and −0.0017 cm−1 K−1, respectively). The C–C–H bend vibration (1184.8 cm−1 at 298 K) also shifts by a small observable amount (ν30K–ν298K = 0.4 cm−1, −0.0014 cm−1 K−1). A distinguishing vibrational mode in COF-300-amine is the C–N stretch of the amine linkage (1259.2 cm−1),18,37,39 a functional group which is not present in COF-300; this mode is notably not observed to shift as a function of temperature. This observation agrees with the work of Chi et al., who found that the mode near 1252 cm−1 within COF-300-amine is invariant under temperatures ranging from 300 K up to 500 K.18
 | ||
| Fig. 5 FTIR spectra at varying temperatures for COF-300-amine (a) and COF-V (b). Peaks marked with an asterisk are from H2O and CO2 contaminants in the pellets and not from the samples under investigation. Spectra from 4000–700 cm−1 are given in Fig. S23 and S24 (ESI†). | ||
The two-dimensional COF-V had even less quantitative peak shifting observed as a function of temperature (Fig. 5b). The two aromatic CC stretches blue shift by 2.1 and 0.9 cm−1 (from 1445.0 and 1516.6 cm−1, respectively, at 298 K) at a rate of −0.0087 and −0.0041 cm−1 K−1, respectively. The room-temperature CN stretch at 1610.3 cm−1 shifts by a small amount (ν30K–ν298K = 0.8 cm−1, −0.0037 cm−1 K−1).40 Both C–C–H bend vibrations at 1013.5 and 827.0 cm−1 remain unshifted as a function of temperature. Peak assignments for the selected vibrational modes discussed above are given in Table 1.
| 298 K | 200 K | 150 K | 100 K | 50 K | 30 K | ν 30K–ν300K | Slope | Mode |
|---|---|---|---|---|---|---|---|---|
| COF-300 | ||||||||
| 1621.9 | 1623.1 | 1623.4 | 1624.0 | 1624.3 | 1624.4 | 2.5 | −0.098 | CN imine stretch |
| 1591.3 | 1592.0 | 1592.6 | 1592.9 | 1593.3 | 1593.4 | 2.2 | −0.0081 | CC aromatic stretch |
| 1223.2 | 1227.7 | 1229.8 | 1231.0 | 1232.7 | 1232.7 | 9.5 | −0.038 | C–C–H imine/aromatic stretch |
| 965.7 | 967.6 | 968.4 | 968.8 | 968.8 | 970.0 | 4.3 | −0.015 | C–C–H bend |
| 873.0 | 874.6 | 875.2 | 875.8 | 876.4 | 876.5 | 3.5 | −0.013 | C–C–H bend |
| COF-300-amine | ||||||||
| 1608.1 | 1609.3 | 1609.9 | 1610.1 | 1610.1 | 1610.5 | 2.4 | −0.0093 | C–N–H bend |
| 1510.9 | 1511.6 | 1511.9 | 1512.2 | 1512.4 | 1512.4 | 1.5 | −0.0061 | CC aromatic stretch |
| 1461.5 | 1461.5 | 1461.5 | 1461.5 | 1462.0 | 1462.1 | 0.6 | −0.0017 | CC aromatic stretch |
| 1259.2 | 1259.2 | 1259.2 | 1259.2 | 1259.2 | 1259.2 | 0.0 | 0.0 | C–N stretch |
| 1184.8 | 1184.8 | 1185.2 | 1185.2 | 1185.2 | 1185.2 | 0.4 | 0.0014 | C–C–H bend |
| COF-V | ||||||||
| 1610.3 | 1611.1 | 1611.1 | 1611.1 | 1611.1 | 1611.1 | 0.8 | −0.0037 | CN stretch |
| 1516.6 | 1517.2 | 1517.5 | 1517.5 | 1517.5 | 1517.5 | 0.9 | −0.0041 | CC aromatic stretch |
| 1445.0 | 1445.8 | 1446.4 | 1447.1 | 1447.1 | 1447.1 | 2.1 | −0.0087 | CC aromatic stretch |
| 1013.5 | 1013.5 | 1013.5 | 1013.5 | 1013.5 | 1013.5 | 0.0 | 0.0 | C–C–H bend |
| 827.0 | 827.0 | 827.0 | 827.0 | 827.0 | 827.0 | 0.0 | 0.0 | C–C–H bend |
Overall, the three-dimensional, imine-linked framework COF-300 showed a greater average blue shift than its amine-linked counterpart COF-300-amine and the two-dimensional framework COF-V. This increase in vibrational frequency in COF-300 could be partly attributed to “pedal motion” experienced by the framework in response to an external stimulus.17,18 Pedal motion has been observed in organic crystals such as stilbenes, azobenzenes, and benzylideneanilines with CC, NN or CN functionalities.17,18,41 In the pedal analogy, phenyl rings act as the pedals of a bicycle while the central double bond acts as the crank arm (Fig. 6a and b). Such pedal motion has been reported to cause a conformational interconversion of organic crystals as a function of temperature.18 Hence, changes in the vibrational frequencies as temperature decreases may indicate conformational change in the pedal motif of COF-300. We note that this pedal motion is likely to occur in tandem with other structural changes throughout the framework, as has been observed for other covalent organic frameworks in response to external stimuli.20
 | ||
| Fig. 6 Illustration of the pedal analogy (a) for motion around the CN functionality in N-benzylideneaniline (b). Depiction of varying dihedral angle in N-benzylideneaniline, from the ground-state configuration (c) to an angle of 20° (d). | ||
The assigned experimental spectra show that the functional groups involved in the pedal motion in COF-300 are the most impacted by temperature change. To better understand this observation, we used quantum-chemistry calculations to explore the effect of the pedal motion on IR peak position in silico. We varied the dihedral angle between the planes of the two “pedals,” or phenyl rings; the energy-optimized ground-state configuration was defined as an angle of 0° (Fig. 6c and d). Fig. 7a shows a portion of the simulated vibrational spectrum for N-benzylideneaniline (a molecular fragment of COF-300 containing the pedal motif) at three different dihedral angles. (The full spectra are plotted in Fig. S14 and S15 (ESI†), and all vibrational frequencies and shifts are in Tables S1 and S2. Line widths were set to 11.7 cm−1 for comparison to experimental spectra). Most of the vibrational modes red shift as a function of increasing dihedral angle, including the CN stretch, which red shifts by more than 11.5 cm−1 as the angle increases from 0° to 20°. When considered in the context of the IR spectra discussed above, we interpret this in silico result to mean that the COF-300 pedal motif exhibits greater dihedral angles at room temperature than at 30 K. As the sample cools to 30 K, the dihedral angle decreases, resulting in a blue shift in the observed vibrational frequencies. Looking macroscopically at the larger framework, this rotation will lead to not only a variation in the frequency in the CN mode but different interactions arising from the rotation of the attached crank arms, thus affecting other vibrational modes.
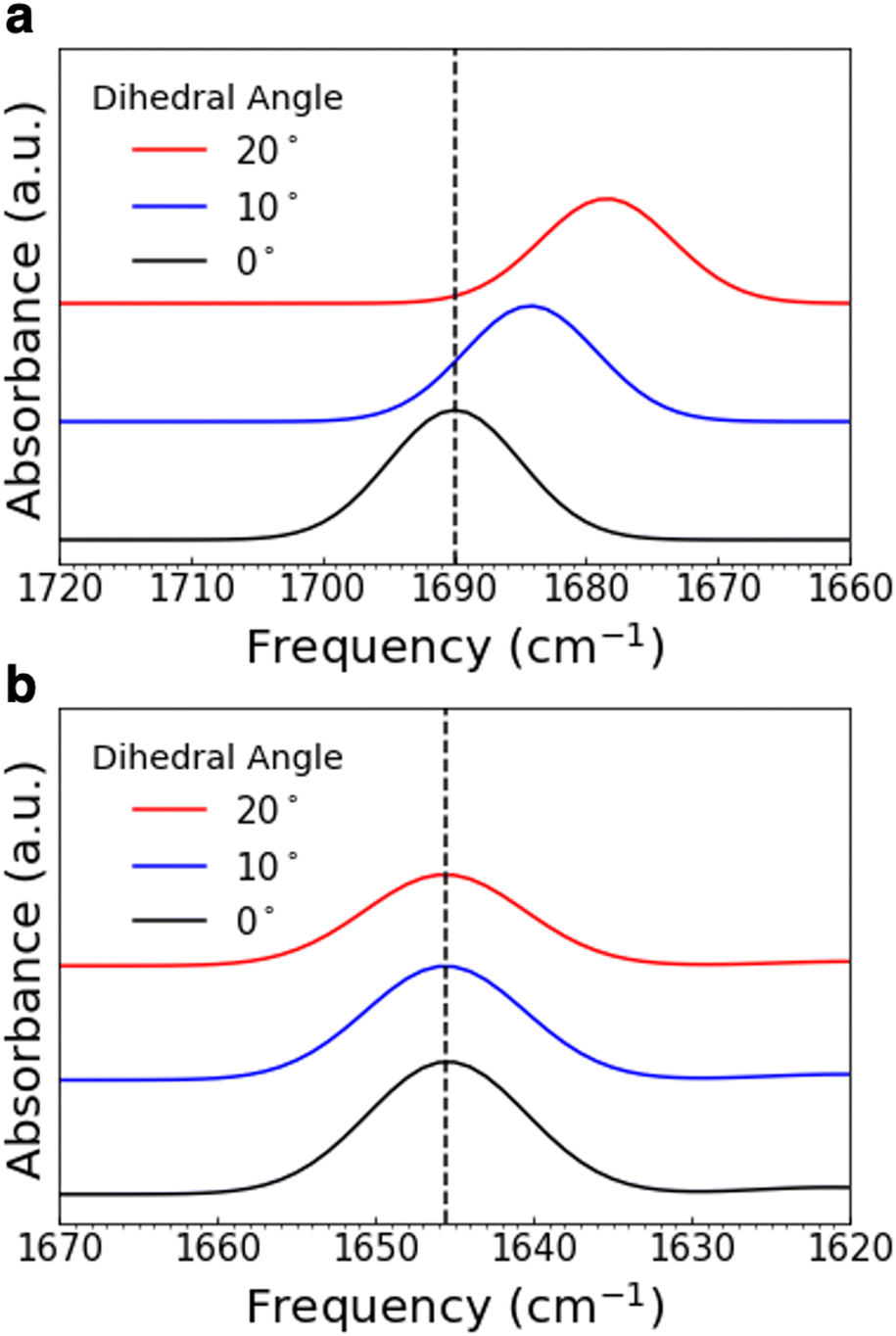 | ||
| Fig. 7 Simulated IR peaks for varying dihedral angles in N-benzylideneaniline (a) and N-benzylaniline (b). Shown are just a portion of the spectrum; the CN stretch in (a) and the C–N–H bend in (b). | ||
In comparison, a fragment of COF-300-amine containing the pedal motif (N-benzylaniline) shows only a slight red shift in the calculated C–N–H bend but substantial shifting among the other calculated peaks, as the dihedral angle increases from 0° to 20° (Fig. 7b and Fig. S15, Table S2, ESI†). This in silico result is in contrast with our experimental spectra, which show that the C–N–H bend shifts by the greatest amount, with other modes affected in only minor ways. These observations imply that the pedal motion is likely not driving any structural change that may occur in COF-300-amine as a function of temperature, consistent with the high-temperature results reported by Chi et al. for this framework.21
The relative lack of pedal motion in COF-300-amine may appear counterintuitive, since the sp3-hybridized atoms in its amine linkage are expected to allow for greater rotation and bond flexibility than the sp2-hybridized atoms in the COF-300 imine linkage. We hypothesize that this bond flexibility allows COF-300-amine to adopt a contracted structure under vacuum; such a contracted structure is stabilized by intra-framework interactions and resistant to temperature-induced structural change. In addition to our IR spectroscopy results, our gas adsorption results also support this hypothesis. The N2 sorption isotherm taken at 77 K shows a drastic decrease in adsorption capacity for COF-300-amine compared to COF-300 (Fig. S7, ESI†); the Brunauer–Emmett–Teller (BET) surface area measured for COF-300 reduced from 1383 m2 g−1 to 13 m2 g−1 in COF-300-amine. Literature reports also support the low N2 uptake of COF-300-amine37,39 and of other frameworks with freely-rotating linkages.42,43 This large loss in surface area is not accompanied by framework degradation, as shown by PXRD and SEM (Fig. S5 and S9, ESI†). Thus, a structural contraction under vacuum that prevents N2 from entering the pores of COF-300-amine may explain the framework's low BET surface area as well as its resistance to temperature-induced pedal motion.
Since the two-dimensional framework COF-V contains the same imine-linked pedal motif as COF-300, it would be reasonable to assume that structural changes driven by pedal motion should occur as the COF-V sample is cooled to cryogenic temperatures. However, our experimental results show little change in the observed spectra as a function of temperature. The defining aspect of two-dimensional covalent organic frameworks is their flat, sheet-like nature stacked together by π–π interactions. The tight packing of these sheets greatly inhibits their structural flexibility along the z-dimension (perpendicular to the planes of the sheets).19 Consequently, changes in dihedral angle associated with pedal motion are hindered by the close inter-layer stacking of two-dimensional frameworks, which likely explains the minor extent of peak shifting we observed experimentally for COF-V.
Overall, our results show that the cooling-induced conformational changes are enhanced for COF-300, reduced for COF-300-amine, and minimized for COF-V. In comparing the design elements of the three frameworks, we note several distinctions: three-dimensional vs. two-dimensional connectivity, high vs. low BET surface area, and imine linkages with rigid sp2 character vs. amine linkages with flexible sp3 character. The interplay between these structural elements determines whether or not a framework experiences major conformational transformations in response to cryogenic cooling. These results can inform the selection of a given framework for cryogenic applications and can guide synthetic chemists in the design of new frameworks that show temperature-dependent behavior.
4. Conclusions
A series of covalent organic frameworks were subjected to cryogenic temperatures as a form of external stimulus. We studied the conformational transformations in these materials by observing the infrared spectra in situ during the cooling process. We observed a greater overall blue shift in the IR spectrum of the three-dimensional, imine-linked framework COF-300, compared to amine-linked COF-300-amine and two-dimensional COF-V. We attribute these observations to the combination of high BET surface area with significant pedal motion about the CN linkages in COF-300. This combination of structural features allows for greater conformational change as COF-300 cools, compared to the frameworks with low surface area or inhibited pedal motion. We conducted quantum-chemistry calculations on molecular fragments of the covalent organic frameworks to probe how IR peak positions change in response to pedal motion in these fragments. This work provides insight into the structural features of covalent organic frameworks that give rise to temperature-dependent IR shifts. Relationships between framework structure and response to cooling can guide future efforts to apply covalent organic frameworks to cryogenic applications, which are particularly relevant for gas storage and separations.
Data availability
The data supporting this article have been included as part of the ESI.† All electronic structure and frequency analysis output files can be accessed via FigShare, DOI: 10.6084/m9.figshare.26510275.v1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Maryland, College Park for funding. We also thank the National Science Foundation (NSF-1726058) for funding a solid-state NMR spectrometer. We acknowledge the support of the Maryland NanoCenter and its AIMLab and the University of Maryland supercomputing resources (https://hpcc.umd.edu) made available for conducting the research reported in this paper. M. K. T. acknowledges funding from the Gordon and Betty Moore Foundation through award #12070. L. G. D. acknowledges that this material is based upon work supported by the U. S. Department of Energy, Office of Science Early Career Research Program, Office of Basic Energy Sciences under Award Number DE-SC0024262.References
- A. P. Cote, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- N. Sinha and Srimanta Pakhira, ACS Appl. Electron. Mater., 2021, 3, 720–732 CrossRef CAS.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- Z. Zhao, Y. Zheng, C. Wang, S. Zhang, J. Song, Y. Li, S. Ma, P. Cheng, Z. Zhang and Y. Chen, ACS Catal., 2021, 11, 2098–2107 CrossRef CAS.
- Y. Yusran, H. Li, X. Guan, D. Li, L. Tang, M. Xue, Z. Zhuang, Y. Yan, V. Valtchev, S. Qiu and Q. Fang, Adv. Mater., 2020, 32, 1907289 CrossRef CAS PubMed.
- Y. Sun, G. I. N. Waterhouse, L. Xu, X. Qiao and Z. Xu, Sens. Actuators, B, 2020, 321, 128501 CrossRef CAS.
- E. Martinez-Perinan, M. Martínez-Fernández, J. L. Segura and E. Lorenzo, Sensors, 2022, 22, 4758 CrossRef CAS PubMed.
- J. M. Vicent-Luna, A. Luna-Triguero and S. Calero, J. Phys. Chem. C, 2016, 120, 23756–23762 CrossRef CAS.
- N. A. Babujohn, A. Eluri and V. P. Nabeela, Chem. Eng. J., 2023, 464, 142459 CrossRef CAS.
- A. Modak, P. Bhanja, M. Selvaraj and A. Bhaumik, Environ. Sci. Nano, 2020, 7, 2887–2923 RSC.
- S. Kandambeth, B. P. Biswal, H. D. Chaudhari, K. C. Rout, S. H. Kunjattu, S. Mitra, S. Karak, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, Adv. Mater., 2016, 29, 1603945 CrossRef PubMed.
- Y. Song, J. Phipps, C. Zhu and S. Ma, Angew. Chem., Int. Ed., 2023, 135, e202216724 CrossRef.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- Y. Yang, A. P. Sandra, A. Idström, C. Schäfer, M. Andersson, L. Evenäs and K. Börjesson, J. Am. Chem. Soc., 2022, 144, 16093–16100 CrossRef CAS PubMed.
- Y. Wang, M. S. Shim, N. S. Levinson, H.-W. Sung and Y. Xia, Adv. Funct. Mater., 2014, 24, 4206–4220 CrossRef CAS PubMed.
- Z.-B. Zhou, H.-H. Sun, Q.-Y. Qi and X. Zhao, Angew. Chem., Int. Ed., 2023, 135, e202305131 CrossRef.
- J. Harada and K. Ogawa, Chem. Soc. Rev., 2009, 38, 2244 RSC.
- H. Chi, Y. Liu, Z. Li, W. Chen and Y. He, Nat. Commun., 2023, 14, 5061 CrossRef CAS PubMed.
- C. Ji, C. Kang, B. Chandra Patra and D. Zhao, CCS Chem., 2023, 1–26 Search PubMed.
- J. Fang, Z. Fu, X. Chen, Y. Liu, F. Chen, Y. Wang, H. Li, Y. Yusran, K. Wang, V. Valtchev, S. Qiu, B. Zou and Q. Fang, Angew. Chem. DOI:10.1002/anie.202304234.
- Y. Chen, Z.-L. Shi, L. Wei, B. Zhou, J. Tan, H.-L. Zhou and Y.-B. Zhang, J. Am. Chem. Soc., 2019, 141, 3298–3303 CrossRef CAS PubMed.
- R. Yan, J. R. Simpson, S. Bertolazzi, J. Brivio, M. Watson, X. Wu, A. Kis, T. Luo, A. R. Hight Walker and H. G. Xing, ACS Nano, 2014, 8, 986–993 CrossRef CAS PubMed.
- H. M. Hill, A. F. Rigosi, S. Krylyuk, J. Tian, N. V. Nguyen, A. V. Davydov, D. B. Newell and A. R. Hight, Phys. Rev. B, 2018, 98, 165109 CrossRef CAS PubMed.
- A. McCreary, J. R. Simpson, T. T. Mai, R. D. McMichael, J. E. Douglas, N. Butch, C. Dennis, R. Valdés Aguilar and A. R. Hight, Phys. Rev. B, 2020, 101, 064416 CrossRef CAS PubMed.
- H. Iturriaga, L. M. Martinez, T. T. Mai, A. J. Biacchi, M. Augustin, A. R. Hight Walker, M. Fathi Sanad, S. T. Sreenivasan, Y. Liu, E. J. G. Santos, C. Petrovic and S. R. Singamaneni, npj 2D Mater. Appl., 2023, 7, 56 CrossRef.
- H. Oh, S. Babu Kalidindi, Y. Um, S. Bureekaew, R. Schmid, R. A. Fischer and M. Hirscher, Angew. Chem., Int. Ed., 2013, 52, 13219–13222 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Chem, 2018, 4, 1696–1709 CAS.
- A. N. Zeppuhar, D. S. Rollins, D. L. Huber, E. A. Bazan-Bergamino, F. Chen, H. A. Evans and M. K. Taylor, ACS Appl. Mater. Interfaces, 2023, 15, 52622–52630 CAS.
- W. Ma, Q. Zheng, Y. He, G. Li, W. Guo, Z. Lin and L. Zhang, J. Am. Chem. Soc., 2019, 141, 18271–18277 CrossRef CAS PubMed.
- I. Gaussian, Gaussian 16 IOps Manual, 2016 Search PubMed.
- P. Carbonniere, T. Lucca, C. Pouchan, N. Rega and V. Barone, J. Comput. Chem., 2005, 26, 384–388 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. Sinha, S. E. Boesch, C. Gu, R. A. Wheeler and A. K. Wilson, J. Phys. Chem. A, 2004, 108, 9213–9217 CrossRef CAS.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS PubMed.
- P. Jin, X. Niu, F. Zhang, K. Dong, H. Dai, H. Zhang, W. Wang, H. Chen and X. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 20414–20422 CrossRef CAS PubMed.
- Z. Wang, Z. Si, D. Cai, L. Shufeng and P. Qin, J. Membr. Sci., 2020, 615, 118466 CrossRef CAS.
- M. Kong, P. Jin, W. Wei, W. Wang, H. Qin, H. Chen and J. He, Microchem. J., 2021, 160, 105650 CrossRef CAS.
- Q. Jiang, Y. Li, X. Zhao, P. Xiong, X. Yu, Y. Xu and L. Chen, J. Mater. Chem. A, 2018, 6, 17977–17981 RSC.
- J. Harada, M. Harakawa and K. Ogawa, Acta Crystallogr. Sect. B, Struct. Sci., 2004, 60, 589–597 CrossRef PubMed.
- X. Liu, J. Li, B. Gui, G. Lin, Q. Fu, S. Yin, X. Liu, J. Sun and C. Wang, J. Am. Chem. Soc., 2021, 143, 2123–2129 CrossRef CAS PubMed.
- X. Liu, Z. Wang, Y. Zhang, N. Yang, B. Gui, J. Sun and C. Wang, J. Am. Chem. Soc., 2024, 146, 11411–11417 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02338b |
| This journal is © the Owner Societies 2024 |