
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic engineering of a new biocatalyst encapsulating [NiFe]-hydrogenases for enhanced hydrogen production†
Qiuyao
Jiang
a,
Tianpei
Li
ab,
Jing
Yang
ac,
Catherine M.
Aitchison
 c,
Jiafeng
Huang
a,
Yu
Chen
a,
Fang
Huang
a,
Qiang
Wang
b,
Andrew I.
Cooper
c and
Lu-Ning
Liu
*ad
c,
Jiafeng
Huang
a,
Yu
Chen
a,
Fang
Huang
a,
Qiang
Wang
b,
Andrew I.
Cooper
c and
Lu-Ning
Liu
*ad
aInstitute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool L69 7ZB, UK. E-mail: luning.liu@liverpool.ac.uk
bState Key Laboratory of Crop Stress Adaptation and Improvement, School of Life Sciences, Henan University, Kaifeng 475004, China
cMaterials Innovation Factory and Department of Chemistry, University of Liverpool, Liverpool L7 3NY, UK
dCollege of Marine Life Sciences, and Frontiers Science Center for Deep Ocean Multispheres and Earth System, Ocean University of China, Qingdao 266003, China
First published on 20th February 2023
Abstract
Hydrogenases are microbial metalloenzymes capable of catalyzing the reversible interconversion between molecular hydrogen and protons with high efficiency, and have great potential in the development of new electrocatalysts for renewable fuel production. Here, we engineered the intact proteinaceous shell of the carboxysome, a self-assembling protein organelle for CO2 fixation in cyanobacteria and proteobacteria, and sequestered heterologously produced [NiFe]-hydrogenases into the carboxysome shell. The protein-based hybrid catalyst produced in E. coli shows substantially improved hydrogen production under both aerobic and anaerobic conditions and enhanced material and functional robustness, compared to unencapsulated [NiFe]-hydrogenases. The catalytically functional nanoreactor as well as the self-assembling and encapsulation strategies provide a framework for engineering new bioinspired electrocatalysts to improve the sustainable production of fuels and chemicals in biotechnological and chemical applications.
Introduction
Rapid human development and increasing energy demand have had a massive impact on the climate, and the search for new sustainable energy supplies to mitigate climate change has become an important and urgent global challenge. Compared to traditional fossil fuels, hydrogen has advantages as a green energy storage medium when generated renewably from water, owing to its higher energy density (∼120 kJ g−1, 3 times more than gasoline or diesel) and clean combustion for a low-carbon future.1,2 As such, developing novel catalysts for hydrogen production from water has become a target in chemistry and biology for researchers and engineers to underpin hydrogen economy and address the social challenges.Many microorganisms have evolved special metalloenzymes, namely hydrogenases, to produce molecular hydrogen from protons and electrons using inexpensive and abundant metals.3 Three major phylogenetical classes of hydrogenases have been identified based on their active site metals: [Fe]-hydrogenases, [FeFe]-hydrogenases and [NiFe]-hydrogenases.4 Given their superb electrocatalytic activities, hydrogenases provide great promise for constructing new hydrogen-evolution catalysts. However, these enzymes have intrinsic limitations, such as high sensitivity to oxygen and pH and poor thermostability.5 To address these issues and enhance hydrogen production, many strategies have been implemented, including integrating hydrogenases into immobilization matrixes6,7 or scaffolding materials.8–11
Intriguingly, the encapsulation principle has been exploited by a variety of microbes in nature. A paradigm of the encapsulating systems is a self-assembling proteinaceous organelle, known as the carboxysome, an anabolic bacterial microcompartment (BMC) found in all cyanobacteria and many proteobacteria.12–18 Carboxysomes encapsulate the key CO2-fixing enzyme, ribulose-1,5-bisphosphate carboxylase oxygenase (Rubisco), using a polyhedral protein shell; unlike virus capsids that do not possess permeability, the carboxysome shell is selectively permeable to HCO3− and protons while diminishing O2 entry and CO2 leakage.19–23 This elaborate bacterial organelle provides elevated levels of CO2 around Rubisco to enhance carbon fixation on the global scale and represents an attractive engineering objective in synthetic biology.24–28 Recent studies have shown that direct encapsulation of O2-sensitive [FeFe]-hydrogenases into the cavity of a synthetic α-carboxysome shell resulted in improved hydrogen production and O2 tolerance of the hybrid biocatalyst, taking advantage of the unique α-carboxysome shell permeability and confinement of cargo enzymes.29
In contrast, [NiFe]-hydrogenases are relatively O2 tolerant and can catalyze H2 oxidation in the presence of O2, and have high H2-evolution activities under anaerobic conditions.30–32 While [NiFe]-hydrogenases are promising candidates as functional catalysts in biotechnological applications,33 optimization of [NiFe]-hydrogenases and their surrounding microenvironment is still needed for improved catalytic performance and robustness.
Here, we generate a new hybrid catalyst by sequestering [NiFe]-hydrogenase-1 from Escherichia coli (EcHyd-1) within the recombinant shell of the α-carboxysome from a chemoautotroph Halothiobacillus neapolitanus. This catalyst exhibits a substantial increase in hydrogen evolution under aerobic and anaerobic conditions compared to unencapsulated EcHyd-1 hydrogenases. We also demonstrate that shell encapsulation results in improved oxygen tolerance and thermostability of functional [NiFe]-hydrogenase cargos in hydrogen production. Our study represents a step towards repurposing self-assembling bio-systems to develop sustainable biocatalysis in biotechnological applications.
Experimental
Construction of recombinant [NiFe]-hydrogenase-1
Genes encoding HyaA and HyaB were amplified from the E. coli BL21(DE3) genome via using CloneAmp HiFi polymerase (Takara, Japan) (Table S1, ESI†). The C-terminus of full-length CsoS2 served as an encapsulation peptide (EP) and was amplified from the pHnCBS1D plasmid (Addgene, US). The EP sequence was fused to the 3′-end of hyaB followed by ligation to the pCDFDuet-1 vector linearized by EcoRI and HindIII to produce the pCDF-hyaB-EP vector. The hyaA gene with the C-terminus fused with the CsoS2 C-terminus was ligated to the pCDF-hyaB-EP vector linearized by NdeI and XhoI to generate pCDF-hyaAB-EP. The resulting PCR products and linearized vectors were purified by using a DNA gel extraction kit (New England BioLabs, UK) following the standard manufacture protocol. All vectors were verified by PCR and DNA sequencing (IDT, US) and transformed into E. coli BL21(DE3) competent cells.Expression of recombinant [NiFe]-hydrogenase-1 and α-carboxysome shells
For the expression of [NiFe]-hydrogenases, E. coli strains containing HyaAB-EP plasmids were grown in LB medium containing 50 μg mL−1 spectinomycin, 0.03 mM (final) ferric ammonium citrate and 0.03 mM (final) nickel chloride monohydrate, and expression was induced by adding 0.05 mM isopropyl β-D-thiogalactopyranoside (IPTG) at OD600 = 0.6. For the co-expression of α-carboxysome shells and mature [NiFe]-hydrogenases, the empty shell expression plasmid was generated as reported,29 including the csoS2, csoS4AB, csoS1ABC and csoS1D genes. The HyaAB-EP plasmid was transformed into E. coli BL21(DE3) competent cells containing Shell. Cells were grown at 37 °C in LB medium supplemented with 50 μg mL−1 spectinomycin, 100 μg mL−1 ampicillin, 0.03 mM (final) ferric ammonium citrate and 0.03 mM (final) nickel chloride monohydrate. Expression of HyaAB-EP was induced at OD600 = 0.6 by adding 0.05 mM IPTG at 25 °C. L-arabinose was added at a final concentration of 1 mM 4 hours after IPTG induction to initiate shell expression.Purification of recombinant HyaAB-Shell assemblies
Cells were harvested by centrifugation at 5000 × g for 10 minutes at 4 °C. The pellets were washed with TMB buffer (10 mM Tris-HCl pH 8.0, 10 mM MgCl2, 20 mM NaHCO3) twice and resuspended in 20 mL of TMB buffer. The resuspended cells with 10% (v/v) CelLytic B cell Lysis reagent (Sigma-Aldrich) were lysed by French press (STANSTED FPG12800 pressure cell homogenizer) following the standard recommend process. Cell debris was removed by initial centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 10 minutes at 4 °C. After centrifugation at 50000 × g for 30 minutes at 4 °C, the supernatant was discarded and the pellet was gently resuspended in 2 mL TMB buffer using a soft brush. A 2 minute spin at 4 °C was performed to remove the insoluble fraction. The soluble pellet fraction was applied onto a step sucrose gradient (10–50%, w/v, solubilized in TMB buffer) for ultracentrifugation at 105000 × g for 35 minutes. Sucrose fractions were separately collected and stored at 4 °C for further analysis.
000 × g for 10 minutes at 4 °C. After centrifugation at 50000 × g for 30 minutes at 4 °C, the supernatant was discarded and the pellet was gently resuspended in 2 mL TMB buffer using a soft brush. A 2 minute spin at 4 °C was performed to remove the insoluble fraction. The soluble pellet fraction was applied onto a step sucrose gradient (10–50%, w/v, solubilized in TMB buffer) for ultracentrifugation at 105000 × g for 35 minutes. Sucrose fractions were separately collected and stored at 4 °C for further analysis.
SDS-PAGE and immunoblot analysis
Protein samples were prepared and then mixed with 4× sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer (250 mM Tris pH 6.8, 8% SDS, 0.2% bromophenol blue, 40% glycerol, 20% mercaptoethanol). After 95 °C heating for 10 minutes, samples were centrifuged at 12000 × g for 2 minutes and then loaded on 15% SDS-PAGE gels to analyze their composition. About 75 μg proteins were loaded in each SDS-PAGE gel well. 3 μL of unstained protein ladder (10–250 kDa from NEB) was loaded as a marker. SDS-PAGE was run at 200 V for 45 minutes with SDS running buffer (25 mM Tris, 192 mM glycine and 1% SDS). The gels were stained in Coomassie blue stain buffer (0.25% Coomassie Brilliant Blue R-250, 20% methanol, 10% acetic acid) and destained by destaining buffer (20% methanol, 10% acetic acid).
Proteins (30 μg) were loaded onto 15% SDS-PAGE gels and then transferred to PVDF (polyvinylidene difluoride) membranes (Bio-Rad). Immunoblot analysis was performed using primary mouse monoclonal anti-Histag (Life Technologies, 69-74-9, UK), primary rabbit polyclonal anti-CsoS1A/B/C (Agrisera, AS142760, dilution 1:5000, US), as well as horseradish peroxidase-conjugated goat anti-mouse IgG secondary antibody (Agrisera, AS111772 dilution 1:10000, US) and anti-rabbit IgG secondary antibody (Agrisera, AS09602, dilution 1:10000, US). Then, the membranes were washed with TBS buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl) and TBST buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% Tween-20). Immunoblot signals were developed by using a Bio-Rad chemiluminescence kit and images were recorded via ImageQuant LAS 4000 software version 1.2.1.119.
Free HyaAB-EP were isolated through a HisTrap HP column (Cytiva, UK), and were loaded on a 15%(v/v) SDS-PAGE gel. HyaB-EP proteins were extracted from SDS-PAGE gels for protein quantification as the reference for the quantification of HyaAB-EP content in the unencapsulated and encapsulated forms, according to previous studies.29 The purified HyaB-EP at various protein concentrations were loaded onto SDS-PAGE gels for immunoblot analysis, which allowed us to generate a standard curve according to the linear relationship between the HyaB-EP protein band intensities and the amount of loaded HyaB-EP proteins. Protein quantification analysis was performed by using ImageJ software (version 1.52 h). For each experiment, at least three biological replicates were examined.
Transmission electron microscopy (TEM)
Isolated protein samples (1–2 mg mL−1 total protein) were stained on carbon grids (Carbon Films on 300-mesh Grids Copper, Agar Scientific) for 40 seconds, followed by staining with 2% (v/v) uranyl acetate (Sigma-Aldrich). The carbon grids were then washed with distilled water and then dried using 0.2 μm filter paper. Images were recorded using an FEI Tecnai G2 Spirit Bio TWIN transmission electron microscope equipped with a Gatan Rio 16 camera. ImageJ was used for the image analysis. Statistical analysis was calculated by using Student's t-test.Dynamic light scattering (DLS) analysis
Briefly, 1 mL (5–10 mg mL−1 total protein) of samples were analyzed by dynamic light scattering (Malvern DLS ZetaSizer) to measure the size distribution and average size of the particles. For each experiment, at least three biological replicates were examined.In vivo dissolved oxygen (DO) measurement
A polarographic DO probe (New BrunswickTM BioFlo/CellGen 115 Fermentor, Eppendorf) was used to measure dissolved oxygen (DO) in strains expressing free HyaAB-EP, HyaAB-Shell for in vivo activity assays. Oxygen-saturated LB medium (100% DO) and sodium dithionite (DT)-treated LB medium (0% DO) were used to calibrate the polarized polarographic DO probe, respectively. The DO levels of strains expressing free HyaAB-EP and HyaAB-Shell were determined before adding IPTG and every four hours after adding IPTG and L-arabinose. For each experiment, at least three biological replicates were examined.In vivo H2-evolution assay
Strains expressing free HyaAB, free HyaAB-EP and HyaAB-Shell were aerobically grown at 37 °C in a 200 mL flask with 50 mL LB medium supplemented with 0.03 mM (final) ferric ammonium citrate and 0.03 mM (final) nickel chloride monohydrate, 50 μg mL−1 spectinomycin, and/or 100 μg mL−1 ampicillin. At OD600 = 0.6, 30 mL culture was transferred to a 50 mL falcon tube with sealed rubber closure (Sigma-Aldrich) and was degassed by 100% nitrogen for 5 mins before the addition of IPTG, L-cysteine, and sodium fumarate for anaerobic treatment. For aerobic treatment, 30 mL culture was transferred to a falcon tube with a sealed rubber closure without a N2 degassing process. 1 mM L-arabinose was added 4 hours after the addition of 0.05 mM IPTG for strains containing HyaAB-Shell. Cells were grown at 25 °C for 16 hours with constant shaking following induction. 1 mL gas samples were taken with a gas-tight syringe and a sample loop was flushed (100 μL) with the sample. The sample loop was then switched and run on a Bruker 450-GC gas chromatograph. The system was equipped with a molecular sieve 13 × 60–80 mesh 1.5 m × 1/8 in. × 2 mm ss column at 50 °C with an argon flow of 40.0 mL min−1. Hydrogen was detected with a thermal conductivity detector referencing against standard gas with a known concentration of hydrogen. For each experiment, at least three biological replicates were examined.In vitro H2-evolution assay
Cells were grown aerobically at 37 °C in a 2 L flask containing 800 mL LB medium with the corresponding antibiotics, 0.03 mM (final) ferric ammonium citrate and 0.03 mM (final) nickel chloride monohydrate, 50 μg mL−1 spectinomycin, and/or 100 μg mL−1 ampicillin. At OD600 = 0.6, the culture was degassed by 100% N2 for 30 min, and then HyaAB-EP expression was induced by 0.05 mM IPTG. For strains containing HyaAB-Shell, 0.05 mM IPTG was added for HyaAB-EP expression. After a 4 hour induction, 1 mM (final) L-arabinose was added for shell expression and the cells were then grown at 25 °C with constant shaking.The whole purification process was performed under anaerobic conditions, followed by in vitro hydrogenase H2-evolution activity assays. For in vitro hydrogenase kinetics assays, the protein amount of HyaAB in the samples containing free HyaAB or HyaAB-Shell were quantified by immunoblot using purified HyaB as the reference (Fig. S6, ESI†). Then, samples (0.5 mL, ∼10 mg mL−1) containing equal amounts of HyaB in TMB buffer were mixed with 100% nitrogen-degassed methyl viologen (MV+) (0–200 mM, final) and DT (500 mM, final) in sealed serum vials (Agilent Technologies) inside anaerobic glove bags. The vials were incubated at 37 °C for 16 hours with constant shaking and were then assayed using a Bruker 450-GC gas chromatograph for hydrogen production. Hydrogenase activity at a range of MV concentrations was plotted and fitted using a standard Michaelis–Menten model. For time series measurement, hydrogen evolution of HyaAB-Shell with 50 mM MV and 500 mM DT was measured every 20 minutes in comparison with free HyaAB-EP as a control. For each experiment, at least three biological replicates were examined.
Oxygen exposure and heat treatment
0.5 mL free HyaAB-EP and HyaAB-Shell samples (∼10 mg mL−1) isolated under anaerobic conditions were exposed to the air at 4 °C for 24 hours, respectively. Then, the samples were transferred into 10 mL sealed serum vials and were degassed by pure nitrogen for 5 minutes. The samples were incubated with 2 mL nitrogen-degassed MV (50 mM in TMB buffer, final) and 0.5 mL DT (500 mM in TMB buffer, final) at 37 °C for 20 minutes and were then subjected to hydrogen evolution assays.For heat treatment, the protein samples (∼10 mg mL−1) were heat treated at 65 °C for 20 minutes under anaerobic conditions, followed by incubation with 2 mL nitrogen-degassed MV (50 mM in TMB buffer, final) and 0.5 mL DT (500 mM in TMB buffer, final) in 10 mL sealed serum vials at 37 °C for 20 minutes. The samples were then subjected to hydrogen evolution assays. For each experiment, at least three biological replicates were examined.
Results and discussion
Generation of [NiFe]-hydrogenase-encapsulated α-carboxysome shells
EcHyd-1 is encoded by a gene operon of six genes (hyaABCDEF).34 HyaA is a small core subunit containing three [Fe–S] clusters and is involved in electron transfer, whereas HyaB is a large core subunit consisting of a bimetallic active site.35–39 HyaC is an integral membrane cytochrome b subunit that is responsible for anchoring EcHyd-1 to the cytoplasmic membrane.40 HyaD, HyaE and HyaF were assumed to be involved in hydrogenase biosynthesis and maturation,3,37,41,42 but their exact roles are still poorly understood. Previous studies have shown that HyaAB-only assemblies exhibit H2-producing activities and there was no significant difference in H2 production between E. coli cells expressing hyaABCDEF and hyaAB only.43,44To create an α-carboxysome shell encapsulating EcHyd-1, we first generated a hyaAB-expressing vector (hyaAB-EP), which can express HyaA and HyaB with both of their C-termini fused with the C-terminus of CsoS2 (Fig. 1A and Fig. S1, ESI†). In our previous studies, we demonstrated that the C-terminus of CsoS2 could serve as an encapsulation peptide (EP) to bind with shell proteins and efficiently recruit non-native cargos, for example fluorescent proteins, algal [FeFe]-hydrogenases, and ferredoxin-NADP+ reductase, into the α-carboxysome shell; without the fusion with the CsoS2 C-terminus, foreign cargo proteins could not be incorporated within the shells.19,29 The hyaAB-EP vector was then transformed into the E. coli BL21(DE3) cells comprising an α-carboxysome shell-expressing vector, which can express α-carboxysome shell proteins (CsoS4A, CsoS4B, CsoS1A, CsoS1B, CsoS1C, CsoS1D, Fig. 1A) resulting in the formation of empty α-carboxysome shells.19,29 Expression of the hyaAB-EP plasmid was induced by adding IPTG for 4 hours before arabinose-induced shell expression, to ensure heterologous production of functional [NiFe]-hydrogenases prior to shell encapsulation (Fig. 1B).
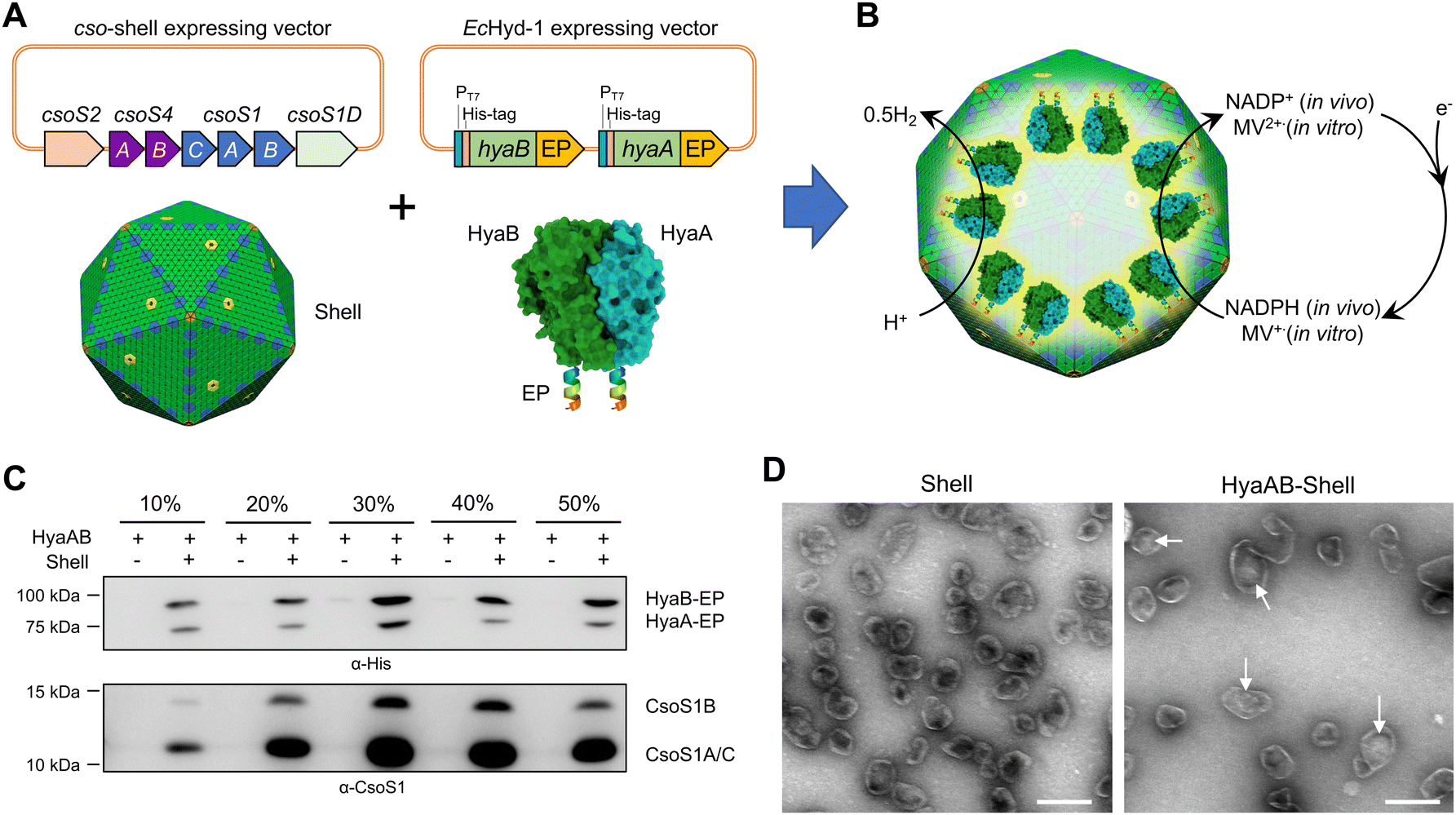 | ||
| Fig. 1 Generation of [NiFe]-hydrogenase-encapsulating catalyst based on the α-carboxysome shell. (A) Genetic organization of the vectors producing synthetic α-carboxysome shells and [NiFe]-hydrogenases (HyaAB). The C-termini of HyaA and HyaB were fused with the CsoS2 C-terminus as the encapsulation peptide (EP) to ensure cargo encapsulation. PDB ID for HyaAB: 3USC. (B) Schematic of the HyaAB-Shell hybrid catalyst comprising the α-carboxysome shell and HyaAB-EP. Hydrogen production by the nanoreactor systems was evaluated by using endogenous NADPH in cells as the electron donor for in vivo assays and methyl viologen (MV+), which was chemically reduced by sodium dithionite (DT), as the electron donor for in vitro tests. Note that the copy number of [NiFe]-hydrogenases depicted in the model does not represent the real content of [NiFe]-hydrogenases encapsulated within the hybrid catalyst, which remains speculative. (C) Immunoblot analysis confirmed the presence of HyaAB and shell proteins in the HyaAB-Shell sample purified from sucrose gradient ultracentrifugation. (D) Electron microscopy (EM) images of isolated empty shells and HyaAB-Shell from 20% sucrose fractions. The potential cargo enzymes in the lumen of the shells are indicated by arrows. Scale bar: 200 nm. | ||
To verify the encapsulation of HyaAB into α-carboxysome shells, we purified recombinant shells containing HyaAB-EP (HyaAB-shell) from E. coli using centrifugation and sucrose gradient ultracentrifugation. The components of HyaAB-Shell, including shell proteins and HyaAB-EP, were detected in the pellet after 50000 × g centrifugation, whereas unencapsulated HyaAB-EP was only present in the supernatant, indicating the formation of HyaAB-Shell assemblies (Fig. S2, ESI†). After sucrose gradient ultracentrifugation, individual sucrose fractions were collected. Immunoblot analysis revealed that HyaA-EP, HyaB-EP, and CsoS1 shell proteins were detectable in 10–50% sucrose fractions, predominantly enriched in the 20% and 30% sucrose fractions (Fig. 1C). In contrast, unencapsulated HyaAB-EP in the absence of shells were not detected in 10–50% sucrose fractions (Fig. 1C), confirming the EP-mediated encapsulation of HyaAB into the shells. Along with previous evidence revealing that CsoS2 C-terminus could drive the encapsulation of [FeFe]-hydrogenases with cofactors and fluorescence proteins,19,29 we provide an efficient encapsulation approach using the CsoS2 C-terminus as an EP to encase foreign cargos within the α-carboxysome shells in synthetic biology.
The HyaAB-shell assemblies at the 20% sucrose fraction were further examined using negative-staining electron microscopy (EM) (Fig. 1D). HyaAB-shells exhibit a polyhedral structure with a mean diameter of ∼105 nm as examined by dynamic light scattering (DLS) (Fig. S3A, ESI†), comparable to native α-carboxysomes from H. neapolitanus, synthetic α-carboxysomes and empty α-carboxysome shells.27–29 Moreover, the shell size gradually increased from 20% to 30% sucrose fractions (Fig. S3B, ESI†).
Shell encapsulation stimulates hydrogen evolution of [NiFe]-hydrogenase
The HyaB subunit of EcHyd-1 carries the [NiFe] active site, whereas HyaA contains [Fe–S] clusters that transfer electrons to the active site of HyaB.40,45,46 Recent studies revealed that HyaB only encased in the bacteriophage P22 capsid was unable to produce high-yield H2,10 confirming the importance of HyaA in hydrogen production. The presence of both HyaA and HyaB provides a promise for producing catalytically active EcHyd-1 within the α-carboxysome shells (Fig. 1C).The H2-evolution activities of the E. coli cells expressing HyaAB-Shell and free HyaAB-EP grown under aerobic or anaerobic conditions were assayed, using endogenous NADPH in E. coli as the electron source (Fig. 1B). First, we measured the levels of dissolved oxygen (DO) in the two different types of cell cultures under aerobic and anaerobic conditions (Fig. 2A). After 16 hour cell culturing in falcon tubes under aerobic conditions, DO dropped from 37.2% to 1.2% (n = 3) for the HyaAB-Shell and from 38.4% to 1.2% (n = 3) for free HyaAB-EP, demonstrating that the two constructs consumed a large amount of oxygen. The final DO levels of all cell types under aerobic conditions were higher than those under anaerobic conditions (remaining constant at ∼0.6%, Fig. 2A).
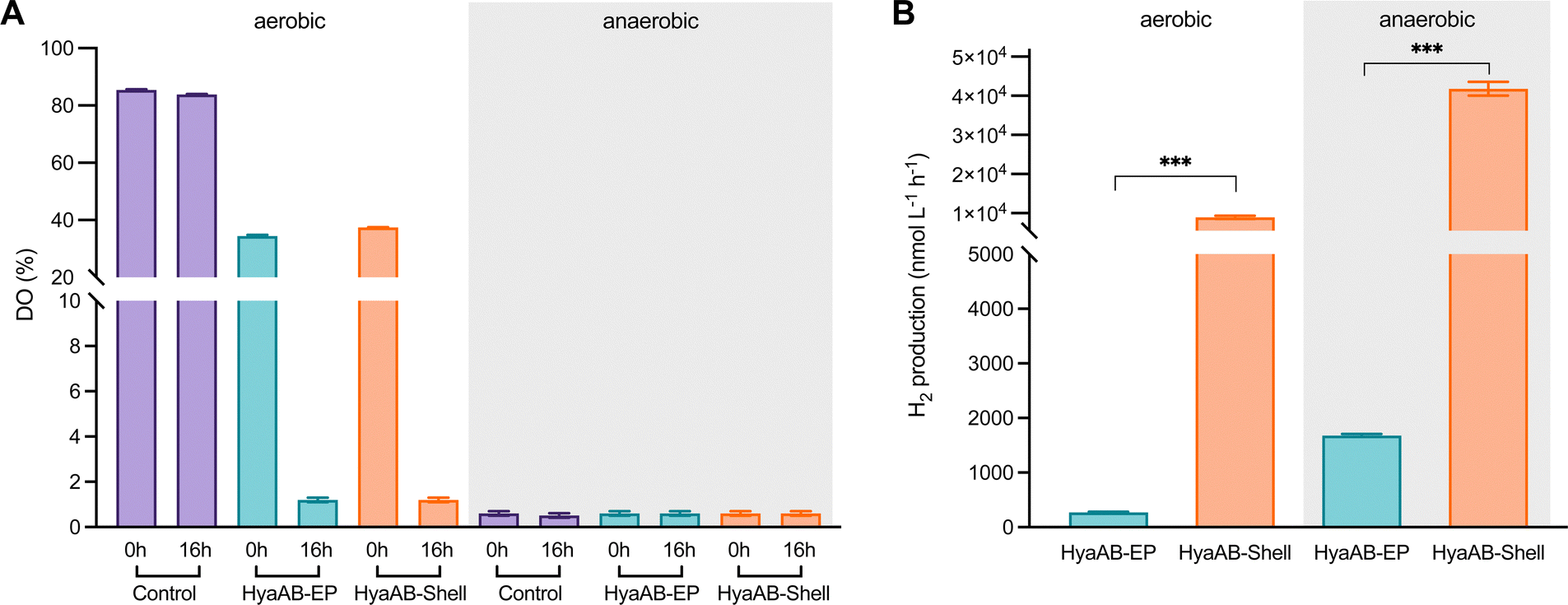 | ||
| Fig. 2 In vivo hydrogen production of E. coli cells expressing HyaAB-Shell and free HyaAB-EP. (A) The levels of dissolved oxygen (DO) in LB medium (control), LB medium containing the E. coli expressing HyaAB-EP or Shells-HyaAB-EP under aerobic and anaerobic conditions during 16-hour induction. Water-saturated DO was set as 100%. The data are presented as the average of nine (or three for control) DO measurements of nine (or three) distinct cell cultures. (B) In vivo hydrogenase activity assays. Hydrogen production of E. coli cells expressing free HyaAB-EP or HyaAB-Shell grown under aerobic (left) and anaerobic (right) conditions was measured using gas chromatography, normalized by inducing 30 mL cultures after 16 hours. ***, p < 0.001, two-tailed unpaired t-test. Error bars represent the standard deviation of the mean of three biological replicates. | ||
In vivo H2-evolution assays at pH 7 using gas chromatography revealed that the H2-production rate of E. coli cells expressing HyaAB-Shell under anaerobic conditions was 41777.5 ± 1753.4 nmol L−1 h−1 (n = 3), more than 23-fold greater than that of cells expressing unencapsulated HyaAB-EP (1,780.1 ± 197.9 nmol L−1 h−1, n = 3) (Fig. 2B and Fig. S4, ESI†). Intriguingly, under aerobic conditions, the H2-production rate of cells producing HyaAB-Shell (8,937.4 ± 414.5 nmol L−1 h−1, n = 3) was ∼33-fold higher than that of cells expressing free HyaAB-EP (269.2 ± 15.2 nmol L−1 h−1, n = 3). Since there was no drastic difference in oxygen consumption between the two cell lines (Fig. 2A), the possibility that the different hydrogenase activities of the two cell constructs were ascribed to their distinct oxygen-consumption capacities can be excluded. Moreover, the hydrogen-production activity of E. coli BL21(DE3) expressing only endogenous [NiFe]-hydrogenases accounts for <20% of that of E. coli BL21(DE3) expressing free HyaAB-EP (Fig. S5, ESI†), suggesting that endogenous [NiFe]-hydrogenases had no significant effect on the overall hydrogen-production performance of E. coli BL21(DE3) producing unencapsulated HyaAB-EP and HyaAB-Shell. Collectively, our results indicate that hydrogen production of EcHyd-1 is greatly enhanced by direct encapsulation of the α-carboxysome shell, consistent with the previous observation of the α-carboxysome shell-based nanoreactor that sequesters [FeFe]-hydrogenases.29 It is likely that the lower O2 levels and enriched cargo enzyme concentrations developed within the shell provided a catalytically favorable microenvironment for EcHyd-1 hydrogenases to catalyze the reversible reduction of protons into hydrogen. In addition, cells producing [NiFe]-hydrogenase-encapsulated shells have a much greater hydrogen-production capacity than cells synthesizing [FeFe]-hydrogenase-encapsulated shells29 under both aerobic and anaerobic conditions, highlighting the remarkable catalytic performance of the [NiFe]-hydrogenase-packaged nanoreactor.
We also performed in vitro H2-evolution assays of free HyaAB-EP and HyaAB-Shell isolated under anaerobic conditions using gas chromatography, based on quantification of the HyaB-EP content (Fig. S6, ESI†). Nitrogen-degassed methyl viologen (MV+) with varying concentrations was used as the electron donor reduced by DT in the assays (Fig. 1B). After 16 hour reaction at 37 °C under anaerobic conditions, the maximum H2-evolution rate of HyaAB-Shell at pH 8 was 543.4 ± 67.7 nmol H2 mg−1 min−1 (n = 3), ∼12-fold greater than that of free HyaAB-EP (47.3 ± 1.6 nmol H2 mg−1 min−1, n = 3) (Fig. 3A). Moreover, the amount of H2 produced by HyaAB-Shell increased linearly as a function of time at 50 mM MV, notably greater than that produced by free HyaAB-EP, signifying the electrocatalytic capabilities of HyaAB-Shell for H2 evolution (Fig. 3B). These results demonstrate explicitly the enhanced H2-production performance of [NiFe]-hydrogenases within the α-carboxysome shell-based catalyst, predominantly taking advantage of the reduced O2 levels and enriched cargo concentrations by shell encapsulation.
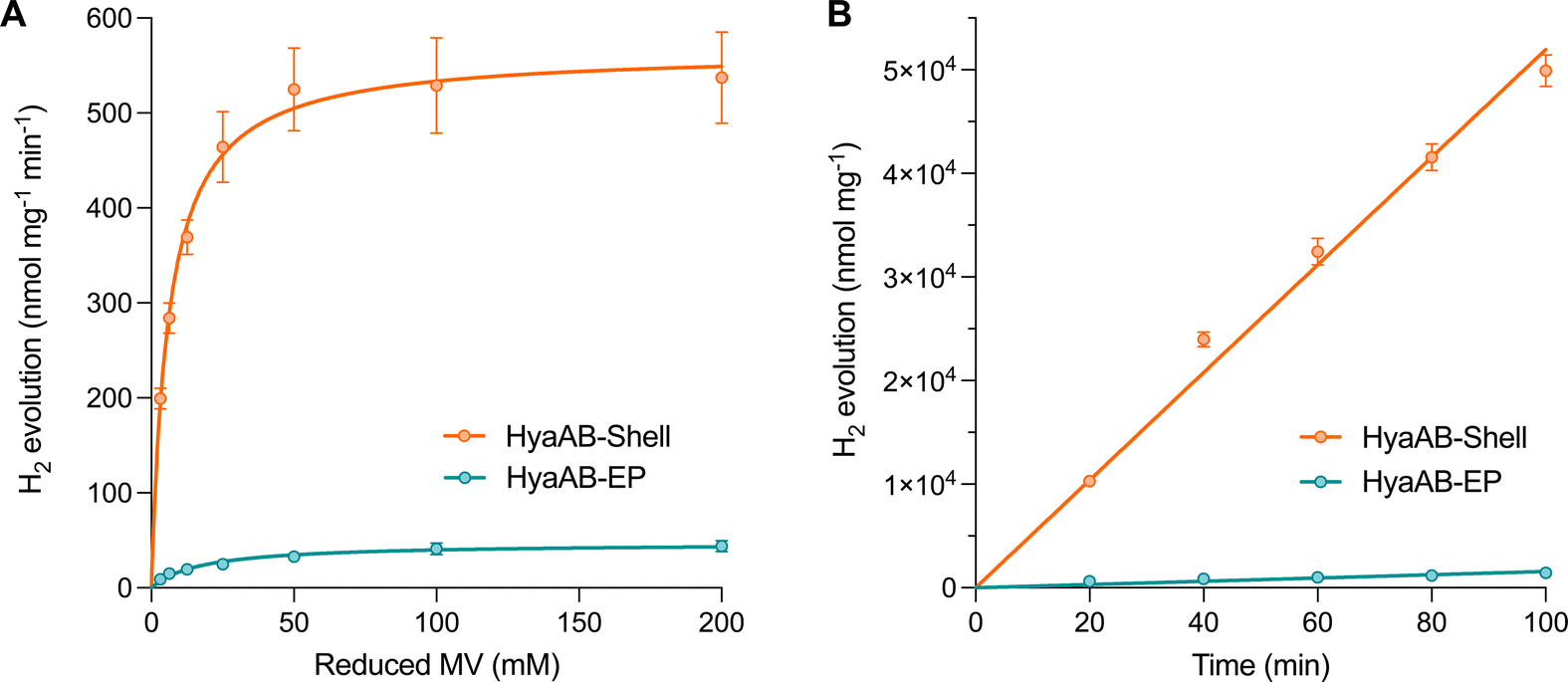 | ||
| Fig. 3 In vitro hydrogen production of the HyaAB-Shell catalyst. (A) Hydrogen production activities of anaerobically purified free HyaAB-EP and HyaAB-Shell at pH 8 as a function of different concentrations of nitrogen-degassed MV as the electron mediator reduced by DT, fitted with Michaelis–Menten kinetics. (B) Kinetic hydrogen production (measured at pH 8) of anaerobically isolated free HyaAB-EP and HyaAB-Shell using MV+ reduced by 500 mM DT as the electron mediator. Error bars show the standard deviation of the mean of three biological replicates. | ||
Shell encapsulation enhances oxygen tolerance and thermostability of [NiFe]-hydrogenase
Compared to the highly O2-sensitive [FeFe]-hydrogenase, [NiFe]-hydrogenase is an O2-tolerant catalyst that retains certain hydrogen turnover activities in the presence of O2.47–51 Our in vivo assays also revealed that the H2-producing rate of free [NiFe]-hydrogenases under anaerobic conditions was ∼6.6-fold greater than that under aerobic conditions (Fig. 2B). Since O2 is the only varying factor in this experiment, our results highlight the dominant effect of O2 inactivation on tuning the catalytic activity of free [NiFe]-hydrogenases and the necessity of protecting [NiFe]-hydrogenases from O2 damage.To examine the O2-tolerance of HyaAB-Shell, we exposed purified HyaAB-Shell and unpackaged HyaAB-EP to ambient air for 24 hours, followed by degassing and H2-evolution assays using 50 mM nitrogen-degassed MV+ reduced by DT as the electron donor (Fig. 4). After O2 exposure, free HyaAB-EP had only ∼13% (4.2 ± 0.3 nmol H2 mg−1 min−1, n = 3) of the H2-evolution activity measured before O2 exposure (32.9 ± 2.7 nmol H2 mg−1 min−1, n = 3). By comparison, HyaAB-Shell retained ∼88% (433.6 ± 8.3 nmol H2 mg−1 min−1, n = 3) of the H2-evolution activity before O2 exposure (491.4 ± 21.0 nmol H2 mg−1 min−1, n = 3) at pH 8 (Fig. 4A and B). These results indicate explicitly that the protein shell could significantly improve the O2 tolerance of encapsulated [NiFe]-hydrogenases. Interestingly, this improvement appears even greater than that of [FeFe]-hydrogenase-packaged shells.29 As [NiFe]-hydrogenase is relatively O2-tolerant compared to [FeFe]-hydrogenase, even low levels of O2 within the shell may substantially impede the H2-evolution activity of [FeFe]-hydrogenases than that of [NiFe]-hydrogenases in O2-tolerance assays. Hence, the higher O2 tolerance of [NiFe]-hydrogenase-shells than that of [FeFe]-hydrogenase-packaged shells might be due to the low O2 but not strictly O2-free microenvironment created within the carboxysome shell and the potentially different packaging efficiencies of the two hydrogenases within the shells. The detailed mechanisms underlying the catalytic improvement of encapsulated hydrogenases and their regulatory factors merit further investigation. Nevertheless, it makes this [NiFe]-hydrogenase-encapsulated catalyst a promising candidate for biohydrogen generation in applications.
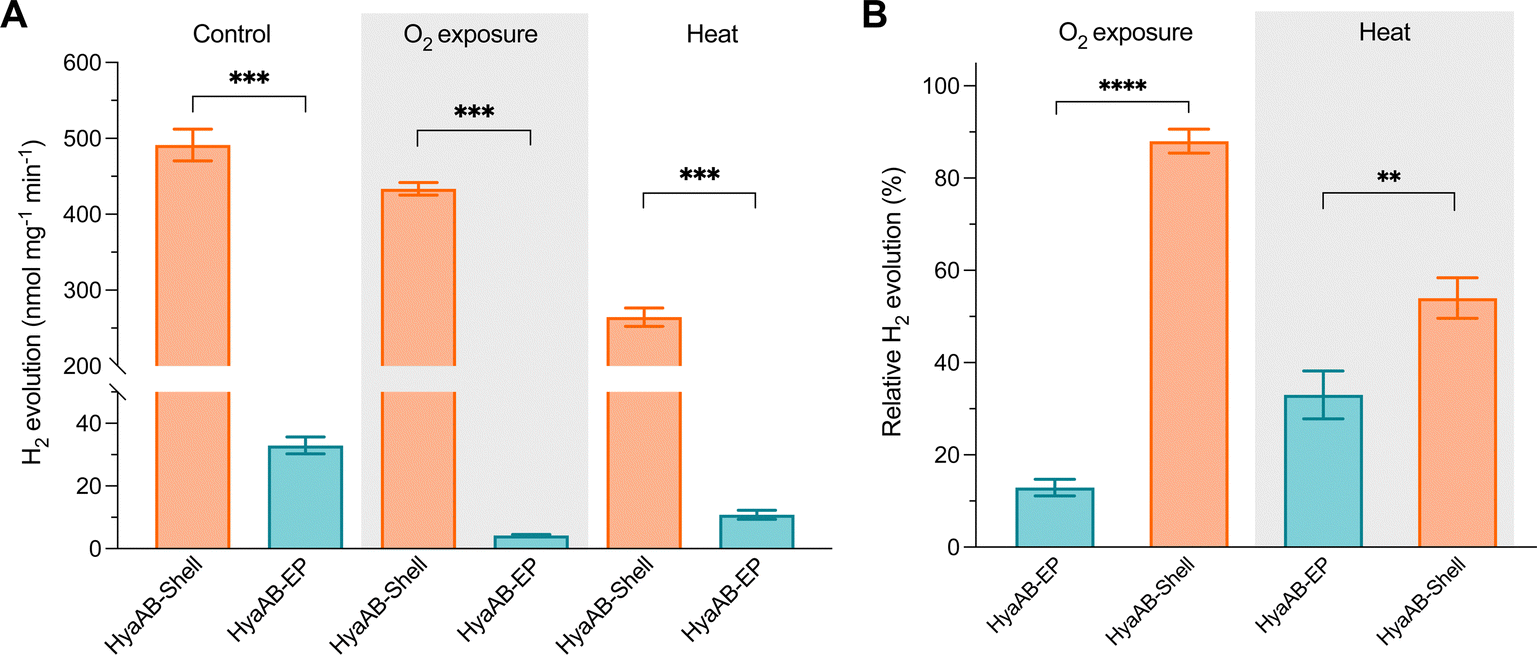 | ||
| Fig. 4 The HyaAB-Shell catalyst shows enhanced oxygen tolerance and thermostability. (A) Hydrogen production activities of isolated free HyaAB-EP and HyaAB-Shell before and after O2 exposure and 65 °C treatment at 50 mM MV. p = 0.0006 for control; p = 0.0001 for O2 exposure, p = 0.0006 for heat treatment (two-tailed unpaired t-test). (B) Relative hydrogen production activities of anaerobically purified free HyaAB-EP and HyaAB-Shell after O2 exposure for 24 hours at 4 °C (**** p < 0.0001, two-tailed unpaired t-test) and for 20min heat treatment under anaerobic conditions at 65 °C (** p = 0.0072, two-tailed unpaired t-test), as a relative percentage of total activities measured under anaerobic conditions in (A). Values represent the mean of three independent biological replicates and standard deviation. | ||
Furthermore, we incubated HyaAB-Shell and free HyaAB-EP at 65 °C for 20 minutes under anaerobic conditions. Free HyaAB-EP showed a ∼67% decline in H2-evolution activity at pH 8 (from 32.9 ± 2.7 nmol H2 mg−1 min−1 to 10.8 ± 1.4 nmol H2 mg−1 min−1, n = 3) (Fig. 4A and B), indicating that high temperature could affect the hydrogenase activity of EcHyd-1, consistent with previous results.10 In contrast, HyaAB-Shell incubated at 65 °C preserved ∼54% of the H2-evolution activity (from 491.4 ± 21.0 nmol H2 mg−1 min−1 to 264.4 ± 12.1 nmol H2 mg−1 min−1 at pH 8, n = 3), indicating that shell encapsulation provides the physical protection to enhance the thermostability and H2-evolution activity of encased [NiFe]-hydrogenases.
Conclusions
In summary, we generate genetic constructs using synthetic biology techniques to reprogramme the α-carboxysome shells as unique nanoreactors that encapsulate functional [NiFe]-hydrogenases for sustainable catalysis. The engineered hybrid biocatalyst demonstrates remarkably increased catalytic rates of hydrogen evolution under aerobic and anaerobic conditions, as well as improved oxygen tolerance and thermostability of [NiFe]-hydrogenase cargos by shell encapsulation for a longer functional lifetime. We also show an efficient encapsulation approach using the C-terminus of CsoS2 as a general EP to sequester foreign cargos within the α-carboxysome shells. Future studies will focus on further optimization of the encapsulation efficiency, cargo packaging, and structural homogeneity of the produced biocatalysts. The hydrogenase-integrating hybrid biocatalysts open new avenues for advancing our understanding of the self-assembly principles of protein organelles, and have enormous potential to recruit various cargo enzymes and multicomponent assemblies, modulate shell size and permeability, and combine inorganic/organic materials for the future development of new electrocatalytically active devices to achieve enhanced performance and robustness in industrial biocatalysis applications.Author contributions
Qiuyao Jiang: conceptualization, methodology, validation, formal analysis, investigation, resources, writing – original draft. Tianpei Li: methodology, formal analysis, writing – original draft. Jing Yang: methodology, investigation. Catherine M. Aitchison: methodology, investigation. Jiafeng Huang: methodology, investigation. Yu Chen: methodology, investigation. Fang Huang: formal analysis, writing – review & editing. Qiang Wang: formal analysis, writing – review & editing. Andrew I. Cooper: supervision, writing – review & editing. Lu-Ning Liu: conceptualization, formal analysis, supervision, visualization, writing – review & editing, funding acquisition.Conflicts of interest
A patent (WO2022074380A1) has been granted for the development and use of the α-carboxysome shell.Acknowledgements
We thank the Liverpool Biomedical Electron Microscopy Unit for technical assistance and provision for microscopic imaging and the Materials Innovation Factory (MIF) for provision of analytical equipment. This work was supported by the National Key R&D Program of China (grant number 2021YFA0909600), the National Natural Science Foundation of China (grant number 32070109), the Biotechnology and Biological Sciences Research Council (BBSRC) (grant numbers BB/V009729/1, BB/M024202/1 to LNL), the Royal Society (grant numbers URF\R\180030, RGF\EA\181061, RGF\EA\180233 to LNL), and the Leverhulme Trust (grant number RPG-2021-286 to LNL). FH was supported by an MRC Harmonised IAA fund.Notes and references
- P. C. Hallenbeck, Int. J. Hydrogen Energy, 2009, 34, 7379–7389 CrossRef CAS.
- A. Mudhoo, T. Förster-Carneiro and A. Sanchez, Crit. Rev. Biotechnol., 2011, 31, 250–263 CrossRef CAS PubMed.
- F. Sargent, Adv. Microb. Physiol., 2016, 68, 433–507 CrossRef CAS PubMed.
- H. Ogata, W. Lubitz and Y. Higuchi, J. Biochem., 2016, 160, 251–258 CrossRef CAS PubMed.
- H. S. Shafaat, O. Rudiger, H. Ogata and W. Lubitz, Biochim. Biophys. Acta, 2013, 1827, 986–1002 CrossRef CAS PubMed.
- P. Mullai, M. K. Yogeswari and K. Sridevi, Bioresour. Technol., 2013, 141, 212–219 CrossRef CAS PubMed.
- S. Krishnan and F. A. Armstrong, Chem. Sci., 2012, 3, 1015–1023 RSC.
- K. Menzel, U. P. Apfel, N. Wolter, R. Ruger, T. Alpermann, F. Steiniger, D. Gabel, S. Förster, W. Weigand and A. Fahr, J. Liposome Res., 2014, 24, 59–68 CrossRef CAS PubMed.
- J. W. Wilkerson, S. O. Yang, P. J. Funk, S. K. Stanley and B. C. Bundy, New Biotechnol., 2018, 44, 59–63 CrossRef CAS PubMed.
- P. C. Jordan, D. P. Patterson, K. N. Saboda, E. J. Edwards, H. M. Miettinen, G. Basu, M. C. Thielges and T. Douglas, Nat. Chem., 2016, 8, 179–185 CrossRef CAS PubMed.
- D. Balestri, Y. Roux, M. Mattarozzi, C. Mucchino, L. Heux, D. Brazzolotto, V. Artero, C. Duboc, P. Pelagatti, L. Marchiò and M. Gennari, Inorg. Chem., 2017, 56, 14801–14808 CrossRef CAS PubMed.
- L. N. Liu, Trends Microbiol., 2022, 30, 567–580 CrossRef CAS PubMed.
- M. Sutter, M. R. Melnicki, F. Schulz, T. Woyke and C. A. Kerfeld, Nat. Commun., 2021, 12, 3809 CrossRef CAS PubMed.
- C. A. Kerfeld, C. Aussignargues, J. Zarzycki, F. Cai and M. Sutter, Nat. Rev. Microbiol., 2018, 16, 277–290 CrossRef CAS PubMed.
- L. N. Liu, M. Yang, Y. Sun and J. Yang, Curr. Opin. Microbiol., 2021, 63, 133–141 CrossRef CAS PubMed.
- B. D. Rae, B. M. Long, M. R. Badger and G. D. Price, Microbiol. Mol. Biol. Rev., 2013, 77, 357–379 CrossRef CAS PubMed.
- F. Huang, W. Kong, Y. Sun, T. Chen, G. F. Dykes, Y. L. Jiang and L. N. Liu, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 17418–17428 CrossRef CAS PubMed.
- Y. Sun, A. J. M. Wollman, F. Huang, M. C. Leake and L. N. Liu, Plant Cell, 2019, 31, 1648–1664 CrossRef CAS PubMed.
- J. Huang, Q. Jiang, M. Yang, G. F. Dykes, S. L. Weetman, W. Xin, H. L. He and L. N. Liu, Biomacromolecules, 2022, 23, 4339–4348 CrossRef CAS PubMed.
- M. Faulkner, I. Szabó, S. L. Weetman, F. Sicard, R. G. Huber, P. J. Bond, E. Rosta and L.-N. Liu, Sci. Rep., 2020, 10, 17501 CrossRef CAS PubMed.
- P. Mahinthichaichan, D. M. Morris, Y. Wang, G. J. Jensen and E. Tajkhorshid, J. Phys. Chem. B, 2018, 122, 9110–9118 CrossRef CAS PubMed.
- Z. Dou, S. Heinhorst, E. B. Williams, C. D. Murin, J. M. Shively and G. C. Cannon, J. Biol. Chem., 2008, 283, 10377–10384 CrossRef CAS PubMed.
- T. Ni, Y. Sun, W. Burn, M. M. J. Al-Hazeem, Y. Zhu, X. Yu, L.-N. Liu and P. Zhang, Nat. Commun., 2022, 13, 4299 CrossRef CAS PubMed.
- S. Frank, A. D. Lawrence, M. B. Prentice and M. J. Warren, J. Biotechnol., 2013, 163, 273–279 CrossRef CAS PubMed.
- W. Bonacci, P. K. Teng, B. Afonso, H. Niederholtmeyer, P. Grob, P. A. Silver and D. F. Savage, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 478–483 CrossRef CAS PubMed.
- B. M. Long, W. Y. Hee, R. E. Sharwood, B. D. Rae, S. Kaines, Y.-L. Lim, N. D. Nguyen, B. Massey, S. Bala, S. von Caemmerer, M. R. Badger and G. D. Price, Nat. Commun., 2018, 9, 3570 CrossRef PubMed.
- Y. Sun, V. M. Harman, J. R. Johnson, T. Chen, G. F. Dykes, Y. Lin, R. J. Beynon and L.-N. Liu, mBio, 2022, 13, e0362921 CrossRef PubMed.
- T. Chen, Y. Fang, Q. Jiang, G. F. Dykes, Y. Lin, G. D. Price, B. M. Long and L. N. Liu, ACS Synth. Biol., 2022, 11, 154–161 CrossRef CAS PubMed.
- T. Li, Q. Jiang, J. Huang, C. M. Aitchison, F. Huang, M. Yang, G. F. Dykes, H. L. He, Q. Wang, R. S. Sprick, A. I. Cooper and L. N. Liu, Nat. Commun., 2020, 11, 5448 CrossRef CAS PubMed.
- M. J. Lukey, M. M. Roessler, A. Parkin, R. M. Evans, R. A. Davies, O. Lenz, B. Friedrich, F. Sargent and F. A. Armstrong, J. Am. Chem. Soc., 2011, 133, 16881–16892 CrossRef CAS PubMed.
- J. Fritsch, P. Scheerer, S. Frielingsdorf, S. Kroschinsky, B. Friedrich, O. Lenz and C. M. Spahn, Nature, 2011, 479, 249–252 CrossRef CAS PubMed.
- N. K. Menon, J. Robbins, J. C. Wendt, K. T. Shanmugam and A. E. Przybyla, J. Bacteriol., 1991, 173, 4851–4861 CrossRef CAS PubMed.
- L. Zhang, G. Morello, S. B. Carr and F. A. Armstrong, J. Am. Chem. Soc., 2020, 142, 12699–12707 CrossRef CAS PubMed.
- J. Robbins, N. K. Menon, J. C. Wendt, K. T. Shanmugam and A. E. Przybyla, J. Bacteriol., 1991, 173, 11 Search PubMed.
- E. J. Brooke, R. M. Evans, S. T. Islam, G. M. Roberts, S. A. Wehlin, S. B. Carr, S. E. Phillips and F. A. Armstrong, Biochemistry, 2017, 56, 132–142 CrossRef CAS PubMed.
- R. M. Evans, E. J. Brooke, S. A. Wehlin, E. Nomerotskaia, F. Sargent, S. B. Carr, S. E. Phillips and F. A. Armstrong, Nat. Chem. Biol., 2016, 12, 46–50 CrossRef CAS PubMed.
- L. Forzi and R. G. Sawers, Biometals, 2007, 20, 565–578 CrossRef CAS PubMed.
- S. Ogo, T. Kishima, T. Yatabe, K. Miyazawa, R. Yamasaki, T. Matsumoto, T. Ando, M. Kikkawa, M. Isegawa, K. S. Yoon and S. Hayami, Sci. Adv., 2020, 6, eaaz8181 CrossRef CAS PubMed.
- A. Volbeda, P. Amara, C. Darnault, J. M. Mouesca, A. Parkin, M. M. Roessler, F. A. Armstrong and J. C. Fontecilla-Camps, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5305–5310 CrossRef CAS PubMed.
- A. Volbeda, C. Darnault, A. Parkin, F. Sargent, F. A. Armstrong and J. C. Fontecilla-Camps, Structure, 2013, 21, 184–190 CrossRef CAS PubMed.
- B. Soboh, L. Adrian and S. T. Stripp, J. Biol. Chem., 2022, 298, 102291 CrossRef CAS PubMed.
- A. Dubini, R. L. Pye, R. L. Jack, T. Palmer and F. Sargent, Int. J. Hydrogen Energy, 2002, 27, 1413–1420 CrossRef CAS.
- C. Pinske, S. Kruger, B. Soboh, C. Ihling, M. Kuhns, M. Braussemann, M. Jaroschinsky, C. Sauer, F. Sargent, A. Sinz and R. G. Sawers, Arch. Microbiol., 2011, 193, 893–903 CrossRef CAS PubMed.
- J. Y. Kim, B. H. Jo and H. J. Cha, Microb. Cell Fact., 2010, 9, 54 CrossRef PubMed.
- B. J. Murphy, F. Sargent and F. A. Armstrong, Energy Environ. Sci., 2014, 7, 8 RSC.
- M. W. Adams and D. O. Hall, Biochem. J., 1979, 183, 11–22 CrossRef CAS PubMed.
- J. A. Cracknell, A. F. Wait, O. Lenz, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20681–20686 CrossRef PubMed.
- R. M. Evans, A. Parkin, M. M. Roessler, B. J. Murphy, H. Adamson, M. J. Lukey, F. Sargent, A. Volbeda, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2013, 135, 2694–2707 CrossRef CAS PubMed.
- L. A. Flanagan, J. J. Wright, M. M. Roessler, J. W. Moir and A. Parkin, Chem. Commun., 2016, 52, 9133–9136 RSC.
- A. A. T. Tatyana and V. Lurinavichene, FEMS Microbiol. Lett., 2001, 202, 5 Search PubMed.
- M. J. Lukey, A. Parkin, M. M. Roessler, B. J. Murphy, J. Harmer, T. Palmer, F. Sargent and F. A. Armstrong, J. Biol. Chem., 2010, 285, 3928–3938 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 and Table S1. See DOI: https://doi.org/10.1039/d2tb02781j |
| This journal is © The Royal Society of Chemistry 2023 |