
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced detoxification of Cr6+ by Shewanella oneidensis via adsorption on spherical and flower-like manganese ferrite nanostructures†
Diana S.
Raie
 ab,
Ioannis
Tsonas
c,
Melisa
Canales
d,
Stefanos
Mourdikoudis
ab,
Konstantinos
Simeonidis
e,
Antonis
Makridis
e,
Dimitrios
Karfaridis
e,
Shanom
Ali
f,
Georgios
Vourlias
e,
Peter
Wilson
f,
Laurent
Bozec
g,
Lena
Ciric
d and
Nguyen Thi
Kim Thanh
*ab
ab,
Ioannis
Tsonas
c,
Melisa
Canales
d,
Stefanos
Mourdikoudis
ab,
Konstantinos
Simeonidis
e,
Antonis
Makridis
e,
Dimitrios
Karfaridis
e,
Shanom
Ali
f,
Georgios
Vourlias
e,
Peter
Wilson
f,
Laurent
Bozec
g,
Lena
Ciric
d and
Nguyen Thi
Kim Thanh
*ab
aBiophysics Group, Department of Physics and Astronomy, University College London, Gower Street, London, WC1E 6BT, UK. E-mail: ntk.thanh@ucl.ac.uk; Web: http://www.ntk-thanh.co.uk
bUCL Healthcare Biomagnetics and Nanomaterials Laboratories, 21 Albemarle Street, London, W1S 4BS, UK
cUCL Electronic and Electrical Engineering, UCL, Gower Street, London, WC1E 7JE, UK
dHealthy Infrastructure Research Group, Department of Civil, Environmental & Geomatic Engineering, UCL, Gower Street, London, WC1E 6BT, UK
eDepartment of Physics, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece
fEnvironmental Research Laboratory, ClinicalMicrobiology and Virology, University College London Hospitals NHS Foundation Trust, London, UK
gFaculty of Dentistry, University of Toronto, Toronto, Ontario, Canada
First published on 16th January 2023
Abstract
Maximizing the safe removal of hexavalent chromium (Cr6+) from waste streams is an increasing demand due to the environmental, economic and health benefits. The integrated adsorption and bio-reduction method can be applied for the elimination of the highly toxic Cr6+ and its detoxification. This work describes a synthetic method for achieving the best chemical composition of spherical and flower-like manganese ferrite (MnxFe3−xO4) nanostructures (NS) for Cr6+ adsorption. We selected NS with the highest adsorption performance to study its efficiency in the extracellular reduction of Cr6+ into a trivalent state (Cr3+) by Shewanella oneidensis (S. oneidensis) MR-1. MnxFe3−xO4 NS were prepared by a polyol solvothermal synthesis process. They were characterised by powder X-ray diffraction (XRD), transmission electron microscopy (TEM), X-ray photoelectron spectrometry (XPS), dynamic light scattering (DLS) and Fourier transform-infrared (FTIR) spectroscopy. The elemental composition of MnxFe3−xO4 was evaluated by inductively coupled plasma atomic emission spectroscopy. Our results reveal that the oxidation state of the manganese precursor significantly affects the Cr6+ adsorption efficiency of MnxFe3−xO4 NS. The best adsorption capacity for Cr6+ is 16.8 ± 1.6 mg Cr6+/g by the spherical Mn0.22+Fe2.83+O4 nanoparticles at pH 7, which is 1.4 times higher than that of Mn0.8Fe2.2O4 nanoflowers. This was attributed to the relative excess of divalent manganese in Mn0.22+Fe2.83+O4 based on our XPS analysis. The lethal concentration of Cr6+ for S. oneidensis MR-1 was 60 mg L−1 (determined by flow cytometry). The addition of Mn0.22+Fe2.83+O4 nanoparticles to S. oneidensis MR-1 enhanced the bio-reduction of Cr6+ 2.66 times compared to the presence of the bacteria alone. This work provides a cost-effective method for the removal of Cr6+ with a minimum amount of sludge production.
1. Introduction
Chromium (Cr) is a common environmental pollutant coming from several industries such as wood preservation,1 leather tanning,2 steel production,3,4 wool dyeing,5 painting,6 refractories,4 lasers,7 and electroplating,8 among others. End-of-life products such as unwanted steel, wood,1 leather and textiles, among other materials are extra sources of Cr leakage in the environment. The release of Cr in the environment was also attributed to mining activities,9 and improper waste treatment associated with industrial processes.10 Various Cr-bearing minerals, including chromite, are available in the soil, and bedrock also releases natural Cr into the environment.10 It mainly occurs in two valence states, which are highly toxic carcinogenic11 Cr6+ and less toxic Cr3+. Various technologies have been developed to tackle the presence of Cr6+, including membranes,12 coagulation,13 photocatalysis,14 electrochemical treatments,13 adsorption15,16 and biological treatments.17,18 Integrating both adsorption and biological reduction of Cr6+ together has been proposed as a promising solution.19 Applying such combined methods can overcome the accessibility of certain technologies,20 using less toxic chemicals and reducing the production of contaminated toxic waste.19 The recovered chromium can be used in metallurgical industries and minimize the contaminated landfill.21,22Microbial reduction of Cr6+ has been regarded as a suitable Cr remediation approach because of being more eco-friendly than the conventional physico-chemical strategies, which are often costly. Recently, many types of bacteria have been reported to detoxify Cr6+ to less toxic Cr3+, including dissimilatory metal-reducing bacteria such as Shewanella oneidensis MR-1.23,24 Under anaerobic conditions, S. oneidensis can use Cr6+ as a terminal electron acceptor,25 however cells exposed to Cr6+ exhibited a loss in their enzymatic activity and cell lysis.26 The bactericidal concentration of Cr6+ was reported to be ∼42–65 mg L−1 for S. oneidensis MR-1.26,27 A lethal effect of heavy metals on the microbes during respiration26,29–31 was considered as a potential limitation for the bio-remediation of Cr6+.26 Compared with physical and chemical materials, the concentration of Cr6+ that can be reduced by bacteria is much lower, and it is a great challenge to improve the efficiency of bioremediation.28
Enhancing the bacterial tolerance to Cr6+ is an effective way to improve the reduction of Cr6+. Zero-valent iron nanoparticles (ZVI NPs) can easily be oxidised to ferric oxides and hydroxides in water. The active surface of ZVI NPs can be decreased due to the attached layers of iron oxides and hydroxides. Shewanella, as iron-reducing bacteria, can reduce the adsorbed Fe3+ to Fe2+, which reverses the oxidation of ZVI NPs, as shown in a review by Dong et al.32 Hematite (α-Fe2O3) particles enhanced the bio-reduction of Cr6+ bio-reduction by S. oneidensis MR-1, but they cause cytotoxicity to such kind of bacteria.33 The reduction of Cr6+ by S. oneidensis was enhanced by goethite (α-FeOOH) and humic acid through the bio-reduction of Fe3+ to Fe2+. The reactivity of magnetite (Fe3O4) was increased by microbial Fe3+ reduction to form Fe2+, which then can reduce Cr6+.34,35
A biocompatible material such as manganese ferrite (MnFe2O4)36 was considered for enhancing microbial respiration of Cr6+. This ferrite was used to accelerate extracellular electron transfer in the microbial fuel cell,37,38 and it showed the highest adsorption capacity among other ferrites for Cr6+.39 The maximum adsorption capacity of MnFe2O4 NPs for Cr6+ was reported to be ranging from 31 to 35 mg g−1.36,39,40
The influence of structural features of MnxFe3−xO4 NPs on Cr6+ adsorption has not been thoroughly explored. The effect of the oxidation state of Mn precursors on the chemical structure, morphological and magnetic properties of MnxFe3−xO4 NPs prepared by scalable polyol solvothermal method has been studied in a few reports41,42 but not in relation to their adsorption efficiency for heavy metals.
Herein, we report syntheses and characterization of the most suitable chemical structure of MnxFe3−xO4 NPs and nanoflowers (NFs) for the best adsorption capacity of Cr6+. The impact of the oxidation states of Mn precursors and variation in Mn doping levels on the chemical structural and morphological characteristics of MnFe2O4 NPs prepared by polyol solvothermal route has been investigated. The nature of Cr6+ adsorption by doped and undoped ferrite NPs and, subsequently, the bio-detoxification of Cr6+ by S. oneidensis have been studied.
2. Results and discussion
2.1 Synthesis of nanomaterials
In polyol synthesis, metal precursors are reduced at a high temperature by alcohols (polyols), which can act as a capping agent, solvent and reductant. Then metal nuclei form, grow and controllably coalesce together to produce the desired particles.43 In such a non-aqueous solvent, the metal oxide NPs were proposed to be formed via two steps. In the first step, solvolysis of the precursor involved an interaction between tetraethylene glycol (TEG) and the selected metal acetylacetonate, causing the generation of metal carboxylate.44 The second step is a condensation reaction in which carboxylate reacts with iron leading to the formation of an oxo-bridge between metal (metal–oxygen–metal clusters) and ultimately resulting in the formation of metal oxide nanocrystals.442.2 Characterization of MnxFe3−xO4 NPs
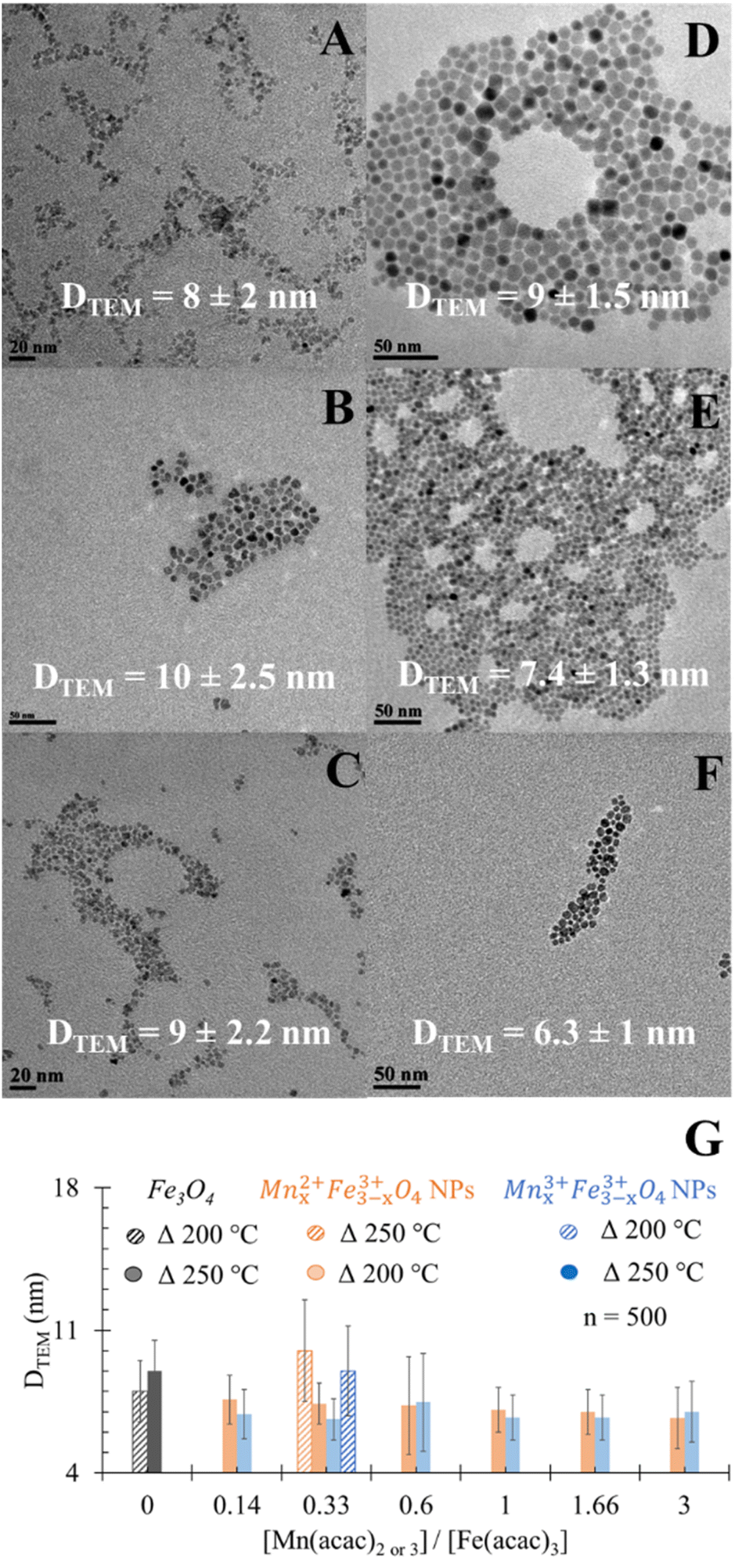 | ||
| Fig. 1 TEM images for the spherical (A) undoped Fe3O4 NPs, (B) MnxFe3−xO4 of precursor ratio [Mn(acac)2]/[Fe(acac)3] = 0.33, (C) [Mn(acac)3]/[Fe(acac)3] = 0.33 prepared at 200 °C as reaction temperatures, (D) undoped Fe3O4 NPs and (E) and (F) MnxFe3−xO4 NPs prepared with the same precursor ratio but at 250 °C. (G) Impact of reaction temperatures on DTEM of MnxFe3−xO4 NPs prepared from [Mn(acac)2]/[Fe(acac)3] = 0.33. *P < 0.05. | ||
Doping Mn had astatistically insignificant change in the DTEM of MnxFe3−xO4 NPs compared to undoped Fe3O4 NPs (Fig. 1D). In the case of using the divalent Mn precursor, a statistically insignificant change in DTEM was observed when increasing the ratio of precursors. This is in agreement with what was reported by Garcia-Soriano et al.45
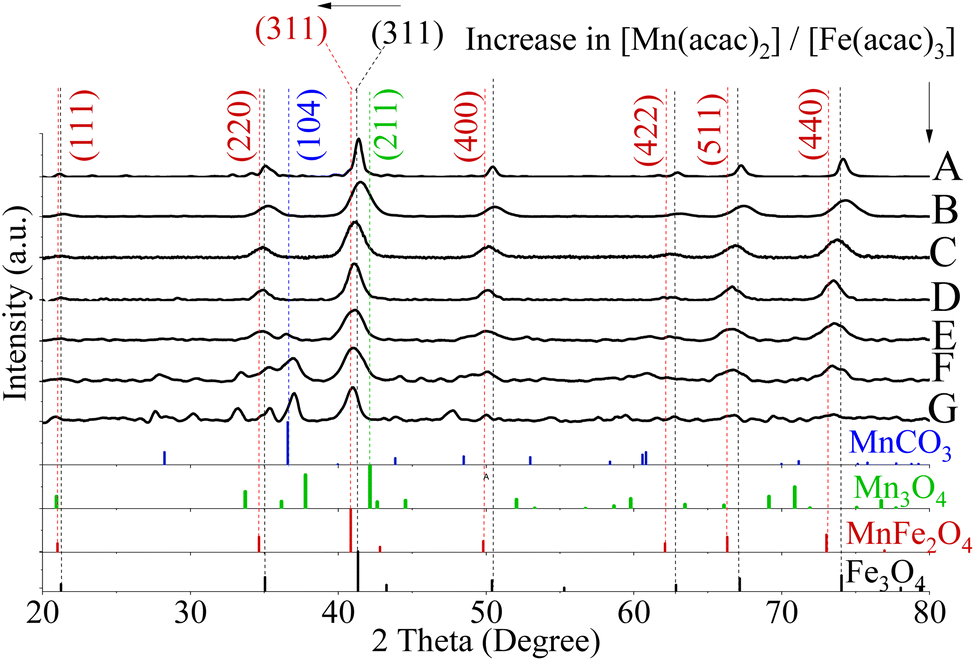 | ||
| Fig. 2 XRD patterns for fcc lattice of Fe3O4 (A) and MnxFe3−xO4 NPs where [Mn(acac)2]/[Fe(acac)3] were 0.14 (B), 0.33 (C), 0.6 (D), 1 (E), 1.66 (F), 3 (G). The horizontal arrow pointed out the shifting in the peak of 311 from the reference Fe3O4 (PDF card no. 01-089-0688) towards the lower diffraction angle of MnFe2O4 (PDF card no. 00-010-0319) in response to the increase in [Mn(acac)2]/[Fe(acac)3]. A secondary phase of MnCO3 (Reference ICDD PDF card no. 00-044-1472) was found for NPs prepared from (1 ≤ [Mn(acac)2 or 3]/[Fe(acac)3] ≤ 3) (E)–(G). The synthesis temperature for all NPs was 250 °C. The vertical arrow indicated the gradual increase in [Mn(acac)2]/[Fe(acac)3] from (A)–(G) Mn3O4 (PDF card no. 01-080-0382). | ||
With an increase in the Mn precursor concentration, the slight broadening of full-width half maximum (FWHM) of the most intense XRD peaks (311), was observed, which implies a reduction in crystal size.50 Calculated size (in diameters) is summarized in Table S1.†
The use of 0.33 ≤ [Mn(acac)2 or 3]/[Fe(acac)3] ≤ 0.6 resulted in the formation of MnFe2O4 with an fcc structure as verified by XRD patterns and presented in Fig. 2B–D and S4A–C.† The XRD peaks appearing matched with ICDD PDF card no. 00-010-0319 of MnFe2O4. The lattice planes correspond to the cubic spinel structure of MnFe2O4 (ICDD card no. 00-010-0319). For NPs prepared using 1 ≤ [Mn(acac)2]/[Fe(acac)3] < 7 in Fig. 2E–G, the distinct additional peaks at 2θ values of 36.8, 36.9° and 37.0°, respectively, indicated the formation of a secondary phase that was indexed to the (104) Miller plane of MnCO3 (ICDD card no. 00-044-1472). In Fig. 2F and G, peaks attributed to the tetraethyleneglycol (TEG) molecule appeared at 2θ equal to 27.8° and 27.6° respectively, as shown in the XRD of TEG compound alone before and after thermal treatment (Fig. S5†) as reported by Vamvakidis et al.49 as well as Khanna and Verma.51 Peaks were noticed at 30.2°, 33.1° which were assigned to MnOOH (ICDD PDF card no. 01-074-1631, data are not shown), and MnO2 (ICDD PDF card no. 00-024-0735, data are not shown) correspondingly. The presence of multiple phases of Mn oxides/hydroxides was attributed to the formation of H2O and Mn2O3 (the products of thermal decomposition of Mn(acac)2),52 which can lead to oxidation of Mn3+ into Mn4+ and hydroxylation of Mn3+ oxides.
The increase in the [Mn(acac)2 or 3]/[Fe(acac)3] led to a slight broadening in the 311 peaks, which indicated a possible alteration of the crystal size,50,53 as determined by measuring the FWHM and summarized in Table S1.†42,53 The calculated crystal size obtained from XRD of samples (6.5–7 and 5–7.5 nm for NPs prepared from divalent and trivalent Mn precursors, respectively, see PDI of crystal size in Fig. S6†) were within the range of the average particle size derived from TEM. Therefore, these NPs were considered to be single crystalline. However, the XRD analysis indicated the presence of MnCO3 for NPs synthesised with precursor ratios in the range of 1 ≤ [Mn(acac)2 or 3]/[Fe(acac)3] ≤ 3, and the crystal sizes determined by XRD were also within the size range observed by TEM.
2.3 Characterization of MnxFe3−xO4 NFs
The preparation of MnxFe3−xO4 NFs was implemented through a modified solvothermal method, and the morphology was precisely regulated by varying the ratios between [Mn(acac)3]/[Fe(acac)3] precursors as well as the reaction temperature as shown in Fig. 3A and B and [Mn(acac)2]/[Fe(acac)3] in Fig. S7.†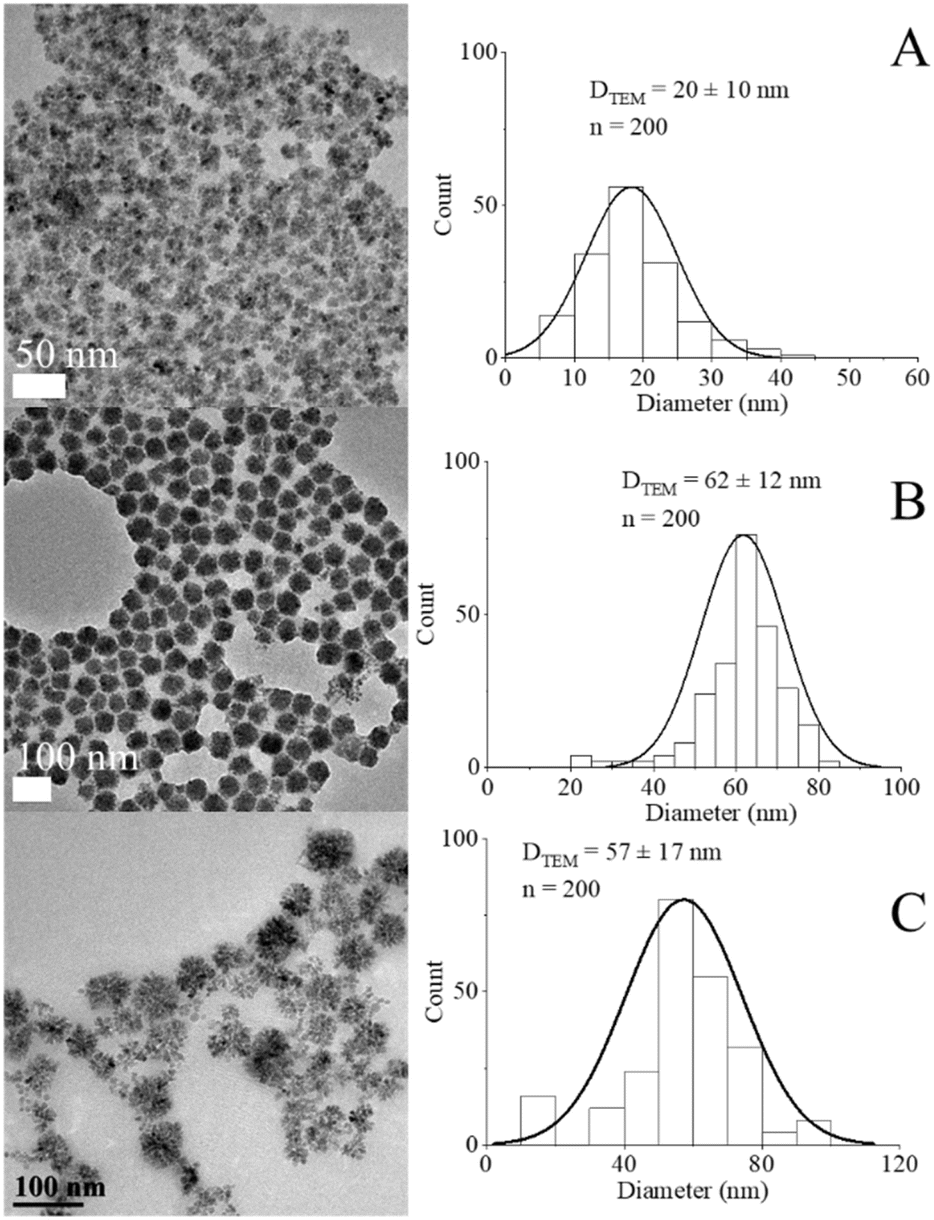 | ||
| Fig. 3 TEM images and histograms for DTEM of NFs prepared from [Mn(acac)3]/[Fe(acac)3] = (A) and (B) 1 & 3, respectively at 200 °C, (C) ratio = 7 at 250 °C. | ||
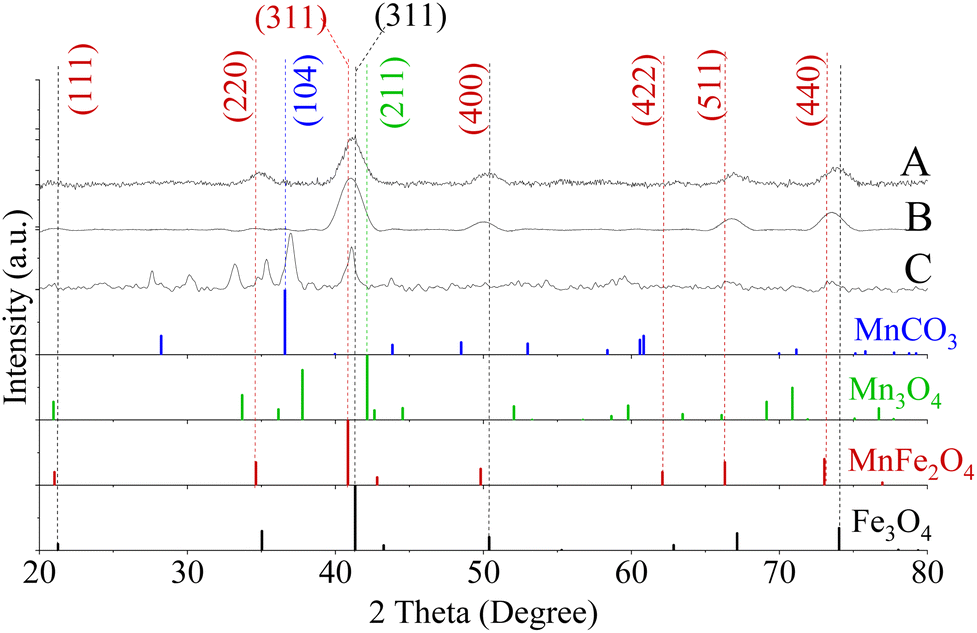 | ||
| Fig. 4 XRD patterns of MnxFe3−xO4 NFs prepared from [Mn(acac)3]/[Fe(acac)3] ratio equal to (A) 1 and (B) 3 at 200 °C, (C) 7 at 250 °C. A secondary phase matched MnCO3 (Reference ICDD PDF card no. 00-044-1472) was found for NFs prepared from a precursor ratio equal to 7 at 250 °C. No detected peaks matched Mn3O4 (PDF card no. 01-080-0382). | ||
In the case of preparing NPs with [Mn(acac)3]/[Fe(acac)3] ratio equal to 7 at 250 °C, the XRD pattern revealed the formation of a polycrystalline material corresponding to a mixture of phases. As shown in Fig. 4D, the peaks at 36.9° (104) and 27.7° (102) diffraction peaks were indexed to MnCO3 (JCPDS card no. 00-044-1472). The 2θ Bragg reflections at 21.5° (111), 35.4° (220), 41.0° (311), 50.1° (400), 66.6° (511), and 73.5° (440) confirmed the formation of MnFe2O4 (JCPDS card no. 00-010-0319). A peak at 27.5° was assigned to TEG51, which was supported by our results, as shown in Fig. S5.† Also, peaks appeared at 2θ equal to 30.1°, 33.2° and 35.3° were related to MnOOH (JCPDS card no. 01-074-1631) and MnO2 (JCPDS card no. 00-024-0735). Our results revealed that the increase in ratios between the used precursors led to the formation of nano-clusters of MnxFe3−xO4, which matched what was reported for MnxFe3−xO4 (ref. 57) and other ferrites by a solvothermal method.58 The inability of nanocluster formation using [Mn(acac)2]/[Fe(acac)3] equal to 7 (Fig. S7†) can be attributed to the relative thermal stability of Mn(acac)2 which limits its decomposition.52
2.4 Elemental analysis of MnxFe3−xO4 NPs
Results of elemental analyses are presented in Fig. 5A, showing a significant positive relationship between the Mn doping level and [Mn(acac)2 or 3]/[Fe(acac)3] ratios. The doping level of Mn in MnxFe3−xO4 NPs was probably the reason behind the small shifts in XRD patterns from the reference peak of Fe3O4 towards a lower diffraction angle of MnFe2O4 when [Mn(acac)2 or 3]/[Fe(acac)3] increases as shown in Fig. 2, S3 and S4.† The variation in the Mn doping levels was not significantly affected by the oxidation state of Mn precursor except in the cases when [Mn(acac)2 or 3]/[Fe(acac)3] ratios were equal to 0.14 and 0.33. The faster thermal decomposition of Mn(acac)3 than Mn(acac)2 (ref. 52) resulted in more Mn-rich NPs that were prepared by the trivalent Mn precursor than those prepared by the divalent Mn precursor.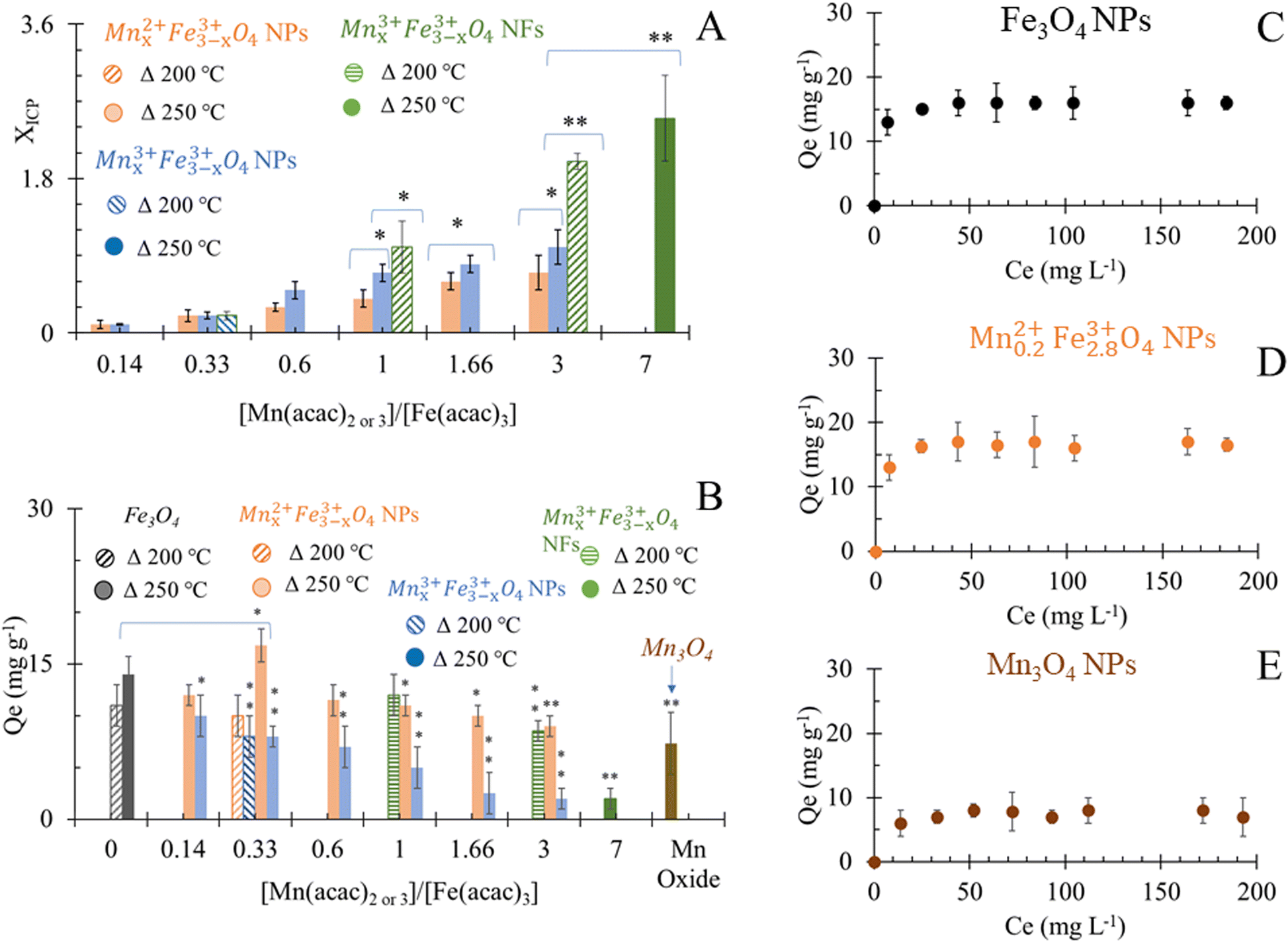 | ||
| Fig. 5 (A) Elemental analysis of MnxFe3−xO4 NS that were prepared at 200 °C and 250 °C. *P < 0.05 and **P < 0.01 in comparison to Mnx3+Fe3−x3+O4 NPs of similar precursor ratios; (B) adsorption capacity of NPs for Cr6+, *P < 0.05 and **P < 0.01 in comparison to Fe3O4 NPs; (C)–(E) adsorption isotherm of Cr6+ by Fe3O4, Mn0.22+Fe2.83+O4 and Mn3O4 NPs respectively. | ||
Overall, at 250 °C, the change in the oxidation state and the ratios between the precursors did not show a variation in the morphology of NPs, but it significantly affected the Mn doping level. While at a synthesis temperature of 200 °C, the oxidation state and the ratios between the precursors affected both the Mn doping level and resulted in different shapes of MnxFe3−xO4 NPs and NFs.
2.5 Functionalisation of NPs and NFs
The advantage of applying a small molecule like citrate as a ligand is that a smaller hydrodynamic radius (DHD) of NPs can be obtained compared to polymeric ligands, which are reflected in hydrodynamic size (Fig. S8†). Yet, the hydrodynamic shell size of citrate-coated NPs was large enough to maintain a physical barrier leading to good dispersibility. The obtained stable dispersions of nano-colloids were attributed to the negative charges induced by the citrate59 as determined by ζ-potentials (Fig. S9†).The most negative value of ζ-potential was observed for MnxFe3−xO4 NPs of [Mn(acac)2 or 3]/[Fe(acac)3] = 3. The colloidal stability was attributed to a weak base (MnCO3), as expected from XRD patterns (Fig. 2E–G and 4A) and the negative charge of citrate. For other MnxFe3−xO4 NPs and NFs, the Mn doping level did not show any crucial impact on their ζ-potentials.
The FTIR measurements, as shown in Fig. S10,† confirmed that TEG ligand was exchanged by trisodium citrate, similarly observed by Chakraborty et al.60 Carboxylates exhibited strong absorptions for infra-red spectrum due to their characteristic asymmetric (at 1620–1560 cm−1) and symmetric carbonyl stretching (1440 to 1310 cm−1). Bands in the region of 1280–1027 cm−1 represented the deformation of C–H.61–63
2.6 Adsorption of Cr6+ by NS
The adsorption of Cr6+ by MnxFe3−xO4 NPs and NFs was studied as a function of the chemical composition and morphology of nanostructures. The quantities of Cr6+ (Qe) adsorbed onto citrate-coated Fe3O4 NPs were estimated to be 14 ± 1.7 mg g−1 at room temperature and pH 7 at equilibrium (Fig. 5B). The smaller size of the NPs presented herein can probably explain the 1.4 fold higher adsorption efficiency compared to that reported by Luther et al.64 In aqueous solution, Cr6+ mainly was present in oxyanion form of chromate (Cr2O7)2− species and formed sphere complexes with iron oxide via surface hydroxyl exchange,15,36,40,65 resulting in the generation of monodentate complexes, with simultaneous desorption of surface hydroxyl groups from the metal oxide surface sites.15,36,40,65 Upon evaluating commercially available Mn3O4 as a control material under the same conditions, the binding capacity of Cr6+ (7 ± 2.6 mg g−1) was significantly lower than to Fe3O4 which was attributed to the physisorption affinity of Cr6+ to such material.66The higher adsorption capacity of NFs in comparison to NPs made of similar [Mn(acac)3]/[Fe(acac)3] ratio (1 and 3) but at different synthesis temperatures (200 °C vs. 250 °C) was attributed to the higher surface area which allows Cr6+ to penetrate into NFs.67 Increasing the doping level of Mn in NFs causes a decrease in Cr6+ adsorption as Mn0.83+Fe2.83+O4 NFs that was prepared from [Mn(acac)3]/[Fe(acac)3] = 1 can adsorb 12 ± 2 mg g−1, while using [Mn(acac)3]/[Fe(acac)3] = 3 resulted in NFs with adsorption capacity equal to 8.5 ± 2 mg g−1.
Under the employed conditions, only Mn0.22+Fe2.83+O4 (x = 0.2), prepared at 250 °C was comparable to adsorption capacity to Fe3O4 NPs (16.8 ± 1.6 vs. 14 ± 1.7 mg g−1). The use of Mn0.22+Fe2.83+O4 NPs improved the Cr6+ adsorption by over 2 fold compared to Mn3O4, as shown in Fig. 5B. The adsorption of chromate anions was due to the formation of weak bonds with MnxFe3−xO4 substrate.40 The adsorption capacity of Cr6+ by stoichiometric MnFe2O4 NPs was reported to be higher than by non-stoichiometric Mn1−xCoxFe2O4 (x = 0.2, 0.4 and 0.6).68 Our results are in agreement with what was stated by Martinez-Vargas et al.69 as non-stoichiometric Mn0.25Fe2.75O4 NPs exhibited the best adsorption capacity to As3+. The surface of Mn0.22+Fe2.83+O4 has been reported to be rich in hydroxyl groups69 which favour Cr6+ adsorption. The colloidal dispersion of our NPs and their small diameter can explain the reason behind their better adsorption capacity compared to other reported MnFe2O4 NPs (15 mg g−1 (ref. 36) and 13.54 mg g−1 (ref. 70)), while it is also comparable with results from other reports (18.02 mg g−1).40 The increase in the Co substitution for iron in magnetite (Fe3−xCoxO4, 0 ≤ x ≤ 1) enhanced the adsorption capacity of NPs to Cr6+ slightly.71 Increasing the zinc content in magnetite (Fe3−xZnxO4, x = 0, 0.25, 0.49) has been reported to initially decrease the Cr removal efficiency, but Fe2.26Zn0.74O4 and Fe2.1Zn0.99O4 led to its improvement.72
Except Mn0.22+Fe2.83+O4, MnxFe3−xO4 NPs prepared from both Mn sources showed an inverse trend for adsorption capacity of Cr6+ with the increase of x in comparison to Fe3O4 NPs. The inverse relationship between Mn concentration in the ferrite composition and adsorption of heavy metals was also observed in the case of arsenic adsorption by MnxFe3−xO4 NPs and was attributed to low binding affinity to the As.69 Introducing Mn into ferrite reduced the adsorption capacity of Fe3O4 to Cr6+ from 15.9 mg g−1 to 8.54–8.9 mg g−1.64,70 It was suggested that the release of Mn cations into the solution as a result of reduction of Cr6+ (ref. 64) alters the surface structure. The decrease in Mn doping in the Mn1−xCoxFe2O4 (x = 0.2, 0.4 and 0.6) induced a progressive, positive impact on the adsorption efficiency of Cr6+. Since Mn2+ ions have larger ionic radii than Co2+ (0.8 Å vs. 0.7 Å), the increase in x turned the overcoming of energy barriers for ion exchange interaction more difficult.68 Given that the physical mechanism of Cr6+ adsorption on the surface of oxide was reported to be a combination of electrostatic interactions between charged oxides and Cr6+ and ion exchange in the aqueous solution,40 the increase in Mn doping level showed a negative impact on Cr6+ adsorption by Mn1−xCoxFe2O4.68 At higher dopant concentration x = 0.8, more CoFe2O4 was proposed to be formed on the surface.68 Considering the larger ionic radii of Mn2+ cation than Fe3+ (0.80 Å vs.49 0.64 (ref. 49)), our results can be explained on the basis of the reverse impact of Mn doping level on the adsorption capacity of MnxFe3−xO4 NPs to Cr6+. Yet, in the case of Mnx3+Fe3−x3+O4, the ionic radius of Mn3+ is smaller than Mn2+ and approximately equal to Fe3+ radius but Mnx3+Fe3−x3+O4 NPs showed lower adsorption capacities than their Mn0.22+Fe2.83+O4 NPs counterparts. The inferiority of Mn3+ in the adsorption of Cr6+ could be attributed to the lower redox potential of Mn3+/Mn2+ (+1.5 V) than Fe3+/Fe2+ (+1.9 V).49 Mn2+ has half-filled 3d orbital ([Ar] 3d5 4S0), which makes it more stable than Mn3+ ([Ar] 3d4 4S0). While, oxidizing Fe2+ ([Ar] 3d6 4S0) of Fe3O4 NPs into more stable Fe3+ ([Ar] 3d5 4S0) is favored and can support the possible redox-based adsorption of Cr6+.
The adsorption of Cr6+ by the selected citrate-coated adsorbents that showed the best Qe at pH 7 at room temperature can be described by Langmuir isotherm model as a function of the initial Cr6+ concentrations (Fig. 5C–E). Hence, the surface of nano-sorbents has homogeneous energy distribution via a monolayer sorption process. The calculated maximum adsorption capacity (Qmax) by Langmuir isotherm model fitted the results of Qe.
2.7 The Raman spectrum of Mn0.22+Fe2.83+O4 NPs
Raman spectroscopy is also a useful tool that provides further structural details.73 Raman spectra of representative Fe3O4 and Mn0.22+Fe2.83+O4 NPs were recorded. The Raman spectrum of Fe3O4 (Fig. S11A†) expressed 5 Raman active modes including 190 (T2g(1)), 340 (Eg), 490 (T2g(2)), 540 (T2g(3)) and 670 cm−1 (A1g). For Mn0.22+Fe2.83+O4 NPs, the broad A1g band involved two modes centered at 595 and 670 cm−1 due to the presence of Mn and Fe cations. The Raman shift at 220 cm−1 in Fig. S11B† showed an induced phase transition at the surface of the Mn0.22+Fe2.83+O4 NPs due to the laser's power.742.8 The oxidation state of Mn and Fe in Mn0.22+Fe2.83+O4 and Mnx3+Fe3−x3+O4 NPs
XPS was utilized to gain insights into the chemical composition and oxidation state of the selected MnxFe3−xO4 NPs, which have either maximum or minimum Cr6+ adsorption capacity (Fig. 5). Binding energies (BE) were used to identify different elements and their valence states. In Fig. S12,† the wide-scan spectra of Mn0.22+Fe2.83+O4 and Mnx3+Fe3−x3+O4 NPs indicate the presence of carbon (C) and oxygen (O) elements besides Mn and Fe. Using the relative area under the deconvoluted XPS bands, a semi-quantitative estimation of the valence states of the elements in the mixed-valence compounds was achieved.The presence of C was identified by BEs of C 1s around 284.6, characteristic energies correspond to C–C, C–O–C, O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and CO bonds which are due to the presence of surfactant. The presence of O in the XPS spectra was attributed to the metal oxide itself, hydroxyl bonded to metal or adsorbed H2O as was expressed by BE of O 1s at 529.7 eV. Other BE appeared at 531.0, 532.3, and 535.3 eV were ascribed to CO and C–O bonds coming from the ligand.
O, and CO bonds which are due to the presence of surfactant. The presence of O in the XPS spectra was attributed to the metal oxide itself, hydroxyl bonded to metal or adsorbed H2O as was expressed by BE of O 1s at 529.7 eV. Other BE appeared at 531.0, 532.3, and 535.3 eV were ascribed to CO and C–O bonds coming from the ligand.
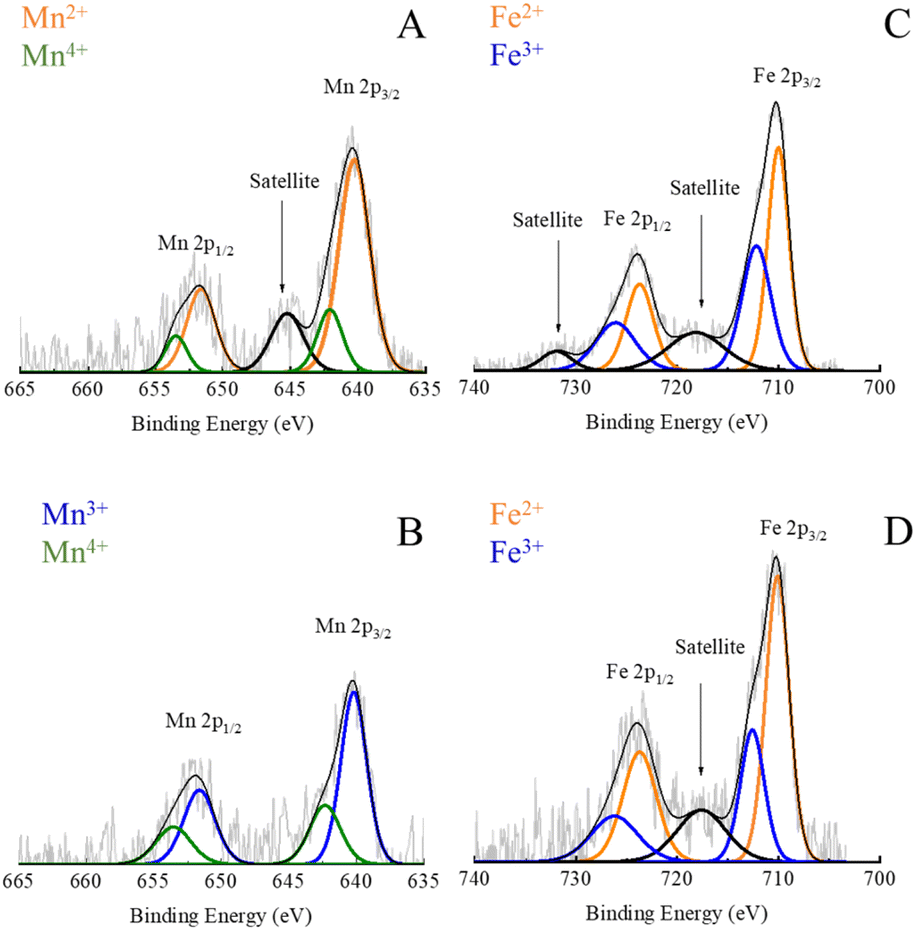 | ||
| Fig. 6 High-resolution XPS spectra of Mn 2p in Mn0.22+Fe2.83+O4 (A); Mn 2p in Mnx3+Fe3−x3+O4 NPs (B); Fe 2p in Mn0.22+Fe2.83+O4 (C) and Fe 2p in Mnx3+Fe3−x3+O4 NPs (D). | ||
Nevertheless, in the case of Mnx3+Fe3−x3+O4 NPs, the BE of Mn 2p was fitted by 4 peaks which are located at 640.2, 642.1, 651.6 and 653.4 eV, as shown in Fig. 6B. The absence of a small satellite peak at 645.2 was reported due to the formation of either Mn3+ or Mn4+.30
A partial oxidation of Mn2+ to Mn3+ has been reported for MnxFe3−xO4 NPs prepared via polyol solvothermal method using Mn(acac)2 precursor42 due to the oxidative atmosphere inside the autoclave.49 Both divalent and trivalent Mn cations were reported to be present in MnxFe3−xO4 NPs with a strong preference for tetrahedral sites and octahedral positions for Mn2+ and Mn3+, respectively.42 Hence, trivalent Mn was supposed to be formed during the synthesis of NPs as suggested by XRD and matched MnCO3 ICDD PDF card no. 00-010-0319. The relatively lower amount of Mn2+ in Mn3+-substituted ferrites could account for the fact that Mnx3+Fe3−x3+O4 NPs showed lower Cr6+ adsorption capacity than Mn2+-substituted ferrites (Fig. 5B).
Mn0.22+Fe2.83+O4 NPs adsorb Cr6+via an ion exchange between the hydroxyl groups on the surface of NPs and chromate (Cr2O7)2− oxyanion.15,36,40,65 The adsorption of Cr6+ on iron oxides/hydroxides was reported to generate inner-sphere coordination complexes,79,80 in which chromates are linked to a central metal atoms (or ions) by covalent bonds. Fe forms monodentate (one covalent bond) and bidentate (two covalent bonds) complexes with chromates.79,80 The inner-sphere complex is strong and non-reversible.79 The reduction of adsorbed Cr6+ to Cr3+ by Fe2+ or Mn2+ resulted in the formation of precipitated Cr(OH)3 or CrxFe1−x(OH)3.72 So, there is a possibility of the presence of Cr6+ and Cr3+ on the surface of NS.
Due to the highest adsorption capacity of Mn0.22+Fe2.83+O4 NPs, this sample was selected to explore its enhancement impact on the bio-reduction of Cr6+ by S. oneidensis MR-1 in comparison to undoped Fe3O4 and Mn3O4 NPs.
2.9 The lethal dose of Cr6+ for the tested Shewanella
Shewanella bacterial species are considered metal-reducing and resistant bacteria.26 In our work, results revealed that the minimum inhibition concentration (MIC) of Cr6+ for the tested wild-type Shewanella (S. oneidensis and S. loihica PV-4, see the molecular identification (Table S4†)) was 60 mg L−1 and 70 mg L−1 respectively, being slightly higher than what was reported previously.26 For S. oneidensis JG1486 and JG3355 (molecular identification at ESI†), MICs were 20 mg L−1 and 5 mg L−1, respectively. The bactericidal effect of Cr6+ was documented because of being taken up by Shewanella intracellularly and caused cell lysis. In fact, the toxic effect of Cr3+ appeared to be associated with extracellular interactions, leading to stress-associated cell morphology and then to a lethal effect.26,81–83 Before reacting with Cr6+, the wild-type S. oneidensis MR-1 and S. loihica PV-4 cells were reported to be regular small rod-shaped with smooth surfaces.26,81–83 Meanwhile, the bacterial cells changed to be atrophic with a shrunken-surface shape and crack formation was also observed after the reaction.26,81,83 However, S. loihica PV-4 cells were reported to be elongated and exhibited a rough surface upon exposure to Cr6+,82 which can explain why S. loihica PV-demonstrated higher resistance and reduction ability for Cr6+. As hazardous metal ions could damage microbial DNA when they entered the cells, extracellular reduction benefitted Shewanella for their survival.82 The low resistance of mutants was due to the inability of bio-reduction of Cr6+ for JG1486,84 but the possibility of the presence of other stress regulators made such mutants more resistant regarding JG3355.85,862.10 Effect of the selected NPs on the viability of Shewanella in response to the sublethal concentration of Cr6+
In the absence of Cr6+, tri-sodium citrate alone, citrate-coated Mn0.22+Fe2.83+O4, Fe3O4 and Mn3O4 can sustain the viability of bacteria. At a sub-lethal concentration of Cr6+, the viability of wild-type bacteria was improved in the tested groups amended by citrate alone (only 10–12%), as illustrated in Fig. 7A. Bencheikh-Latmani et al.87 explained a similar observation as a result of the complexation between the product of bio-reduction (Cr3+) and citrate, which consequently limits the availability of the toxic metal to bacterial cells.87 In response to Cr6+ toxicity, Mn0.2Fe2.8O4, Fe3O4 and Mn3O4 NPs improved the viability of S. oneidensis JG1486 strain by 3.3, 2.5, 1.3 folds, and of S. oneidensis JG5533 strain by 1.2, 0.5, 0.2 folds, respectively.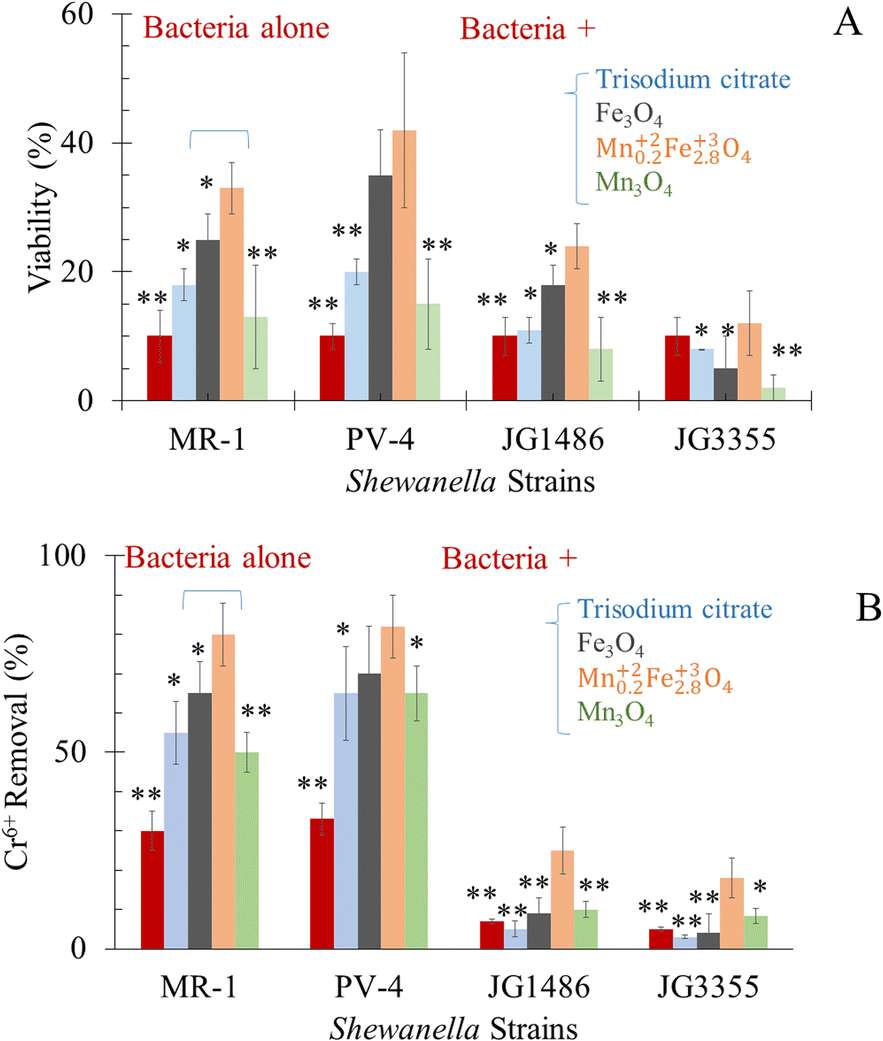 | ||
| Fig. 7 In the presence of the tested agents (A) viability of tested Shewanella under sublethal dose of Cr6+ (B) removal of Cr6+ by Shewanella strains. *P < 0.05 and **P < 0.01 in relation to the impact of Mn0.22+Fe2.83+O4 NPs in each data set separately. | ||
2.11 Bio-reduction of Cr6+ by tested Shewanella
For safe removal of Cr6+, such hexavalent cations should be detoxified by reduction to Cr3+. The capability of S. oneidensis83 to respire Cr6+ was affected by the initial concentration of the heavy metal.83,88,89 These results were credited for the chromate dose-dependent toxicity, which causes growth and viability inhibition.83,88 This occurred in the presence of Cr6+ alone83,88,89 or in the presence of goethite and humic acid34 or ferric oxyhydroxide mediators.83,88 Hence, our experiments were designed at a high concentration of Cr6+, i.e. sub-lethal dose.Our results (Fig. 7B) revealed a significant drop in the concentration of Cr6+ in media supplemented by both wild-type of Shewanella which included the strain of interest (S. oneidensis MR-1) and positive control (S. loihica PV-4); this was attributed to the respiration of Cr6+ into Cr3+ form81,90,91 or bio-sorption92,93 by bacterial cells. The drop in the concentration of Cr6+ in the medium exposed to S. oneidensis JG1486 and JG3355 was significantly lower than those supplemented by both wild type bacteria.
The tested Shewanella oxidized lactate (electron source), and the liberated electrons are transferred via the respiratory chain to be directed to an externally available terminal electron acceptor (Cr6+). The redox potential of Cr6+ (1.33 V vs. standard hydrogen electrode; SHE) has been reported to be higher than the redox potential of oxygen (1.23 V) and the electron source (−0.19 V).94 So, Cr6+ was considered a favourable electron acceptor for bacteria in the process of respiration as bacteria gain more energy.94
Cr6+ can be reduced extracellularly82 and also transported into the cell interior and then reduced in the cytoplasm.82 The ability of Shewanella to transfer electrons to metal ions was known to take place via one of four porin–cytochrome conduits; the MtrCAB complex,95 the MtrFED complex,96 the DmsEFA dimethyl sulfoxide reductase system97 and the SO4359–SO4360 system.96 The superiority of S. loihica PV-4 in the respiration of Cr6+ in our experiment was thanks to their higher content of c-type cytochrome genes in the metal reductase-containing locus than S. oneidensis MR-1.98S. oneidensis JG1486 (ΔmtrCAB/ΔmtrFED/ΔomcA/ΔdmsE/ΔSO4360/ΔcctA/ΔrecA) lacks the responsible genes for extracellular metal reduction.84 The recombination between the expression of outer membrane cytochromes (controlled by lac promoters) and periplasmic electron carriers was stopped by the deletion of RecA gene.84 Such mutants showed the lowest removal of Cr6+ as a result of the inability of bioreduction.84S. oneidensis JG3355 lacked both ClpX and ClpP genes.86 The role of ClpXP has been revealed for regulating Fe2+ stress in anaerobic bacteria86 and stress regulation (ClpP) in response to 24 h Cr6+ exposure.85 Therefore, the inability of S. oneidensis JG3355 to respire metal could be the result of losing the bacterial viability as was reported before29–31 and presented in Fig. 7A.
2.12 Enhancement of respiration of Cr6+ by Shewanella in the presence of selected materials
The respiration of Cr6+ (at sub-lethal concentration) was improved in the tested groups supplied by citrate alone (only 0.83 folds), as illustrated in Fig. 7B. Bencheikh-Latmani et al.87 explained a similar observation as a result of the possible complexation between the product of bio-reduction (Cr3+) and citrate, which consequently limits the availability of the toxic metal to bacterial cells.87In the presence of a sublethal concentration of Cr6+, the alive cell extent was the highest in the group of citrate coated Mn0.22+Fe2.83+O4, Fe3O4 and Mn3O4 (in descending order). The presence of Mn0.22+Fe2.83+O4 as adsorbent was beneficial to microbial survival, which was positively related to enhanced Cr6+ bio-reduction by 2.5–3.6 folds. The increase in the percentage of bacterial viability may be attributed to the adsorption of Cr6+ by NPs, which led to the decrease of stress on the strains themselves. In addition, the possible continuous adsorption–desorption rate of Cr6+ was based on Langmuir adsorption isotherm of the equilibrium between the adsorbate and adsorbent system (Fig. 5C–E).
The presence of manganese in the chemical structure of NPs improved the antioxidant activity and, in turn, the viability of cells and the ability to respire metal.99 Mn2+ ions can act as antioxidants which helps enzymatic systems to act against oxidative stress. For Fe-rich and Mn-poor cells such as S. oneidensis MR-1, death at low doses of ionizing radiation might not be caused by DNA damage inflicted during irradiation but instead by the release of Fe2+ and the subsequently formed toxic by-product of energy-metabolism after irradiation.100
The electron transfer from cells to the acceptor101 occurred via redox cycling of the electron-donating and accepting functional groups via direct electron transfer through NPs. The affinity of MnFe2O4 NPs to bind proteins on the bacterial outer membrane can improve the contact area between a single bacterium cell and an external electron acceptor.37 There are some explanations for NP-enhanced bio-reduction of Cr6+ to Cr3+by S. oneidensis MR-1; however, the exact mechanism is not fully unravelled.102 NPs can act as a bridge between the bacterial cell and Cr6+ to promote electron transfer.102 Mn0.22+Fe2.83+O4 NPs can adsorb Cr6+via ion exchange40,103 and covalent bonding of Cr6+ on their surfaces.36 The adsorption of Cr6+ on the surface of NPs and its reduction to Cr3+ decreases the availability and toxicity of Cr6+, which improves the efficiency of microbial respiration.90,104 Since MnFe2O4 NPs have electrochemical properties,37,38 they can link S. oneidensis MR-1 with Cr6+ as an electron mediator from the cell to Cr6+, a terminal electron acceptor. In MnFe2O4, the existence of Mn and Fe in different oxidation states facilitates the redox processes on the NP surface.103 Finally, S. oneidensis MR-1 can reduce Fe3+of Mn0.22+Fe2.83+O4 NPs to Fe2+, which can further reduce Cr6+ to Cr3+.90 The bio-genic Fe2+ can reduce Cr6+ leading to releasing Fe3+ into the medium and the dissolution of NPs.88
3. Experimental section
3.1 Materials
All chemicals were used as received without further purification.3.2 Synthetic methodology
MnxFe3−xO4 nanostructures (NS) were prepared by a polyol solvothermal synthetic procedure42,49 with some modifications. The impact of the oxidation state of Mn precursors, i.e. Mn(acac)2 and Mn(acac)3, the molar ratio between [Mn precursor] to [Fe(acac)3] and reaction temperature on the nanoparticle properties were studied. Based on previous experience from our research group, using 15 wt%/vol as a total dissolved precursor concentration resulted in NPs with narrow size distribution.46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 v/v of acetone, followed by ethanol and water three times for each solvent. Then, the nanomaterials were ready for characterization and functionalization.
:10 v/v of acetone, followed by ethanol and water three times for each solvent. Then, the nanomaterials were ready for characterization and functionalization.
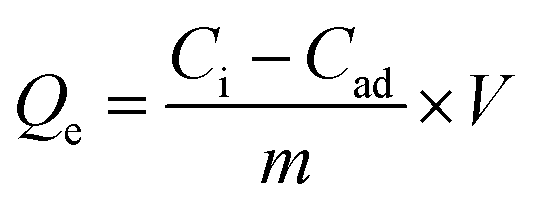 | (1) |
The adsorption isotherms of Cr6+ by the selected Mn0.22+Fe2.83+O4 NPs were studied due to their superior Qe. The isotherm was measured at room temperature by varying the initial Cr6+ concentration from 10–250 mg L−1 at pH 7 for 6 h contact time. The concentration of the adsorbents was adjusted to 1 mg mL−1. For Mn0.22+Fe2.83+O4, the mass of the adsorbent was calculated in respect to both Fe and Mn fractions which were 0.68 and 0.05, respectively. Adsorption isotherms were fitted by both Langmuir and Freundlich models.108
 | (2) |
4. Conclusions
Adsorption of hexavalent chromium (Cr6+) on manganese ferrite (MnxFe3−xO4) nanostructures enhanced the bio-detoxification of Cr6+ by S. oneidensis MR-1. A synthetic platform for achieving the most suitable chemical structure of MnxFe3−xO4 nanoparticles (NPs) and nanoflowers (NFs) acting as Cr6+ adsorption agents was presented. At 250 °C, both divalent or trivalent manganese precursors formed spherical NPs, whereas, at 200 °C, nanoflowers were obtained using a trivalent precursor. Mn0.22+Fe2.83+O4 NPs that were prepared from divalent manganese precursor showed the highest Cr6+ adsorption capacity (16.8 ± 1.6 mg g−1) and led to 3.3 times improvement in the viability of S. oneidensis MR-1 in the presence of Cr6+ and 2.66 times an enhancement in Cr6+ bio-detoxification. This will open up a new venue of research using nanomaterials for boosting the bio-reduction of Cr6+ using bacteria.Author contributions
N. T. K. T. and L. C. devised and coordinated the project and provided resources. D. S. R. designed and did most of the experiments and wrote the manuscript. I. T. assisted in particle synthesis and data analysis. S. A., P. W. and M. C. helped with the microbiology work. N. T. K. T. and S. M. provided expertise, corrected the manuscript and helped to acquire funding. K. S., A. M. and D. K. carried out XPS characterization, processed data and corrected the manuscript. G. V. provided resources for characterization. L. B. helped to acquire funding.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
D. S. R. acknowledges funding from Newton Mosharafa scholarship given to Egyptian Petroleum Research Institute. The authors acknowledge Jeffrey A. Gralnick at University of Minnesota, for kindly providing S. oneidensis JG1486 and JG3355. The authors would like to thank EPSRC (EP/M015157/1) for financial support. We thank UCL Grand Challenges and UCL Small Grant for funding. Adam Strange was acknowledged for participation in writing UCL Small Grant application. We thank Ana Alvarez Prendes for contributing in the data analysis of some TEM images. Thithawat Trakoolwilaiwan is acknowledged for the ICP analysis. Rachel Duan is thanked for some functionalization of NPs. We acknowledge Chris Howard and Szymon Bartus at UCL, for using Raman spectroscopy facility.Notes and references
- A. S. Jones, J. Marini, H. M. Solo-Gabriele, N. M. Robey and T. G. Townsend, Waste Manage., 2019, 87, 731–740 CrossRef CAS PubMed.
- S. Famielec, Materials, 2020, 13, 1533–1545 CrossRef CAS PubMed.
- B. Wang, B. Liu, J. Gu and M. A. J. Somers, Surf. Coat. Technol., 2022, 438, 128408–128422 CrossRef CAS.
- H. Tang, Z. Peng, F. Gu, L. Yang, W. Tian, Q. Zhong, M. Rao, G. Li and T. Jiang, Ceram. Int., 2021, 47, 10809–10818 CrossRef CAS.
- J. Xing and M. T. Pailthorpe, J. Soc. Dyers Colour., 2000, 116, 91–93 CAS.
- Z. Wang, C. Bao, K. Yan, Y. Song and W. Li, J. Appl. Polym. Sci., 2021, 138, 1–10 Search PubMed.
- A. Sennaroglu and Y. Morova, Appl. Phys. B: Lasers Opt., 2022, 128, 1–25 CrossRef.
- A. Bratovcic, H. Buksek, C. Helix-Nielsen and I. Petrinic, Chem. Eng. J., 2022, 431, 133918–133927 CrossRef CAS.
- V. Bolaños-Benítez, E. D. van Hullebusch, J. L. Birck, J. Garnier, P. N. L. Lens, M. Tharaud, C. Quantin and Y. Sivry, Chem. Geol., 2021, 561, 120000–120011 CrossRef.
- M. A. Islam, M. J. Angove and D. W. Morton, Environ. Nanotechnol., Monit. Manage., 2019, 12, 100267–100287 Search PubMed.
- International Agency for Research on Cancer, List of Classifications–IARC Monographs on the Identification of Carcinogenic Hazards to Humans, 2020, vol. 1 Search PubMed.
- N. A. Awang, W. N. W. Salleh, A. F. Ismail, N. Yusof, F. Aziz and J. Jaafar, Ind. Eng. Chem. Res., 2019, 58, 720–728 CrossRef CAS.
- A. D. Villalobos-lara, F. Alvarez, Z. Gami, R. Navarro and J. M. Peralta-hern, Chemosphere, 2021, 264, 128491–128499 CrossRef CAS PubMed.
- F. I. El-Dib, D. E. Mohamed, O. A. A. El-Shamy and M. R. Mishrif, Egypt. J. Pet., 2020, 29, 1–7 CrossRef.
- Z. Q. Id, W. Dong, Y. Chen, G. Dong, S. Zhu, Y. Yu and D. Bian, PLoS One, 2020, 15, 1–17 Search PubMed.
- V. E. Pakade, T. Tavengwa and L. M. Madikizela, RSC Adv., 2019, 9, 26142–26164 RSC.
- A. B. Mpofu, O. O. Oyekola and P. J. Welz, J. Cleaner Prod., 2021, 296, 126490–126504 CrossRef CAS.
- R. Hern, V. Y. Mena-cervantes, E. Ruiz-baca, E. E. Neri-torres, I. Chairez, S. M. García-solares and J. Vazquez-arenas, J. Environ. Chem. Eng., 2021, 9, 104626–104637 CrossRef.
- G. Wu, F. Wan, H. Fu, N. Li and H. Gao, J. Bacteriol., 2015, 197, 3563–3572 CrossRef CAS PubMed.
- L. Cheng, R. He, D. Min, W. Li, D. Liu and H. Yu, ACS ES&T Engg, 2021, 1, 842–850 Search PubMed.
- F. Asdrubali, F. D. Alessandro and S. Schiavoni, Sustainable Mater. Technol., 2020, 4, 1–17 Search PubMed.
- A. Zuazua-ros, M. Vidaurre-arbizu and P. Silvia, J. Cleaner Prod., 2021, 291, 125960–125971 CrossRef.
- M. Naveenkumar and K. Senthilkumar, Biomass Bioenergy, 2021, 149, 106082–106089 CrossRef CAS.
- H. Gang, C. Xiao, Y. Xiao, W. Yan, R. Bai, R. Ding, Z. Yang and F. Zhao, Environ. Int., 2019, 127, 94–102 CrossRef CAS PubMed.
- A. Elahi and A. Rehman, J. King Saud Univ., Sci., 2019, 31, 1005–1013 CrossRef.
- D. L. Parker, P. Borer and R. Bernier-Latmani, Front. Microbiol., 2011, 2, 1–14 Search PubMed.
- M. Kheirabadi, R. Mahmoodi, N. Mollania and M. Kheirabadi, Int. J. Environ. Sci. Technol., 2020, 17, 143–152 CrossRef CAS.
- X. Tang, Y. Huang, Y. Li, L. Wang, X. Pei, D. Zhou, P. He and S. S. Hughes, Ecotoxicol. Environ. Saf., 2021, 208, 111699–111711 CrossRef CAS PubMed.
- A. Carra, P. W. Villalta, J. He, X. Yao, R. J. Hamers, S. Balbo, Z. Vivian and C. L. Haynes, Chem. Sci., 2020, 11, 11244–11258 RSC.
- S. L. Mitchell, N. V Hudson-Smith, M. S. Cahill, B. N. Reynolds, S. D. Frand, C. M. Green, C. Wang, M. N. Hang, R. T. Hernandez and R. J. Hamers, Chem. Sci., 2019, 10, 9768–9781 RSC.
- M. N. Hang, I. L. Gunsolus, H. Wayland, E. S. Melby, A. C. Mensch, K. R. Hurley, J. A. Pedersen, C. L. Haynes and R. J. Hamers, Chem. Mater., 2016, 28, 1092–1100 CrossRef CAS.
- H. Dong, L. Li, Y. Lu, Y. Cheng, Y. Wang, Q. Ning, B. Wang, L. Zhang and G. Zeng, Environ. Int., 2019, 124, 265–277 CrossRef CAS PubMed.
- C. Qu, S. Qian, L. Chen, Y. Guan, L. Zheng, S. Liu, W. Chen, P. Cai and Q. Huang, Environ. Sci. Technol., 2019, 53, 8147–8156 CrossRef CAS PubMed.
- K. Zhang, N. Li, P. Liao, Y. Jin, Q. Li, M. Gan, Y. Chen, P. He, F. Chen, M. Peng and J. Zhu, Environ. Pollut., 2021, 286, 117227–117236 CrossRef CAS PubMed.
- A. Sundman, A. L. Vitzthum, K. Adaktylos-surber, A. I. Figueroa, G. van der Laan, B. Daus, A. Kappler, J. M. Byrne, G. Van Der Laan, B. Daus, A. Kappler and J. M. Byrne, J. Hazard. Mater., 2020, 384, 121450–121456 CrossRef CAS PubMed.
- B. Eyvazi, A. Jamshidi-zanjani and A. Khodadadi, Environ. Pollut., 2020, 265, 113685–113695 CrossRef CAS PubMed.
- Y. Ma, X. Wu, Z. Shi, X. Li, S. Qian, X. Sun, W. Sun, C. Guo and C. M. Li, ACS Sustainable Chem. Eng., 2022, 10, 3355–3362 CrossRef CAS.
- S. Khilari, S. Pandit, J. L. Varanasi, D. Das and D. Pradhan, ACS Appl. Mater. Interfaces, 2015, 7, 20657–20666 CrossRef CAS PubMed.
- J. Hu, I. M. C. Lo and G. Chen, Sep. Purif. Technol., 2007, 56, 249–256 CrossRef CAS.
- J. Hu, I. M. C. Lo and G. Chen, Langmuir, 2005, 21, 11173–11179 CrossRef CAS PubMed.
- M. Li, Q. Gao, T. Wang, Y. S. Gong, B. Han, K. S. Xia and C. G. Zhou, Mater. Des., 2016, 97, 341–348 CrossRef CAS.
- X. Lasheras, M. Insausti, J. M. De La Fuente, I. Gil De Muro, I. Castellanos-Rubio, L. Marcano, M. L. Fernández-Gubieda, A. Serrano, R. Martín-Rodríguez, E. Garaio, J. A. García and L. Lezama, Dalton Trans., 2019, 48, 11480–11491 RSC.
- O. Antonoglou and C. Dendrinou-Samara, in Reducing Agents in Colloidal Nanoparticle Synthesis, 2021, pp. 51–72 Search PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- D. García-Soriano, R. Amaro, N. Lafuente-Gómez, P. Milán-Rois, Á. Somoza, C. Navío, F. Herranz, L. Gutiérrez and G. Salas, J. Colloid Interface Sci., 2020, 578, 510–521 CrossRef PubMed.
- R. Hachani, M. Lowdell, M. Birchall, A. Hervault, D. Mertz, S. Begin-Colin and N. T. B. D. K. Thanh, Nanoscale, 2016, 8, 3278–3287 RSC.
- D. S. Mathew and R. S. Juang, Chem. Eng. J., 2007, 129, 51–65 CrossRef CAS.
- H. Sharifi Dehsari, V. Ksenofontov, A. Möller, G. Jakob and K. Asadi, J. Phys. Chem. C, 2018, 122, 28292–28301 CrossRef.
- K. Vamvakidis, M. Katsikini, G. Vourlias, M. Angelakeris, E. C. Paloura and C. Dendrinou-Samara, Dalton Trans., 2015, 44, 5396–5406 RSC.
- S. A. Ahmed, Results Phys., 2017, 7, 604–610 CrossRef.
- L. Khanna and N. K. Verma, Phys. B, 2013, 427, 68–75 CrossRef CAS.
- F. Branda, A. Buri, A. Marotta and S. Saiello, Thermochim. Acta, 1984, 93, 65–68 Search PubMed.
- K. Vamvakidis, D. Sakellari, M. Angelakeris and C. Dendrinou-Samara, J. Nanopart. Res., 2013, 15, 1743–1753 CrossRef.
- S. Fu, R. Yang, J. Ren, J. Liu, L. Zhang, Z. Xu, Y. Kang and P. Xue, ACS Nano, 2021, 15, 11953–11969 CrossRef CAS PubMed.
- J. Ge, Y. Hu, M. Biasini, W. P. Beyermann and Y. Yin, Angew. Chem., Int. Ed., 2007, 46, 4342–4345 CrossRef CAS PubMed.
- H. Gavilán, E. H. Sánchez, M. E. F. Brollo, L. Asín, K. K. Moerner, C. Frandsen, F. J. Lázaro, C. J. Serna, S. Veintemillas-Verdaguer, M. P. Morales and L. Gutiérrez, ACS Omega, 2017, 2, 7172–7184 CrossRef PubMed.
- Y. Xie, C. Tian, W. Chen, C. Wu, Z. Liu, P. Ning, H. Deng and Z. Lin, Environ. Sci.: Nano, 2019, 6, 1406–1417 RSC.
- S. Xuan, F. Wang, Y. J. Wang, C. Yu and K. C. Leung, J. Mater. Chem., 2010, 20, 5086–5094 RSC.
- A. Qureashi, A. H. Pandith, A. Bashir, T. Manzoor, L. A. Malik and F. A. Sheikh, Surf. Interfaces, 2021, 23, 101004–101019 CrossRef CAS.
- I. Chakraborty, D. Majumder, S. Talukdar, S. Roy and K. Mandal, Surf. Interfaces, 2017, 9, 154–159 CrossRef CAS.
- D. Wyrzykowski and L. Chmurzyński, J. Therm. Anal. Calorim., 2010, 102, 61–64 CrossRef CAS.
- M. Matzapetakis, N. Karligiano, A. Bino, M. Dakanali, C. P. Raptopoulou, V. Tangoulis, A. Terzis, J. Giapintzakis and A. Salifoglou, Inorg. Chem., 2000, 39, 4044–4051 CrossRef CAS PubMed.
- M. Justi, M. P. de Freitas, J. M. Silla, C. A. Nunes and C. A. Silva, J. Mol. Struct., 2021, 1237, 130405–130417 CrossRef CAS.
- S. Luther, N. Brogfeld, J. Kim and J. G. Parsons, J. Colloid Interface Sci., 2013, 400, 97–103 CrossRef CAS PubMed.
- W. G. Gao, X. C. Liu and M. F. Chen, RSC Adv., 2017, 7, 41011–41016 RSC.
- Y. Cantu, A. Remes, A. Reyna, D. Martinez, J. Villarreal, H. Ramos, S. Trevino, C. Tamez, A. Martinez, T. Eubanks and J. G. Parsons, Chem. Eng. J., 2014, 254, 374–383 CrossRef CAS PubMed.
- H. Kumar, K. L. Maurya, A. K. Gehlaut, D. Singh, S. Maken, A. Gaur and S. Kamsonlian, Appl. Water Sci., 2020, 10, 1–10 CrossRef.
- K. Ahalya, N. Suriyanarayanan and V. Ranjithkumar, J. Magn. Magn. Mater., 2014, 372, 208–213 CrossRef CAS.
- S. Martinez-Vargas, A. I. Martínez, E. E. Hernández-Beteta, H. H.-F. O. F. Mijangos-Ricardez, V. Vázquez-Hipólito, C. Patiño-Carachure and J. López-Luna, J. Mater. Sci., 2017, 52, 6205–6215 CrossRef CAS.
- J. Wang, Q. Xu, W. Yin, J. Hou, S. Wang and X. Wang, Ecotoxicol. Environ. Saf., 2021, 217, 112209–112216 CrossRef CAS PubMed.
- Y. Li, G. Wei, C. Zhang, X. Liang, W. Chu, H. He, J. W. Stucki, L. Ma, X. Lin and J. Zhu, Sci. Total Environ., 2019, 656, 400–408 CrossRef CAS PubMed.
- J. Zhang, C. Zhang, G. Wei, Y. Li, X. Liang, W. Chu, H. He, D. Huang, J. Zhu and R. Zhu, J. Colloid Interface Sci., 2017, 500, 20–29 CrossRef CAS PubMed.
- N. Antonatos, D. Bouša, S. Shcheka, S. M. Beladi-Mousavi, M. Pumera and Z. Sofer, Inorg. Chem., 2019, 58, 10227–10238 CrossRef CAS PubMed.
- M. Testa-Anta, M. A. Ramos-Docampo, M. Comesaña-Hermo, B. Rivas-Murias and V. Salgueiriño, Nanoscale Adv., 2019, 1, 2086–2103 RSC.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- Z. Zhang, Y. Wang, Q. Tan, Z. Zhong and F. Su, J. Colloid Interface Sci., 2013, 398, 185–192 CrossRef CAS PubMed.
- I. Desai, M. N. Nadagouda, M. Elovitz, M. Mills and B. Boulanger, Mater. Sci. Energy Technol., 2019, 2, 150–160 Search PubMed.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- J. Han, M. Kim and H. M. Ro, Environ. Chem. Lett., 2020, 18, 631–662 CrossRef CAS.
- J. Xie, X. Gu, F. Tong, Y. Zhao and Y. Tan, J. Colloid Interface Sci., 2015, 455, 55–62 CrossRef CAS PubMed.
- C. Ri, J. Tang, F. Liu, H. Lyu and F. Li, J. Environ. Sci., 2022, 113, 12–25 CrossRef CAS PubMed.
- G. Wang, B. Zhang, S. Li, M. Yang and C. Yin, Bioresour. Technol., 2017, 227, 353–358 CrossRef CAS PubMed.
- A. Mohamed, L. Yu, Y. Fang, N. Ashry, Y. Riahi, I. Uddin, K. Dai and Q. Huang, Chemosphere, 2020, 247, 125902–125914 CrossRef CAS PubMed.
- D. Coursolle and J. A. Gralnick, Front. Microbiol., 2012, 3, 1–11 Search PubMed.
- K. Chourey, M. R. Thompson, J. Morrell-Falvey, N. C. VerBerkmoes, S. D. Brown, M. Shah, J. Zhou, M. Doktycz, R. L. Hettich and D. K. Thompson, Appl. Environ. Microbiol., 2006, 72, 6331–6344 CrossRef CAS PubMed.
- B. D. Bennett, K. E. Redford, J. A. Gralnick and A. Gralnick, J. Bacteriol., 2018, 200, 1–14 CrossRef CAS PubMed.
- R. Bencheikh-Latmani, A. Obraztsova, M. R. Mackey, M. H. Ellisman and B. M. Tebo, Environ. Sci. Technol., 2007, 41, 214–220 CrossRef CAS PubMed.
- X. Liu, G. Chu, Y. Du, J. Li and Y. Si, World J. Microbiol. Biotechnol., 2019, 35, 1–8 CrossRef CAS PubMed.
- S. Viamajala, B. M. Peyton, R. K. Sani, W. A. Apel and J. N. Petersen, Biotechnol. Prog., 2004, 20, 87–95 CrossRef CAS PubMed.
- A. Mohamed, B. Sun, C. Yu, X. Gu, N. Ashry, Y. Riahi, K. Dai and Q. Huang, J. Environ. Chem. Eng., 2021, 9, 105096–105106 CrossRef CAS.
- R. Elmeihy, X. C. Shi, P. L. Tremblay and T. Zhang, Chemosphere, 2021, 263, 128281–128289 CrossRef CAS PubMed.
- J. Cheng, J. Gao, J. Zhang, W. Yuan, S. Yan, J. Zhou, J. Zhao and S. Feng, Water, Air, Soil Pollut., 2021, 232, 1–14 CrossRef.
- Y. Xiao, C. Xiao and F. Zhao, Front. Environ. Sci. Eng., 2020, 14, 1–11 CrossRef.
- U. Schröder, Phys. Chem. Chem. Phys., 2007, 9, 2619–2629 RSC.
- D. J. Richardson, J. N. Butt, J. K. Fredrickson, J. M. Zachara, L. Shi, M. J. Edwards, G. White, N. Baiden, A. J. Gates, S. J. Marritt and T. A. Clarke, Mol. Microbiol., 2012, 85, 201–212 CrossRef CAS PubMed.
- J. S. McLean, P. D. Majors, C. L. Reardon, C. L. Bilskis, S. B. Reed, M. F. Romine and J. K. Fredrickson, J. Microbiol. Methods, 2008, 74, 47–56 CrossRef CAS PubMed.
- J. A. Gralnick, H. Vali, D. P. Lies and D. K. Newman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4669–4674 CrossRef CAS PubMed.
- G. J. Newton, S. Mori, R. Nakamura, K. Hashimoto and K. Watanabe, Appl. Environ. Microbiol., 2009, 75, 7674–7681 CrossRef CAS PubMed.
- I. L. Gunsolus, M. N. Hang, N. V Hudson-Smith, J. T. Buchman, J. W. Bennett, D. Conroy, S. E. Mason, R. J. Hamers and C. L. Haynes, Environ. Sci.: Nano, 2017, 4, 636–646 RSC.
- D. Ghosal, M. V. Omelchenko, E. K. Gaidamakova, V. Y. Matrosova, A. Vasilenko, A. Venkateswaran, M. Zhai, H. M. Kostandarithes, H. Brim, K. S. Makarova, L. P. Wackett, J. K. Fredrickson and M. J. Daly, FEMS Microbiol. Rev., 2005, 29, 361–375 CAS.
- J. Qin, L. Qian, J. Zhang, Y. Zheng, J. Shi, J. Shen and C. Ou, Chemosphere, 2021, 263, 128048–128057 CrossRef CAS PubMed.
- Y. Yin, C. Liu, G. Zhao and Y. Chen, J. Hazard. Mater., 2022, 440, 165187–165211 Search PubMed.
- J. Hu, I. M. C. Lo and G. Chen, Sep. Purif. Technol., 2007, 56, 249–256 CrossRef CAS.
- H. Cheng, Z. Jing, L. Yang, A. Lu, G. Ren and J. Liu, Geochim. Cosmochim. Acta, 2021, 305, 19–32 CrossRef CAS.
- M. G. Fortune and W. B. Mellon, Ind. Eng. Chem., Anal. Ed., 1938, 10, 60–64 CrossRef.
- A. Sanchez-Hachair and A. Hofmann, C. R. Chim., 2018, 21, 890–896 CrossRef CAS.
- P. F. Urone, Anal. Chem., 1955, 27, 1354–1355 CrossRef CAS.
- M. A. Al-ghouti and D. A. Da, J. Hazard. Mater., 2020, 393, 122383–122404 CrossRef CAS PubMed.
- L. Xu, H. Chen, M. Canales and L. Ciric, J. Microbiol. Methods, 2019, 164, 105670–105676 CrossRef CAS PubMed.
- N. Wurzler, J. David, R. Wagner, M. Dimper, D. Lützenkirchen-hecht and O. Ozcan, Corros. Sci., 2020, 174, 108855–108862 CrossRef CAS.
- E. D. Kees, C. E. Levar, S. P. Miller, D. R. Bond, J. A. Gralnick and A. M. Dean, Commun. Biol., 2021, 4, 1–9 CrossRef PubMed.
- C. Bankier, Y. Cheong, S. Mahalingam, M. Edirisinghe, G. Ren, E. Cloutman-Green and L. Ciric, PLoS One, 2018, 13, 1–13 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00691j |
| This journal is © The Royal Society of Chemistry 2023 |