
Sequential administration of virus-like particle-based nanomedicine to elicit enhanced tumor chemotherapy†
Chufan
Wang
a,
Cheng
Xiao
a,
Yurong
Chen
b,
Yao
Li
 a,
Qiang
Zhang
a,
Wenjun
Shan
c,
Yulin
Li
d,
Shengli
Bi
e,
Yunlong
Wang
d,
Xiumin
Wang
*b and
Lei
Ren
*af
a,
Qiang
Zhang
a,
Wenjun
Shan
c,
Yulin
Li
d,
Shengli
Bi
e,
Yunlong
Wang
d,
Xiumin
Wang
*b and
Lei
Ren
*af
aKey Laboratory of Biomedical Engineering of Fujian Province University/Research Center of Biomedical Engineering of Xiamen, Department of Biomaterials, College of Materials, Xiamen University, Xiamen 361005, P. R. China
bSchool of Pharmaceutical Sciences, Xiamen University, Xiamen 361102, P. R. China
cDepartment of Pharmacology, College of Pharmacy, Army Medical University (Third Military Medical University), Chongqing 400038, P. R. China
dHenan Bioengineering Research Center, Zhengdong New District, Zhengzhou, China
eChinese Center for Disease Control & Prevention, Institute Viral Disease Control & Prevention, Beijing, P. R. China
fState Key Lab of Physical Chemistry of Solid Surfaces, Xiamen University, Xiamen 361005, P. R. China
First published on 21st December 2022
Abstract
Protein cages have played a long-standing role in biomedicine applications, especially in tumor chemotherapy. Among protein cages, virus like particles (VLPs) have received attention for their potential applications in vaccine development and targeted drug delivery. However, most of the existing protein-based platform technologies are plagued with immunological problems that may limit their systemic delivery efficiency as drug carriers. Here, we show that using immune-orthogonal protein cages sequentially and modifying the dominant loop epitope can circumvent adaptive immune responses and enable effective drug delivery using repeated dosing. We genetically modified three different hepadnavirus core protein derived VLPs as delivery vectors for doxorubicin (DOX). These engineered VLPs have similar assembly characteristics, particle sizes, and immunological properties. Our results indicated that there was negligible antibody cross-reactivity in either direction between these three RGD-VLPs in mice that were previously immunized against HBc VLPs. Moreover, the sequential administration of multiple RGD-VLP-based nanomedicine (DOX@RGD-VLPs) could effectively reduce immune clearance and inhibited tumor growth. Hence, this study could provide an attractive protein cage-based platform for therapeutic drug delivery.
1. Introduction
During recent decades, nanomaterial-based drug delivery systems (DDSs) have been extensively explored as a means to improve properties, such as solubility, bioavailability, diffusivity, pharmacokinetics, biodistribution, absorption, stability, targeting, and controlled release of drugs, which often leads to improved therapeutic performance and safety compared to the traditional drug delivery systems.1,2 Different types of nano-sized carriers, such as liposomes, polymeric nanomicelles, dendrimers, and inorganic nanoparticles/nanowires, have been developed for various drug-delivery applications.3–7 Despite significant academic progress in these synthetic drug nanocarriers, only a limited number of advanced therapeutic strategies have resulted in successful clinical translation because of their non-degradability, potential toxicity, and lack of biological motifs.8,9 One promising approach is the use of natural carrier systems to be therapeutically exploited which may be highly effective in addressing these challenges.10Among natural carrier systems, protein cage-based drug carriers have been founded on the premise of exhibiting characteristics from both protein inherent properties (biodegradability, biocompatibility, high abundance, generally low cost, and ease of modification) and the advantages of nanoparticles (improved bioavailability, controlled release, and encapsulation of drug molecules), thereby proving to be a better alternative to adapt and improve the pharmacokinetic and pharmacodynamic properties of the various types of drug molecules.11,12 Virus-like particles (VLPs), self-assembled through viral coat proteins, but devoid of genetic information and thus safe, have emerged as one kind of empty protein cages.13,14 Various VLPs (such as hepatitis B virus, human papillomavirus, bacteriophage Qbeta, cowpea mosaic virus, and tobacco mosaic virus derived VLPs), with similar characteristics to a virus capsid (highly ordered structure, precise surface topologies, virus receptor binding, and host cell entry), have been well studied for delivering materials including DNA, RNA, drugs, and bioimaging agents.15,16 For facilitating specific cell targeting and efficient packaging of the target molecules, the surface and inside of VLPs have been genetically and chemically modified, respectively.17,18 In our previous works, we have developed an array of engineered VLPs (∼30 nm) by inserting sets of functional groups within the major immune region (MIR) domains of hepatitis B virus core protein (HBc) to load drugs (doxorubicin and phenytoin), bioimaging substances (indocyanine green), nucleic acids, tumor antigens (OVA, gp-100), and inorganic nanoparticles (Fe3O4 NPs and Cu2S NPs), respectively.19–25 Our results indicated that these HBc VLP-based carriers with lower toxicity, biodegradability, and ability to bypass the blood–brain barrier and release cargo in a controlled manner could be a promising nanomedicine approach.
However, the immunogenicity of protein cages is one of the major considerations for both their safety and efficacy when used as drug delivery nanocarriers.26,27 Many efforts have been devoted to elucidating the bio–nano interactions, and lower undesirable immunogenic responses to materials in a complex biological system. For example, pegylation and glycosylation have already been widely used to decrease immunogenicity by shielding immunogenic epitopes, while maintaining the native conformation of protein cages.28,29 In addition, Nguyen et al. reported that phosphatidylserine could convert an immunogen to a tolerogen, thereby reducing unwanted immune responses.30 In our previous works, we also found that the insertion of foreign protein segments within MIR domains could decrease the immunogenicity of HBc VLPs which are extremely powerful immunogens.31 Immunological researchs have shown that despite the high degree of affinity and specificity raised by the antibodies against the antigen, they can exhibit cross-reactivity with disparate antigens. It is believed that antibody cross-reactivity arises because of the existence of similar epitopes on different antigens.32 For this reason, the sequential use of immune-orthogonal protein cages in which amino acid sequences are extremely diverse could evade the antibody cross-reactivity and allow subsequent protein cages to avoid neutralization by existing antibodies.33 For example, Billaud et al. found that the woodchuck hepatitis core antigen (WHcAg) did not have significant cross-immunity with the HBcAg at the antibody level, and then they successfully used WHc VLPs as a heterogeneous B cell epitope carrier for the treatment of chronic hepatitis B.34
To avoid an undesired immune response elicited by repeated dosing and improve the target activity, in this study, we constructed three structural similar VLPs by genetic insertion of the RGD peptide within the MIR domain of HBcAg, WHcAg, and duck hepatitis B virus core antigen (DHBcAg).35 They have similar assembly characteristics, particle size, and immunological properties. Compared to using a single VLP, sequential administration of different RGD-modified VLPs might elicit only a weak antigen-specific immune response, which could dramatically reduce antibody mediated clearance and enhance the therapeutic effect. Furthermore, DOX was encapsulated into the cavity of RGD-modified VLPs via a disassembly–reassembly pathway for tumor chemotherapy, and the therapeutic efficacy was quantitatively identified by in vivo anti-tumor experiments (Scheme 1). More importantly, this approach could provide a novel multiple VLP drug delivery strategy and demonstrates that sequential administration with immune-orthogonal VLPs could address the “immune clearance” problem that has hindered the protein cage-based platform technology.
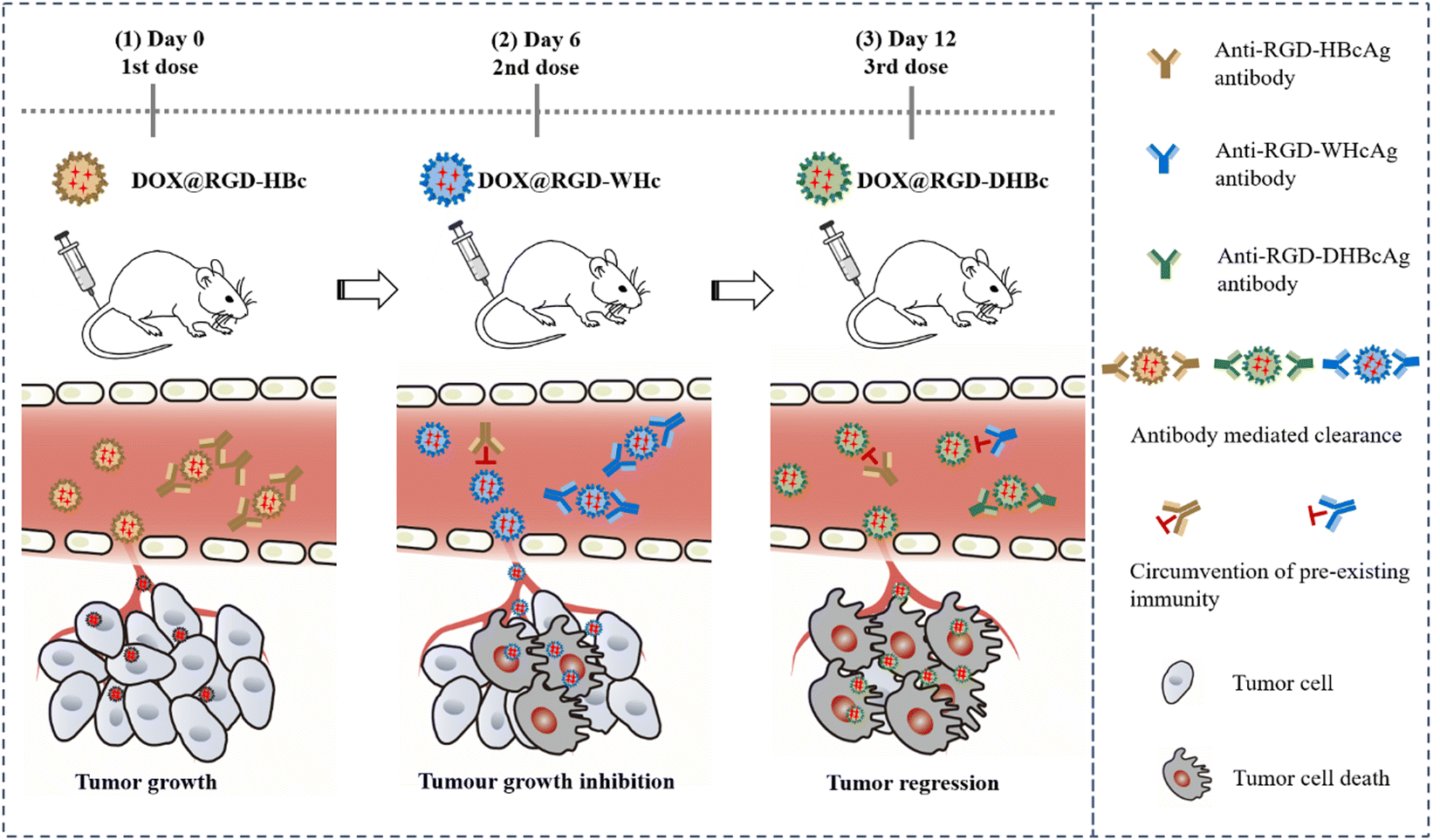 | ||
| Scheme 1 Scheme of sequential administration with immune-orthogonal VLPs to circumvent the immune response and targeted delivery of chemotherapeutic drugs for tumor treatment. | ||
2. Experimental
2.1. Preparation of RGD-VLPs
RGD sequence accompanied with glycine-rich linkers (GTSGSSGSGSGGSGSGGGG) were respectively inserted on the surface (between residues 78 and 81) of the truncated HBc and WHc and beside the surface (between the residues 113 and 114) of the truncated DHBc. Based on this design, the target gene was purchased from Shanghai Generay Biotech Co., Ltd. The three different plasmid pET43.1(a)-RGD-VLPs, as an expression vector, was transformed into E. coli BL21(DE3). We refer to published articles for expression, extraction, and purification of a variety of RGD-VLPs.19 To examine the molecular weight of RGD-VLPs, the purified VLPs were separated by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, Bio-Rad, USA).2.2. Preparation and characterization of DOX@RGD-VLPs
The DOX loaded RGD-VLPs were prepared as follows. The purified RGD-VLPs were reversibly disassembled with 6 M urea at 25 °C for 1 h. After the dissociation, an appropriate amount of sulfobutyl-beta-cyclodextrin and adriamycin hydrochloride was added and the solution was gently stirred for 2 h in the dark. Sulfobutyl-β-cyclodextrin can increase the amount of DOX carried by RGD-VLPs. Then, the reassembly of VLPs was conducted by dialysis in an assembling buffer at 4 °C and free drug was removed by ultrafiltration. To calculate the loading capacity, the protein concentration was determined by bicinchoninic acid assay.2.3. Characterization of RGD-VLPs and DOX@RGD-VLPs
The morphology of VLPs with or without DOX encapsulation was observed by transmission electron microscopy (TEM, Hitachi HT-7800, Japan) with an acceleration voltage of 100 kV. Besides, the diameter distribution of VLPs was obtained by counting at least 10 particles in TEM images and the average particle size in water suspension was measured by dynamic laser scattering (DLS, Malvern Nano-ZS, UK).2.4. Cell lines and mice
Mouse colon carcinoma CT26 was purchased from American Type Culture Collection. Roswell Park Memorial Institute's medium (RPMI-1640, Cellgro) containing 10% fetal bovine serum was used for cell culture at 37 °C under a 5% CO2 atmosphere. Male Balb/c white mice (21 ± 2 g) were purchased from Xiamen University Laboratory Animal Center and all mice experiments were carried out in accordance with the Institutional Animal Care and Use Committee approved procedure at Xiamen University.2.5. Immunogenicity of RGD-VLPs
Each group of mice (n = 5) was subcutaneously immunized with HBc, RGD-HBc, RGD-WHBc, and RHD-DHBc, once a week, for antibody production. The first immunization time was day 0, and blood was collected from the tails of mice on day 7, 14, 21, and 28, and sera were collected for antibody titer experiments. Anti-VLPs IgG antibodies in murine sera were measured by ELISA in 96-well plates by using the homologous or heterologous core proteins (1 μg per well). The specific experimental steps are as follows: 10 μg mL−1 of VLPs in coating buffer (1.59 g Na2CO3, 2.93 g NaHCO3 in 1 L distilled water) were coated on the plate at 4 °C overnight. Wells were washed and blocked with 2% BSA in PBS containing 0.1% Tween-20 (PBS-T) for 2 h at 37 °C. Mouse sera (diluted in PBS) were incubated in wells for 1 h at 37 °C, followed by washing with PBS-T. Then, goat anti-mouse IgG-HRP was added in. Wells were washed again with PBS-T before the addition of tetramethylbenzidine solution. The data are expressed as antibody titers representing the reciprocals of the highest dilutions of sera required to yield an optical density at 492 nm (OD492) that is seen with an equal dilution of non-immunization sera.2.6. Cellular uptake of RGD-VLPs in vitro
To assess the cellular uptake of Cy5.5 labeled RGD-VLPs, CT-26 cells were cultured on glass coverslips in six-well plates at a density of 3 × 105 cells per well. After the cells were attached to the wall, fresh 1640 medium containing Cy 5.5, RGD-HBc, RGD-WHBc, and RHD-DHBc were added. After incubation for 1 hour, the medium was removed, and cells were washed 3 times in PBS. After fixation in 1 mL paraformaldehyde for 20 minutes, the nuclei were stained with DAPI. Then staining images were observed and captured using a confocal microscope (Zeiss LSM 880, Germany). The Cy5.5 fluorescence intensity in cells reflected the amount of RGD-VLPs taken up by CT-26 cells.2.7. Hemolysis assays
Fresh mouse blood was collected in anticoagulant tubes with 1% heparin sodium. The red blood cells were separated, purified, and diluted to 5.0 × 107 mL for the hemolysis assays. Various concentrations of RGD-HBc VLPs, RGD-WHc VLPs and RGD-DHBc VLPs were incubated with 500 μL of RBC at 37 °C for 3 h. After centrifugation at 500×g for 5 min at 4 °C, the absorbance of the supernatants from each group was measured using a microplate reader (INFINITE m200, TECAN Group Ltd, Switzerland) at 540 nm. RBCs that had been treated with ultra-pure water were used as a positive control, and the release rate of hemoglobin was set at 100%.2.8. In vivo biodistribution analysis of RGD-VLPs
Tumor-bearing mice were administered Cy5.5 labeled RGD-VLPs (50 μL, 2 mg mL−1) by intravenous injection. Images of the mice were captured using an IVIS Spectrum Imaging System (IVIS Lumina II, PE, USA) at 0, 3, 6, 12, and 24 h time points. Regions of interest were circled around the body, and the optical intensity (in total scaled counts per second) was read by the IVIS software. After 24 h, all of the mice were euthanized. Tumors as well as major organs were harvested and subjected to ex vivo imaging. The fluorescence intensity was analyzed by the IVIS software.2.9. Evaluation of anti-tumor effect
Male Balb/c mice (6–8 weeks) were injected subcutaneously with 0.1 mL of CT26 cell suspension (1 × 106). When tumors grew up to an average volume of ≈80 mm3, the mice were treated by tail vein injection. The dose of DOX was 3 mg kg−1, which was administered once every 3 days for a total of 6 administrations. The tumor growth rate was studied by monitoring the tumor volume (calculated by formulation (1)) and body weight every other day to evaluate the therapeutic efficacy in vivo.| V = Length × Width2/2 | (1) |
The mice were euthanized on day 18 after treated by DOX@RGD-VLPs. The serum and tissues (including heart, liver, spleen, lung, kidney, and tumor) were collected for further histological examination and antibody titer experiments.
2.10. Statistical analysis
Quantitative results were presented as mean ± standard error of mean (sem). Statistical differences among groups were checked by one-way ANOVA followed by Duncan multiple comparisons test. A probability value p < 0.05 was accepted as statistically significant.3. Results and discussion
3.1. Preparation and characterization of various tumor-targeting RGD-VLPs
The RGD peptide for tumor targeting and two glycine-rich linkers were genetically inserted onto the major immune region of wtHBcAg, WHcAg, and DHBcAg. The molecular weight of wtHBc, RGD-HBc, and RGD-WHc recombinant protein was about 20 kDa and RGD-DHBc protein was about 25 kDa, which can be observed in the SDS-PAGE analysis (Fig. 1a).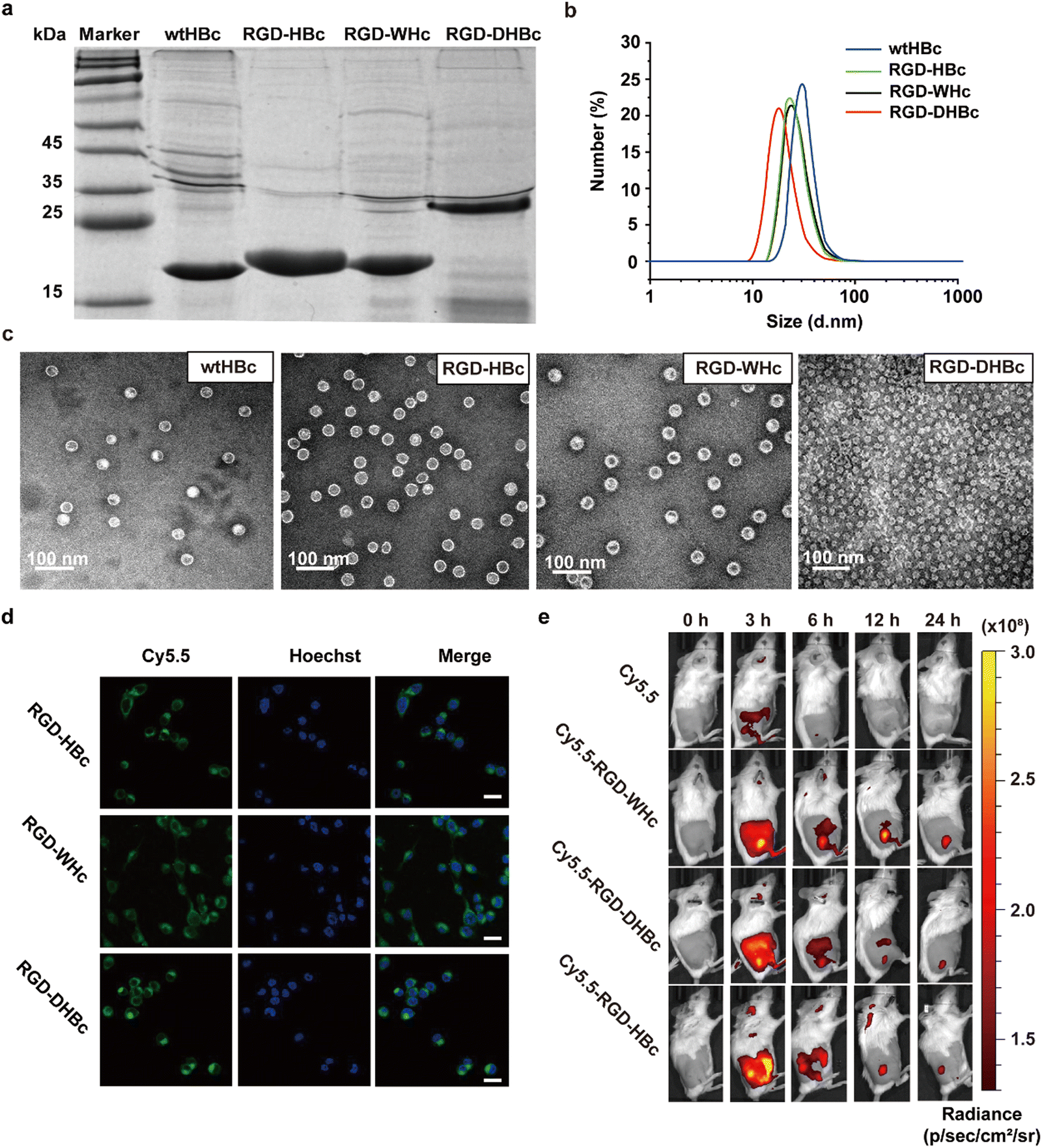 | ||
| Fig. 1 Characterization of purified VLPs. (a) SDS-PAGE analysis of various VLPs: Marker (lane 1), wtHBc (lane 2), RGD-HBc (lane 3), RGD-WHc (lane 4), RGD-DHBc (lane 5). wt, wild type. (b) DLS analysis of various VLPs. (c) Representative TEM images of various VLPs. (d) Confocal laser scanning microscopy images of CT-26 cells incubated with RGD-VLPs. CT-26 cells were stained using Hoechst and RGD-VLPs were labeled using Cy5.5. Scale bar, 20 μm. (e) In vivo fluorescence images after tail intravenous injection of Cy5.5, RGD-HBc, RGD-WHc, and RGD-DHBc. The images were taken at the indicated time interval, and the circles indicated the tumor site. | ||
As shown in the TEM images and DLS analysis (Fig. 1b and c), these VLPs expressed as well-defined spherical nanoparticles, and the diameter of wtHBc, RGD-HBc, and RGD-WHc was about 32 nm (Table S1, ESI†). Interestingly, although the molecular weight of RGD-DHBc protein was larger than the other three VLPs, its diameter was smaller, about 24 nm, which is in agreement with the literature.34 These results confirmed that the insertion of RGD peptide onto HBc, WHc, and DHBc protein did not disturb the nanocage structure and self-assembly ability. In our previous studies, we have proved that RGD modified HBc VLPs showed a time-dependent intracellular accumulation in U87MG cells more rapidly than unmodified HBc VLPs. Similarly, in this work, we observed that Cy5.5 labeled RGD-WHc and RGD-DHBc VLPs could also effectively be taken up by CT-26 cells. (Fig. 1d). All in all, these tumor-targeting RGD-VLPs could be used for drug delivery.
To validate the tissue distribution and tumor-targeting properties of various RGD-VLPs in vivo, the mice challenged by CT-26 tumors were intravenously injected with these Cy5.5 labeled RGD-VLPs and observed via an IVIS Spectrum Imaging System at the indicated time points post administration. As shown in Fig. 1e and Fig. S1a (ESI†), these RGD-VLPs were primarily accumulated in the tumor tissue within 3 h and the fluorescence intensity was observed until 24 h after injection. Meanwhile, these RGD-VLPs obviously gathered in the liver at 3 h and the intensity of the fluorescence decreased over time, indicating that they were eliminated from the liver. Consistent with in vivo imaging results, ex vivo biodistribution studies demonstrated sustained accumulation of RGD-VLPs in tumors and no fluorescence was observed in the liver and kidney for 24 h following injection (Fig. S1b, ESI†). Moreover, more than half of these RGD-VLPs could be cleared from the blood after 24 h with a faster rate than free cy5.5 which also suggested RGD-VLPs could be more highly enriched in organs and tumor tissue (Fig. S1c, ESI†). It was thus suggested that the active targeting capability of RGD was a favorable factor to make RGD-VLPs accumulate in tumor tissues much more than they do in normal tissues.
3.2. Comparative immunogenicity and antigenicity of various core proteins
HBcAg is an extremely powerful immunogen and its unique properties are closely related to its structural characterization. To determine whether the immunogenicity is reducible by modifying MIR domains on each VLPs, we performed immunogenicity–antigenicity studies with mice by comparing HBcAg with the engineered core proteins derived from different hosts at the antibody level. Therefore, Balb/c mice were subcutaneously immunized with wtHBcAg, RGD-HBcAg, RGD-WHcAg, and RGD-DHBcAg (Fig. 2a). As shown in Fig. 2b, the antibody produced against each core protein gradually increased with the number of s.c. immunizations, which means that all four core proteins could elicit stronger antibody responses by repeated administration of homologous protein. After the fourth immunization, the wtHBcAg appeared to be more immunogenic than the other three core proteins, followed by RGD-DHBcAg, RGD-WHcAg, and RGD-HBcAg. The RGD-HBcAg has the lowest humoral immune response to Balb/c mice, which indicated that inserting the RGD targeting peptide onto the MIR of HBcAg can effectively reduce its immunogenicity and the same conclusion was also applicable to RGD-DHBcAg and RGD-WHcAg.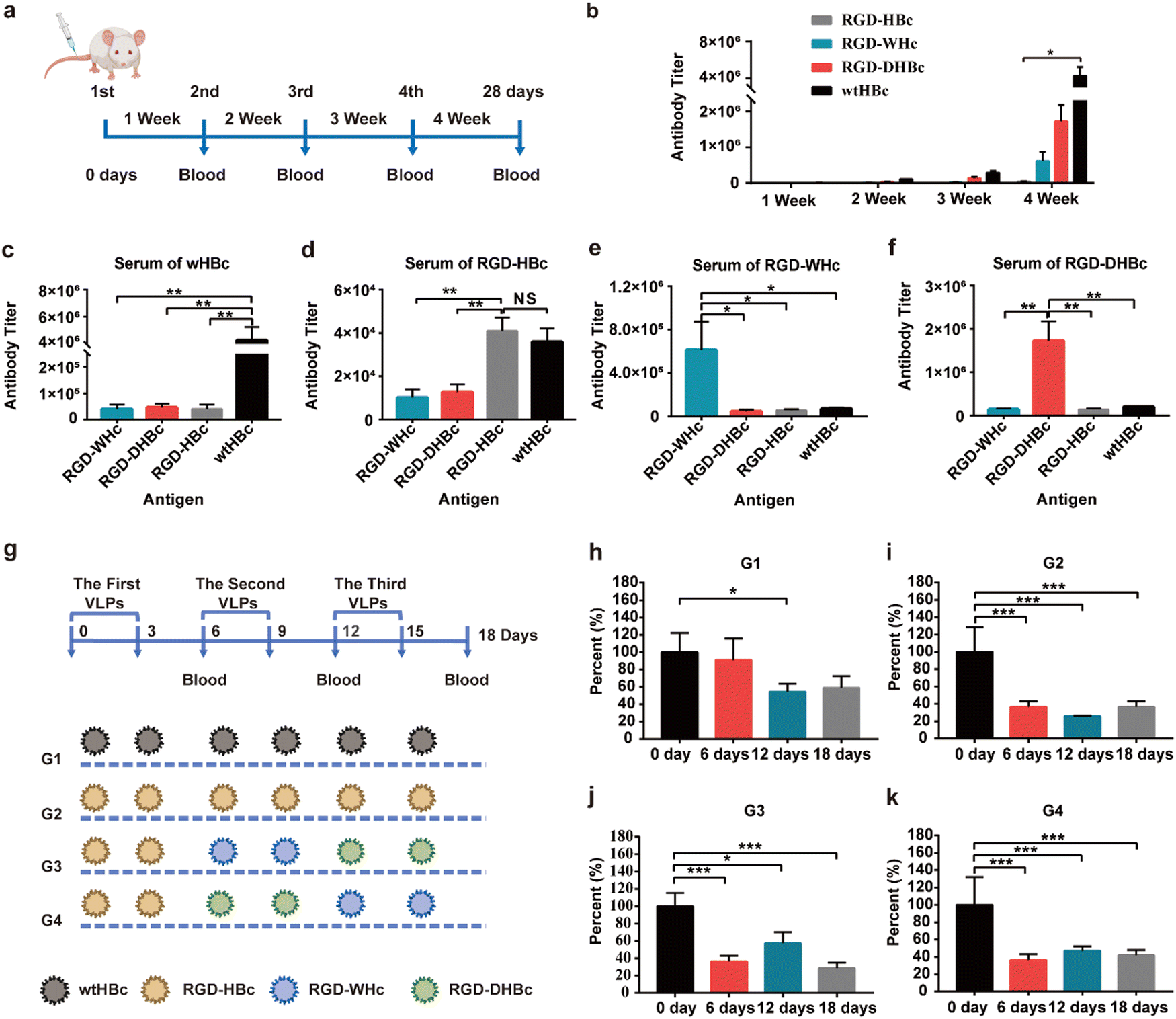 | ||
| Fig. 2 Immunogenicity and antigenicity of various core proteins. (a) Scheme of various core proteins immunized plan: Mice (n = 5) were immunized (s.c.) with various VLPs (4 μg g−1) on day 0, 7, 14, and 21, and serum was collected after 7 days of immunization. (b) VLPs-specific IgG antibody titers (reciprocal serum dilution) of immunized mice. (c)–(f) Cross-reactivity of anti-VLPs antibodies. Primary antisera (week 4) were analyzed for IgG reactivity with the homologous antigen used for immunization as well as the other three heterologous antigens. (g) Scheme of various core proteins sequential immunized plan: Mice (n = 5) with high wtHBcAg special IgG titer were immunized (i.v.) with different VLPs (4 μg g−1) on day 0, 3, 6, 9, 12, and 15. Mice sera were collected on day 0, 6, 12, and 18wt, wild type. (h)–(k) Mice sera were analyzed by solid-phase ELISA for anti-HBcAg antibody titer. The result was relative to the percentage of anti-HBc of 5 mice in each group on day 0, which was expressed as mean ± SEM. The statistical significance of the results was analyzed and indicated: * p < 0.05, ** p < 0.01, *** p < 0.0001, NS stands for no significant difference. | ||
The primary antisera (depicted in Fig. 2a) were analyzed for cross-reactivity between the various hepatitis virus core proteins (Fig. 2c–f). The results of antibody titers showed that RGD-HBc, RGD-WHc, and RGD-DHBc VLPs had no significant cross-immunity with heterologous core antigens immunized mouse sera. It is suggested that when VLPs formed by these core proteins are used as carriers in vivo, the antibodies produced against themselves would not significantly affect the transport efficiency of other immune-orthogonal VLPs. Namely, the RGD-WHc, RGD-DHBc, and RGD-HBc core proteins had no significant cross-immunity with the polyclonal anti-HBc antibodies produced against wtHBcAg. These results thus indicated that these core proteins would be useful in patients with high antibody titers of wtHBcAg, such as patients with chronic hepatitis B. The antibody cross-reactivity in the direction from RGD-HBcAg to wtHBcAg was especially obvious, but the level was low and the effect was less severe when using. The main reason is that, except the main immune region, there are other B cell epitopes shared between RGD-HBcAg and wtHBcAg, so the polyclonal anti-RGD-HBc antibodies cross-reacted with wtHBcAg. It's worth noting that there was low but still detectable antibody cross-reactivity from wtHBcAg to RGD-HBcAg. However, when one considers that the dominant B-cell epitope on wtHBcAg that resides in the main immune region at the tip of the structural spikes was truncated in RGD-HBcAg, the data seems reasonable.35 However, if attention is paid to the sequential treatment with different VLPs, the application of RGD-HBc will not be affected.
The other independent method of determining the degrees of cross-reactivity between these heterologous capsid antigens is the measurement of antibody specificities in sera from HBc-infected mice. To establish the HBc-infected mice model, Balb/c mice were subcutaneously immunized with wtHBcAg once a week for four weeks and the final titers reached 5 × 105 (1/dilution) (Fig. S2, ESI†). The preexisting anti-HBc antibody, elicited by repeated dosing or in individuals previously infected with HBV, may contribute to the immune clearance and significantly reduce the delivery efficiency of HBc VLPs in humans.36 As described in Fig. 2g, the sequential treatment with different VLPs was carried out in four different ways (G1, G2, G3, and G4) and sequential serum samples from different VLPs immunized HBc-infected mice on day 6, 12, and 18 were tested for anti-HBcAg antibody specificities. Anti-HBcAg antibody was obviously decreased on day 6 after intravenous injection of RGD-HBc, RGD-WHc, and RGD-DHBc in G2, G3, and G4 while there was no striking difference in G1 (Fig. 2g–h and k). Thus, it can be concluded that anti-HBcAg antibody cross-reacted on RGD-WHc, RGD-DHBc, and RGD-HBc core proteins minimally, and these tumor-targeting VLPs can be used as a drug delivery platform and circumvent the “preexisting immunity” problem that is inherent in the use of protein cages for human vaccine development.
3.3. Preparation and characterization of DOX@RGD-VLPs
In our previous studies, the genetically modified RGD-HBc VLPs possessed the advantages of high targeting efficiency to the tumor and low toxicity to normal tissues.19 The use of HBc proteins from woodchucks and Pekin ducks as drug carriers has not been explored. As illustrated in Fig. 3a and b, these three different DOX@VLPs were expressed as monodisperse with a relatively uniform particle size. The average particle size in a water suspension of DOX@RGD-HBc and DOX@RGD-WHc was about 30 ± 3.4 nm, and the particle size of DOX@RGD-DHBc VLPs was 25.27 ± 2.52 nm (Table S1, ESI†). Both the morphologies and size of RGD-HBc, RGD-WHc VLPs and RGD-DHBc with DOX loaded were comparable to unloaded VLPs, indicating that DOX packaging did not affect their capsid structures basically.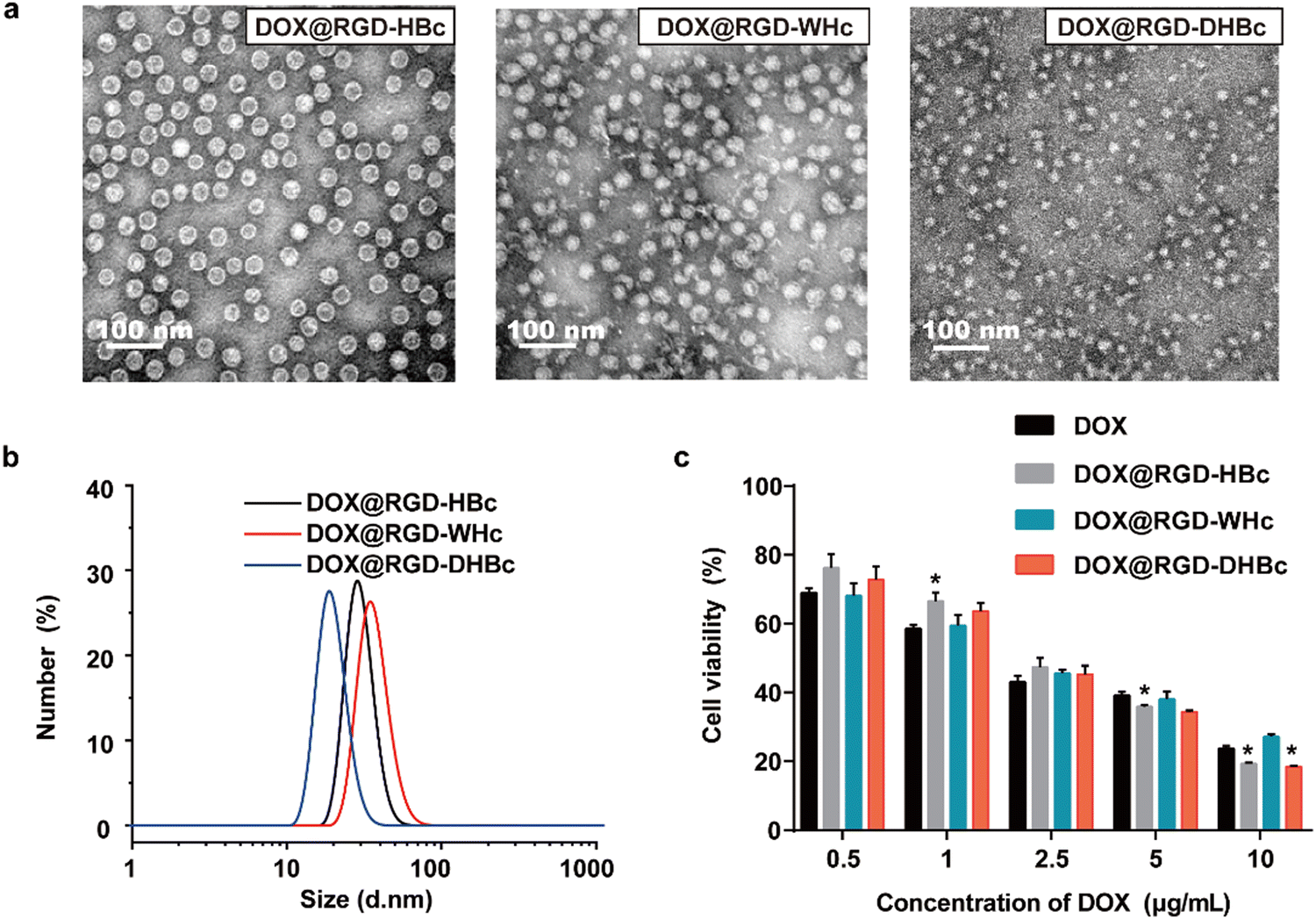 | ||
| Fig. 3 Characterization of DOX@VLPs. (a) TEM images of DOX@VLPs. (b) Diameter distribution of DOX@VLPs. (c) Cytotoxic effects of DOX@VLPs at different DOX concentrations on CT26 cells after 24 h co-incubation at 37 °C (Mean ± SEM, n = 5). | ||
Before clinical application, it is vitally important to test cytotoxicity in vitro. To test the hemocompatibility of various VLPs, hemolysis assays were performed in vitro using red blood cells (RBC). As shown in Fig. S4a and b (ESI†), very high concentrations of RGD-VLPs (50 μM) induced minimal hemoglobin release (less than 4%, n = 3). In addition, an MTT assay was performed with different concentrations of RGD-VLPs and DOX@VLPs, to respectively evaluate the impact of genetically modified VLPs and DOX on the viability of CT-26 cells. After incubation for 24 h, the cell viability of CT-26 cells was more than 80% in the highest concentration of various RGD-VLPs (250 μg mL−1) (Fig. S3, ESI†), indicating that these RGD-VLPs had high biocompatibility and low toxicity as drug delivery carriers. Besides, the cytotoxicity of three DOX@VLPs was also evaluated on CT-26 cells in the same way. Three DOX@VLPs exhibited concentration-dependent cancer cell killing, and the half maximum inhibiting concentration (IC50 value) was about 2.5 μg mL−1, which means that RGD-VLPs can be used as drug delivery tools to deliver DOX and inhibit the growth of tumor cells (Fig. 3c).
3.4. Evaluation of antitumor activity of DOX@RGD-VLPs
We next evaluated the therapeutic effect of multiple VLPs with or without sequential administration in vivo by using Balb/c mice xenografted CT-26 tumors. The detailed sequential administration procedure is shown in Fig. 4a. From the results of the tumor growth curve, we could find out that, compared with the PBS group, the multiple DOX@VLPs sequential treatment group had the best tumor suppression effect which was more pronounced than the DOX@RGD-HBc VLP treated group (Fig. 4b and c). Notably, after 12 days of treatment, only the sequential treatment group demonstrated a significantly decreased tumor growth rate. We speculated that this phenomenon might be caused by the humoral immune response. After repeated administration of DOX@RGD-HBc VLPs, anti-RGD-HBcAg antibodies, which could affect the efficiency of drug delivery of DOX@RGD-HBc VLPs, were generated in large amounts. During the treatment period, the body weight stability of each group was good (Fig. 4d), which reflects the better biocompatibility and active targeting capability of the VLPs nano drug delivery system in vivo.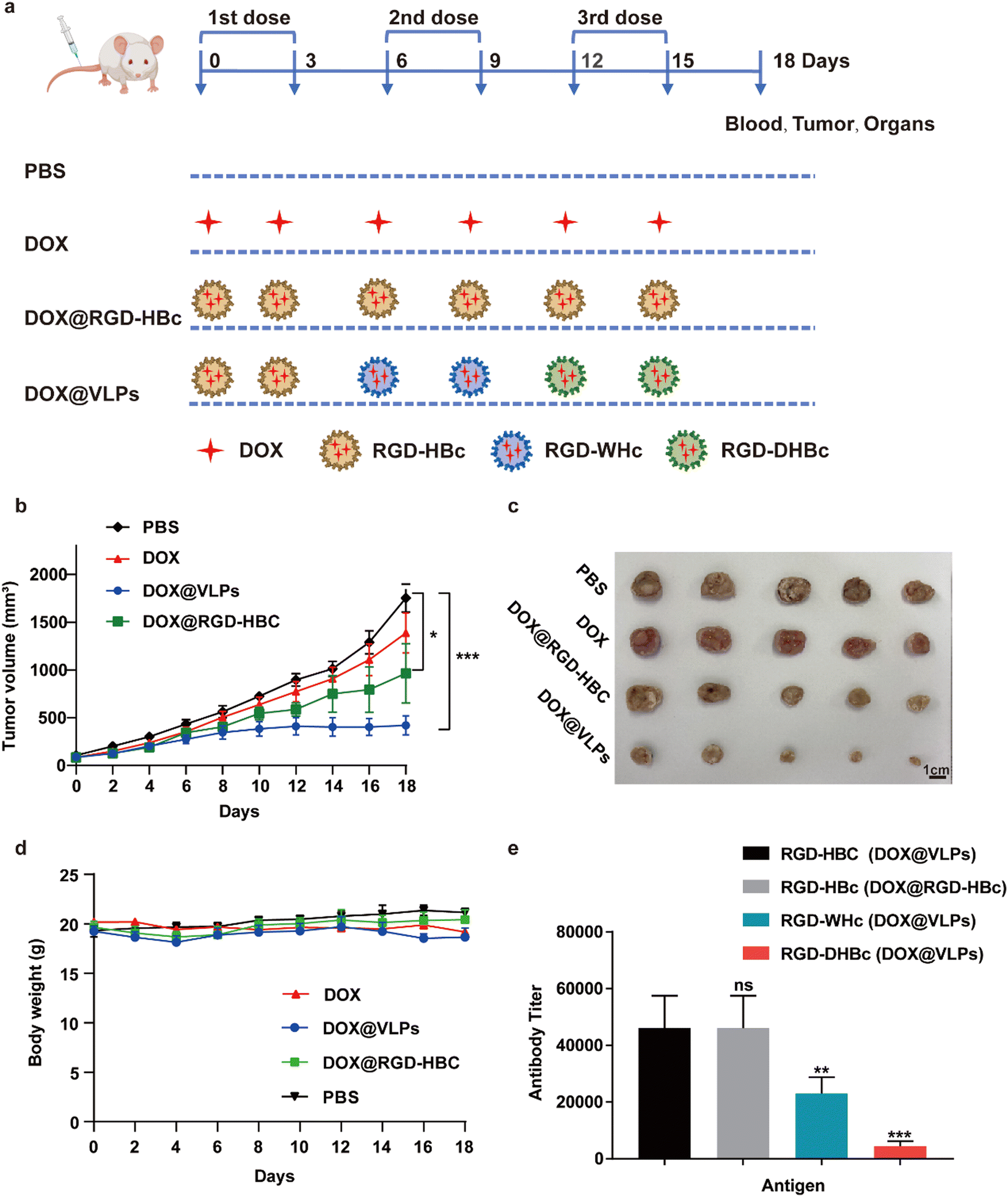 | ||
| Fig. 4 Evaluation of antitumor activity in vivo. (a) Schematic illustration of the administration scheme for tumor-bearing mice. Tumor-bearing mice (n =5) were given intravenous administration (DOX dose was at 3 mg kg−1) or the corresponding volume of PBS every 3 days. The Combination group received intravenous injection of DOX@RGD-HBc on day 0 and 3, injection of DOX@RGD-WHc on day 6 and 9, and injection of DOX@RGD-DHBc on day 12 and 15. The mice were sacrificed on day 18 and the tumors, organs and sera of mice were collected after treatment. (b) The CT26 tumor (n = 5) growth curves after intravenous injection of different formulations of DOX at a dose of 3 mg kg−1. (c) Photographic images of tumors from CT26 tumor-bearing mice on day 18 after treatment (n = 5). (d) The body weight variation of CT26 tumor-bearing mice during treatment (n = 5). € VLPs-specific IgG antibody titers of CT26 tumor-bearing mice (n = 5) on day 15. The statistical significance of the results was analyzed and indicated: * p < 0.05, ** p < 0.01, *** p < 0.001. | ||
The serum of each group of mice was collected after treatment and the antibody titers of RGD-HBc, RGD-WHc, and RGD-DHBc in the DOX@RGD-HBc treatment group and DOX@VLPs sequential treatment group were detected using ELISA (Fig. 4e). Since DOX@RGD-HBc was administrated in both groups of mice, the antibody titers in their serum were high. While in the multiple DOX@VLPs group, anti-RGD-WHc and anti-RGD-DHBc antibody titers were low, only around 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 040 and 4480 (1/dilution). The changes in antibody titers reflected the insignificant cross-immunogenicity of these heterogenous VLPs and they could be used in sequence to improve the delivery efficiency.
040 and 4480 (1/dilution). The changes in antibody titers reflected the insignificant cross-immunogenicity of these heterogenous VLPs and they could be used in sequence to improve the delivery efficiency.
Even though the apparent relative lack of toxicity in vitro and low cross-immunity in vivo made these RGD-VLPs attractive carriers for sequential treatment, evaluation of their systematic toxicity in vivo is still meaningful for their further extended application. By inhibiting the synthesis of DNA and RNA, DOX has a good killing effect on hematologic and solid tumors, but long-term use of doxorubicin is prone to cardiotoxicity.37 Therefore, the major organs and tumors of the sequentially treated mice were analyzed by H&E staining (Fig. S5, ESI†). Compared with the PBS treatment group, the H&E staining of sliced organs, especially the heart, showed no significant damage and toxic side effects in mice treated with different formulations, and this result indicated that sequential administration of various RGD-VLPs could effectively reduce the cardiotoxicity of DOX.
4. Conclusions
HBc VLPs are promising protein cages for efficient drug delivery; however, the existing HBc VLP-based platform technology is plagued with immunological problems that may limit its full potential as a drug carrier. In this work, our main aim was to assess the possibility of using immune-orthogonal RGD-VLPs sequentially to circumvent the immune response and targeted delivery of chemotherapeutic drugs for tumor treatment. Our results suggest that the genetical insertion of RGD onto the main immunogenic loop regions can significantly reduce the immunogenicity of HBcAg and these structurally similar VLPs had no significant cross-immunity. In addition, these monodisperse well-defined RGD-VLPs have good biocompatibility and could be used as efficient drug carriers to deliver Doxorubicin. The results of in vivo treatment of tumor-bearing mice showed that compared with using DOX or DOX@RGD-HBc singly, the sequential treatment with different DOX@RGD-VLPs could further inhibit tumor progression and metastasis. Overall, our finding that sequential administration of immune-orthogonal virus-like particle-based nanomedicine could lower antibody-mediated immune clearance and improve anti-tumor effect has the potential to shape the design of future clinical therapies.Author contributions
Chufan Wang and Cheng Xiao contributed equally to this work. Chufan Wang, Cheng Xiao, Yurong Chen, Qiang Zhang, Xiumin Wang and Lei Ren conceived and designed the experiments. Chufan Wang, Cheng Xiao and Yurong Chen performed the experiments. Chufan Wang, Cheng Xiao, Qiang Zhang, Yurong Chen and Yao Li collected and analyzed the data. Xiumin Wang, Yulin Li, Shengli Bi, Wenjun Shan and Yunlong Wang provided suggestions and technical support on the project. Chufan Wang, Cheng Xiao, Yurong Chen and Lei Ren prepared the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (U1904206, 31870994, 32271469), and the Fujian Provincial Science and Technology Department Guiding Project (2022Y0001).References
- J. M. V. Makabenta, A. Nabawy, C. H. Li, S. Schmidt-Malan, R. Patel and V. M. Rotello, Nat. Rev. Microbiol., 2021, 19(1), 23–36 CrossRef CAS PubMed.
- A. R. Kirtane, M. Verma, P. Karandikar, J. Furin, R. Langer and G. Traverso, Nat. Nanotechnol., 2021, 16(4), 369–384 CrossRef CAS PubMed.
- S. Shah, V. Dhawan, R. Holm, M. S. Nagarsenker and Y. Perrie, Adv. Drug Delivery Rev., 2020, 154, 102–122 CrossRef PubMed.
- J. Wang, J. J. Nie, P. Guo, Z. Yan, B. Yu and W. Bu, J. Am. Chem. Soc., 2020, 142(6), 2709–2714 CrossRef CAS PubMed.
- H. Kheraldine, O. Rachid, A. M. Habib, A. E. Al Moustafa, I. F. Benter and S. Akhtar, Adv. Drug Delivery Rev., 2021, 178, 113908 CrossRef CAS PubMed.
- F. Scaletti, J. Hardie, Y. W. Lee, D. C. Luther, M. Ray and V. M. Rotello, Chem. Soc. Rev., 2018, 47, 3421–3432 RSC.
- Y. Z. Long, M. Yu, B. Sun, C. Z. Gu and Z. Fan, Chem. Soc. Rev., 2012, 41(12), 4560–4580 RSC.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- E. P. Stater, A. Y. Sonay, C. Hart and J. Grimm, Nat. Nanotechnol., 2021, 16(11), 1180–1194 CrossRef CAS PubMed.
- J. Wang, Y. Li and G. Nie, Nat. Rev. Mater., 2021, 6(9), 766–783 CrossRef CAS PubMed.
- N. Habibi, A. Mauser, Y. Ko and J. Lahann, Adv. Sci., 2022, 9(8), 2104012 CrossRef PubMed.
- E. J. Lee, N. K. Lee and I.-S. Kim, Adv. Drug Delivery Rev., 2016, 106, 157–171 CrossRef CAS PubMed.
- Y. H. Chung, H. Cai and N. F. Steinmetz, Adv. Drug Delivery Rev., 2020, 15, 6214–6235 Search PubMed.
- S. Nooraei, H. Bahrulolum, Z. S. Hoseini, C. Katalani, A. Hajizade, A. J. Easton and G. Ahmadian, J. Nanobiotechnol., 2021, 19(1), 59 CrossRef CAS PubMed.
- N. F. Steinmetz, S. Lim and F. Sainsbury, Biomater. Sci., 2020, 8(10), 2771–2777 RSC.
- M. O. Mohsen, D. E. Speiser, A. Knuth and M. F. Bachmann, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12(1), e1579 CAS.
- M. O. Mohsen, G. Augusto and M. F. Bachmann, Immunol. Rev., 2020, 296(1), 155–168 CrossRef CAS PubMed.
- S. Mobini, M. Chizari, L. Mafakher, E. Rismani and E. Rismani, Front Immunol., 2020, 11, 2074 CrossRef CAS PubMed.
- W. Shan, D. Zhang, Y. Wu, X. Lv, B. Hu, X. Zhou, S. Ye, S. Bi, L. Ren and X. Zhang, Nanomedicine, 2018, 14(3), 725–734 CrossRef CAS PubMed.
- J. Zhao, Z. S. Ye, J. Yang, Q. Zhang, W. J. Shan, X. M. Wang, Z. X. Wang, S. F. Ye, X. Zhou, Z. C. Shao and L. Ren, Biomaterials, 2020, 240, 119849 CrossRef CAS PubMed.
- W. Shan, R. Chen, Q. Zhang, J. Zhao, B. Chen, X. Zhou, S. Ye, S. Bi, L. Nie and L. Ren, Adv. Mater., 2018, 30(28), 1707567 CrossRef PubMed.
- W. Shan, H. Zheng, G. Fu, C. Liu, Z. Li, Y. Ye, J. Zhao, D. Xu, L. Sun, X. Wang, X. L. Chen, S. Bi, L. Ren and G. Fu, Nano Lett., 2019, 19(3), 1719–1727 CrossRef CAS PubMed.
- Q. Zhang, W. Shan, C. Ai, Z. Chen, T. Zhou, X. Lv, X. Zhou, S. Ye, L. Ren and X. Wang, Nanotheranostics, 2018, 2(1), 87–95 CrossRef PubMed.
- Q. R. Jia, D. Y. Li, Q. Zhang, S. F. Ye, Z. Xi, X. M. Wang, W. J. Shan and L. Ren, J. Mater. Sci., 2019, 54(20), 13255–13264 CrossRef CAS.
- K. Cheng, T. Du, Y. Li, Y. Qi, H. Min, Y. Wang, Q. Zhang, C. Wang, Y. Zhou, L. Li, S. Ye, X. Zhou, S. Bi, J. Yang and L. Ren, ACS Appl. Mater. Interfaces, 2020, 12(48), 53682–53690 CrossRef CAS PubMed.
- V. Jawa, L. P. Cousens, M. Awwad, E. Wakshull, H. Kropshofer and A. S. De Groot, Clin. Immunol., 2013, 149(3), 534–555 CrossRef CAS PubMed.
- F. Mingozzi and K. A. High, Blood, 2013, 122(1), 23–36 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- Q. Zhang, L. Li, Q. Lan, M. Li, D. Wu, H. Chen, Y. Liu, D. Lin, W. Qin, Z. Zhang, J. Liu and W. Yang, Crit. Rev. Food Sci. Nutr., 2019, 59(15), 2506–2533 CrossRef CAS PubMed.
- N. H. Nguyen, F. Y. Glassman, R. K. Dingman, G. N. Shenoy, E. A. Wohlfert, J. G. Kay, R. B. Bankert and S. V. Balu-Iyer, Sci. Rep., 2021, 11(1), 17853 CrossRef CAS PubMed.
- R. Chen, S. Huang, T. Lin, H. Ma, W. Shan, F. Duan, J. Lv, J. Zhang, L. Ren and L. Nie, Nat. Nanotechnol., 2021, 16(4), 455–465 CrossRef CAS PubMed.
- D. Jain and D. M. Salunke, Biochem. J., 2019, 476, 433–447 CrossRef CAS PubMed.
- A. M. Moreno, N. Palmer, F. Alemán, G. Chen, A. Pla, N. Jiang, W. Leong Chew, M. Law and P. Mali, Nat. Biomed. Eng., 2019, 3(10), 806–816 CrossRef CAS PubMed.
- J.-N. Billaud, D. Peterson, F. Schödel, A. Chen, M. Sallberg, F. Garduno, P. Goldstein, W. McDowell, J. Hughes, J. Jones and D. Milich, J. Virol., 2005, 79(21), 13641–13655 CrossRef CAS PubMed.
- M. Nassal, I. Leifer, I. Wingert, K. Dallmeier, S. Prinz and J. Vorreiter, J. Virol., 2007, 81(23), 13218–13229 CrossRef CAS PubMed.
- J.-N. Billaud, D. Peterson, B. O. Lee, T. Maruyama, A. Chen, M. Sallberg, F. Garduño, P. Goldstein, J. Hughes, J. Jones and D. Milich, Vaccine, 2007, 25(9), 1593–1606 CrossRef CAS PubMed.
- A. Pugazhendhi, T. Edison, B. K. Velmurugan, J. A. Jacob and I. Karuppusamy, Life Sci., 2018, 200, 26–30 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb02163c |
| This journal is © The Royal Society of Chemistry 2023 |