
Synthesis, kinetic studies, and atom transfer reactivity of [2Fe–2E] model compounds†
Erwin A.
Weerawardhana
a,
Matthias
Zeller
b and
Wei-Tsung
Lee
 *a
*a
aDepartment of Chemistry and Biochemistry, Loyola University Chicago, Chicago, Illinois 60660, USA. E-mail: wlee5@luc.edu
bDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA
First published on 1st June 2023
Abstract
The synthesis of [2Fe–2E] (E = S and Se) complexes supported by an N-alkyl,N′-aryl-β-diketiminate ligand is described. The [2Fe–2S] model compound has an unusually long Fe⋯Fe distance, but the equivalent distance in the [2Fe–2Se] compound is comparable to those reported in the literature. These model compounds display varying electron transfer capabilities, which were elucidated through electronic spectroscopy and electrochemical analysis. Kinetic data and activation parameters suggest the formation mechanism of the [2Fe–2S] compound. Additionally, to probe the possible sulfur-transfer mechanism of [2Fe–2S] clusters, atom transfer reactivity was pursued.
Introduction
Iron–sulfur clusters ([mFe–nS], where m and n = 2–4) form redox active, multiple-metal cofactors in the active sites of many enzymes. These clusters are often viewed as special inorganic cofactors and elicit a variety of biological functions, including electron transfer, sensing, and regulation.1–4 A majority of the functions of such clusters stem from their redox reactivity. The electronic structure of iron–sulfur clusters has been widely studied and it was found that the oxidation state of iron centers commonly ranges from +2 to +3,1,5 which contrasts with the higher oxidation states found in heme systems. A common structural feature of these clusters is that each iron center is coordinated by four donor atoms (N-donor or S-donor) in a tetrahedral environment.1Although many functions of iron–sulfur clusters are considered well-understood, there are two newly emerging interests in this area: (1) the exploration of new biological functions of iron–sulfur clusters, and (2) understanding the electronic origins of the chemistry of iron–sulfur versus iron–selenium clusters and how these may influence or determine enzyme function and mechanism. For example, the sulfur-atom transfer involving an iron–sulfur cluster in the last step of the biotin biosynthetic pathway has long been debated.6–11 In addition, several studies have shown that sulfur atoms can be substituted by selenium in iron–sulfur clusters while retaining structural integrity.12–14 Studying iron–selenium clusters or related model compounds could yield valuable information as to why S has been universally selected to function in these clusters over Se at the active sites of enzymes.15,16
Researchers have extensively explored structure–function relationships of naturally occurring iron–sulfur clusters. Synthetic model iron sulfur compounds can provide additional insight into the complexity of the natural systems. While continuous efforts have led to the successful synthesis of [3Fe–4S] and [4Fe–4S] model clusters,17,18 methods to make and study [2Fe–2E] (where E = S and Se) models are less developed.11–14 The production of synthetic [2Fe–2S/Se] clusters with supporting chalcogenide ligand and its effect on spectroscopy and reduction potentials of natural systems has been reported.9 Due to the coordination arrangement, some bidentate ligands have been successfully demonstrated to mimic the [2Fe–2S] active site of enzymes.19–24 More recently, bidentate β-diketiminates were used to synthesize [2Fe–2S] clusters owing to their high tunability as supporting ligands.25–27 [2Fe–2Se] model systems supported by the nitrogen donor ligands are however rather rare (Scheme 1).23,28,29
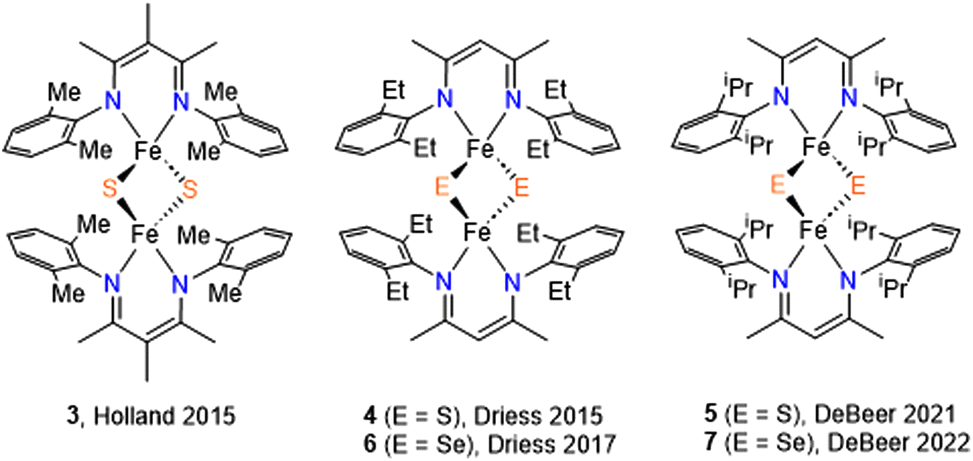 | ||
| Scheme 1 [2Fe–2S] and [2Fe–2Se] model compounds supported by β-diketiminate ligands. | ||
We recently reported an asymmetric N-alkyl,N′-aryl-β-diketiminate (L1) ligand and its low-valent iron complex30 as a suitable candidate for preparing [2Fe–2E] model compounds. Herein we report the synthesis of [2Fe–2E] model compounds 1 and 2, which were characterized by X-ray crystallography, 1H-NMR spectroscopy, and electrochemical studies. In the context of the recent advances in [2Fe–2E] model compounds, our objective was to gain a deeper understanding of their formation from low-valent iron precursors and elemental sulfur. Thus, we conducted kinetic experiments to investigate the kinetic isotopic effect and activation parameters involved in the reaction of an Fe(I) complex with elemental sulfur, resulting in the formation of 1. Moreover, in light of the unusually long Fe⋯Fe distance noted in 1, further investigations were performed to explore the atom transfer reactions.
Experimental section
Materials and methods
All manipulations were performed under a nitrogen atmosphere using standard Schlenk techniques or in an M. Braun UNIlab Pro glovebox. Glassware was dried at 150 °C overnight. Diethyl ether, n-pentane, tetrahydrofuran, and toluene were purified using a pure process technology solvent purification system. Before use, an aliquot of each solvent was tested with a drop of sodium benzophenone ketyl in THF solution. All reagents were purchased from commercial vendors and used as received. (L1)Fe(cod) (cod = 1,5-cyclooctadiene) was prepared according to a literature procedure.301H NMR data were recorded on a Varian Inova 500 MHz spectrometer at 22 °C. Resonances in the 1H NMR spectra are referenced to residual C6D5H at δ = 7.16 ppm. Solution magnetic susceptibilities were determined using the Evans method.31 UV-vis spectra were recorded on an Agilent Cary 8454 UV-vis spectrophotometer equipped with a Unisoku Scientific Instruments Cryostat USP-203B for various temperature experiments. An IR spectrum of 1 was recorded on a Jasco FT-IR spectrometer (model 4600) equipped with an ATR diamond probe head. Elemental analysis was conducted by Midwest Microlab, LLC (Indianapolis, IN).Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 080), 327 (sh) (19800), 430 (5900), 448 (sh) (5400), 545 (6440). Anal. Calcd for C50H66Fe2N4S2: C, 66.81; H, 7.40; N, 6.23. Found: C, 66.90; H, 7.24; N, 6.18.
080), 327 (sh) (19800), 430 (5900), 448 (sh) (5400), 545 (6440). Anal. Calcd for C50H66Fe2N4S2: C, 66.81; H, 7.40; N, 6.23. Found: C, 66.90; H, 7.24; N, 6.18.
Cautious: When handling and heating concentrated H2SO4, it is imperative to exercise utmost caution given its strong corrosive properties and potential hazards.
560), 434 (2880), 613 (3000). Anal. Calcd for C50H66Fe2N4Se2: C, 60.13; H, 6.59; N, 5.72. Found: C, 60.31; H, 6.44; N, 5.83.
Electrochemistry
Cyclic voltammetry was conducted using a CH-Instruments electrochemical analyzer (model 620E), employing a 3 mm glassy carbon working electrode, a silver wire pseudo-reference electrode, and a platinum coiled wire counter electrode. All measurements were performed using THF solutions, containing 1 mM analyte and 0.1 M N(Bu)4PF6 as the supporting electrolyte. The potentials were referenced to a ferrocene/ferrocenium redox couple.Crystallography
Data were collected using a Rigaku Rapid II curved image plate diffractometer with Cu-Kα radiation (λ = 1.54178 Å). Single crystals were mounted on a Mitegen micromesh mount using a trace of Fomblin oil and cooled in situ to at 100 K. Data were processed using HKL3000 and the data were corrected for absorption and scaled using Scalepack.33 The space groups were assigned using XPREP within the SHELXTL suite of programs,34,35 were solved by direct methods using ShelXS and refined by full matrix least squares against F2 with all reflections using Shelxl2018 using the graphical interface ShelXle.36 H atoms attached to carbon atoms were positioned geometrically and constrained to ride on their parent atoms, with carbon hydrogen bond distances of 0.95 Å for alkene and aromatic C–H, 1.00 and 0.98 Å for aliphatic C–H and CH3, respectively. Uiso(H) values were set to a multiple of Ueq(C) with 1.5 for CH3 and 1.2 for C–H groups respectively.Kinetic measurements
Pseudo-first-order reaction conditions were employed for all kinetic studies, using 20-fold and greater excesses of 32S8 or 34S8. Reactions were monitored by UV-Vis spectroscopy. Due to the low solubility of S8, low concentrations of (L1)Fe(cod) were used in kinetic experiments. Typically, 3 mL of a toluene solution containing the (L1)Fe(cod) complex (0.3 μmol) in a sealable cuvette under N2 was placed in a temperature-controlled cryostat. Upon temperature equilibration, a toluene solution of 32S8 or 34S8 was added rapidly via syringe. Spectra were taken at 30 s to 45 min intervals. The reaction was followed by monitoring the increase in one of the ligand-to-metal charge transfer (LMCT) absorption bands used to indicate the formation of 1. Nonlinear least-squares fitting of the absorbance at 545 nm using a first-order model yielded the pseudo-first-order rate constants, kobs. The transition state parameters were determined over a temperature range of −40 to −70 °C and via an Eyring plot, which shows a linear relationship. S/Se atom transfer reactions were performed by mixing (L1)Fe(cod) complex (9.5 μmol) with an excess of S8 or Se8 and PPh3 in C6D6 at 40 °C and monitored by 1H NMR spectroscopy. The formation of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3 and SePPh3 were quantified with respect to hexamethylbenzene (internal standard). Each parameter reported represents the average value of duplicate or triplicate determinations.
PPh3 and SePPh3 were quantified with respect to hexamethylbenzene (internal standard). Each parameter reported represents the average value of duplicate or triplicate determinations.
Results and discussion
Syntheses
1 was synthesized by the reaction of S8 with (L1)Fe(cod) at room temperature (Scheme 2). Immediately a color change of the solution from green to dark purple was observed. However, when treating Se8 with (L1)Fe(cod) to yield 2, the reaction occurred at a much slower rate with a gradual color change to teal over the course of several hours. Note that reactions run at lower temperature did not produce significantly improved yields. At room temperature, the 1H NMR spectra of compounds 1 and 2 display characteristics of paramagnetism, manifesting as broadening and up-field shifted peaks (−4.5 and −6.0 ppm for 1 and 2, respectively), despite most of the resonances lying within the diamagnetic region. The β-methyl protons are responsible for the paramagnetic chemical shifts, which can also be observed in other [2Fe–2S] model compounds containing β-diketiminate ligands (−0.4 ppm for 3 and −8.1 ppm for 4).26,27 The magnetic properties of synthetic [2Fe–2E] model compounds are primarily determined by strong antiferromagnetic coupling between the two high-spin S = 5/2 Fe3+ ions.19–21,23,24 The presence of an antiferromagnetic coupled dimer was evident by measuring the magnetic moment of 1 and 2 in C6D6 which were found to be by Evan's method 1.2 and 1.3μB, respectively. The values agree with those of other reported [LFe(μ-E)]2 complexes.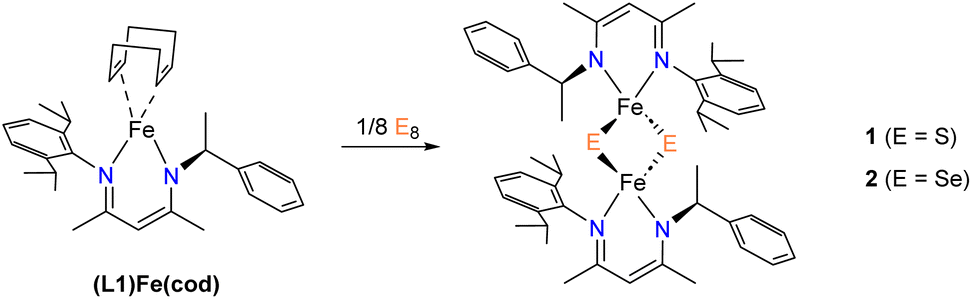 | ||
| Scheme 2 Synthesis of 1 and 2. | ||
Crystal structures of 1 and 2
Single crystals suitable for X-ray structure determination were obtained for complex 1 and 2. The ORTEP diagrams of 1 and 2 are given in Fig. 1 and selected bond lengths and angles are summarized in Table 1 and Table S1.† The solid-state structures confirm complexes 1 and 2 as binuclear iron complexes, where each iron center is coordinated by two N atoms from a β-diketiminate ligands and two bridged S atoms (1) or Se atoms (2) (Fig. 1). The molecular geometry of the metal ion in 1 (τ4 = 0.847) and 2 (τ4 = 0.932) is best described as distorted tetrahedral. For 1, the average Fe–N distances of 1.989(7) Å agrees with those of other β-diketiminate supported complexes of [2Fe–2S] clusters bearing high-spin Fe(III) centers.26,27 However, the average bond distances of Fe–S (2.377(3) Å) and inter-atomic distance of Fe⋯Fe (3.360(2) Å) are much longer than any of the reported values for other synthetic [2Fe–2S] model compounds. On the other hand, the average bond distance of Fe–Se (2.317(7) Å) and inter-atomic distance of Fe⋯Fe (2.750(3) Å) in 2 are only slightly longer than those in [(Pipiso)Fe(μ-Se)]2 (Pipiso = [(DipN)2C(cis-2,6-Me2NC5H8)]−, (Dip = 2,6-C6H3iPr2). Interestingly, the Fe⋯Fe distance in [(Pipiso)Fe(μ-E)]2 (E = O, S, Se) reflects the size of bridging chalcogenide ligands, but the opposite trend was found for 1 and 2.23 Therefore, by comparing the crystal structures of 1 and 2 the unusual Fe⋯Fe distance found in 1 can be attributed to the steric effect of methyl groups pointing toward the aryl ring.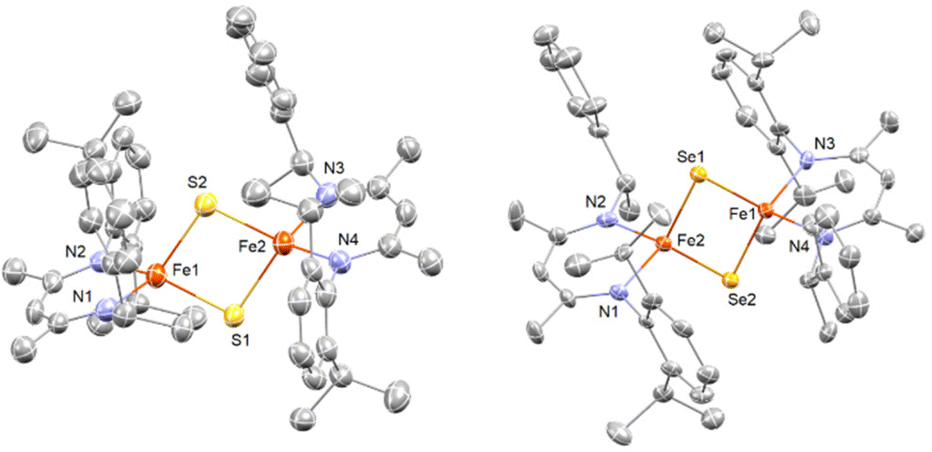 | ||
| Fig. 1 X-ray crystal structures of 1 (left) and 2 (right). Thermal ellipsoids shown at 50% probability, hydrogen atoms and solvate molecules (toluene, pentane) omitted for clarity. Color key: orange = Fe, blue = N, gray = C, yellow = S, gold = Se. | ||
a
| 1 (E = S) |
326 |
427 |
525 |
2 (E = Se) |
629 |
|
|---|---|---|---|---|---|---|
| a Numbers in parentheses are standard uncertainties in the last significant figures. Atoms are labeled as indicated in Fig. 1. b Torsion angle is defined by a dihedral angle between two N–Fe–N planes. | ||||||
| Fe–N | 1.989(7) | 1.973(5) | 2.004(1) | 2.028(2) | 1.983(5) | 1.996(2) |
| Fe–E | 2.377(3) | 2.204(2) | 2.194(1) | 2.210(1) | 2.317(1) | 2.318(1) |
| ∠N–Fe–N | 94.6(3) | 97.7(2) | 93.23(6) | 91.64(8) | 96.1(3) | 92.6(1) |
| ∠E–Fe–E | 90.07(9) | 105.49(7) | 104.72(2) | 100.82(3) | 107.23(8) | 107.46(2) |
| ∠Fe–E–Fe | 89.83(9) | 74.51(6) | 75.28(2) | 79.18(2) | 72.77(7) | 72.55(2) |
| Fe⋯Fe | 3.360(2) | 2.669(1) | 2.679(1) | 2.816(1) | 2.750(3) | 2.743(1) |
| N–Fe–Fe–Nb | 11.17 | 2.44 | 8.43 | 1.93 | 1.92 | 5.66 |
| τ 4 | 0.816 | 0.910 | 0.891 | 0.895 | 0.932 | 0.913 |
The Fe⋯Fe distance for most [2Fe–2S] model complexes are typically in a small range of 2.69–2.82 Å, which are smaller than protein [2Fe–2S] clusters.19–24,26,27,37,38 It is known that a short Fe⋯Fe distance is one of the contributions for strong antiferromagnetic coupling between two Fe(III) centers.39 However, diamagnetism of complex 1 suggests antiferromagnetic coupling occurring in solution although the Fe⋯Fe distance is rather long in solid state. In comparison of [2FeIII–2S] with the super-reduced cluster, [2FeII–2S], the difference of their Fe⋯Fe distances is <0.1 Å.21,24 Furthermore, long Fe⋯Fe distances (3.2 to 3.4 Å) are often observed in thiolate-bridged complexes [2FeII-2SR],40–42 which may imply that complex 1 is a [2FeII-2SH] cluster compound and the hydrogen atoms were not resolved by X-ray crystallography. However, this possibility can be ruled out since no characteristic S–H stretching vibrations were observed in 1.
In addition, although some monosulfido-bridged Fe(II) complexes are diamagnetic,43,44 most [2FeII-2SR] complexes are paramagnetic with large magnetic moments.40–42 Hence, 1 is most likely a [2FeIII–2S] model compounds with an unusual long Fe–Fe distance resulting from the steric repulsion between the two β-diketiminate ligands. The steric profiles were also suggested by the τ4 values among all [2Fe–2S] model complexes supported by β-diketiminate ligands (Table 1). In addition, as the steric bulkiness increases, the torsion angle between the two chelated faces (N–Fe–N) increases (Table 1 and Fig. S1†).
Electrochemical and UV–vis studies of complex 3 and 4
Cyclic voltammetry of complex 1 (Fig. 2) suggests there are two quasi-reversible events occurring, while there is only one quasi-reversible event observed for complex 2 (Fig. S2†). These events indicate the electron transfer is a diffusion controlled process which was confirmed by the linear relationship in the plot of peak current vs. square root of the scan rate (Fig. 2 inset). The first quasi-reversible event of 1 appears at −1.68 V and the second is at −2.13 V. However, the peak height of the first event is twice as large as the second event. This suggests the first event may involve two times the number of electrons with respect to the second event. To understand the number of electrons transferred based on the voltammograms, titration of complex 1 with a chemical reductant, bis-pentamethylcyclopentadienylcobalt(II) (CoCp*2) was monitored using UV-vis spectroscopy. Since the second quasi-reversible event of 1 is at −2.13 V, we can use this chemical reductant, whose redox potential is −1.94 V, to investigate only the first electron transfer process at −1.61 V.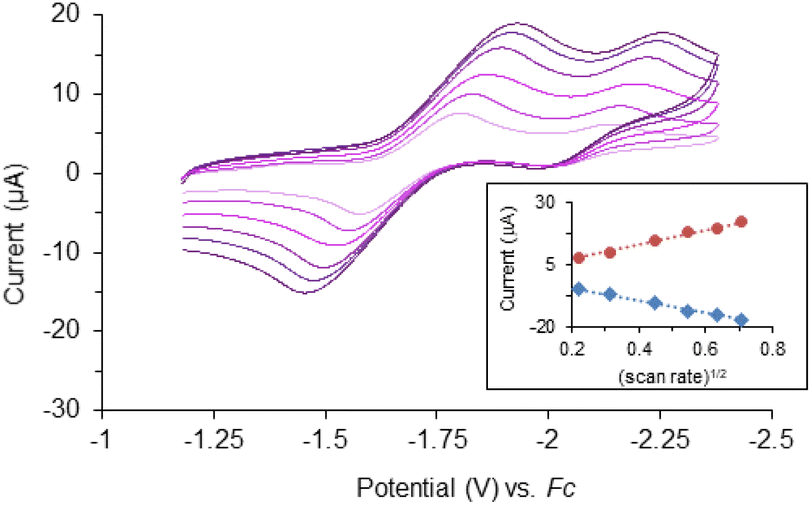 | ||
Fig. 2 Overlaid cyclic voltammograms of 1 (1 mM) in THF with 0.1 M N(Bu)4PF6. Scan rates = 50, 100, 200, 300, 400, 500 mV s−1 referenced to a ferrocene/ferrocenium redox couple (Fc). Inset: plot of cathodic peak current ( , R2 = 0.9953) and anodic peak current ( , R2 = 0.9953) and anodic peak current ( , R2 = 0.9962) versus square root of scan rate. , R2 = 0.9962) versus square root of scan rate. | ||
Complex 1 shows three major LMCT bands (300, 431, and 545 nm) which are consistent with those observed in other model compounds and Rieske-type proteins with the oxidation state of both iron ions being 3+.23,29,45,46 Complex 2, on the other hand, exhibits two charge-transfer bands that are bathochromically shifted compared to 1 (Fig. S3†). The red-shift of the LMCT bands is consistent with the trend of ligand field transition energy of the bridging atoms. These features diminished upon titration with reductant, CoCp*2 (Fig. 3). Further addition of the chemical reductant past two equivalents did not show any additional change. Therefore, the 2:1 (CoCp*2: 1) molar ratio suggests the first quasi-reversible event of 1 involves two electrons and implies the second event is a one-electron process. The single quasi-reversible event of complex 2 is at −1.91 V and could represent the transfer of one or two electrons. Similarly, titration of 2 with CoCp*2 was monitored by using UV-vis spectroscopy at λmax = 614 nm, which diminished and did not show any change after addition of 1 equiv. of CoCp*2 (Fig. S4†). Therefore, this 1:1 (CoCp*2: 2) molar ratio suggests that the single quasi-reversible event of 2 involves one electron. Attempts to reduce 1 were encouraged by the quasi-reversible redox events and relatively stable in THF solution upon reduction. However, the reactions led to intractable products through many trials under different conditions (e.g. variety of reductants, solvents, and temperatures). Although several reduced species supported by other bidentate nitrogen-donor ligands were successfully synthesized and reported,21,24,26,27 the futile attempts for isolating the reduction products of 1 could be due to the unusually long Fe⋯Fe distance.
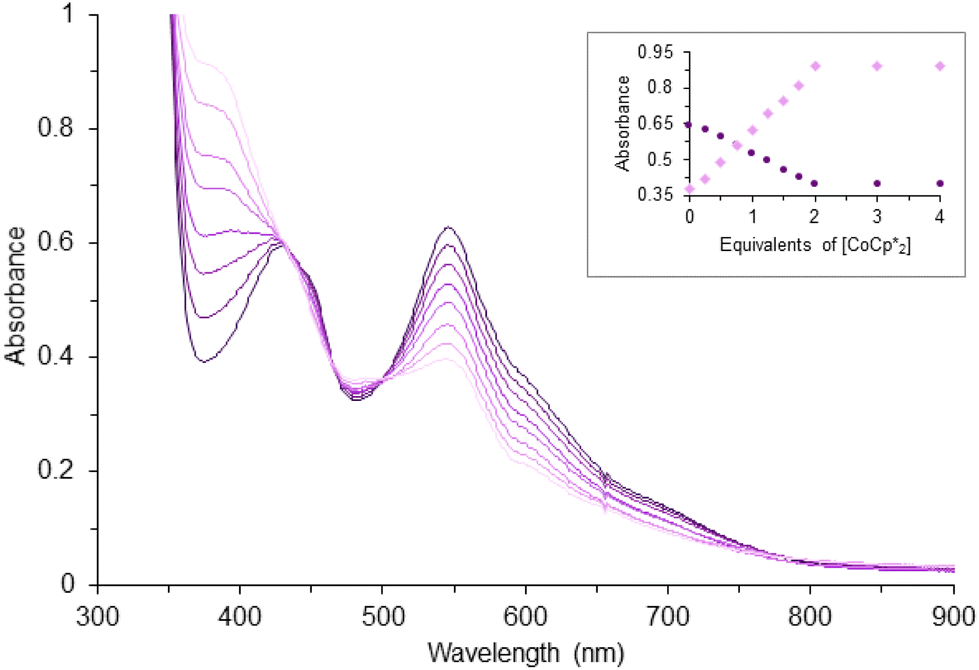 | ||
Fig. 3 Reduction of 0.1 mM 1 in THF by addition of CoCp*2 in increments of 0.25 equiv. as monitored by UV-vis spectroscopy (path length, 1.0 cm). Inset: corresponding changes of the absorbance at 389 ( ) and 545 ( ) and 545 ( ) nm. ) nm. | ||
Kinetic studies of the formation of 1
Since the synthesis of [2Fe–2S] model compounds are not always straightforward, with attempts often yielding homoleptic complexes, L2Fe (L = bidentate ligands), an alternative procedure was discovered by the oxidation of elemental sulfur with mono- or di-nuclear Fe(I) precursors.23,24,26 Therefore, to gain insights into the formation of 1, we carried out kinetic studies for the activation of elemental sulfur with the low-valent Fe complex, (L1)Fe(cod). When the observed rate constants kobs are plotted as a function of the concentration of S8, a linear dependence that, within error, passes through the origin is observed (Fig. S5 and S6†). These data indicate a bimolecular reaction between (L1)Fe(cod) and S8. When comparing the rates for reactions conducted under the same conditions with 34S8, the kinetic isotopic effect (KIE) of k32S8/k34S8 = 1.02. The normal KIE value suggests that the rate-determining step is likely the cleavage of the S–S bond, but we cannot completely rule out the possibility of experimental errors based on this value. More insights into the formation mechanism of 1 from (L1)Fe(cod) and 32S8 was gained from temperature-dependence kinetic experiments. An Eyring analysis over a temperature range of −40 to −70 °C gave the activation parameters Δ‡H = 17.0 ± 0.6 kJ mol−1 and Δ‡S = −155 ± 8 J mol−1 K−1 which correspond to an associated Δ‡G298 K = 63.2 ± 0.8 kJ mol−1 (Fig. S7†). The positive enthalpy of activation presumably implies cleavage of the S–S bond in the transition state. The large negative entropy of activation suggests the reaction proceeds via an associative mechanism and could be due to the formation of a highly ordered transition state where a partially broken S–S bond of the S8 molecule chelates the Fe center.Sulfur atom transfer studies
Albeit still under debate, sulfur atom transfer reactions in enzymes with [2Fe–2S] clusters may play an important role in metabolic reactions e.g. biotin synthase transforms dethiobiotin to biotin. Considering the long Fe–S bond length, 1 could be a suitable candidate for an S-atom transfer reaction. When treating 1 with PPh3 at 40 °C, the formation of SPPh3 was observed by 1H NMR spectroscopy. This result has encouraged us to further study the potential catalytic S-atom transfer reaction. However, due to the limited solubility of S8, it was not possible to obtain consistent kinetic data when using excess (10-fold) amounts of S8. Thus, the only viable approach to determine whether the iron complex catalyzes the S-atom transfer reaction is to obtain the overall yields of SPPh3 from reaction of 1 with triphenyl phosphine. Direct reaction of S8 with PPh3 was expected to be a much slower process. Since Fe(I) species are considered to be generated after transferring an S atom from 1, we tested if catalytic atom transfer is possible by mixing an Fe(I) precursor, (L1)Fe(cod), with excess S8 and PPh3. The mixture was maintained at 40 °C for one hour and the formation of SPPh3 was confirmed and quantified (yield = 70%) by 31P and 1H NMR spectroscopy.47 Conversely, in the absence of (L1)Fe(cod), only 11% conversion was observed. Scheme S1† presented the proposed reaction mechanism, whereby mixing (L1)Fe(cod) with S8 resulted in the immediate formation of 1, which subsequently reacted with PPh3 to generate SPPh3. The residual two-coordinate Fe(I) species could form species A through binding with an excess amount of triphenylphosphine ligand(s). Note that the two-coordinate Fe(I) species or A may act as an active catalyst, or alternatively, the regeneration of (L1)Fe(cod) could occur through ligand substitution of free cyclooctadiene and triphenylphosphine ligand(s). The Se atom transfer reaction, under the same conditions, was slower, regardless of the presence (12%) or absence (5%) of (L1)Fe(cod).
Conclusions
This study reports two [2Fe–2E] model compounds, [(L1)Fe(μ-S)]2 (1) and [(L1)Fe(μ-Se)]2 (2), supported by a chiral diketiminate ligand. The presence of steric hindrance in 1 is believed to be the cause of its remarkably elongated Fe⋯Fe distance, which is thought to have played a role in the failure to synthesize reduced [2Fe–2S] compounds. 1 is capable of transferring three electrons whereas a single electron redox event is observed in 2. For the formation of 1, kinetic analysis suggests that the cleavage of the S–S bond is the rate-determining step and the reaction proceeds via an associative mechanism. The highly efficient S-atom transfer reaction could be attributed to the unusually long Fe–S bond length in 1, and these results imply that S-atom transfer reactions may occur in biological relevant [2Fe–2S] clusters. The ongoing research is to gain further insight into the reaction mechanism of E-atom transfer reactions with the goal of facilitating the efficient synthesis of chiral E-containing compounds by utilizing these [2Fe–2E] model complexes as catalysts.Author contributions
Erwin A. Weerawardhana: writing original draft, synthesis and characterization of compounds. Matthias Zeller: crystallography. Wei-Tsung Lee: experimental design, supervision, data validation, manuscript review, and editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by start-up funds (Loyola University Chicago). The X-ray diffractometers were funded by National Science Foundation through the Major Research Instrumentation Program under grants no. CHE 1625543 (Purdue University). We thank Dr Daniel J. McElheny (University of Illinois at Chicago) for acquiring 31P-NMR spectra.References
- H. Beinert, R. H. Holm and E. Münck, Iron-sulfur clusters: nature's modular, multipurpose structures, Science, 1997, 277, 653–659 CrossRef CAS PubMed.
- J. C. Crack, J. Green, A. J. Thomson and N. E. L. Brun, Iron–Sulfur Clusters as Biological Sensors: The Chemistry of Reactions with Molecular Oxygen and Nitric Oxide, Acc. Chem. Res., 2014, 47, 3196–3205 CrossRef CAS PubMed.
- E. L. Mettert and P. J. Kiley, Fe-S proteins that regulate gene expression, Biochim. Biophys. Acta, 2015, 1853, 1284–1293 CrossRef CAS PubMed.
- J. A. Santos, N. Alonso-García, S. Macedo-Ribeiro and P. J. Pereira, The unique regulation of iron-sulfur cluster biogenesis in a Gram-positive bacterium, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E2251–E2260 CrossRef CAS PubMed.
- W. R. Hagen, in Adv. Inorg. Chem, ed. R. Cammack, Academic Press, 1992, vol. 38, pp. 165–222 Search PubMed.
- A. P. Bali, D. Lennox-Hvenekilde, N. Myling-Petersen, J. Buerger, B. Salomonsen, L. S. Gronenberg, M. O. A. Sommer and H. J. Genee, Improved biotin, thiamine, and lipoic acid biosynthesis by engineering the global regulator IscR, Metab. Eng., 2020, 60, 97–109 CrossRef CAS PubMed.
- F. Berkovitch, Y. Nicolet, J. T. Wan, J. T. Jarrett and C. L. Drennan, Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme, Science, 2004, 303, 76–79 CrossRef CAS PubMed.
- B. T. Bui, D. Florentin, F. Fournier, O. Ploux, A. Méjean and A. Marquet, Biotin synthase mechanism: on the origin of sulphur, FEBS Lett., 1998, 440, 226–230 CrossRef CAS PubMed.
- K. J. Gibson, D. A. Pelletier and I. M. Turner Sr., Transfer of sulfur to biotin from biotin synthase (BioB protein), Biochem. Biophys. Res. Commun., 1999, 254, 632–635 CrossRef CAS PubMed.
- J. T. Jarrett, Biotin synthase: enzyme or reactant?, Chem. Biol., 2005, 12, 409–410 CrossRef CAS PubMed.
- C. Sirithanakorn and J. E. Cronan, Biotin, a universal and essential cofactor: synthesis, ligation and regulation, FEMS Microbiol. Rev., 2021, 45, 1–18 Search PubMed.
- S. B. Yu, G. C. Papaefthymiou and R. H. Holm, Comprehensive iron-selenium-thiolate cluster chemistry, Inorg. Chem., 1991, 30, 3476–3485 CrossRef CAS.
- J. G. Reynolds and R. H. Holm, Core chalcogenide atom substitution reactions of [Fe2X2(SR)4]2− and [Fe4X4(SR)4]2−,3− clusters (X = sulfur, selenium), Inorg. Chem., 1981, 20, 1873–1878 CrossRef CAS.
- J. M. Moulis and J. Meyer, Characterization of the selenium-substituted 2[4Fe-4Se] ferredoxin from Clostridium pasteurianum, Biochemistry, 1982, 21, 4762–4771 CrossRef CAS PubMed.
- T. M. Buscagan, J. T. Kaiser and D. C. Rees, Selenocyanate derived Se-incorporation into the nitrogenase Fe protein cluster, eLife, 2022, 11, e79311 CrossRef CAS PubMed.
- J. T. Henthorn, R. J. Arias, S. Koroidov, T. Kroll, D. Sokaras, U. Bergmann, D. C. Rees and S. DeBeer, Localized Electronic Structure of Nitrogenase FeMoco Revealed by Selenium K-Edge High Resolution X-ray Absorption Spectroscopy, J. Am. Chem. Soc., 2019, 141, 13676–13688 CrossRef CAS PubMed.
- F. Fabrizi de Biani and P. Zanello, The competition between chemistry and biology in assembling iron-sulfur derivatives. Molecular structures and electrochemistry. Part IV. {[Fe3S4](SγCys)3} proteins, Inorg. Chim. Acta, 2017, 455, 319–328 CrossRef CAS.
- P. Zanello, The competition between chemistry and biology in assembling iron–sulfur derivatives. Molecular structures and electrochemistry. Part V. {[Fe4S4](SCysγ)4} proteins, Coord. Chem. Rev., 2017, 335, 172–227 CrossRef CAS.
- A. Albers, T. Bayer, S. Demeshko, S. Dechert and F. Meyer, A Functional Model for the Rieske Center: Full Characterization of a Biomimetic N-Ligated [2Fe-2S] Cluster in Different Protonation States, Chem. – Eur. J., 2013, 19, 10101–10106 CrossRef CAS PubMed.
- J. Ballmann, X. Sun, S. Dechert, E. Bill and F. Meyer, Relatively stable N-ligated [2Fe2S]2+ clusters with dipyrromethane capping ligands, J. Inorg. Biochem., 2007, 101, 305–312 CrossRef CAS PubMed.
- A. Albers, S. Demeshko, K. Pröpper, S. Dechert, E. Bill and F. Meyer, A Super-Reduced Diferrous [2Fe–2S] Cluster, J. Am. Chem. Soc., 2013, 135, 1704–1707 CrossRef CAS PubMed.
- A. Albers, S. Demeshko, S. Dechert, E. Bill, E. Bothe and F. Meyer, The Complete Characterization of a Reduced Biomimetic [2Fe-2S] Cluster, Angew. Chem., Int. Ed., 2011, 50, 9191–9194 CrossRef CAS PubMed.
- L. Fohlmeister, K. R. Vignesh, F. Winter, B. Moubaraki, G. Rajaraman, R. Pöttgen, K. S. Murray and C. Jones, Neutral diiron(III) complexes with Fe2(μ-E)2 (E = O, S, Se) core structures: reactivity of an iron(I) dimer towards chalcogens, Dalton Trans., 2015, 44, 1700–1708 RSC.
- Q. Liang, J. C. DeMuth, A. Radović, N. J. Wolford, M. L. Neidig and D. Song, [2Fe–2S] Cluster Supported by Redox-Active o-Phenylenediamide Ligands and Its Application toward Dinitrogen Reduction, Inorg. Chem., 2021, 60, 13811–13820 CrossRef CAS PubMed.
- R. G. Castillo, A. W. Hahn, B. E. Van Kuiken, J. T. Henthorn, J. McGale and S. DeBeer, Probing Physical Oxidation State by Resonant X-ray Emission Spectroscopy: Applications to Iron Model Complexes and Nitrogenase, Angew. Chem., Int. Ed., 2021, 60, 10112–10121 CrossRef CAS PubMed.
- M. E. Reesbeck, M. M. Rodriguez, W. W. Brennessel, B. Q. Mercado, D. Vinyard and P. L. Holland, Oxidized and reduced [2Fe–2S] clusters from an iron(I) synthon, J. Biol. Inorg. Chem., 2015, 20, 875–883 CrossRef CAS PubMed.
- S. Yao, F. Meier, N. Lindenmaier, R. Rudolph, B. Blom, M. Adelhardt, J. Sutter, S. Mebs, M. Haumann, K. Meyer, M. Kaupp and M. Driess, Biomimetic [2Fe-2S] Clusters with Extensively Delocalized Mixed-Valence Iron Centers, Angew. Chem., Int. Ed., 2015, 54, 12506–12510 CrossRef CAS PubMed.
- J. T. Henthorn, G. E. Cutsail, T. Weyhermüller and S. DeBeer, Stabilization of intermediate spin states in mixed-valent diiron dichalcogenide complexes, Nat. Chem., 2022, 14, 328–333 CrossRef CAS PubMed.
- C. Panda, P. W. Menezes, C. Walter, S. Yao, M. E. Miehlich, V. Gutkin, K. Meyer and M. Driess, From a Molecular 2Fe-2Se Precursor to a Highly Efficient Iron Diselenide Electrocatalyst for Overall Water Splitting, Angew. Chem., Int. Ed., 2017, 56, 10506–10510 CrossRef CAS PubMed.
- E. A. Weerawardhana, A. Pena, M. Zeller and W.-T. Lee, Synthesis and characterization of iron and cobalt complexes with an asymmetric N-alkyl,N′-aryl-β-diketiminate ligand, Inorg. Chim. Acta, 2017, 460, 29–34 CrossRef CAS.
- E. M. Schubert, Utilizing the Evans Method with a Superconducting NMR Spectrometer in the Undergraduate Laboratory, J. Chem. Educ., 1992, 69, 62 CrossRef CAS.
- J. Ebels, S. Spirk and R. Pietschnig, Presented in part at the Proceedings of the 10th International Electronic Conference on Synthetic Organic Chemistry, Basel, Switzerland, 1–30 November 2006, 2006.
- Z. Otwinowski and W. Minor, in Methods Enzymol, Academic Press, 1997, vol. 276, pp. 307–326 Search PubMed.
- SHELXTL, 6.14; Bruker Advanced X-ray Solutions, Bruker AXS Inc., Madison, Wisconsin, USA, 2000–2003 Search PubMed.
- G. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- J. Ballmann, S. Dechert, S. Demeshko and F. Meyer, Tuning Electronic Properties of Biomimetic [2Fe-2S] Clusters by Ligand Variations, Eur. J. Inorg. Chem., 2009, 2009, 3219–3225 CrossRef.
- J. Ballmann, X. Sun, S. Dechert, B. Schneider and F. Meyer, A convenient ligand exchange pathway to [2Fe–2S] ferredoxin analogues, Dalton Trans., 2009, 4908–4917, 10.1039/B901242G.
- J. J. Mayerle, R. B. Frankel, R. H. Holm, J. A. Ibers, W. D. Phillips and J. F. Weiher, Synthetic analogs of the active sites of iron-sulfur proteins. Structure and properties of bis(o-xylyldithiolato-μ2-sulfidoferrate (3)), an analog of the 2Fe-2S proteins, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 2429–2433 CrossRef CAS PubMed.
- D. Sellmann, J. Utz and F. W. Heinemann, Transition-Metal Complexes with Sulfur Ligands. 132. Electron-Rich Fe and Ru Complexes with [MN2S3] Cores Containing the New Pentadentate Ligand ‘N2H2S3′2− (=2,2′-Bis(2-mercaptophenylamino)diethyl Sulfide(2−)), Inorg. Chem., 1999, 38, 459–466 CrossRef CAS PubMed.
- L. Wang, M. Gennari, F. G. Cantú Reinhard, S. K. Padamati, C. Philouze, D. Flot, S. Demeshko, W. R. Browne, F. Meyer, S. P. de Visser and C. Duboc, O2 Activation by Non-Heme Thiolate-Based Dinuclear Fe Complexes, Inorg. Chem., 2020, 59, 3249–3259 CrossRef CAS PubMed.
- D. K. Mills, Y. M. Hsiao, P. J. Farmer, E. V. Atnip, J. H. Reibenspies and M. Y. Darensbourg, Applications of the N2S2 ligand, N,N’-bis(mercaptoethyl)-1,5-diazacyclooctane (BME-DACO), toward the formation of bi- and heterometallics: [(BME-DACO)Fe]2 and [(BME-DACO)NiFeCl2]2, J. Am. Chem. Soc., 1991, 113, 1421–1423 CrossRef CAS.
- C.-Y. Lu and W.-F. Liaw, Formation Pathway of Roussin's Red Ester (RRE) via the Reaction of a {Fe(NO)2}10 Dinitrosyliron Complex (DNIC) and Thiol: Facile Synthetic Route for Synthesizing Cysteine-Containing DNIC, Inorg. Chem., 2013, 52, 13918–13926 CrossRef CAS PubMed.
- S. E. Creutz and J. C. Peters, Diiron Bridged-Thiolate Complexes That Bind N2 at the FeIIFeII, FeIIFeI, and FeIFeI Redox States, J. Am. Chem. Soc., 2015, 137, 7310–7313 CrossRef CAS PubMed.
- D. Coucouvanis, A. Salifoglou, M. G. Kanatzidis, A. Simopoulos and V. Papaefthymiou, Dimeric complexes containing the disulfidodiiron(2+) cores coordinated by non-sulfur containing terminal ligands. The crystal and molecular structures of the tetraethylammonium salts of the [Fe2S2(o,o’-C12H8O2)2]2− and [Fe2S2(C4H4N)4]2− anions, J. Am. Chem. Soc., 1984, 106, 6081–6082 CrossRef CAS.
- M. E. Konkle, S. K. Muellner, A. L. Schwander, M. M. Dicus, R. Pokhrel, R. D. Britt, A. B. Taylor and L. M. Hunsicker-Wang, Effects of pH on the Rieske Protein from Thermus thermophilus: A Spectroscopic and Structural Analysis, Biochemistry, 2009, 48, 9848–9857 CrossRef CAS PubMed.
- T. A. Albright, W. J. Freeman and E. E. Schweizer, Nuclear magnetic resonance studies. IV. Carbon and phosphorus nuclear magnetic resonance of phosphine oxides and related compounds, J. Org. Chem., 1975, 40, 3437–3441 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2252043 and 2252044. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi00728f |
| This journal is © the Partner Organisations 2023 |