
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing magneto-ionic effects in cobalt oxide films by electrolyte engineering†
Sofia
Martins
 *a,
Zheng
Ma
a,
Xavier
Solans-Monfort
b,
Mariona
Sodupe
b,
Luis
Rodriguez-Santiago
b,
Enric
Menéndez
a,
Eva
Pellicer
*a and
Jordi
Sort
ac
*a,
Zheng
Ma
a,
Xavier
Solans-Monfort
b,
Mariona
Sodupe
b,
Luis
Rodriguez-Santiago
b,
Enric
Menéndez
a,
Eva
Pellicer
*a and
Jordi
Sort
ac
aDepartament de Física, Universitat Autònoma de Barcelona, E-08193 Cerdanyola del Vallès, Spain. E-mail: CarlaSofia.Martins@uab.cat; Eva.Pellicer@uab.cat
bDepartament de Química, Universitat Autònoma de Barcelona, E-08193 Cerdanyola del Vallès, Spain
cInstitució Catalana de Recerca i Estudis Avançats (ICREA), Pg. Lluís Companys 23, E-08010 Barcelona, Spain
First published on 25th November 2022
Abstract
Electric-field-driven ion motion to tailor magnetic properties of materials (magneto-ionics) offers much promise in the pursuit of voltage-controlled magnetism for highly energy-efficient spintronic devices. Electrolyte gating is a relevant means to create intense electric fields at the interface between magneto-ionic materials and electrolytes through the so-called electric double layer (EDL). Here, improved magneto-ionic performance is achieved in electrolyte-gated cobalt oxide thin films with the addition of inorganic salts (potassium iodide, potassium chloride, and calcium tetrafluoroborate) to anhydrous propylene carbonate (PC) electrolyte. Ab initio molecular dynamics simulations of the EDL structure show that K+ is preferentially located on the cobalt oxide surface and KI (when compared to KCl) favors the accumulation of positive charge close to the surface. It is demonstrated that room temperature magneto-ionics in cobalt oxide thin films is dramatically enhanced in KI-containing PC electrolyte at an optimum concentration, leading to 11-fold increase of generated magnetization and 35-fold increase of magneto-ionic rate compared to bare PC.
New conceptsVoltage control of magnetism (VCM) through electric field-induced ion motion, referred to as magneto-ionics, is a cutting-edge research topic since it holds the promise to revolutionize current strategies to enhance energy efficiency in magnetically actuated devices (e.g., micro-electro-mechanical systems, magnetic logics, spintronics, or neuromorphic computing). Modifying the film composition/microstructure or the working conditions are the typical approaches followed in the literature to boost magneto-ionics. However, at present, room temperature oxygen magneto-ionics is still too slow for relevant applications. Increased O2− motion speeds and smaller threshold voltages are highly desirable. Here, we demonstrate that O2− motion in electrolyte-gated cobalt oxide films can be significantly enhanced by suitable ‘electrolyte engineering’ in films of a few nm in thickness. In particular, inorganic salts (e.g., KI, KCl and (Ca(BF4)2), have been added to anhydrous propylene carbonate (PC) to modulate the ionic strength and, in turn, the electric field at the cobalt oxide/electrolyte interface by means of the built-in electric double layer. For KI-containing PC, a 35-fold increase of the magneto-ionic rate is observed compared to plain PC and, importantly, under a relatively low bias value (<−1.5 V), much lower than the voltage values needed so far to induce magneto-ionics in this kind of systems. |
Introduction
Advancements in further downsizing the next-generation spintronic devices while minimizing their Joule power dissipation have been remarkable in the last decades.1,2 Voltage control of magnetism (VCM) promises great potential to reduce the energy demands of spintronics. As such, magnetoelectric (ME) effects, related to the coupling between magnetic and electric behaviors in matter, have been proposed as one of the most promising avenues to realize the manipulation of magnetic properties by an electric field. This can be accomplished, to some extent, in ME multiferroics, where ferroelectricity and magnetism coexist and are usually coupled. Nevertheless, several challenges must be overcome, such as the scarcity of intrinsic single-phase ME materials, the weak coupling between electric polarization and magnetization at room temperature, and the clamping effects in the case of ME composites (or heterostructured multiferroics). Alternatively, voltage-driven ion motion to tune magnetism (i.e., magneto-ionics) has recently attracted great attention thanks to its ability to considerably modulate magnetic properties, in a permanent manner.3 In recent years, magneto-ionics has been demonstrated in a wide variety of materials, including both, metals and semiconductors.4Electrolyte-gating through the formation of an electric double layer (EDL) has shown to be a very convenient means to tailor the magnetic properties of magneto-ionic systems. These systems work as electrochemical capacitors (EC), i.e., supercapacitors, thereby involving the formation of an EDL between the solid material and the electrolyte. At least three types of ECs can be distinguished, namely electrical double layer capacitors (EDLC), pseudocapacitors, and a combination of both.5,6 Whatever the type, ECs refer to a system capable to accumulate electrical charges at the surface of an electrode material in contact with an electrolyte. When voltage is applied to a magneto-ionic material, the electric field causes changes in its saturation magnetization (MS) or coercivity (HC), among other magnetic properties, which can be finely tuned based on electrostatic and/or electrochemical mechanisms.7,8
According to the different models put in place to explain the concept of EDLs formed at the interface between planar solids and aqueous solutions, the simplest theory states that EDLs form to neutralize the surface charge on the solid material by ions of opposite sign (Helmholtz model). The more elaborated Gouy–Chapman–Stern model (GCS) is currently widely used to describe EDLs. It assumes the occurrence of two layers, one fixed layer next to the electrode surface (Stern layer or Helmholtz region) of opposite charge, and a diffuse layer, wherein ions can move freely. The Stern layer, in turn, is divided into the inner Helmholtz plane (IHP) and the outer Helmholtz plane (OHP). The former has approximately one single ion thickness. Compared to aqueous solutions, studies on the structure of EDL in organic solvents are more limited. Feng et al. examined a system made of tetraethylammonium tetrafluoroborate (TEABF4) salt in acetonitrile (CH3CN) and found remarkable contact adsorption of TEABF4 ions to the surface of the electrode and strong layering and orientational ordering of acetonitrile molecules until approx. 1.1 nm into the bulk electrolyte.9 The EC based on interfacial ion-modulation has attracted much interest because of the giant charge-carrier density accumulation achieved because of the large EDL capacitance. Under electrolyte gating, permanent changes at the surface and the bulk of electrode material are possible, whenever chemical reactions are involved.
Materials such as carbon, metal oxides and nitrides, and conducting polymers have been used in an EC.5,6 Metal oxides/nitrides such as Co3O410–12 and CoN13,14 are very appealing for magneto-ionics, since they not only store energy like electrostatic carbon materials but can also undergo electrochemical reactions under voltage actuation, eventually leading to a fully reversible transformation from a paramagnetic state (OFF) to a ferromagnetic state (ON), and vice versa. This is explained by oxygen and nitrogen motion, respectively, under voltage actuation, the concurrent creation of atomic defects, and the consequent generation of magnetic moment arising from metallic Co.10–14
The maximum electric field achievable in ECs is limited by the breakdown voltage of the electrolyte. Four primary classes of electrolyte solutions have been used in EC: organic, aqueous, polymeric and ionic liquid electrolytes. Among organic electrolytes, ACN and propylene carbonate (PC) are the most widely used solvents. ACN presents the ability to solvate large amounts of salts but it is toxic, while PC is more environmentally friendly, offers a wide electrochemical window and operating temperature, as well as conductivity of 1–2 × 10−8 Ohm−1 cm−1.5,15,16 Indeed, PC is a dipolar protic solvent with high dielectric constant (64.40 at 25 °C) and a large dipole moment (4.81 D) which has been considered an ideal structureless dielectric to solubilize strong electrolytes for a variety of applications. Since PC has a relatively large dielectric constant, dissolved ionic compounds will be extensively dissociated.
In spite of the achievements with VCM of Co oxide films, room temperature magneto-ionics is still too slow for relevant applications. Even though sub-10 s magneto-ionics has recently been demonstrated by reducing the Co oxide thickness down to 5 nm,12 further improvements are required to enable magneto-ionics in real devices. In addition, the applied voltages required to observe a remarkable change in the magnetic properties of cobalt oxide were rather large, of the order of −50 V. Recently, we showed that magneto-ionics depends on the electrical properties of the target materials.4 Here we demonstrate that not only the electrical properties of cobalt oxide are important, but also the composition of the electrolyte. More specifically, its ionic strength also plays a key role in magneto-ionics. In the past years, various salts and additives have been added to solvents to increase the ionic strength of electrolytes for a variety of purposes.17–19 As the salt concentration is increased, more ions can accumulate in the compact layer of the EDL, reducing the presence of free solvent molecules in the vicinity of the electrode. As a result, the performance of energy storage systems improves.17,20 This strategy could well work for magneto-ionics, paving the way for higher charge accumulation at the surface of the solid material and, hence, more intense electric fields. To date, different electrolytes have been tested for voltage-driven ion migration and, in some cases, salts were added to the solvent (e.g. LiPF6 in dimethyl carbonate/ethylene carbonate mixture) to observe the effects.7,21,22 In spite of the intensive research carried out in the last decade, studies with complex PC formulations are lacking.
Herein, we have investigated the impact of the addition of inorganic salts, namely potassium iodide (KI), potassium chloride (KCl) and calcium tetrafluoroborate (Ca(BF4)2), to anhydrous PC on the magneto-ionic effects induced in 15 nm-thick Co oxide films. The solubility of KI and KCl in PC at room temperature was reported by Peruzzi et al.23 and that of Ca(BF4)2 in PC was investigated by Forero-Saboya et al.17 (see Experimental section). KCl was primarily chosen as it is the electrolyte support of choice in many electrochemical processes taking place in aqueous solutions. KI was selected as it showed higher solubility in PC and simply implied the replacement of Cl− by I−. Finally, Ca(BF4)2 was chosen since it is typically dissolved in alkyl carbonates like PC in calcium ion batteries.24 Our work reveals that improvements in oxygen ion motion25–27 in cobalt oxide are accomplished by adding these salts to PC. Specifically, the fastest magneto-ionic rate is observed when using PC + 2.5 × 10−4 M KI electrolyte, which is 35 times larger than the rate observed for bare PC electrolyte under −1.5 V. Moreover, upon biasing Co oxide for one hour, the same PC + 2.5 × 10−4 M KI electrolyte results in the largest generation of magnetization. The process is highly reversible for all added salts since the generated magnetism can be reduced after applying a voltage of opposite polarity (i.e., +1.5 V). The magneto-ionic response of the cobalt oxide films was further studied at varying concentrations of KI dissolved in PC. Our results show that improved magneto-ionics can be achieved by conveniently tuning the EDL strength via liquid electrolyte engineering.
Results and discussion
To investigate the magneto-ionic behavior of 15 nm thick Co oxide films in different electrolyte solutions, a home-made electrolytic cell, sketched in Fig. 1(a), was used which allowed to externally polarize the material while recording its magnetic response under in-plane configuration on a vibrating sample magnetometer (VSM). As illustrated in Fig. 1(b), an electrokinetic potential is created at the solid/electrolyte interface under no external electric field applied, considering that EDLs form whenever an electronic conducting material is placed in an electrolyte containing mobile ions.3 This is of course a very simplistic cartoon because the cobalt oxide film is sketched as if it acquires negative charge while positive charges gather together at the solid/electrolyte interface, which is not necessarily the case. Under electrolyte gating (Fig. 1(c)), specifically when cobalt oxide is negatively polarized, positive ions migrate towards the interface and PC molecules reorganize and rearrange, thus strongly causing a modification of the EDL structure.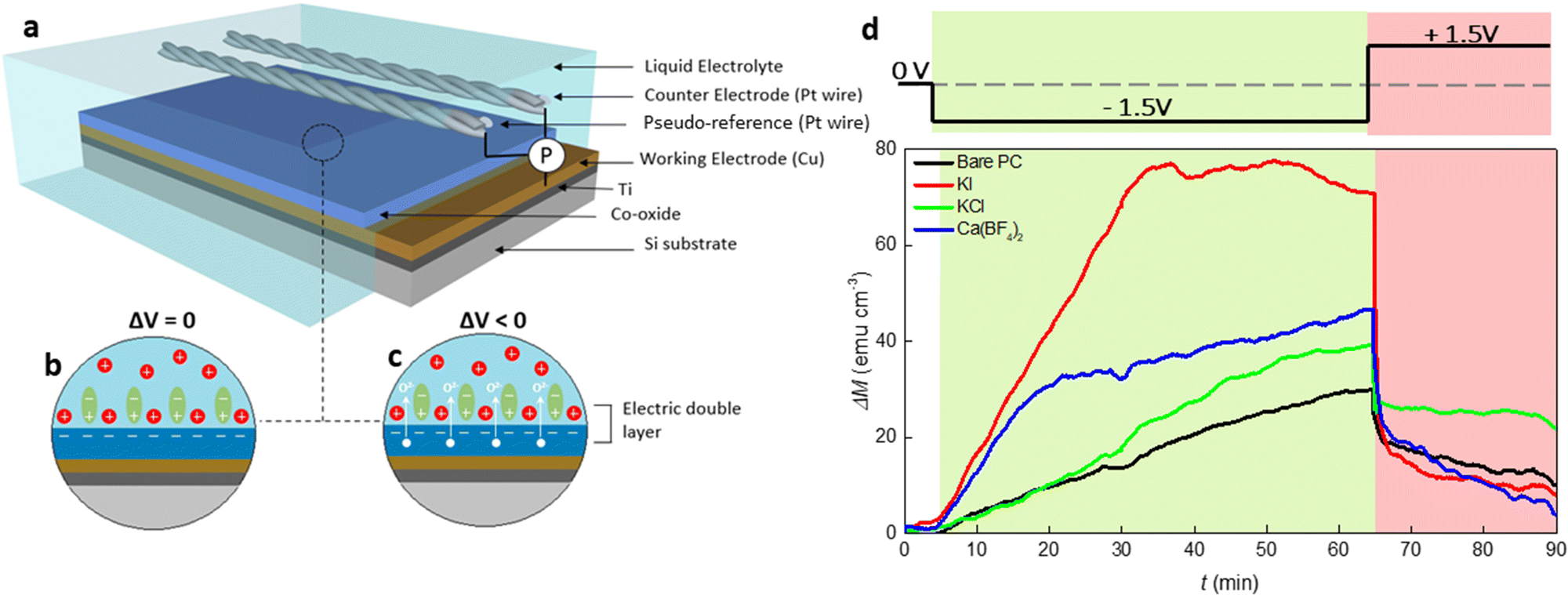 | ||
| Fig. 1 Magneto-ionic characterization of Co oxide under electrolyte-gating. (a) Schematics of the electrolyte gate process under voltage actuation of a Co oxide film. Possible structure of the EDL under (b) no applied electric field (electrostatic forces), and (c) external polarization (electrochemical reaction). (d) Time evolution of magnetization (ΔM) for bare PC and PC-containing KI, KCl and Ca(BF4)2 salts at equal salt concentration (2.5 × 10−5 M), at −1.5 V for 60 min followed by +1.5 V (as schematically shown in the upper panel). Magnetic measurements were done in-plane while applying an external magnetic field of 10 KOe. | ||
The magnetic moment of cobalt oxide in different electrolyte solutions was measured at room temperature under a 10 kOe magnetic field, where the generated ferromagnetic response is saturated. The external voltage was applied in situ during the magnetic measurements. Fig. 1(d) presents the time evolution of magnetization change (ΔM − t) for the films actuated in bare PC and PC containing 2.5 × 10−5 M KI, KCl and Ca(BF4)2 electrolytes. In the first stage of the measurements, ΔM continuously increases to around 30 emu cm−3 for the bare PC electrolyte while ΔV = −1.5 V is applied for 60 min (i.e., t = 65 min) (see the green shaded region in Fig. 1(d)). This indicates the appearance of ferromagnetism in the cobalt oxide. In the second stage (i.e., after applying +1.5 V for 25 min), ΔM sharply drops due to the reversal of the bias polarity, and reaches around 10 emu cm−3. The occurrence of a ferromagnetic-paramagnetic (ON–OFF) transition in the oxide films is related to voltage-induced oxygen ion motion.10 Several observations can be made from the ΔM − t curves for electrolytes with different inorganic salts. First, for the PC + KCl electrolyte, ΔM increases at a similar rate as the bare PC one until t = 20 min, and it then grows faster and approaches 40 emu cm−3 at t = 65 min, representing a 33% increase compared to bare PC. Importantly, the initial ΔM change is significantly enhanced with the additions of KI and Ca(BF4)2 into PC, as reflected by the larger slopes of the blue and red curves for t < 20 min and t < 35 min, respectively. Furthermore, the maximum ΔM values at t = 65 min are much higher in the electrolytes containing KI and Ca(BF4)2 than for bare PC and PC + KCl. Remarkably, the maximum ΔM for PC + 2.5 × 10−5 M KI electrolyte is twice higher than that for bare PC (70 emu cm−3versus 30 emu cm−3). Finally, the sudden decrease of ΔM when applying voltage of opposite polarity is observed for all the electrolytes. In particular, ΔM drops from around 70 emu cm−3 to 10 emu cm−3 during the time interval from t = 65 min to 70 min for the electrolyte made of PC + 2.5 × 10−5 M KI. These observations indicate that magneto-ionic effects in cobalt oxide can be substantially improved by electrolyte engineering. It is worth mentioning that the relatively small gating voltage employed in the current study (1.5 V) can produce similar magneto-ionic responses as previous works where we used applied voltages of the order of 25 V or higher for similar Co oxide film thicknesses.11,12 This decrease in the threshold voltage needed to induce changes in the magnetic properties of the Co oxide can be directly linked to the addition of inorganic salts into PC. Interestingly, although one would expect KCl and KI to yield similar results upon negatively biasing the cobalt oxide film, ΔM was far higher for KI.
To get atomistic insights on the initial cobalt oxide – propylene carbonate + KX (X = Cl, I) interface, ab initio molecular dynamics (AIMD) simulations at 300 K were carried out. Co3O4 was considered for the simulations. Energies were computed at the PBE-D3+U (Ueff = 3.0 eV) without applying any external potential. The statistical analysis was performed on the last 8 (6 depending on the structure) ps of each simulation (see Computational details). We paid attention on the location of KX (X = Cl or I) and thus, two different initial structures were considered for each salt (Fig. S1 of the ESI†). In one case, the two ions were directly interacting with the surface at distances around 2 Å (KXAds). In the other, the salt was fully solvated by PC molecules (KXSol). Fig. 2 reports: (a) one representative snapshot of the simulation leading to the most favorable conformation for each salt KClAds and KISol AIMD, (b) the variation of the surface–ion distances in the simulations leading to the most favorable situation and (c) the energetics of the four AIMD. The variation of the potassium–surface distance, the anion–surface distance and a representative snapshot of the four simulations can be found in Fig. S2, S3 and S4 of the ESI.†
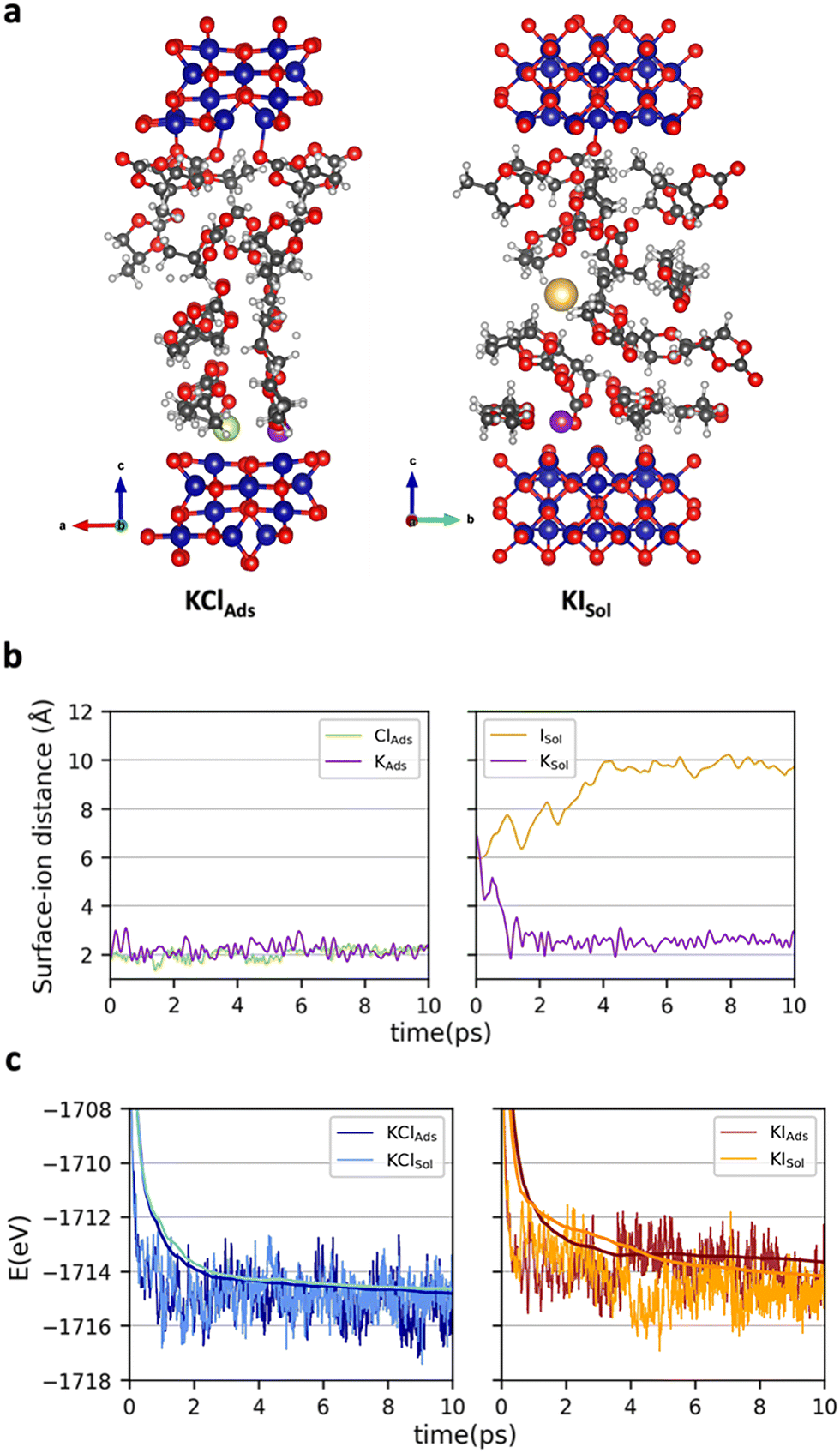 | ||
| Fig. 2 (a) Representative snapshots of the most favorable KClAds and KISol AIMD simulations; variation along the AIMD simulation of the (b) surface–ion distance (K, Cl and I) at the most favorable conformation and (c) energy. Label code: Co (blue), O (red), C (black), H (white), K (purple), Cl (green), I (pale orange). | ||
The AIMD simulations starting with the two ions adsorbed on the surface evolved without any relevant variation. All ions remain attached at the surface with average ion···surface distances of 2.2, 2.1 and 2.4 Å for K+, Cl− and I−, respectively. In these simulations no significant charge separation is generated, suggesting that almost no positive charge will be accumulated on the Co3O4 surface. The simulations starting with the two ions in the solution (KClSol and KISol) showed a larger reorganization. Regardless of the counter ion, K+ rapidly becomes adsorbed on the surface with K···surface average distances that are only slightly higher than in the KXAds simulation (2.5–2.6 Å). In contrast, the anions remain in the solution. The Cl− anion moves around the initial structure with an average surface–Cl− distance of 6.5 Å. Iodide prefers to move further away from the surface, the resulting surface–anion distances becoming about 9.8 Å. That is, the KXSol simulations lead to a structure defining an EDL, with a compact positive charge accumulation on Co3O4. Comparison between the energetics of the KXAds and KXSol simulations rises the different behavior of the anions. For KCl, the configuration in which the two ions are directly adsorbed on the surface is slightly more stable (ΔG = 0.2 eV) than the structure defining the double layer. In contrast, for KI, there is a large preference (ΔG = −1.0 eV) for the conformation that accumulates positive charge on the surface. This can be attributed to the largest ionic radius of I− when compared to Cl− that makes all electrostatic interactions (and particularly that with the surface) weaker. While the AIMD simulations are carried out without applying an external potential, one can expect that upon externally biasing Co3O4 layer with negative voltages, the structure accumulating positive charge on the surface would be favored with respect to that with the two ions close to the surface but without modifying the trends between the two ions. Consequently, simulations shows that KI favors the structure accumulating more positive charge over Co3O4 and, in turn, generates a stronger electric field.
In order to further optimize the magneto-ionic response for the KI-containing PC electrolyte, the room temperature magnetic moment as a function of biasing time were recorded with a 10 kOe field and varying KI concentrations (from 2.5 × 10−6 M to 2.5 × 10−3 M). Upon biasing for 1 hour, the ΔM first increases as the KI concentration increases from 2.5 × 10−6 M to 2.5 × 10−4 M, and then it decreases with further addition of KI. At 2.5 × 10−3 M KI, the oxide film peeled off, possibly because of the strong electrochemical reactions taking place (see the dashed curve in Fig. 3(a)). Indeed, the solution acquired a yellow color, indicating the formation of I2 gas at the Pt counter electrode, and the subsequent formation of I3− according to the reaction:
| I2(g) + I(aq)− = I(aq)3− | (1) |
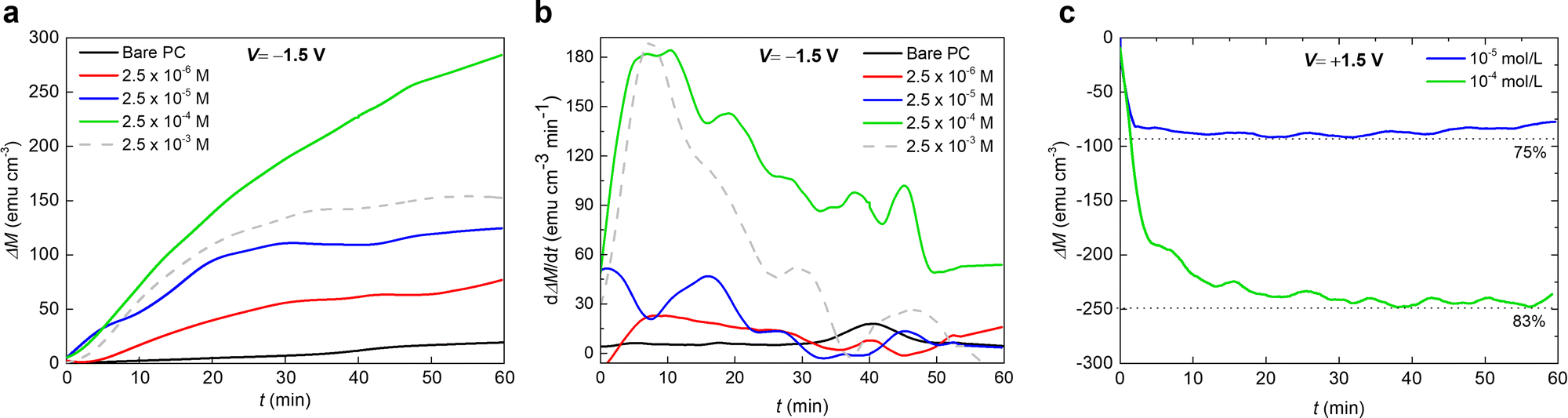 | ||
| Fig. 3 Impact of KI concentration in PC on Co oxide magneto-ionics. (a) Magnetization (ΔM) as a function of time t for PC with different KI concentrations under electrolyte-gating at −1.5 V, while applying an external in-plane magnetic field of 10 KOe, and (b) the respective magneto-ionic rate, dΔM/dt. (c) Magnetization (ΔM) depletion as a function of time t for the two best performing KI concentrations under an opposite bias (+1.5 V), while applying an external in-plane magnetic field of 10 KOe. | ||
Remarkably, after applying −1.5 V for 1 hour, the maximum ΔM for 2.5 × 10−4 M KI electrolyte (≈280 emu cm−3) is more than one order of magnitude larger than that for bare PC (≈25 emu cm−3). The achieved magneto-ionic rate (i.e., the rate at which the ferromagnetic signal from metallic Co is generated) is drastically enhanced from 5 emu cm−3 min−1 for bare PC to 184 emu cm−3 min−1 for PC + 2.5 × 10−4 M KI (see Fig. 3(b)). Moreover, partial recovery under an opposite bias of +1.5 V was observed for the best performing KI concentrations of 2.5 × 10−4 M and 2.5 × 10−5 M (Fig. 3(c)). Room temperature hysteresis loops of the voltage-actuated cobalt oxide films confirm the effect of the KI concentration in PC on the ON–OFF ferromagnetic switching for this system (Fig. 4). This corroborates that magneto-ionic effects have a strong dependence on the KI concentration.
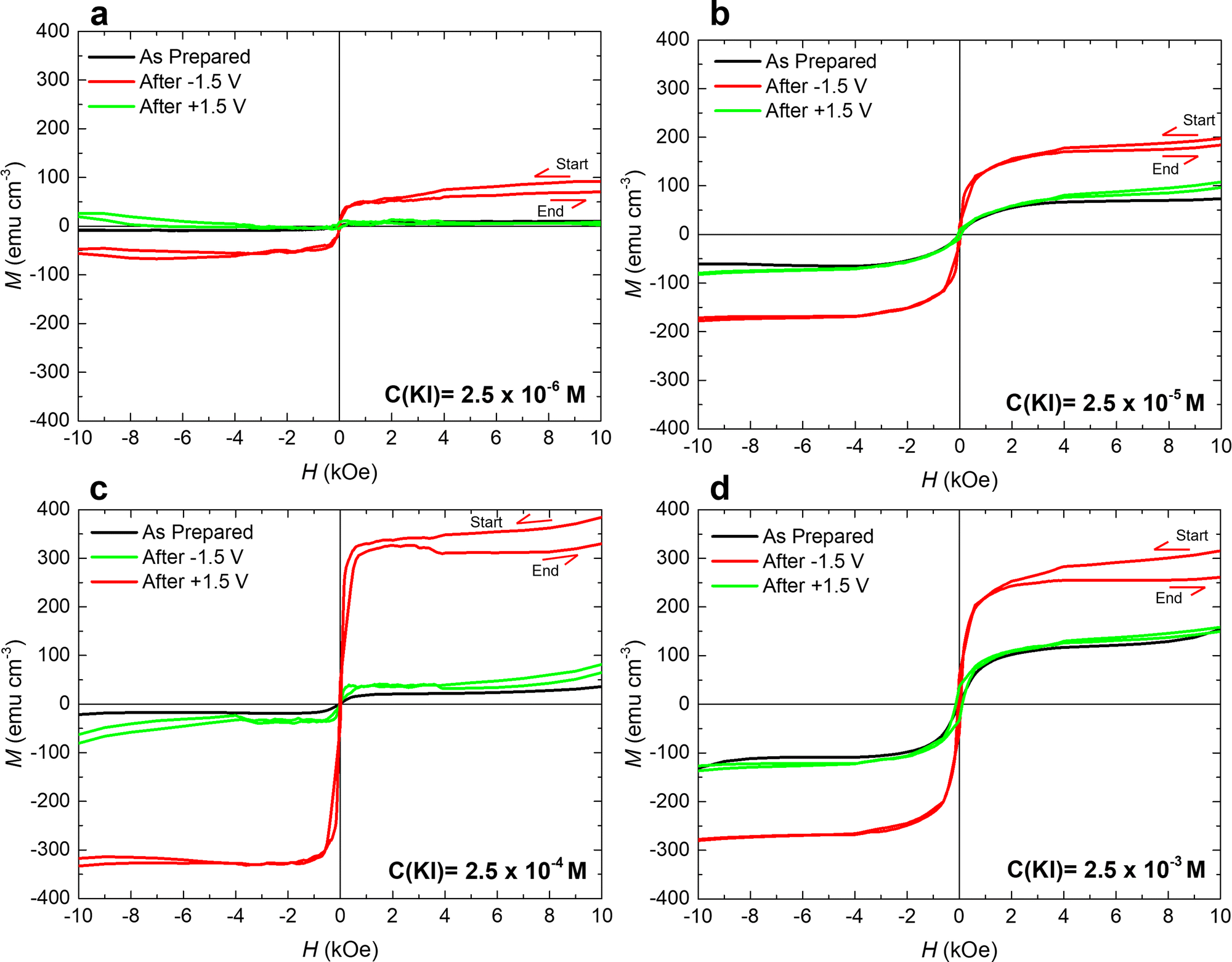 | ||
| Fig. 4 Magnetization (M) vs. applied magnetic field (H) loops of the investigated cobalt oxide films actuated using different concentrations of KI dissolved in PC. Panels a–d show hysteresis loops corresponding to as-grown states (in black), after applying ΔV = −1.5 V for 1 h (in red) and after subsequently applying +1.5 V (in green). All loops were acquired using a vibrating sample magnetometer with magnetic field applied in-plane. Note that the red and green loops were recorded at 0 V just after switching off the negative or positive voltage, respectively. The red loops are open at the high magnetic field region (at positive saturation) because the system exhibits a slight recovery (slight decrease of M with time) once electrolyte gating is stopped. | ||
To gain information on the chemical environment of the cobalt oxide film surface, XPS analyses were carried out before and after ME experiments. The Co 2p core-level XPS spectra of as-prepared sample, and samples treated using bare PC and PC + 2.5 × 10−4 M KI are shown in Fig. 5. For the as-prepared sample (see Fig. 5(a)), the two major peaks at 780.1 eV and 795.9 eV match Co 2p3/2 and Co 2p1/2 binding energies, respectively, of CoO and/or Co3O4 phases.12,28,29 The satellite structure at higher binding energies can be ascribed to the shake-up process of the Co2+ species in the high-spin state. Aside from these four peaks, Fig. 5(b) clearly shows two shoulders (indicated by the downward arrows) on the lower binding energy side of the major peaks for bare PC-treated sample. The best fitting of the spectrum results in two additional XPS peaks located at 778.3 eV and 793.3 eV, which are the characteristic binding energies of Co 2p3/2 and Co 2p1/2, respectively, for metallic Co.2 This unambiguously confirms the reduction of cobalt oxides into Co upon negative voltage actuation in bare PC electrolyte. Interestingly, the relative intensities of these additional peaks and, importantly, at the expense of the oxide Co 2p peaks, become dominant in the case of the film electrolyte-gated in PC + 2.5 × 10−4 M KI. This indicates the formation of significant amounts of metallic Co and, accordingly, higher ΔM as seen in ME experiments. Moreover, it is instructive to point out that there are no observable traces of iodine in the survey spectrum on PC + 2.5 × 10−4 M KI-treated sample (not shown), ruling out the possibility of formation of magnetic iodine-containing compounds. Overall, the formation of metallic Co upon voltage application can be written as:
| CoO → Co + VÖ | (2) |
| Co3O4 → 3Co + 4VÖ | (3) |
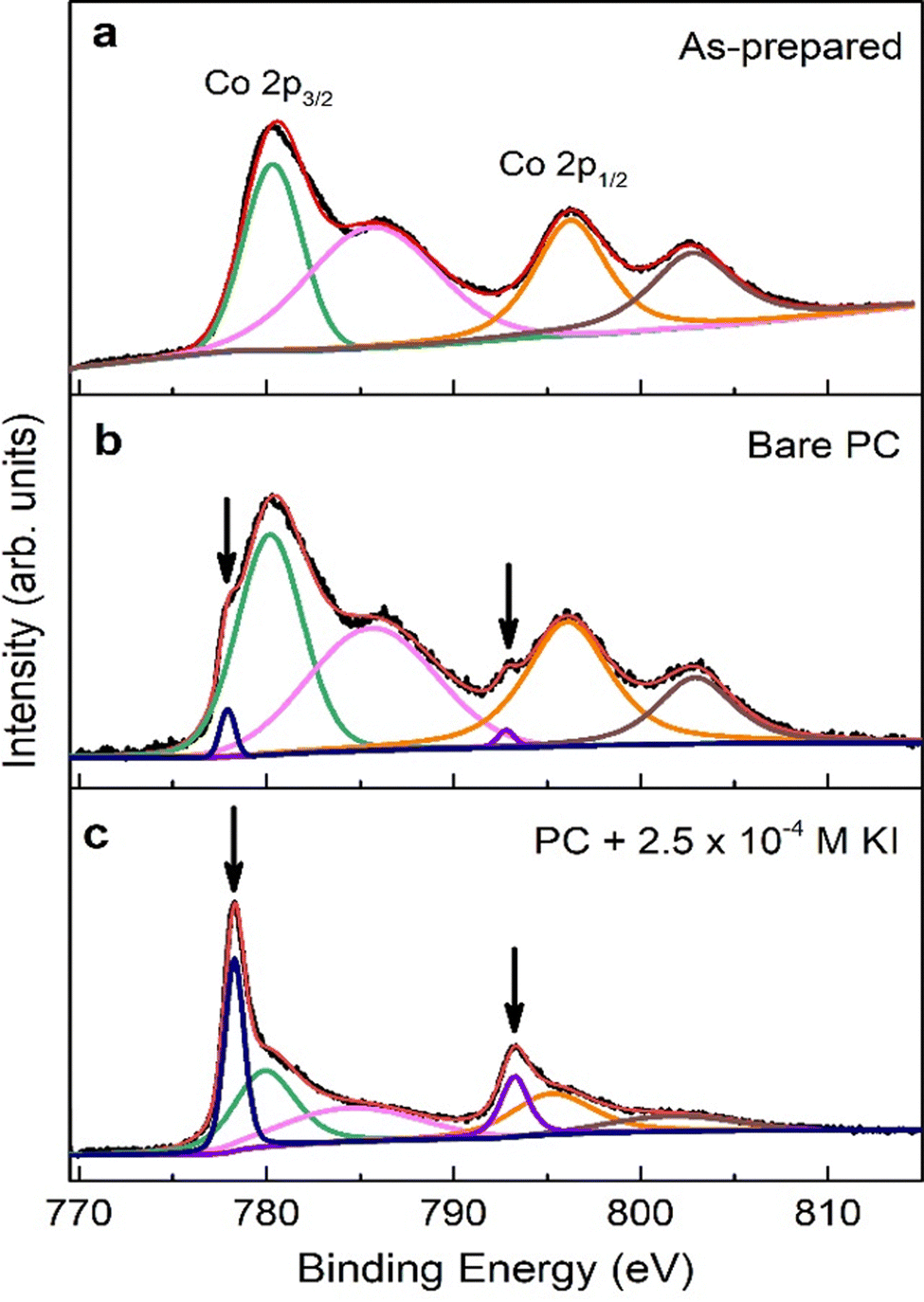 | ||
| Fig. 5 X-ray photoelectron spectra of Co 2p1/2 and Co 2p3/2 for (a) as-prepared, (b) treated in bare PC and (c) PC + 2.5 × 10−4 M KI samples. The downward arrows point to the Co 2p3/2 and Co 2p1/2 peaks of metallic Co (in blue and purple in (b) and (c)). Additionally, green and orange correspond to Co2+, and pink and brown to Co3+. Solid black lines are experimental data, whereas solid red lines refer to fitting spectra. | ||
To further examine the reversibility of the magneto-ionic effects of the cobalt oxide film in the PC +2.5 × 10−4 M KI electrolyte, voltage pulses of −1.35 V and +1.35 V (of 10 min each) were alternated, and clear changes are observed in ΔM running in parallel to voltage actuation (Fig. 6). Note that up to 5 magneto-ionic cycles could be obtained with these applied voltage pulses.
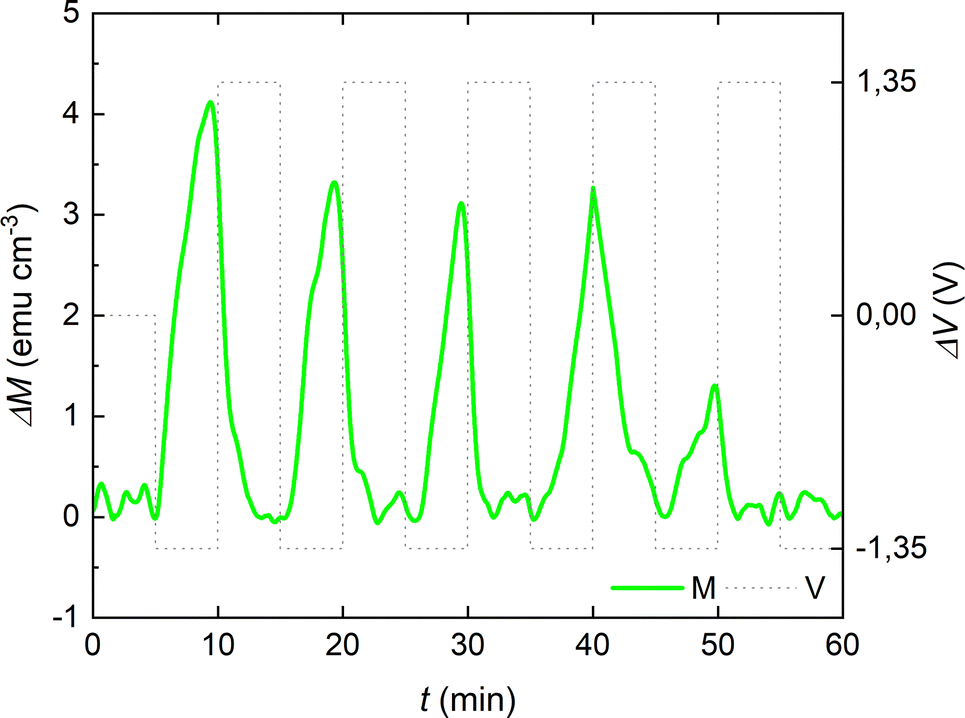 | ||
| Fig. 6 Magneto-ionic cyclability of the 15 nm-thick cobalt oxide film in PC with 2.5 × 10−4 M KI under −1.35 V/+1.35 V voltage pulses of 10 min duration each, assessed from in-plane magnetic measurements while applying a magnetic field of 10 kOe. | ||
Conclusions
Here we have shown that magneto-ionic motion in 15 nm-thick Co oxide films can be dramatically enhanced by proper electrolyte engineering. Faster magneto-ionic rates and larger generation of magnetization are achieved upon the addition of inorganic salts (KI, KCl, and Ca2(BF4)2) to PC. In this way, the ionic strength of the medium is increased, thereby causing a higher electric field strength at the Co oxide/electrolyte interface upon biasing. As a result, oxygen motion within the material is favored, thereby enabling the rapid and more extensive formation of metallic cobalt. In particular, potassium iodide at a concentration of 2.5 × 10−4 M brings about a 35-fold increase of magneto-ionic rate and one order of magnitude larger variation in magnetization than for bare PC under −1.5 V biasing for 1 h. AIMD simulations suggest that a more effective EDL would build up for KI compared to KCl. More importantly, the onset potential at which significant changes in the magnetic properties are observed is dramatically decreased when salts are added to PC. Changes are partially reversible under the application of voltage of opposite polarity for 1 hour, and cyclability was observed under voltage pulses of 10 min.Experimental
Sample preparation
15 nm-thick cobalt oxide films were grown by reactive sputtering at room temperature on B-doped [100]-oriented Si wafers (0.75 mm thick), previously sputtered with an adhesion/seed layer of 20 nm Ti/60 nm Cu. The depositions were performed with an AJA International, Inc. Magnetron Sputtering System ATC Orion with a base pressure of 5 × 10−8 Torr. Argon was used as carrier gas for the Co precursor and O2 plasma as oxygen source. The target to substrate distance was around 10 cm and the sputtering rate about 23 nm min−1. Co oxide was grown in a 9% O2/91% Ar (% given from flow rates of the different gases) atmosphere under a total pressure of 3 × 10−3 Torr. Prior to that, the adhesive (Ti) and seed (Cu) layers were also grown using the same experimental setup but in an inert Ar atmosphere, and then partially masked to use the remaining uncoated surface for electrical connection in subsequent magnetoelectric experiments.Electrolyte preparation
The electrolyte solutions were prepared from propylene carbonate (from Sigma-Aldrich). PC was initially treated with metallic Na to remove traces of water, leading Na+ and OH− ions behind. Later, varying amounts of KI, KCl and Ca(BF4)2 were added in excess to the PC-based solution taking into account the solubility limits of these salts in PC at room temperature (s(KI) = 0.221 ± 0.020 mol kg−1,23s(KCl) = (3.67 ± 0.40) × 10−4 mol kg−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 and way larger for Ca(BF4)2 (ca. 0.1 M)).17 The corresponding supernatants were analyzed by ICP and further diluted with PC-based solution to the final concentrations considered in this study.
23 and way larger for Ca(BF4)2 (ca. 0.1 M)).17 The corresponding supernatants were analyzed by ICP and further diluted with PC-based solution to the final concentrations considered in this study.
Compositional measurements
Inductively coupled plasma mass spectrometry (ICP-MS)
The concentration of the salts dissolved in PC were determined by ICP-MS. Specifically, the K+ concentration (for KI and KCl dissolved in PC) and Ca2+ concentration (for Ca2(BF4)2 dissolved in PC) were determined on an Agilent (7900 model) spectrophotometer. Before analysis, the aliquots were diluted in HNO3 1% (v/v).Magnetoelectric characterization
Magnetic measurements were carried out in-plane at room temperature in a vibrating sample magnetometer from Micro Sense (LOT-Quantum Design) under electrolyte gating using an external power supply (Agilent B2902A). Voltages were applied between the Cu seed-layer beneath the Co oxide film, which acted as a working electrode. and a platinum wire which served as a counter electrode (Fig. 1(a)). An additional platinum wire was introduced into the cell to act as a pseudo-reference electrode. The generated magnetization (M) was studied for Na-treated PC to which different salts (KI, KCl, Ca(BF4)2 had been added.Computational details
The ab initio molecular dynamics (AIMD) were performed at 0 V potential using the PBE density functional level of theory30 with the Hubbard-like term (U), as defined by Dudarev et al.31 The Ueff value was fixed to 3.0 eV based on test calculations reported in Table S1 (ESI†). Moreover, the empirical D3 correction32 was also included to account for dispersion forces. The ionic cores were described with the PAW pseudopotentials and the valence electrons were represented with a plane-wave basis set with an energy cutoff of 500 eV. The first Brillouin zone was sampled with a (2,2,1) Monkhorst–Pack K-point mesh. The simulations were carried out within the NVT ensemble (T = 300 K), using the Nosé–Hoover thermostat.33,34 The time step was set to 1fs, and the simulations were allowed to evolve until 10 ps were achieved. All calculations were performed with the spin polarized formalism as implemented in VASP 5.4 package.35,36The Co3O4-propylene interface was represented with a 4-layers thick slab model of the (110) facet where 13 propylene carbonate molecules were added in the vacuum space between images to reproduce the solvent density at room temperature (r = 1.2 g cm−3). Furthermore, a KX (X = Cl or I) unit was included to represent the dissolved salt. Two initial starting configurations were considered for each salt (Fig. S2, ESI†): (a) the two ions are initially located on the surface, directly interacting with Co3O4, and (b) the ions are solvated by propylene carbonate molecules and thus located at about 6 Å from the surface. Further details can be found in the supporting information.
Author contributions
E. P., J. S. and E. M. had the original idea and led the investigation. S. M., E. P. and J. S. designed the experiments. S. M. and Z. M. synthesized the cobalt oxide layers. S. M. and Z. M. carried out the magnetoelectric measurements and S. M., E. P., J. S. and E. M. analyzed the data. X. S.-M., M. S. and L. R.-S. carried out the AIMD simulations. Z. M performed and analyzed the XPS measurements. All authors discussed the results and commented on the article. The article was written by S. M., E. P., J. S and E. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon 2020 research and innovation programme BeMAGIC under the Marie Sklodowska-Curie grant agreement No 861145. X. S.-M. and E. M. are Serra Húnter Fellows. Partial financial support from the Spanish Government (PID2020-116844RB-C21, PDC2021-121276-C31 and PID2020-112715GB-I00), the Generalitat de Catalunya (2017-SGR-292 and 2017-SGR-1323), the Red Española de Supercomputación (QHS-2022-2-0029) is also acknowledged.References
- Y. Shiota, T. Nozaki, F. Bonell, S. Murakami, T. Shinjo and Y. Suzuki, Nat. Mater., 2012, 11, 39–43 CrossRef CAS.
- C. Song, B. Cui, F. Li, X. Zhou and F. Pan, Prog. Mater. Sci., 2017, 87, 33–82 CrossRef.
- C. Leighton, Nat. Mater., 2019, 18, 13–18 CrossRef.
- J. De Rojas, J. Salguero, A. Quintana, A. Lopeandia, M. O. Liedke, M. Butterling, A. G. Attallah, E. Hirschman, A. Wagner, L. Abad, J. L. Costa-Krämer, J. Sort and E. Menéndez, Phys. Rev. Appl., 2021, 16, 034042 CrossRef.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- P. Sharma and T. S. Bhatti, Energy Convers. Manage., 2010, 51, 2901–2912 CrossRef.
- C. Navarro-Senent, A. Quintana, E. Menéndez, E. Pellicer and J. Sort, APL Mater., 2019, 7, 030701 CrossRef.
- M. Nichterwitz, S. Honnali, M. Kutuzau, S. Guo, J. Zehner, K. Nielsch and K. Leistner, APL Mater., 2021, 9, 030903 CrossRef CAS.
- G. Feng, J. Huang, B. G. Sumpter, V. Meunier and R. Qiao, Phys. Chem. Chem. Phys., 2010, 12, 5468–5479 RSC.
- A. Quintana, E. Menéndez, M. O. Liedke, M. Butterling, A. Wagner, V. Sireus, P. Torruella, S. Estradé, F. Peiró, J. Dendooven, C. Detavernier, P. D. Murray, D. A. Gilbert, K. Liu, E. Pellicer, J. Nogues and J. Sort, ACS Nano, 2018, 12, 10291–10300 CrossRef CAS PubMed.
- J. de Rojas, A. Quintana, A. Lopeandía, J. Salguero, J. L. Costa-Krämer, L. Abad, M. O. Liedke, M. Butterling, A. Wagner, L. Henderick, J. Dendooven, C. Detavernier, J. Sort and E. Menéndez, Adv. Funct. Mater., 2020, 30, 2003704 CrossRef CAS.
- S. Martins, J. de Rojas, Z. Tan, M. Cialone, A. F. Lopeandía, J. Herrero-Martín, J. Costa-Kramer, E. Menéndez and J. Sort, Nanoscale, 2022, 14, 842–852 RSC.
- J. de Rojas, A. Quintana, A. Lopeandía, J. Salguero, B. Muñiz, F. Ibrahim, M. Chshiev, A. Nicolenco, M. O. Liedke, M. Butterling, A. Wagner, V. Sireus, L. Abad, C. J. Jensen, K. Liu, J. Nogués, J. L. Costa-Krämer, E. Menéndez and J. Sort, Nat. Commun., 2020, 11, 5871 CrossRef CAS PubMed.
- J. De Rojas, J. Salguero, F. Ibrahim, M. Chshiev, A. Quintana, A. Lopeandia, M. O. Liedke, M. Butterling, E. Hirschmann, A. Wagner, L. Abad, J. L. Costa-Krämer, E. Menéndez and J. Sort, ACS Appl. Mater. Interfaces, 2021, 13, 30826–30834 CrossRef PubMed.
- A. González, E. Goikolea, J. A. Barrena and R. Mysyk, Renewable Sustainable Energy Rev., 2016, 58, 1189–1206 CrossRef.
- B. Worths, Int. Union Pure Appl. Chem., 1977, 49, 885–892 Search PubMed.
- J. D. Forero-Saboya, E. Marchante, R. B. Araujo, D. Monti, P. Johansson and A. Ponrouch, J. Phys. Chem. C, 2019, 123, 29524–29532 CrossRef PubMed.
- X. Chen, X. Shen, T. Z. Hou, R. Zhang, H. J. Peng and Q. Zhang, Chem, 2020, 6, 2242–2256 Search PubMed.
- L. Zhang and Y. Chen, Energy Mater., 2021, 1–9 Search PubMed.
- J. Zheng, J. A. Lochala, A. Kwok and Z. D. Deng, Adv. Sci. News, 2017, 1700032 CrossRef PubMed.
- A. Molinari, H. Hahn and R. Kruk, Adv. Mater., 2019, 31, 1806662 CrossRef PubMed.
- K. Leistner, Curr. Opin. Electrochem., 2020, 25, 100636 CrossRef.
- N. Peruzzi, P. Lo Nostro, B. W. Ninham and P. Baglioni, J. Solution Chem., 2015, 44, 1224–1239 CrossRef.
- A. M. Melemed, A. Khurram and B. M. Gallant, Batteries Supercaps, 2020, 3, 570–580 CrossRef PubMed.
- L. Cao, O. Petracic, X.-K. Wei, H. Zhang, T. Duchoň, F. Gunkel, A. Koutsioubas, K. Zhernenkov, K. Z. Rushchanskii, H. Hartmann, M. Wilhelm, Z. Li, Y. Xie, S. He, M. L. Weber, K. Veltruská, A. Stellhorn, J. Mayer, S. Zhou and T. Brückell, Small, 2021, 2104356 CrossRef.
- Q. Wang, Y. Gu, S. Yin, Y. Sun, W. Liu, Z. Zhang, F. Pan and C. Song, Nanoscale, 2021, 13, 18256–18266 RSC.
- Q. Wang, Y. Gu, W. Zhu, L. Han, F. Pan and C. Song, Adv. Funct. Mater., 2021, 31, 2106765 CrossRef.
- H. Xiong, Y. Zhang, K. Liew and J. Li, J. Mol. Catal. A: Chem., 2005, 231, 145–151 CrossRef.
- C. D. Wanger, W. M. Riggs, L. E. Davis and J. F. G. E. M. Moulder, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer, John Wiley & Sons, Ltd, Eden Prairie, 1978 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- S. Dudarev and G. Botton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00340f |
| This journal is © The Royal Society of Chemistry 2023 |