
Non-nucleophilic electrolyte with non-fluorinated hybrid solvents for long-life magnesium metal batteries†
Yue
Sun
a,
Yuhang
Wang
b,
Liwei
Jiang
a,
Dejian
Dong
a,
Wanwan
Wang
a,
Jun
Fan
 b and
Yi-Chun
Lu
*a
b and
Yi-Chun
Lu
*a
aElectrochemical Energy and Interfaces Laboratory, Department of Mechanical and Automation Engineering, The Chinese University of Hong Kong, Shatin, N.T., 999077, Hong Kong, SAR, China. E-mail: yichunlu@mae.cuhk.edu.hk
bDepartment of Materials Science and Engineering, City University of Hong Kong, Kowloon 999077, Hong Kong, SAR, China
First published on 7th December 2022
Abstract
Non-nucleophilic and non-fluorinated compounds are the most important class of solvents to enable sustainable rechargeable magnesium (Mg) batteries; however, they suffer from poor stability due to the formation of an unstable solid–electrolyte-interphase (SEI). Here, we design a hybrid-solvent electrolyte by dispersing a non-nucleophilic Mg(TFSI)2–MgCl2-1,2-dimethoxyethane (DME) solution in a non-fluorinated weakly coordinating solvent (tetrahydrofuran, THF) to prevent the decomposition of DME and form a stable SEI. This strategy significantly improved the cycle life of a Mg anode from 24 to 4000 hours (in Mg–Mg symmetric cells) and realized a stable cycle life of Mg–Mo6S8 full cells for more than 2300 hours with an average CE of 99.96%. The non-fluorinated weakly coordinating solvent THF suppressed uneven Mg growth and contributed to the formation of a polymeric SEI, which filled the cracks of the pristine SEI, preventing side reactions and passivation. This strategy revealed the critical role of weakly coordinating solvents in stabilizing the Mg anode for reversible Mg batteries.
Broader contextRechargeable magnesium batteries are high-energy, safer, and more cost-effective alternatives to lithium metal batteries thanks to Mg's high volumetric capacity, low theoretical potential, and earth-abundance. However, the difficulty in stabilizing the Mg metal with low-cost electrolytes impeded their practical applications. Non-nucleophilic and non-fluorinated compounds are the most important class of solvents to enable sustainable rechargeable magnesium batteries. However, it is challenging to simultaneously realize sufficient ion association and reduction stability with a single solvent. In this study, we design a hybrid-solvent electrolyte by dispersing a non-nucleophilic Mg(TFSI)2–MgCl2-1,2-dimethoxyethane (DME) solution in a non-fluorinated weakly coordinating solvent (tetrahydrofuran, THF) to prevent the decomposition of DME and form a stable SEI without leading to salt precipitation. This strategy significantly improved the cycle life of both the Mg anode and Mg–Mo6S8 full cells thanks to the suppression of uneven Mg growth and formation of a polymeric SEI. This strategy revealed the critical role of weakly coordinating solvents in stabilizing the Mg anode for reversible Mg batteries. |
Introduction
Growing energy demands and the widespread need for renewable power sources call for high-energy storage systems beyond lithium-ion batteries (LIBs). Rechargeable magnesium batteries (RMBs) are high-energy, safer, and more cost-effective alternatives to LIBs and lithium metal batteries (LMBs) thanks to Mg's high volumetric capacity (3832 mA h cm−3),1 low theoretical potential (−2.37 V vs. SHE)2 and earth-abundance.3 However, the difficulty in stabilizing the Mg metal with commercial electrolytes has impeded their practical implementation.4–6 Due to the low theoretical potential of the Mg anode (−2.37 V vs. SHE), most electrolytes and contaminants (especially moisture) readily decompose on the Mg anode.2 Furthermore, the Mg2+ cations with high charge density raise the ion migration barrier across the solid electrolyte interface (SEI).7 Therefore, surface films generated by conventional electrolytes containing strongly coordinating ionic salts (such as Mg(ClO4)2 and Mg(BF4)2) and solvents such as carbonates and nitriles are highly resistive, leading to large voltage hysteresis (>2.0 V) even under low current densities (∼0.01 mA cm−2).4,6,8 The presence of impurities also induces parasitic reactions or accelerates the electrolyte decomposition, resulting in inferior electrochemical performance or irreversibility of Mg anodes.2,9 A series of nucleophilic magnesium organochloroaluminates (summarized in Table S1, ESI†) with water-reactive scavengers (such as phenylmagnesium chloride and di-n-butylmagnesium) show remarkable stability against the Mg anode.10,11 Nevertheless, the organochloroaluminate species show low oxidation stability (<2.8 V vs. Mg/Mg2+)2 and may render a nucleophilic attack towards electrophilic cathodes, including carbonyl-based polymers and conversion cathodes, limiting their utilization in high-voltage RMBs.2,12Mitigating parasitic reactions and passivation without nucleophilic components remains a non-trivial task. Polymeric or halogen-rich artificial solid electrolyte interfaces (SEIs) successfully stabilize Mg anode in passivating or water-containing electrolytes. However, the preformed SEI may gradually dissolve in electrolytes or crack owing to the volume change of electrodes, resulting in large voltage hysteresis (0.5–1.2 V) during prolonged cycles.13–15 Therefore, the design of electrolytes with intrinsic stability against the Mg anode is of vital importance. Non-nucleophilic weakly coordinating salts (summarized in Table S1, ESI†) with relatively high ionic conductivity (4.0–11.0 mS cm−1) were designed,12,16 including magnesium tetrakis(hexafluoroisopropyloxy)borate, Mg[B(Ohfip)4]2,17,18 magnesium closo-carbadodecaborate, Mg(CB11H12)2,19,20 magnesium perfluorinated tert-butoxide (Mg(pftb)2),21 and magnesium perfluorinated pinacolatoaluminate (MgFPA).21–23 Nevertheless, most weakly coordinating Mg salts are not commercially available after the first weakly coordinating anion was reported in 1984,24 which is due to complex synthesis procedures, expensive reagents, or low stability under ambient conditions.16,18,25
Solvent design is an effective method to improve the electrochemical performance and enable the utilization of commercial ionic Mg salts, such as magnesium bis(trifluoromethanesulfonimide) (Mg(TFSI)2) and magnesium trifluoromethanesulfonate (Mg(OTf)2). However, only a handful of studies were carried out to provide insights into the contribution of solvents due to the difficulty in screening solvents with both high solvating power and reduction stability. 1,2-Dimethoxyethane (DME) was the most widely utilized solvent for non-nucleophilic electrolytes thanks to its relative reduction stability, low viscosity, and solvating power. However, DME failed to generate an effective SEI.26,27 Co-solvents with higher dielectric constants, such as trimethyl phosphate (TMP),28 were utilized to suppress the decomposition of DME via loosening the coordination. However, the electron-withdrawing groups of solvents with high dielectric constants inevitably sacrificed the reduction stability of electrolytes.29,30 Methoxyethyl-amines with slightly higher dielectric constant but stronger Lewis basicity,31 were utilized to reduce the voltage hysteresis by lowering reorganization energy.31 Nevertheless, the increasing radius of solvation shells lowered ∼40% of ionic conductivity compared with the pristine DME-dominated solvation shells.31 The strongly coordinating co-solvents sacrifice the reductive stability or facile migration of Mg2+–DME clusters, thus failing to achieve long-period cycling (100–300 hours).28,31,32 Consequently, there still a lack of cost-effective strategies to stabilize the Mg anode during prolonged cycling and universal principles to guide the design of high-performance and low-cost non-nucleophilic electrolytes.
Here, we report a non-fluorinated weakly coordinating co-solvent, tetrahydrofuran, THF, to suppress the decomposition of DME while retaining the desirable properties of DME. This strategy significantly improved the cycle life of the Mg anode from 24 to 4000 hours (in Mg–Mg symmetric cells) and realized a stable cycle life of Mg–Mo6S8 full cells for more than 2300 hours with an average CE of 99.96% without reactive/nucleophilic impurity scavengers. The enhancement mechanism was revealed by scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), atomic force microscopy (AFM), in situ Fourier-transform infrared (FT-IR) spectroscopy, and density functional theory (DFT) calculations, suggesting that THF suppressed uneven Mg growth and contributed to the formation of polymeric SEI, preventing side reactions and passivation. This strategy revealed the critical role of weakly coordinating solvents in a stabilizing Mg anode for reversible Mg batteries.
Results and discussion
Structure of hybrid-solvent electrolyte
Ideal solvents for high-performance RMBs should satisfy two requirements: (1) strong solvating power to solubilize Mg salts; and (2) effective protective ability towards Mg metal. However, these are difficult to be met with a single solvent.26 DME, was widely utilized to facilitate the dissociation of Mg salts via multi-dentate coordination with Mg2+ cations (Fig. 1(a), left).26 It benefits ion dissociation and migration under electric field but leads to voltage hysteresis and short cycle life (Fig. 1(a), left).23,28,31 Cyclic ethers benefit the formation of protective films on metal anode via ring-opening reactions,33 but resulted in inferior ion dissociation12 because of their monodentate coordinating properties (Fig. 1(a), right).34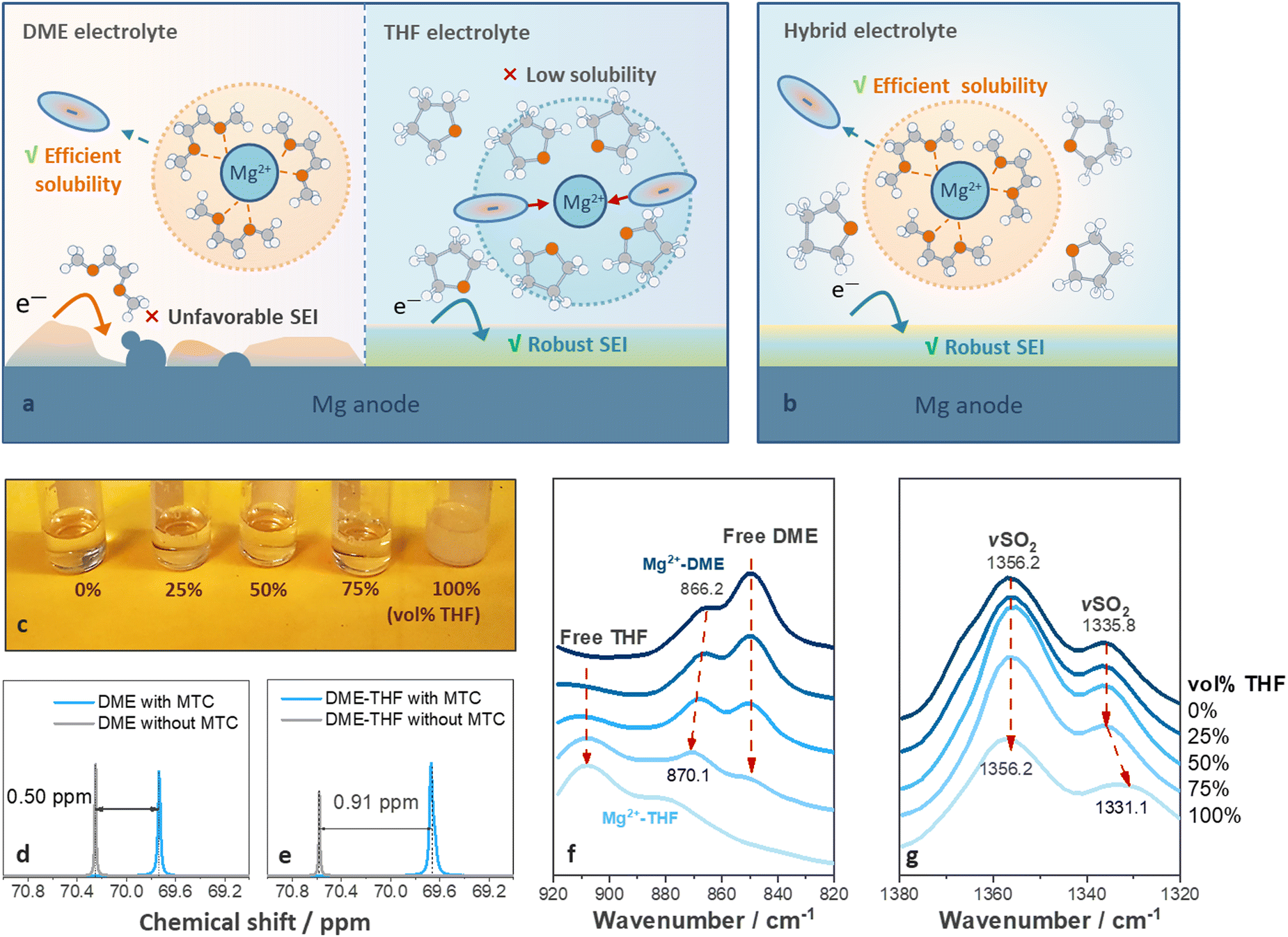 | ||
| Fig. 1 Schematic illustration of structure of (a) single-solvent and (b) hybrid electrolytes. (c) Digital photographs of DME, DME-THF (25–75 vol% THF), and THF electrolytes. 13C NMR spectra of methylene carbons in DME (d) without and (e) with THF (75 vol% THF). (f) and (g) Ex situ FT-IR spectra of DME, DME-THF (25–75 vol% THF), and THF electrolytes. | ||
We hypothesized that a hybrid-solvent electrolyte comprising facile-migrating DME-rich solvation shells surrounded by weakly coordinating cyclic ethers can boost the SEI formation without leading to ion association (Fig. 1(b)). Mg(TFSI)2–2MgCl2 (MTC) was selected as the supporting salt owing to its commercial availability and relatively high stability under ambient conditions.35,36 THF was chosen as a co-solvent because it exhibits a similar dielectric constant (THF ∼ 7.5, DME ∼ 7.3) while slightly lower solvating power compared with DME (THF ∼ 2.8, DME ∼ 6.4), ensuring the miscibility without disturbing the coordination between DME and Mg2+.37,38 As shown in Fig. 1(c), THF fails to fully dissolve MTC (solubility limit ∼0.033 M) but homogeneous solutions can be obtained via diluting 0.8 M MTC/DME solutions with 0–75 vol% THF (Fig. 1(c) and experimental methods, ESI†). In contrast, 2-methyltetrahydrofuran (2-MeTHF) with a low dielectric constant (6.97)39 led to the formation of two immiscible phases (Fig. S1a, ESI†), while 1,3-dioxolane (DOL) with low solvating power (1.0) resulted in the precipitation of MTC salts (Fig. S1b, ESI†).
Ex situ Fourier transform infrared (FT-IR) spectroscopy was carried out to investigate the structure of hybrid-solvent electrolytes. The characteristic peaks related to solvents in the 920–820 cm−1 region,40 and those related to anion coordination in 1380–1320 cm−1 (SO2 stretching, νSO2)41 regions were investigated. As shown in Fig. 1(f), increasing the ratio of THF leads to blue shifts of the Mg2+-coordinated solvent (866.2 cm−1 to 870.1 cm−1) towards the Mg2+-coordinated THF, suggesting that THF has been incorporated into the solvation structure. The peak fitting for each component is included in Fig. S2 (ESI†). 13C NMR results further confirmed the results obtained from FT-IR spectra. After adding MTC salts, the chemical shifts of methylene carbons shifted upfield, and the changes reflected the average between coordinating and free solvents.31 Therefore, a larger change indicated that more DME entered the solvation shell.31 As shown in Fig. 1(d) and (e), the change of chemical shifts related to methylene carbons in DME increased from 0.50 to 0.91 ppm in the presence of THF, indicating that less DME was freed from the solvation shell in the DME-THF electrolytes.31
As for the peaks related to anion coordination in the 1380–1320 cm−1 region (Fig. 1(g)), the position of peaks kept the same (1335.8 cm−1) in the spectra of DME and DME-THF hybrid electrolytes (0 ≤ vol% THF ≤ 75%). However, a redshift of νSO2 peak to 1331.1 cm−1 was observed if THF served as the single solvent (Fig. 1(e)), revealing that the concentration of ion pairs increased42 if DME was excluded from the solvation shell.
Therefore, the film-forming THF molecules exhibit the unique behavior of being associated with the hybrid electrolytes without leading to the formation of unstable ion pairs and sacrificing the ionic conductivity (Fig. S3, ESI†).
Electrochemical performance
Coulombic efficiency (CE) was firstly measured with Mg–Cu cells (the operating protocol is shown in Experimental methods, ESI†) to determine the optimal composition of DME-THF electrolytes. As shown in Fig. 2(a) and (b), Mg–Cu cells with DME show a low CE (95.2%). With the increasing volumetric ratio of THF, the CE was significantly enhanced and achieved the maximum value (98.8%) when vol% THF reached 75% (Fig. 2(a), (b) and Fig. S4, ESI†), indicating that maximizing the concentration of THF in the DME-THF hybrid electrolytes benefits the reversibility of the Mg anode thanks to the film-forming ability of cyclic ethers (will be further discussed in the following sections). It is noted that MTC/THF electrolyte only achieved an inferior CE equal to 90.3% owing to the poor solubility of Mg salts and ionic conductivity (Fig. 2(a) and Fig. S4, ESI†). The improved performance of DME-THF cells compared to single-solvent ones confirmed the hypothesis that facile-migrating DME-rich solvation shells surrounded by weakly coordinating THF can boost the formation of the stable SEI without leading to ion association. The CE enhancement (96.3%) achieved by DME-DOL electrolytes further proved the effectiveness of cyclic ethers to ameliorate the compatibility between the DME and Mg anode (Fig. S5, ESI†), but DOL with a low solvating power is less effective than THF owing to the insufficient ion dissociation. Therefore, cyclic ethers exhibited similar dielectric constants/solvating powers with THF and enhanced film-forming ability are promising to further improve the performance of MTC/DME electrolytes.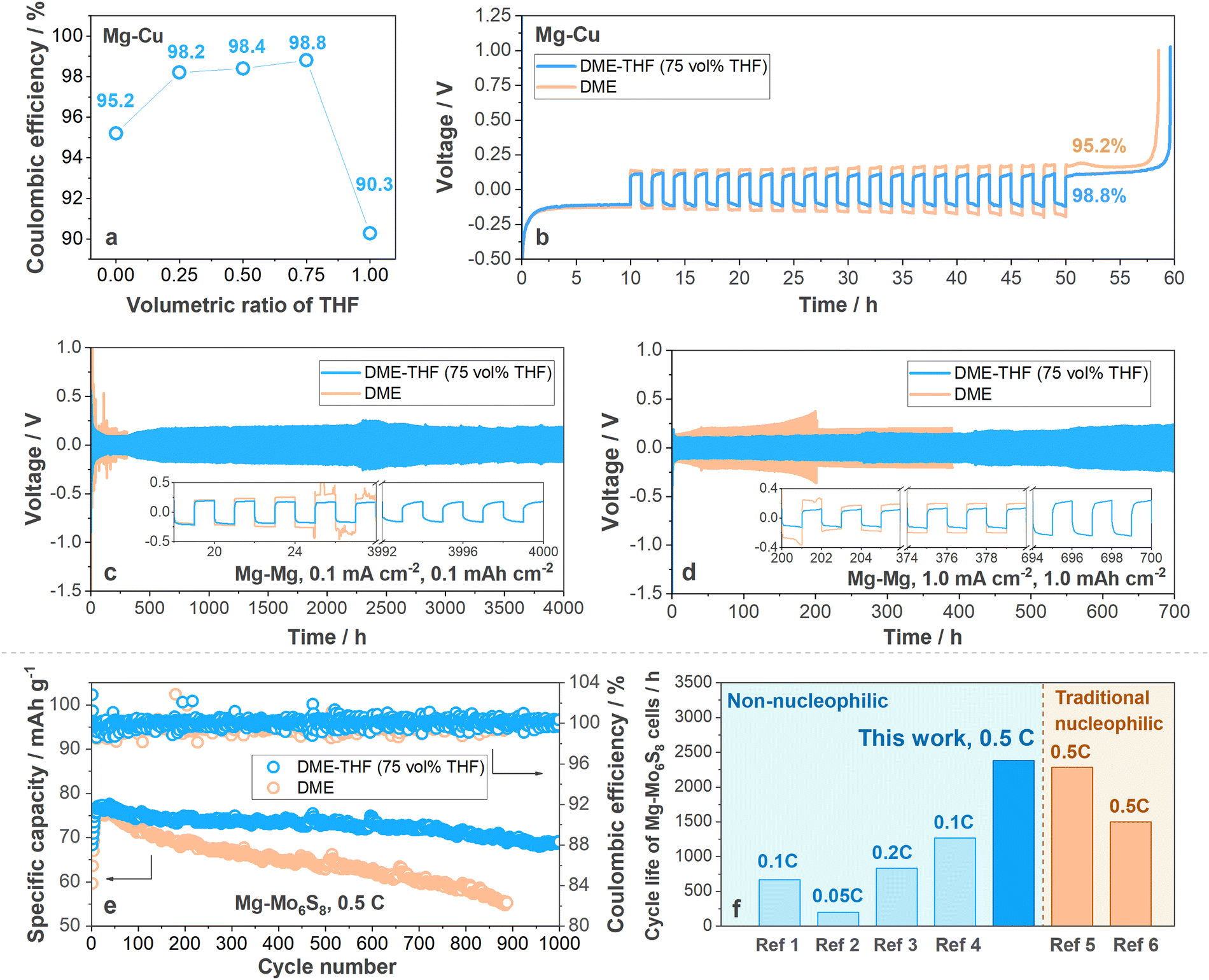 | ||
| Fig. 2 (a) Coulombic efficiency varied with the volumetric ratio of THF. The value was measured with Mg–Cu cells under 1.0 mA cm−2, 1.0 mA h cm−2. (b) Voltage profiles of Mg–Cu cells with DME-THF (75 vol% THF, blue line) and DME (yellow line) electrolytes under 1.0 mA cm−2, 1.0 mA h cm−2. Cycling stability of Mg–Mg symmetric cells with DME (yellow line)/DME-THF (75 vol% THF, blue line) at (c) 0.1 mA cm−2 for 0.1 mA h cm−2 and at (d) 1.0 mA cm−2 for 1.0 mA h cm−2. (e) Cycling stability of Mg–Mo6S8 cells under 0.5 C with areal capacity equal to 5.0 mg cm−2 (1.0 C = 128 mA g−1). (f) Cycle life and rate performance of Mg–Mo6S8 cells with Mg(TFSI)2–2MgCl2 (MTC) in DME-THF, representative electrolytes based on commercial non-nucleophilic salts, and nucleophilic electrolytes. ref. 1: Mg(HMDS)2–4MgCl2 in THF;45 ref. 2: Mg(TFSI)2 in DME-diglyme;46 ref. 3: MgCl2–AlCl3–Mg(TFSI)2 in THF;47 ref. 4: Mg(OTf)2–MgCl2 in DME;40 ref. 5: MgAlCl2BuEt2 in THF;10 ref. 6: PhMgCl–AlCl3 in THF.48 | ||
The long-term cycling stability of the Mg anode in DME and DME-THF (75 vol% THF) electrolytes was further evaluated with Mg–Mg symmetric cells. As shown in Fig. 2(c) and (d) (yellow line), Mg–Mg symmetric cells with the DME electrolyte suffer from a short cycle life owing to the occurrence of short circuits. Under a low current density and areal capacity (0.1 mA cm−2, 0.1 mA h cm−2), a soft short circuit occurred after 24 hours cycling (Fig. 2(c), yellow line). Although elevating the current density and areal capacity to 1.0 mA cm−2, 1.0 mA h cm−2 can guide the formation of the smaller Mg nucleus43,44 and extend the cycle life to 200 hours (Fig. 2(d), yellow line), the DME cells suffer from a rapid polarization build-up from 138 mV to 375 mV and eventually short-circuited after 200 hours cycling. It is noted that the short-circuited Mg–Mg cells usually demonstrated higher polarization than Li–Li cells because Mg deposits were usually covered by resistive passivation film before penetrating the separator.13,49–51 As shown in Fig. 2(c) and (d) (blue line), the addition of THF not only extended the cycle life of Mg–Mg symmetric cells to 4000 hours under 0.1 mA cm−2, 0.1 mA h cm−2 and 700 hours under 1.0 mA cm−2, 1.0 mA h cm−2 but also suppressed the build-up of polarization. Besides the MTC-DME electrolytes, the cycle life of DME-THF electrolytes also outperforms other commercial MgCl2-based complexes in previous reports (Fig. S6 and Table S2, ESI†).
The success of DME-THF cells in stabilizing the Mg anode also benefits the realization of long-life full cells. To evaluate the electrochemical performance of DME and DME-THF electrolytes, the Mo6S8 cathode is selected to couple with the Mg anode because of its prolonged cycle life.10 All the cells were assembled with a piece of Mg foil (thickness ∼100 μm), Mo6S8 cathode (∼5.0 mg cm−2) and tested under 0.5 C (1.0 C = 128 mA g−1). 100 μl electrolyte was added into each cell to rinse the quartz fiber separator. Capacity loss was observed in both DME and DME-THF cells at the 1st cycle because of the irreversible ion trapping in the Mo6S8 host (Fig. S7, ESI†);52 therefore the 1st cycle was excluded in the following discussion. As shown in Fig. 2(e), Fig. S8, and S9a (ESI†), DME cells suffer from a short cycle life owing to the build-up of polarization. The discharge capacity of DME cells achieved 59.6 mA h g−1 under 0.5 C at the 2nd cycle and reached the maximum specific capacity (76.3 mA h g−1) at the 15th cycle. However, DME cells suffer from a severe capacity decay, which gradually decreased to 69.2 mA h g−1 after 150 cycles (357 hours) and 55.4 mA h g−1 after 887 cycles (1890 hours), demonstrating low capacity retention of ∼90.6% and 72.6% (Fig. 2(e)). Although the polarization of DME cells reduced to 0.28 V during first 30 cycles owing to the “conditioning” process,2,53,54 the value augmented to 0.65 V rapidly during the 30–887th cycles (Fig. S8 and S9a, ESI†). In contrast to DME cells, the capacity decay and polarization build-up were mitigated with the addition of THF. As shown in Fig. 2(e), the specific discharge capacity of DME-THF cells achieved 63.6 mA h g−1 at the 2nd cycle and gradually increased to 76.7 mA h g−1 at the 30th cycle. After 1000 cycles (2383 hours), DME-THF cells achieved a high capacity retention of ∼90.0% with CE equal to 99.96%. The enhanced electrochemical performance was contributed by the mitigated polarization build-up. According to Fig. S8 and S9b (ESI†), the polarization of DME-THF cells increased only slightly from 0.28 V (30th cycle) to 0.37 V (887th cycle) and 0.39 V (1000th cycle). As shown in Fig. 2(e) and (f), and Table S3 (ESI†), the cycling stability of Mg–Mo6S8 cells with the DME-THF hybrid electrolyte outperformed other electrolytes based on commercial non-nucleophilic Mg salts and even achieved enhanced cycling stability compared to conventional nucleophilic electrolytes. However, Mg–Mo6S8 cells still demonstrated an insufficient N/P ratio owing to the sluggish Mg2+ insertion.20 Further improvements using new cathode materials are needed to make practical Mg full cells (state-of-the-art <1.0 mA h cm−2).55
Morphology of cycled Mg anode
To understand the enhancement in cycle life contributed by the THF co-solvent, scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) were firstly conducted to investigate the morphology of cycled electrodes. According to Fig. 3(a), Cu electrodes cycled in DME electrolytes (charged to 1.0 V vs. Mg/Mg2+ before test) were covered by large amounts of irregular deposits which could not be recovered during the stripping process. According to EDS results (Fig. 3(c) and Fig. S10, ESI†), 42.2% Mg, 18.0% O, 1.6% F, and 38.3% Cu were detected on the Cu cathode, indicating that the major components of residues were unreacted metallic Mg0 and O or F-containing surface film induced by contaminants or electrolyte decomposition.7 The formation of irregular deposits on the Cu cathode was attributed to the porous and dendritic morphology of Mg anodes induced by DME electrolytes (Fig. 3(e)), leading to inferior electric contacts (Fig. S11, ESI†). During the dissolution process, the irregular Mg deposited from the DME electrolyte readily detached and resulted in a short circuit mentioned above. In contrast, a compact and highly crystalline structure was observed on the Mg anode cycled with the DME-THF electrolyte (Fig. 3(f) and Fig. S11, ESI†). Thanks to the enhanced electric contacts between Mg deposits (Fig. S11, ESI†), the isolation of Mg was mitigated (Fig. 3(b)). Compared with the DME electrolyte, the elemental concentration of Mg left on the Cu electrode significantly reduced from 42.2% to 12.9% (Fig. 3(d) and Fig. S10, ESI†), proving that the addition of THF can significantly suppress the isolation of Mg deposits during Mg stripping. | ||
| Fig. 3 SEM image of Mg residues on Cu cathodes induced by (a) DME and (b) DME-THF electrolytes. EDS mapping of Mg residues on Cu cathodes induced by (c) DME and (d) DME-THF electrolytes. SEM image of Mg anode cycled with (e) DME and (f) DME-THF electrolytes. (g) Nyquist plots of electrochemical impedance spectroscopy (EIS) over 1.5 Hz–1.0 MHz for Mg–Mg symmetric cells after 50 cycles. The AC oscillation amplitude is 10 mV. | ||
The interplay between Mg growth and electrolyte properties was revealed by electrochemical impedance spectroscopy (EIS). According to Fig. 3(g), the semicircles at high frequencies were attributed to the interfacial impedance and those at middle frequencies originated from the charge-transfer impedance.31,56 The slope lines at low frequencies were related to the diffusion impedance.31,56 As shown in Fig. 3(g), the Nyquist plot of Mg–Mg symmetric cell with DME electrolytes after 50 cycles only shows a small semi-circle at high frequencies (1.2 kHz–1.0 MHz) with a long slope line (1.5 Hz–1.2 kHz). The semicircle at middle frequencies was not obvious because the small charge-transfer impedance may be associated with the interfacial impedance, and the long slope line indicated that Mg reactions were dominated by the diffusion process in DME electrolytes. The rapid consumption of Mg2+ cations in the vicinity of anodes and the scarce supply induced by sluggish diffusion resulted in the non-uniform growth of Mg deposits.43,57 In contrast, the Nyquist plot of theMg–Mg symmetric cell with the DME-THF electrolyte shows a semicircle at high frequencies (4.0 kHz–1.0 MHz) for interfacial impedance, a semicircle at middle frequencies (7.6 Hz–4.0 kHz) for charge-transfer impedance, and a short slope line at low frequencies (1.5–7.6 Hz). Accordingly, deposition/stripping in DME-THF electrolytes show a charge-transfer controlled feature, suppressing the depletion of Mg2+ cations and leading to the formation of compact and highly crystalline Mg deposits (Fig. 3(g)).43,57 The uniform Mg deposition not only mitigates the Mg0 loss but also prevents the fracture of surface films to suppress side reactions induced by electrolytes. Therefore, although THF-induced surface films may be resistive, the DME-THF cells still exhibited smaller polarization (Fig. 2(b)–(d) and Fig. S8, ESI†) because of uniform Mg deposition and smaller diffusion impedance.
Characterization of interfacial chemistry
To understand interfacial chemistry, the composition of the surface film was studied by X-ray photoelectron spectroscopy (XPS). The C1s, F1s, and S2p spectra of Mg anodes cycled with DME and DME-THF electrolytes are shown in Fig. 4(a)–(d) and Fig. S12 (ESI†), respectively. The decomposition of both DME and DME-THF electrolytes induced similar species on the top surface of the Mg anode (0 s, before sputtering), including MgF2 (∼685.2 eV),58 C–F (∼688.2 eV, 292.7 eV),9,59 MgSO3 (167.0 eV, 168.2 eV),60 MgSx (164.1 eV, 165.3 eV, 162.0 eV, 163.2 eV),60,61 C–H/C–C (284.8 eV),40 C–O (286.6 eV, 532.5 eV),40 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.0 eV)62 and MgCO3 (290.0 eV).63 MgF2 originated from the decomposition of TFSI−,40 while C–F groups originated from either the decomposition or adsorption of TFSI− anions.40 C–H/C–C, C–O, and CO were attributed to the decomposition/adsorption of DME/THF.40 MgCO3 is related to the chemical reactions between contaminants and the Mg anode or electrochemical reactions with solvents.2,40 After 50 s sputtering, the concentration of organic components (C–H/C–C, C–O, CO) on the Mg anode cycled with DME electrolyte only slightly decreased and that of inorganic components (MgCO3 and MgF2) was nearly unchanged (Fig. 4(a), (c) and (e)). Therefore, the decomposition of DME tends to generate thick SEI, indicating the inferior ability of the surface film to suppress electron leakage.64,65 As for the Mg anode cycled with DME-THF electrolytes, the intensity of all the peaks originated from the decomposition of solvents significantly decreased after 50 s sputtering, indicating that DME-THF electrolytes enabled a more efficient electron-blocking film with reduced thickness (Fig. 4(b), (d) and (e)). The concentration evolution of MgF2 along sputtering depth shows an opposite trend compared with the solvent-induced components, suggesting that the decomposition of TFSI− occurred during the early stage but was suppressed after the completion of the film-forming process.
O (288.0 eV)62 and MgCO3 (290.0 eV).63 MgF2 originated from the decomposition of TFSI−,40 while C–F groups originated from either the decomposition or adsorption of TFSI− anions.40 C–H/C–C, C–O, and CO were attributed to the decomposition/adsorption of DME/THF.40 MgCO3 is related to the chemical reactions between contaminants and the Mg anode or electrochemical reactions with solvents.2,40 After 50 s sputtering, the concentration of organic components (C–H/C–C, C–O, CO) on the Mg anode cycled with DME electrolyte only slightly decreased and that of inorganic components (MgCO3 and MgF2) was nearly unchanged (Fig. 4(a), (c) and (e)). Therefore, the decomposition of DME tends to generate thick SEI, indicating the inferior ability of the surface film to suppress electron leakage.64,65 As for the Mg anode cycled with DME-THF electrolytes, the intensity of all the peaks originated from the decomposition of solvents significantly decreased after 50 s sputtering, indicating that DME-THF electrolytes enabled a more efficient electron-blocking film with reduced thickness (Fig. 4(b), (d) and (e)). The concentration evolution of MgF2 along sputtering depth shows an opposite trend compared with the solvent-induced components, suggesting that the decomposition of TFSI− occurred during the early stage but was suppressed after the completion of the film-forming process.
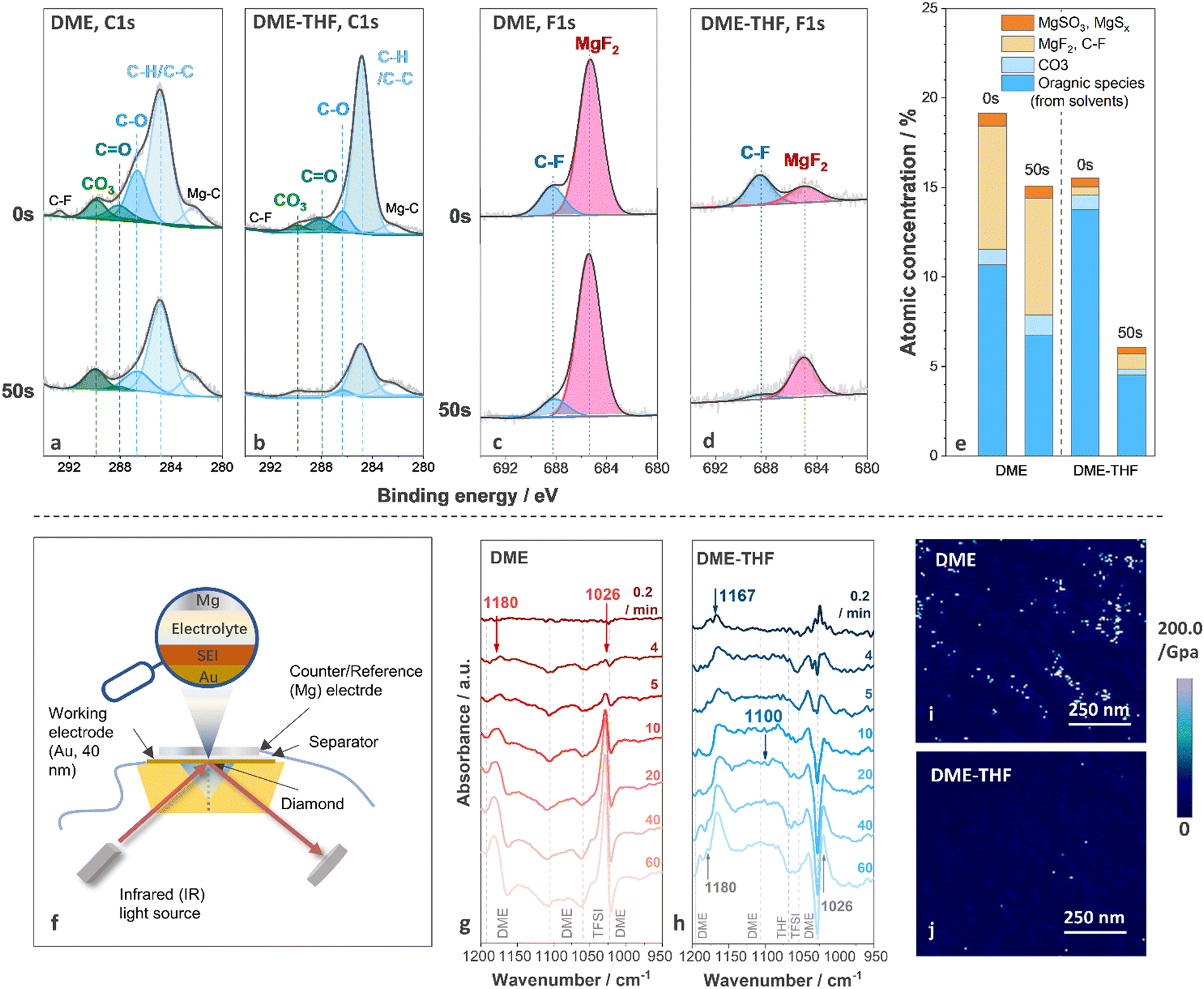 | ||
| Fig. 4 (a), (b) C1s and (c), (d) F1s spectra of the Mg electrode cycled in DME electrolyte and DME-THF electrolyte with sputtering time equal to 0 s and 50 s. (e) The atomic concentration of surface species obtained from XPS spectra. (f) Cell configuration for in situ FT-IR test. In situ FT-IR spectra of Mg deposited from (g) DME and (h) DME-THF electrolytes under 0.25 mA cm−2. Young's modulus mapping of Mg electrode cycled in (i) DME electrolyte and (j) DME-THF electrolyte. | ||
The decomposition products of solvents were further identified by in situ FT-IR. FT-IR spectra were collected during Mg deposition under 0.25 mA cm−2 with a thin Au film (40 nm) as the working electrode (Fig. 4(f)). The spectra collected before Mg deposition were selected as the background, therefore the positive peaks indicated the appearance of decomposition products or adsorbed species, and the negative peaks were related to the disappeared and desorbed species. Because of the strong C–O stretching signal of ethers, spectra in 1200–950 cm−1 region were probed. After 4 min Mg deposition from the DME electrolyte, two positive peaks located at 1026 cm−1 and 1180 cm−1, and four negative peaks located at 1023 cm−1, 1060 cm−1, 1105 cm−1, and 1192 cm−1 emerged (Fig. 4(g)). The positive peaks at 1026 cm−1 and 1180 cm−1 were related to the emergence of Mg(OCH3)266 formed via the C–O bond cleavage of DME molecules.67 The negative peaks located at 1023 cm−1, 1105 cm−1, and 1192 cm−1 are attributed to the consumption/desorption of DME molecules, and the peak located at 1060 cm−1 originated from the consumption/desorption of TFSI− (Fig. S13, ESI†). After 60 min deposition, Mg(OCH3)2 and electrolyte peaks intensified while no newly formed species were detected. According to Fig. 4(h), the addition of THF weakens the peaks originating from Mg(OCH3)2 and led to the formation of poly(tetrahydrofuran), PTHF, via ring-opening polymerization,68 which was revealed by the two positive peaks located at 1167 cm−1 (C–C stretching) and 1100 cm−1 (C–O–C antisymmetric stretching).69 The C–O–C stretching peak is not as obvious as the C–C stretching peak because it overlapped with the negative DME peak at 1105 cm−1. The XPS and in situ FT-IR results confirmed that THF cosolvent altered the composition of surface films and suppressed the decomposition of DME and passivating TFSI−. Therefore, PTHF was a candidate material for a high-performance artificial SEI thanks to the success of PTHF-based polymer electrolytes in supporting Mg2+ transport.70,71 Furthermore, PTHF-based artificial SEI with lower spatial concentration of oxygen atoms may achieve higher ion transport than PEO-based counterparts72 if a novel strategy was applied to fabricate a homogeneous thin film.
The mechanical properties of a SEI generated through electrochemical reactions were observed via atomic force microscopy (AFM). Inorganic surface species usually exhibited a much higher Young's modulus (MgF2 ∼ 130.0 GPa,73,74 MgO, 270–330 GPa75) than that of polymeric/organic species (<10.0 GPa),76 thus the organic-rich and inorganic-rich areas can be readily distinguished by Young's modulus mapping. As shown in Fig. 4(i), discrete organic-rich and inorganic-rich areas were investigated on the Mg anode cycled with DME electrolytes. Although inorganic species promise to reinforce the SEI, their non-uniform spatial distribution may aggravate the inhomogeneity of ion flux, resulting in severe interfacial and morphological instability. Furthermore, the sluggish Mg2+ diffusion across a thick inorganic layer (proved by XPS spectra). As shown in Fig. 4(i), discrete organic-rich and inorganic-rich areas were investigated on the Mg anode cycled with DME electrolytes. Although inorganic species promise to reinforce the SEI, their non-uniform spatial distribution may aggravate the inhomogeneity of ion flux, resulting in severe interfacial and morphological instability. Furthermore, the sluggish Mg2+ diffusion across a thick inorganic layer (proved by XPS spectra) may impede the Mg anode reaction.77 According to previous reports, the migration barriers of Mg2+ in MgF2 is 1123 meV, exceeding the maximum tolerable migration barrier (980 meV) to ensure sufficient Mg2+ diffusion rate under 60 °C with the thickness of surface film ∼1 nm.77 Accordingly, discrete and thick surface films generated by DME electrolytes are unfavorable for Mg anodes.
The uneven surface film was also generated by chemical reactions between the Mg anode and DME electrolytes. As shown in Fig. S14a, c (ESI†), after soaking the Mg anode in the DME electrolyte for 240 hours, the surface of Mg metal was covered by an uneven passivation film (dark area). The presence of the F element in the dark area confirmed that the surface film originated from electrolyte decomposition (Fig. S14e, ESI†). Accordingly, discrete SEI can be generated by DME electrolytes even without the volume change of electrodes, leading to an inferior protection ability against parasitic reactions.78 In contrast, the addition of film-forming THF can homogenize the surface film via both electrochemical and chemical reactions. As shown in Fig. 4(j) and Fig. S14 (ESI†), both AFM and SEM images revealed that the surface film generated by DME-THF electrolytes exhibited a continuous and homogeneous feature, ensuring a more effective protective film to block the parasitic reactions induced by non-nucleophilic electrolytes.
Solvent alignment on anode surfaces
To reveal the origin of divergent SEI homogeneity, density functional theory (DFT) calculations were carried out to understand the alignment of solvent molecules on anode surfaces at a molecular level. Two surface species, Mg and MgO, were considered and surfaces with the lowest energy (Mg (0001) and MgO (100)) were selected (details are shown in Computational details). Three adsorption configurations were considered for the Mg (0001) surface and four configurations were considered for the MgO (100) surface (Fig. S15–S18, ESI†) to evaluate the most favorable adsorption geometries for each system, and the optimal geometries were shown in Fig. 5(a) and (b). The adsorption energy of DME on MgO (100) (−0.595 eV) is much higher than that on Mg (0001) (−0.282 eV), suggesting that DME will preferentially adsorb on MgO (100) surface (Fig. 5(c)). As shown in Fig. 5(a), DME molecules adsorbed on Mg (0001) surface with a mono-dentate adsorption pattern but demonstrated a bi-dentate adsorption pattern on the MgO (100) surface; thus the adsorption energy on MgO (100) surface nearly doubled (Fig. 5(c)). Compared with DME molecules, THF shows similar adsorption energy on Mg (0001) (−0.458 eV) and MgO (100) (−0.567 eV) because the adsorption pattern was less likely to vary with surface conditions (Fig. 5(b) and (c)). Based on the DFT calculations and experimental results, the interaction mechanism was summarized in Fig. 5(d). According to previous reports, the as-received Mg foil is covered by a native oxide layer induced by manufacturing process, which cannot be totally removed by polishing.2 Furthermore, the impurities in non-nucleophilic electrolytes also led to the formation of the passivation film.9 Therefore, DME molecules with selective adsorption properties tend to accumulate on specific surface species instead of uniformly covering the anodes. In contrast, the nonselective adsorption feature of THF ensures the uniform precipitation of decomposition products, thus help in filling cracks in the discrete surface film generated by DME electrolytes and mitigate the side reactions and passivation induced by anions and impurities (Fig. 5(d)).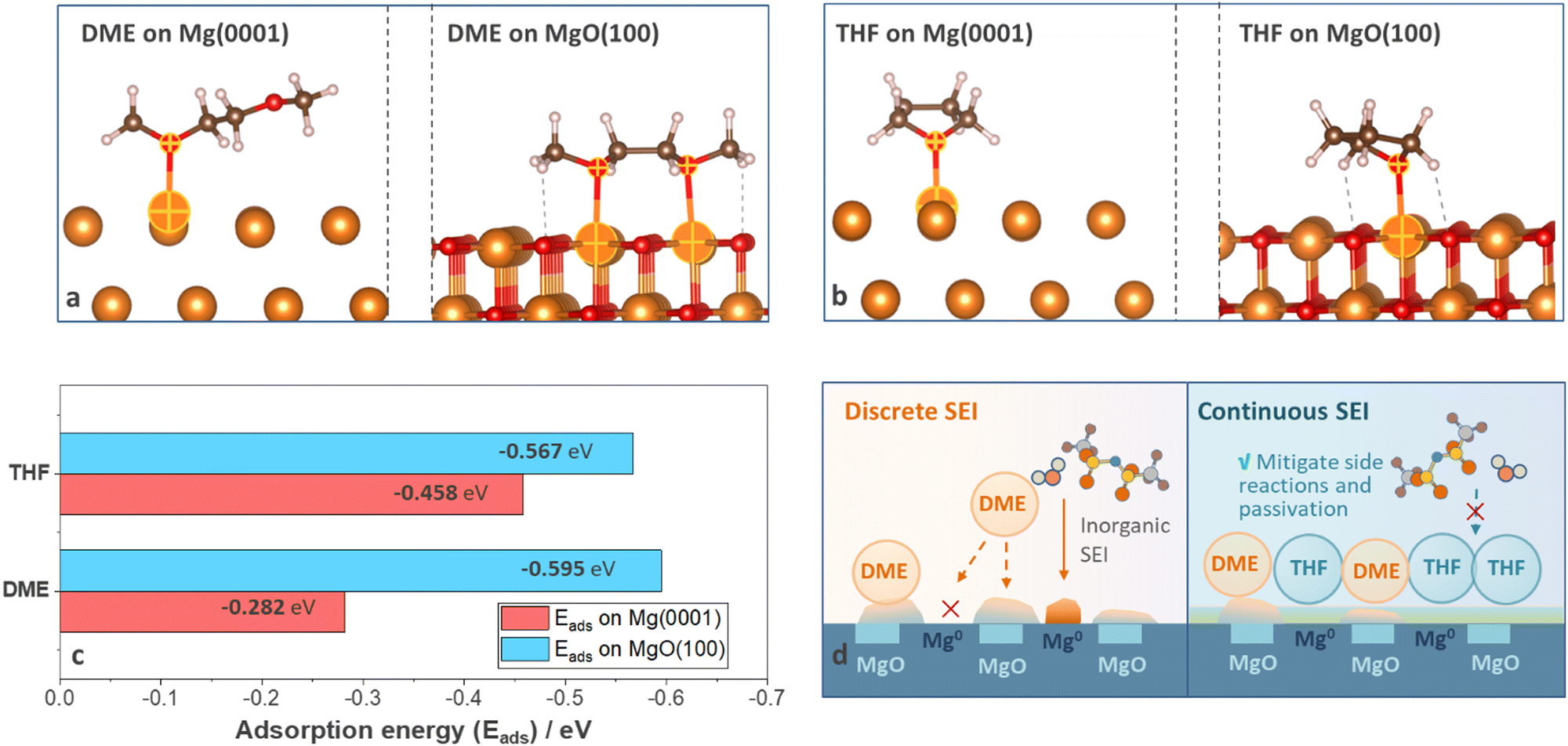 | ||
| Fig. 5 Optimal adsorption geometries of (a) DME and (b) THF on Mg (0001) (left) and MgO (100) (right) surfaces. (c) Adsorption energy (Eads) of DME and THF on Mg (0001) and MgO (100) surfaces calculated based on the optimal adsorption geometries shown in (a) and (b). (d) Schematic illustrations of DME and THF molecules alignment on anode surface covered by native oxide layer. | ||
Conclusions
In summary, we design a hybrid-solvent electrolyte by dispersing a non-nucleophilic DME solution in a non-fluorinated weakly coordinating solvent (tetrahydrofuran, THF) to prevent the decomposition of DME and form a stable SEI. This strategy significantly improved the cycle life of the Mg anode from 24 to 4000 hours (in Mg–Mg symmetric cells) and realized a stable cycle life of Mg–Mo6S8 full cells for more than 2300 hours with an average CE of 99.96% without reactive/nucleophilic impurity scavengers. SEM and EIS results revealed that free THF not only retains the facile ion migration under electric field but also facilitates the ion diffusion driven by concentration gradients, enabling a highly crystalline and compact morphology of Mg anode to mitigate the fracture of surface films. Furthermore, XPS and in situ FT-IR results confirmed that the film-forming THF altered the composition of surface films, realizing a more effective protective film to suppress the electrolyte-induced parasitic reactions. AFM results and DFT calculations revealed the enhancement mechanism of THF, suggesting that THF fills the cracks of DME-induced discrete SEI thanks to the uniform alignment on anode surfaces. This strategy provided a universal methodology to enable the utilization of weakly coordinating solvents and stabilize Mg anode in accessible Mg electrolytes for practical RMBs.Author contributions
Y. S. and Y. L. designed and conceived the experiments. Y. W. and J. F. conducted the DFT calculations. L. J. synthesized the cathodes. D. D. conducted the FT-IR test. Y.S. conducted the XPS, AFM, and electrochemical tests. W. W. conducted the SEM and EDS tests. Y. S and Y. L. wrote the manuscript. Y. L. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from National Natural Science Foundation of China (51922114) and a grant from Research Grant Council (Collaborative Research Fund C1002-21GF).Notes and references
- H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Nat. Commun., 2011, 2, 427 CrossRef PubMed.
- R. Attias, M. Salama, B. Hirsch, Y. Goffer and D. Aurbach, Joule, 2019, 3, 27–52 CrossRef.
- W. Guo, S. Chen, N. Hussain, Y. Cong, Z. Liang and K. Chen, Plant Signaling Behav., 2015, 10, e992287 CrossRef PubMed.
- Z. Lu, A. Schechter, M. Moshkovich and D. Aurbach, J. Electroanal. Chem., 1999, 466, 203–217 CrossRef.
- J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941–5950 RSC.
- P. Bonnick and J. Muldoon, Adv. Funct. Mater., 2020, 30, 1910510 CrossRef.
- H. Kuwata, M. Matsui and N. Imanishi, J. Electrochem. Soc., 2017, 164, A3229–A3236 CrossRef CAS.
- S.-B. Son, T. Gao, S. P. Harvey, K. X. Steirer, A. Stokes, A. Norman, C. Wang, A. Cresce, K. Xu and C. Ban, Nat. Chem., 2018, 10, 532–539 CrossRef CAS.
- Y. Yu, A. Baskin, C. Valero-Vidal, N. T. Hahn, Q. Liu, K. R. Zavadil, B. W. Eichhorn, D. Prendergast and E. J. Crumlin, Chem. Mater., 2017, 29, 8504–8512 CrossRef.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724 CrossRef.
- O. Mizrahi, N. Amir, E. Pollak, O. Chusid, V. Marks, H. Gottlieb, L. Larush, E. Zinigrad and D. Aurbach, J. Electrochem. Soc., 2008, 155, A103 CrossRef.
- R. Mohtadi, O. Tutusaus, T. S. Arthur, Z. Zhao-Karger and M. Fichtner, Joule, 2021, 5, 581–617 CrossRef.
- Y. Zhao, A. Du, S. Dong, F. Jiang, Z. Guo, X. Ge, X. Qu, X. Zhou and G. Cui, ACS Energy Lett., 2021, 2594–2601 CrossRef.
- R. Lv, X. Guan, J. Zhang, Y. Xia and J. Luo, Natl. Sci. Rev., 2020, 7, 333–341 CrossRef PubMed.
- J. Zhang, X. Guan, R. Lv, D. Wang, P. Liu and J. Luo, Energy Storage Mater., 2020, 26, 408–413 CrossRef.
- W. Ren, D. Wu, Y. NuLi, D. Zhang, Y. Yang, Y. Wang, J. Yang and J. Wang, ACS Energy Lett., 2021, 3212–3220 CrossRef.
- Z. Zhao-Karger, M. E. Gil Bardaji, O. Fuhr and M. Fichtner, J. Mater. Chem. A, 2017, 5, 10815–10820 RSC.
- T. Mandai, ACS Appl. Mater. Interfaces, 2020, 12, 39135–39144 CrossRef.
- O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson and Y. V. Sevryugina, Angew. Chem., Int. Ed., 2015, 54, 7900–7904 CrossRef.
- H. Dong, O. Tutusaus, Y. Liang, Y. Zhang, Z. Lebens-Higgins, W. Yang, R. Mohtadi and Y. Yao, Nat. Energy, 2020, 5, 1043–1050 CrossRef CAS.
- J. Xiao, X. Zhang, H. Fan, Y. Zhao, Y. Su, H. Liu, X. Li, Y. Su, H. Yuan, T. Pan, Q. Lin, L. Pan and Y. Zhang, Adv. Mater., 2022, 2203783 CrossRef CAS.
- K. Tang, A. Du, S. Dong, Z. Cui, X. Liu, C. Lu, J. Zhao, X. Zhou and G. Cui, Adv. Mater., 2020, 32, 1904987 CrossRef CAS.
- K. Tang, A. Du, X. Du, S. Dong, C. Lu, Z. Cui, L. Li, G. Ding, F. Chen, X. Zhou and G. Cui, Small, 2020, 16, 2005424 CrossRef CAS.
- H. Nishida, N. Takada, M. Yoshimura, T. Sonoda and H. Kobayashi, Bull. Chem. Soc. Jpn., 1984, 57, 2600–2604 CrossRef CAS.
- J. Luo, Y. Bi, L. Zhang, X. Zhang and T. L. Liu, Angew. Chem., Int. Ed., 2019, 131, 7041–7045 CrossRef.
- M. Li, C. Wang, Z. Chen, K. Xu and J. Lu, Chem. Rev., 2020, 120, 6783–6819 CrossRef CAS PubMed.
- U. Pal, G. M. A. Girard, L. A. O’Dell, B. Roy, X. Wang, M. Armand, D. R. MacFarlane, P. C. Howlett and M. Forsyth, J. Phys. Chem. C, 2018, 122, 14373–14382 CrossRef CAS.
- W. Zhao, Z. Pan, Y. Zhang, Y. Liu, H. Dou, Y. Shi, Z. Zuo, B. Zhang, J. Chen, X. Zhao and X. Yang, Angew. Chem., Int. Ed., 2022, 134, e202205187 Search PubMed.
- R. Gond, W. van Ekeren, R. Mogensen, A. J. Naylor and R. Younesi, Mater. Horiz., 2021, 8, 2913–2928 RSC.
- Q. Zheng, Y. Yamada, R. Shang, S. Ko, Y.-Y. Lee, K. Kim, E. Nakamura and A. Yamada, Nat. Energy, 2020, 5, 291–298 CrossRef CAS.
- S. Hou, X. Ji, K. Gaskell, P. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Science, 2021, 374, 172–178 CrossRef CAS PubMed.
- S. Fan, G. M. Asselin, B. Pan, H. Wang, Y. Ren, J. T. Vaughey and N. Sa, ACS Appl. Mater. Interfaces, 2020, 12, 10252–10260 CrossRef CAS.
- R. Hu, H. Qiu, H. Zhang, P. Wang, X. Du, J. Ma, T. Wu, C. Lu, X. Zhou and G. Cui, Small, 2020, 16, 1907163 CrossRef CAS.
- R. Mohtadi, M. Matsui, T. S. Arthur and S.-J. Hwang, Angew. Chem., Int. Ed., 2012, 124, 9918–9921 CrossRef.
- T. Gao, S. Hou, F. Wang, Z. Ma, X. Li, K. Xu and C. Wang, Angew. Chem., Int. Ed., 2017, 129, 13711–13715 CrossRef.
- N. Pour, Y. Gofer, D. T. Major and D. Aurbach, J. Am. Chem. Soc., 2011, 133, 6270–6278 CrossRef CAS.
- C. Su, M. He, R. Amine, Z. Chen and K. Amine, Angew. Chem., Int. Ed., 2018, 57, 12033–12036 CrossRef CAS PubMed.
- Z. Li, Y. Zhou, Y. Wang and Y.-C. Lu, Adv. Energy Mater., 2019, 9, 1802207 CrossRef.
- N.I.o. Standards, Technology, NIST.
- D.-T. Nguyen, A. Y. S. Eng, M.-F. Ng, V. Kumar, Z. Sofer, A. D. Handoko, G. S. Subramanian and Z. W. Seh, Cell Rep. Phys. Sci., 2020, 1, 100265 CrossRef CAS.
- J. Clarke-Hannaford, M. Breedon, T. Rüther, P. Johansson and M. J. S. Spencer, Chem. – Eur. J., 2021, 27, 12826–12834 CrossRef PubMed.
- A. Bakker, S. Gejji, J. Lindgren, K. Hermansson and M. M. Probst, Polymer, 1995, 36, 4371–4378 CrossRef.
- J. Eaves-Rathert, K. Moyer, M. Zohair and C. L. Pint, Joule, 2020, 4, 1324–1336 CrossRef.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef PubMed.
- C. Liao, N. Sa, B. Key, A. K. Burrell, L. Cheng, L. A. Curtiss, J. T. Vaughey, J.-J. Woo, L. Hu and B. Pan, J. Mater. Chem. A, 2015, 3, 6082–6087 RSC.
- S.-Y. Ha, Y.-W. Lee, S. W. Woo, B. Koo, J.-S. Kim, J. Cho, K. T. Lee and N.-S. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 4063–4073 CrossRef PubMed.
- Y. He, Q. Li, L. Yang, C. Yang and D. Xu, Angew. Chem., Int. Ed., 2019, 58, 7615–7619 CrossRef PubMed.
- M. Mao, Z. Lin, Y. Tong, J. Yue, C. Zhao, J. Lu, Q. Zhang, L. Gu, L. Suo, Y.-S. Hu, H. Li, X. Huang and L. Chen, ACS Nano, 2020, 14, 1102–1110 CrossRef PubMed.
- B.-C. Yu, K. Park, J.-H. Jang and J. B. Goodenough, ACS Energy Lett., 2016, 1, 633–637 CrossRef.
- B. Wu, J. Lochala, T. Taverne and J. Xiao, Nano Energy, 2017, 40, 34–41 CrossRef.
- M. S. Ding, T. Diemant, R. J. Behm, S. Passerini and G. A. Giffin, J. Electrochem. Soc., 2018, 165, A1983–A1990 CrossRef.
- C. Ling and K. Suto, Chem. Mater., 2017, 29, 3731–3739 CrossRef.
- S.-J. Kang, H. Kim, S. Hwang, M. Jo, M. Jang, C. Park, S.-T. Hong and H. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 517–524 CrossRef PubMed.
- S. He, J. Luo and T. L. Liu, J. Mater. Chem. A, 2017, 5, 12718–12722 RSC.
- Y. Sun, Q. Zou, W. Wang and Y.-C. Lu, ACS Energy Lett., 2021, 6, 3607–3613 CrossRef.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 RSC.
- D. Briggs, Handbook of X-Ray and Ultraviolet Photoelectron Spectroscopy, Heyden & Son, 1977 Search PubMed.
- F. Gunn, P. Ghosh, M. Maciejczyk, J. Cameron, D. Nordlund, S. Krishnamurthy, T. Tuttle, P. Skabara, N. Robertson and A. Ivaturi, J. Mater. Chem. C, 2021, 9, 16226–16239 RSC.
- Y. Wang, T. Wang, D. Dong, J. Xie, Y. Guan, Y. Huang, J. Fan and Y.-C. Lu, Matter, 2022, 5, 162–179 CrossRef CAS.
- Q. Zou, Y. Sun, Z. Liang, W. Wang and Y. Lu, Adv. Energy Mater., 2021, 11, 2101552 CrossRef CAS.
- M. Estrada, V. V. Costa, S. Beloshapkin, S. Fuentes, E. Stoyanov, E. V. Gusevskaya and A. Simakov, Appl. Catal., A, 2014, 473, 96–103 CrossRef.
- F. Sebastián Jr., TOSURSJ, 2011, 3, 1–14 CrossRef.
- H. Dou, X. Zhao, Y. Zhang, W. Zhao, Y. Yan, Z.-F. Ma, X. Wang and X. Yang, Nano Energy, 2021, 86, 106087 CrossRef.
- F. Single, B. Horstmann and A. Latz, J. Electrochem. Soc., 2017, 164, E3132–E3145 CrossRef.
- G. V. Zhuang, H. Yang, B. Blizanac and P. N. Ross, Electrochem. Solid-State Lett., 2005, 8, A441–A445 CrossRef.
- D. Aurbach, M. L. Daroux, P. W. Faguy and E. Yeager, J. Electrochem. Soc., 1988, 135, 1863–1871 CrossRef.
- M. Odziemkowski, M. Krell and D. E. Irish, J. Electrochem. Soc., 1992, 139, 3052–3063 CrossRef.
- Y. Matsui, T. Kubota, H. Tadokoro and T. Yoshihara, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 2275–2288 CrossRef.
- A. Du, H. Zhang, Z. Zhang, J. Zhao, Z. Cui, Y. Zhao, S. Dong, L. Wang, X. Zhou and G. Cui, Adv. Mater., 2019, 31, 1805930 CrossRef.
- P. Wang, J. Trück, J. Häcker, A. Schlosser, K. Küster, U. Starke, L. Reinders and M. R. Buchmeiser, Energy Storage Mater., 2022, 49, 509–517 CrossRef.
- D. G. Mackanic, W. Michaels, M. Lee, D. Feng, J. Lopez, J. Qin, Y. Cui and Z. Bao, Adv. Energy Mater., 2018, 8, 1800703 CrossRef.
- M. J. Weber, Handbook of optical materials, CRC Press, 2018 Search PubMed.
- E. D. Palik, Handbook of optical constants of solids, Academic Press, 1998 Search PubMed.
- Goodfellow, AZoM.
- W. Martienssen and H. Warlimont, Springer Handbook of Condensed Matter and Materials Data, Springer, Berlin Heidelberg, 2005 Search PubMed.
- T. Chen, G. Sai Gautam and P. Canepa, Chem. Mater., 2019, 31, 8087–8099 CrossRef.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee03235j |
| This journal is © The Royal Society of Chemistry 2023 |