
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of annulated benzothiadiazole derivatives and their application as medium band gap acceptors in organic photovoltaic devices†
Xiantao
Hu‡
 a,
Ram
Datt‡
b,
Qiao
He
a,
Panagiota
Kafourou
a,
Harrison
Ka Hin Lee
bc,
Andrew J. P.
White
a,
Wing Chung
Tsoi
*b and
Martin
Heeney
*a
a,
Ram
Datt‡
b,
Qiao
He
a,
Panagiota
Kafourou
a,
Harrison
Ka Hin Lee
bc,
Andrew J. P.
White
a,
Wing Chung
Tsoi
*b and
Martin
Heeney
*a
aDepartment of Chemistry and Centre for Processable Electronics, Imperial College London, White City Campus, London W12 0BZ, UK. E-mail: m.heeney@imperial.ac.uk
bSPECIFIC, Faculty of Science and Engineering, Swansea University, Bay Campus, Fabian Way, Swansea SA1 8EN, UK. E-mail: w.c.tsoi@swansea.ac.uk
cDepartment of Physics, City University of Hong Kong, Kowloon, Hong Kong SAR, China
First published on 8th June 2022
Abstract
Two 2,1,3-benzothiadiazole derivatives annulated with 2-(1,3-dithiol-2-ylidene)malonitrile in the 4,5-positions were prepared by nucleophilic substitution reactions of a fluorinated precursor. The resulting derivatives were coupled to an electron rich indacenodithiophene core to afford two new non-fullerene acceptors (NFAs). Both NFAs exhibited identical absorption spectra in solution, but significant differences in the solid state as a result of the end group fluorination. Both materials performed as electron acceptors in solar cell blends with donor polymers. The resulting devices exhibited high open circuit voltages over 1 V under 1 sun illumination, as a result of their high lying LUMO levels. Devices based on the fluorinated acceptor demonstrated reasonable performance under low light conditions, with power conversion efficiencies up to 13.7%.
Introduction
The design and development of new organic semiconducting materials continues to attract tremendous interest and such materials find application in a wide range of areas including organic solar cells (OSCs), organic thin-film transistors (OTFT), photodetectors, etc.1,2 In most of these applications the molecular energetics and band gap are one crucial element influencing their performance, together with control of their solid-state packing.3 As such, the development of novel conjugated building blocks is important to allow the fine-tuning of material properties. This is especially the case for highly electron deficient units, in which the library of widely utilised building blocks tends to be much smaller than that of the electron rich analogues.4One area where electron deficient building blocks have found particular utility has been in the development of acceptor–donor–acceptor (A–D–A) based organic materials.5 These have found particular interest as the electron transporting (or n-type) material in bulk-heterojunction (BHJ) OSCs, where their use as so-called non-fullerene acceptors (NFA's) has fuelled significant increases in device efficiency and stability.6 A–D–A type materials have also found utility as n-type semiconductors in OTFT applications,7 as well as emissive materials exhibiting thermally allowed delayed fluorescence (TADF).8 In this latter area, localisation of the frontier molecular orbital on the D and A components is an important design consideration. In A–D–A materials, the energetics can be tuned by appropriate choice of the D and A components, with the molecular HOMO (highest occupied molecular orbital) largely determined by the electron rich D, and the LUMO (lowest unoccupied molecular orbital) by the acceptor, A.9,10 Thus, the nature of A plays an important role in material performance in many applications.
One acceptor unit which has found wide utility in many organic materials is 2,1,3-benzothiadiazole (BT), which can be thought of as an analogue of naphthalene in which one of the benzo rings is replaced by an electron deficient thiadiazole ring.11 The relatively high electron affinity of benzothiadiazole has seen it used extensively as the acceptor co-monomer in a range of donor–acceptor polymers,12,13 where it is typically linked via the 4,7-positions, as well as an acceptor end-group in NFA materials.7,14
There has been a large interest in the energetics of the BT system. One approach has been to change the heteroatom in the thiadiazole ring. For example, moving to 2,1,3-benzoxadiazole can lead to an increase in polymer band gap when it is used in donor–acceptor polymers, and a corresponding increase in device voltage when such materials are utilised in BHJ PV devices.15,16 Similarly moving to benzoselenadiazole,17 and benzotriazole18 has been extensively investigated. Alternative approaches to modify the electronics of BT have focussed largely on functionalisation of the benzo ring of the BT system, with most examples functionalising the 5 and/or 6 positions in order to allow polymerisation through the 4,7-positions. For example, the inclusion of fluorine groups has been shown to increase both ionisation potential (IP) and electron affinity (EA) by approximately the same amount,19,20 whereas cyanation has a much larger influence on the EA than the IP.7,21 Functionalisation with electron donating groups such as amines has the opposite effect.22 Such functionalisation also influences the backbone planarization of the polymer and its solid state packing.
Another important approach to modify the energetics of BT has been to heteroannulate additional rings to the BT core.11 Many of these annulations have been performed at the 5,6-positions, affording for example [1,2,5]thiadiazolo[3,4-g]quinoxaline and benzo[1,2-c:4,5-c′]bis[1,2,5]thiadiazole (benzobis-thiadiazole, BBT). The polymerisation of such monomers has led to polymers with very low band gaps, as well as interesting properties such as ambipolar charge transport and high-spin ground state structures.23–26 The heteroannulation serves to modify the electron properties as well as extend the size of the delocalised units, potentially facilitating intermolecular interactions of delocalised systems.27 Despite the large interest in heteroannulation, almost all reports explore functionalisation of the BT unit exclusively in the 5,6-position.11 Given the potential of BT as an endgroup in A–D–A systems28–30 we were interested in developing approaches to tune the behaviour of the BT unit by heteroannulation in the 4,5-positions. In particular, we were interested in extending the conjugation of the BT ring by appending an electron-withdrawing 2-(1,3-dithiol-2-ylidene)malonitrile group. Annulation with 2-(1,3-dithiol-2-ylidene)malonitrile has been previously reported for a range of naphthalene diimide derivates,31–34 resulting in materials displaying excellent n-type transistor behaviour. More recently it has been appended to the 5,6-position of the BT ring, affording monomers which were incorporated into a donor–acceptor polymers exhibited thermochromic behaviour and ambipolar charge transport.35 However, annulation in the 4,5-positions has not been reported to the best of our knowledge.
We were interested in annulating the 4,5-position of BT as it offers an approach to tune the acceptor strength. This is particularly important in tuning band gap for applications such as indoor energy harvesting, where OSC's could be potentially used to power a variety of sensors and circuits for internet of things (IOTs) applications.36 The ideal band gap for such applications is wider than that for outdoor energy harvesting, and there is a requirement to develop active acceptor materials which have a good absorption match with the visible emission spectra of indoor light sources such as fluorescent lamp, halogen, and light emitting diodes.
Here we report an expedient synthesis to such annulated electron acceptors, with and without an additional fluorine substituent in the 6-position. We attach both acceptors to an electron rich indacenodithiophene (IDT) core and report the optoelectronic properties and photovoltaic device performance of the resulting A–D–A materials. We investigate their performance as medium band gap non-fullerene small molecule acceptors in OSCs under 1 sun as well as under indoor light, with blends affording high open circuit voltages (VOC) above 1 V and reasonable power output under fluorescent lamps.
Results and discussion
Our approach to annulate the 2-(1,3-dithiol-2-ylidene)malonitrile group to the BT ring was based on the good nucleophilicity of the 2,2-dicyanoethylene-1,1-dithiolate anion and the fact the nucleophilic aromatic substitution reactions are known to readily occur on fluorinated BT cores.37,38 Thus treatment of commercially available 4,7-dibromo-5-fluoro-2,1,3-benzothiadiazole (2a) with sodium 2,2-dicyanoethene-1,1-bis(thiolate) (1) in DMSO afforded reasonable yields of compound 3a (BTSCN), resulting from double substitution of the fluorine and the adjacent bromine group (Scheme 1). Applying the same reaction conditions to the difluorinated starting material (2b) resulted in the formation of 3b (FBTSCN) rather than any product resulting from the displacement of both fluorine groups. Typically, a fluoride is a better leaving group than a bromide in a nucleophilic aromatic substitution (SNAr) reaction, with the presence of an ortho fluoride particularly activating.39 Therefore, the product resulting from difluoride substitution might have been expected but is not observed under our conditions. We propose 3b is formed from an initial substitution of one of the fluorides with a thiolate to form the thioether in the expected manner. The second ring closing displacement occurs preferentially at the bromide in the 4-position, rather than the fluoride in the 6-position, because the resulting Meisenheimer complex can delocalise over two positions in the former versus one position in the latter without disrupting the aromaticity of the fused thiadiazole ring. Such a trend is similar to the reactivity observed in SNAr ring-closing reactions of fluorinated naphthalene rings.40,41 Attack at the 4-position also benefits from the established activating effect of a meta-fluorine substituent.42 The regiochemistry of the substitution was established by a combination of NMR spectroscopy and single-crystal x-ray diffraction (vide infra).The donor core was prepared by alkylation of commercially available indacenodithiophene (IDT) with n-octyl bromide, followed by lithiation with n-BuLi and subsequent reaction with trimethyltin chloride to give compound 5.7 Stille coupling with BTSCN and FBTSCN using catalytic Pd(PPh3)4 gave BTSCN-IDT and FBTSCN-IDT in 65% and 67% isolated yield. The structure of both materials was confirmed by a combination of NMR spectroscopy and mass spectroscopy (see ESI†). Both materials were soluble in common organic solvents such as dichloromethane and chloroform. They also exhibited excellent thermal stability, with the onset of degradation occurring in excess of 350 °C by thermogravimetric analysis (TGA) under nitrogen atmosphere (Fig. S22, ESI†), which exceeds the temperature requirement for device fabrication.
Single crystals of both FBTSCN and BTSCN were grown from dichloromethane/cyclohexane to further confirm their structure. FBTSCN crystalized in the orthorhombic space group Pbca (Fig. 1a and Fig. S24, S25, ESI†), whereas the non-fluorinated BTSCN crystalized in a monoclinic space group P21/c (Fig. 1c and Fig. S23, ESI†). Viewing along the c-axis in the unit cell, the FBTSCN-IDT molecules pack in a lamellar type arrangement, with a number of close contacts between individual molecules within layers (Fig. 1b). The individual FBTSCN molecules are almost completely co-planar, but removing the fluorine group in BTSCN results in the 2-(1,3-dithiol-2-ylidene)malonitrile groups bending slightly out-of-plane with a dihedral angle of 14.66° to the benzothiadiazole core (Fig. 1d).
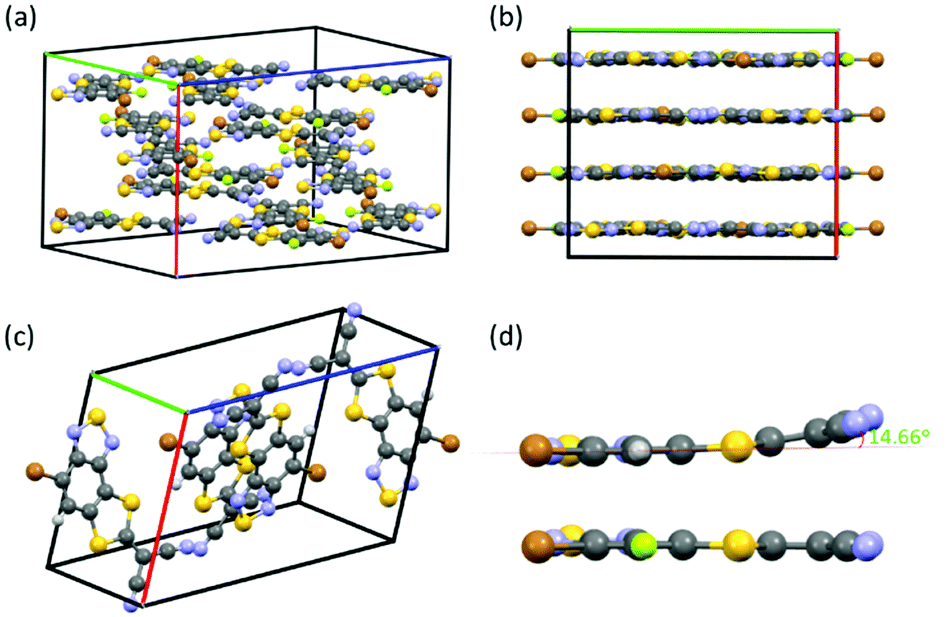 | ||
| Fig. 1 Unit cell and view along c-axis of FBTSCN (a and b) and BTSCN (c) respectively. (d) Dicyanomethylene torsion in BTSCN (top) versus FBTSCN (bottom), viewed along the molecular long axis. | ||
Theoretical calculation
Theoretical calculations were performed to investigate the frontier molecular orbital electron distribution and molecular geometry of FBTSCN-IDT and BTSCN-IDT, using density functional theory (DFT) with B3LYP/6-31G(d,p) basis set.43–45 The octyl chain was replaced by methyl groups to simplify the calculations. The frontier molecular distribution is accordant with the different electronic properties of each building unit, with the HOMO mainly distributed over the conjugated core, while the LUMO localizes largely on BT unit (Fig. 2). The involvement of the 2-(1,3-dithiol-2-ylidene)malonitrile appears to be mainly inductive, since the LUMO wavefunction does not fully extend over the malonitrile groups. The HOMO/LUMO of FBTSCN-IDT and BTSCN-IDT were predicted to be −5.74/−3.65 eV and −5.57/−3.43 eV, respectively. These calculations agree with previous studies that fluorination results in a lowering of both frontier molecular orbitals with respect to the non-fluorinated.46,47 A potential energy scan was conducted to study the conformation preference of the end-group to the central IDT core. Thus, the dihedral angle between BT unit and adjacent IDT core was fixed, while allowing the rest of molecule to relax to their minimum energy conformation. The results are shown in Fig. S26 (ESI†), and demonstrate that both materials exhibit a preference for an anti-confirmation (with respect to the sulfur atom in the BT and IDT) and are almost completely coplanar (dihedral angle of 0.06° and 0.20° for FBTSCN-IDT and BTSCN-IDT respectively).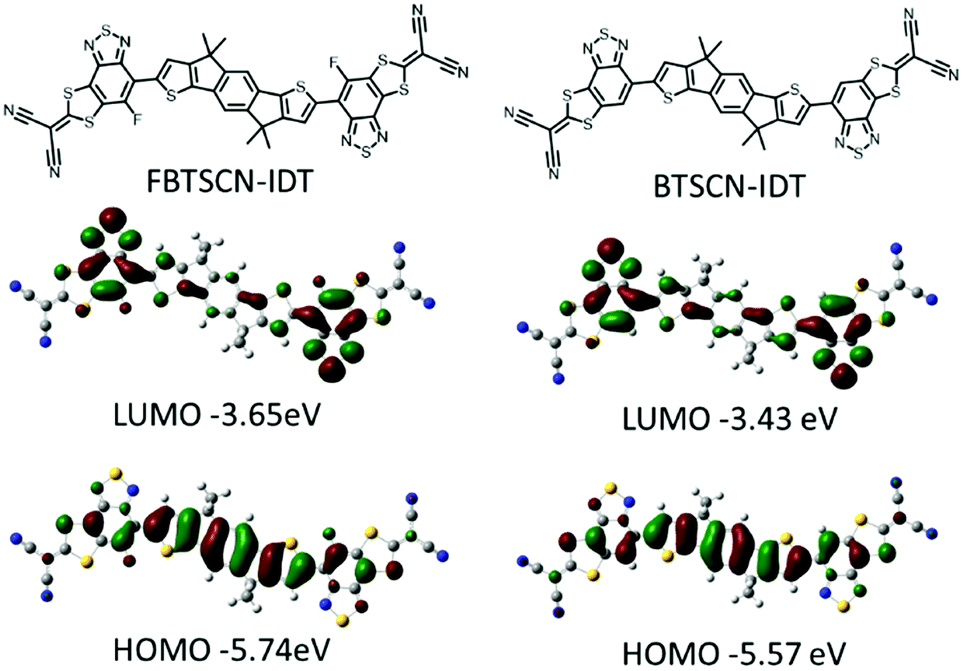 | ||
| Fig. 2 Optimized geometry and frontier molecular orbitals of FBTSCN-IDT and BTSCN-IDT. | ||
Optical and electrochemical properties
The optical properties of both materials were investigated in dichloromethane solution and as thin films (Fig. 3a). BTSCN-IDT in dichloromethane exhibits a strong absorption in the region from 460 nm to 680 nm with a maximum extinction coefficient of 6.3 × 104 mol L−1 cm−1 at 570 nm. Upon moving to the solid state, the maximum absorption peak red-shifts by 44 nm to 614 nm, with an absorption onset of 690 nm corresponding to an optical bandgap of 1.80 eV. The red shift is likely due to the enhanced planarity caused by intermolecular interactions of BTSCN-IDT in film. Fluorination has a minimal effect in the solution state, with FBTSCN-IDT exhibiting an almost identical absorption to the non-fluorinated material, in agreement with similar band gap calculated by DFT. However upon moving to the solid state, the differences are stark, with the maximum absorption peak red-shifting by 100 nm to 660 nm, with an absorption onset at 729 nm corresponding to an optical bandgap of 1.70 eV. The fluorinated materials also exhibit a much broader absorption profile. This suggests significant differences in the solid-state ordering as a result of fluorination, which is consistent with the changes in planarity observed by XRD for the endgroups. Previous studies have highlighted that fluorination often promotes inter- and intramolecular interactions via a range of noncovalent interactions.48–50 Moreover FBTSCN-IDT shows a narrower optical band gap (1.70 eV) and a higher maximum extinction coefficient (9.71 × 104 mol L−1 cm−1) compared to BTSCN-IDT.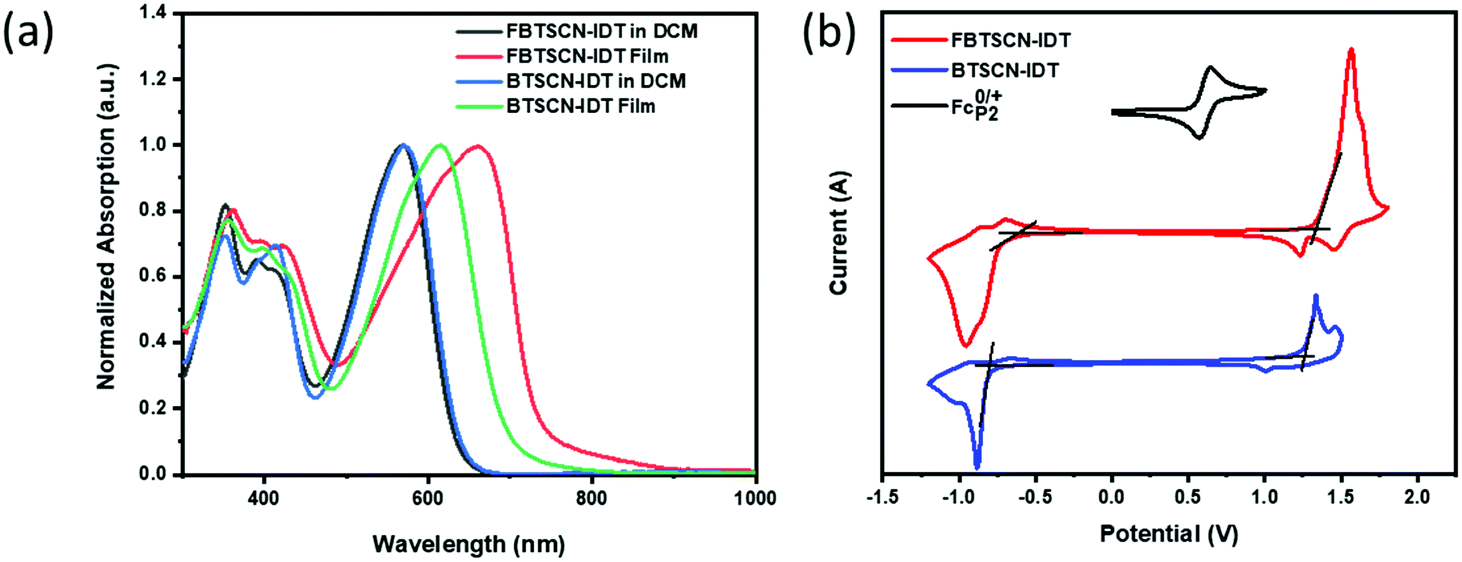 | ||
| Fig. 3 (a) UV-vis absorption spectrum of FBTSCN-IDT and BTSCN-IDT in dichloromethane and film. (b) Cyclic voltammetry curves of FBTSCN-IDT and BTSCN-IDT. Measurements were taken in acetonitrile/[n-Bu4N]PF6 solutions (0.1 mol L−1). Scan rate: 0.1 V s−1. | ||
Cyclic voltammetry was used to investigate the electrochemical properties of both materials as thin films (Fig. 3b). Oxidation results in non-reversible peak, with an onset at 1.16 V and 1.13 V for FBTSCN-IDT and BTSCN-IDT respectively. HOMO levels were therefore estimated to be −5.65 and −5.62 eV for FBTSCN-IDT and BTSCN-IDT respectively based on the ferrocene/ferrocenium couple (EHOMO/LUMO = −[4.8 + Eox/reonset − E0/+Fc] eV). The LUMO energy levels were calculated by the reduction onset with −3.81/−3.67 eV for FBTSCN-IDT and BTSCN-IDT respectively. The slightly downshifted energy levels of FBTSCN-IDT results from the strong electron-withdrawing ability of fluorine, which is in good agreement with DFT theoretical calculation results. Overall, the experimental results and computational modelling are in agreement that the fusion of the 2-(1,3-dithiol-2-ylidene)malonitrile group has a moderate electron withdrawing effect on the BT end-group, resulting in a relatively wide band gap acceptor material.
OSC fabrication and characterization under 1 sun and indoor lighting
Inverted OSC devices were utilized to investigate the performance of FBTSCN-IDT and BTSCN-IDT as NFA small molecule acceptors using an ITO/ZnO/active layer/MnO3/Ag structure (Fig. S27, ESI†), where ZnO and MoO3 are electron and hole transport layers, respectively. Their performance was investigated under both AM 1.5G as well as typical indoor lighting conditions. The measurement procedure is summarized in the ESI.† Based on the measured energy levels of the two NFA's, an initial screen of suitable donor polymers was performed, using PTQ-10/PBDB-T/PM6/PM751–54 (Fig. S27, ESI†). Structurally similar PBDB-T and PM6 donors gave the best initial performance, and we further concentrated on PM6 due to its well-matched energy levels and slightly better performance (Table S1, ESI†). Devices were optimized using various blend ratios, solvents and thermal annealing treatments. Table S2 (ESI†) summarizes the device optimization outcomes. The current density–voltage (J–V) curves of the optimal devices based on FBTSCN-IDT and BTSCN-IDT are shown in Fig. 4a, and their corresponding photovoltaic parameters are summarized in Table 1. PM6/BTSCN-IDT-based devices showed a modest PCE of 1.9%, albeit coupled with a high VOC of 1.16 V, benefiting from the high-lying LUMO energy level of BTSCN-IDT. Fluorination has a remarkable effect on performance, with devices based on PM6/FBTSCN-IDT achieving a much improved PCE of 6.3%, mainly from a much increased short circuit current (JSC) of 9.7 mA cm−2 and a higher fill factor (FF) of 61.5%, under similar coating conditions using chlorobenzene. A slight loss in open circuit voltage was observed, in agreement with the lower lying LUMO, but the device still exhibits a voltage over 1 V. The performance of the fluorinated acceptor is relatively independent of coating solvent, with low boiling chloroform achieving a similar performance to high boiling chlorobenzene. The non-fluorinated blend exhibits a greater variability, with improved performance observed with chloroform, which may relate to the improved solubility of the acceptor in this solvent compared to CB.55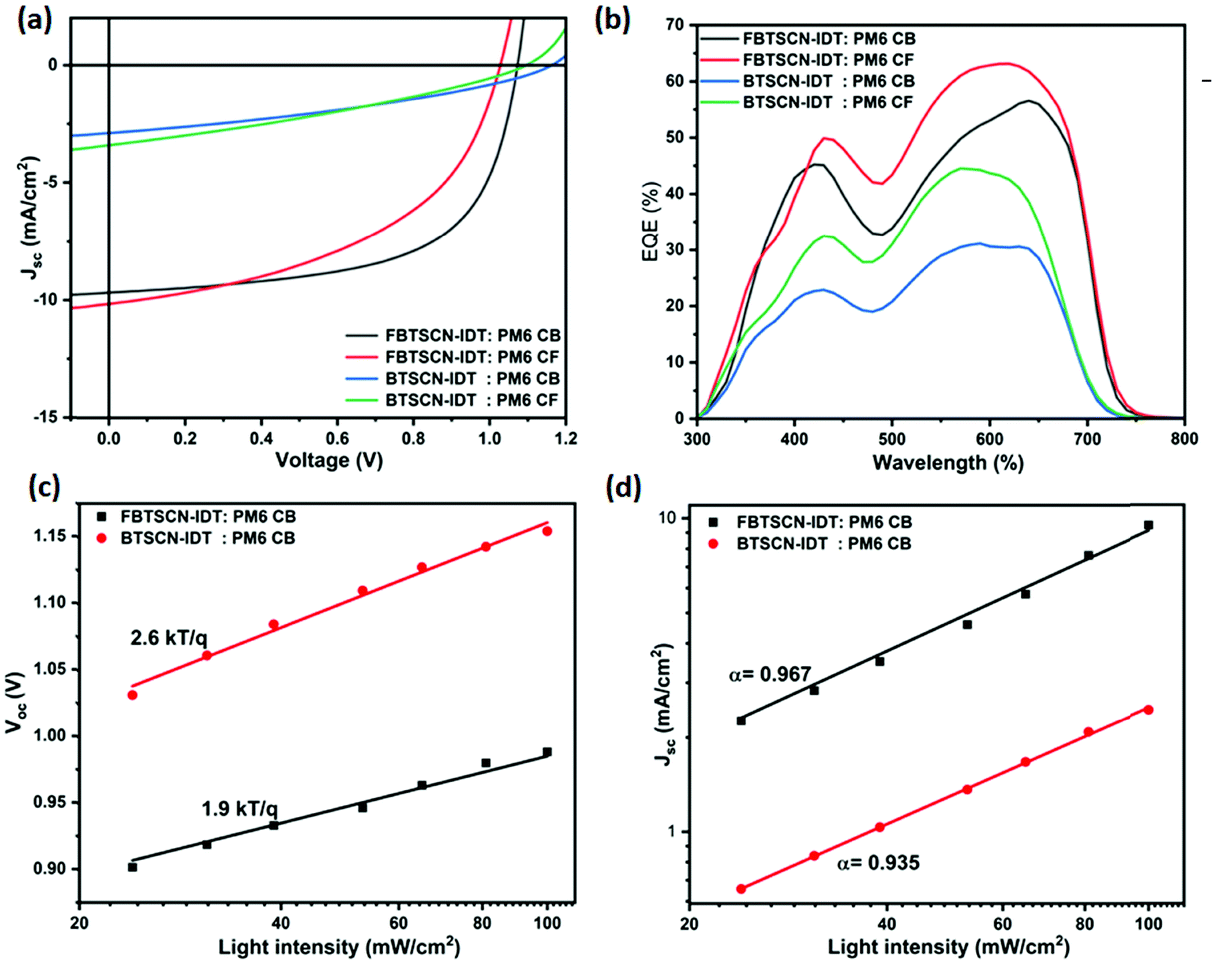 | ||
| Fig. 4 J–V characteristics curves (a) and EQE spectra (b) of FBTSCN-IDT/BTSCN-IDT:PM6 blend system devices, under 1 sun. Light intensity versus (c) VOC and (d) JSC of best performance FBTSCN-IDT and BTSCN-IDT molecules-based devices. | ||
| Active layer | Thermal annealing (°C) | V OC (V) | J sc (mA cm−2) | J calc (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|---|---|
| a OSCs performance obtained from the best devices. b OSCs performance averaged of 4 devices with their standard deviations. c Current density measured by using EQE instrument. | ||||||
| PM6:FBTSCN-IDT, chlorobenzene | As-cast | 1.04b ± 0.024(1.07)a | 9.3b ± 0.34(9.70)a | 9.49c | 57.66b ± 3.92(61.5)a | 5.69b ± 0.61(6.3)a |
| PM6:FBTSCN-IDT, chloroform | 140 | 1.00b ± 0.002(1.01)a | 10.7b ± 0.61(11.40)a | 10.99c | 51.24b ± 0.79(52.03)a | 5.57b ± 0.32(5.90)a |
| PM6:BTSCN-IDT, chlorobenzene | As-cast | 1.11b ± 0.041(1.16)a | 4.71b ± 0.33(5.04)a | 4.97c | 33.93b ± 1.01(34.94)a | 1.77b ± 0.12(1.9)a |
| PM6:BTSCN-IDT, chloroform | 140 | 1.01b ± 0.012(1.03)a | 7.0b ± 0.77(7.80)a | 6.83c | 44.82b ± 2.47(47.79)a | 3.18b ± 0.49(3.6)a |
External quantum efficiency (EQE) measurements were performed to determine the spectral origin of the photocurrent and verify the JSC values for optimal BTSCN-IDT and FBTSCN-IDT-based devices. The measured JSC agrees well with that calculated by integrating the EQE current densities (Jcal) (Table 1). The EQE correlates well with the blend absorption spectra, confirming photocurrent generation across the blend absorption range, with high values observed between 500–700 nm where the maximum blend absorption occurs (Fig. 4b and Fig. S30, ESI†).
The charge carrier transport properties of FBTSCN-IDT and BTSCN-IDT molecules were studied to correlate to the photovoltaic device performance (Table S3 and Fig. S31, ESI†). The electron and hole mobility were measured by using electron-only and hole-only configurations of ITO/ZnO/active layer/Ca/Al, and ITO/PEDOT:PSS/active layer/MoO3/Ag, respectively. The space charge-limited current (SCLC) model was used to fit the J–V characteristics of the devices, as described in the ESI.† The hole mobility (μh) of FBTSCN-IDT:PM6 CB, FBTSCN-IDT:PM6 CF, BTSCN-IDT:PM6 CB, and BTSCN-IDT:PM6 CF blends were 1.82 × 10−4, 2.71 × 10−4, 1.72 × 10−5, and 2.05 × 10−4 cm2 V−1 s−1, respectively, and these values are comparable to those reported in the literature for PM6 donor.56,57 The comparatively low hole mobility of BTSCN-IDT:PM6 CB films may result from the poor solubility of the acceptor in CB, with a rough film observed. The electron mobility (μe) of FBTSCN-IDT:PM6 CB, FBTSCN-IDT:PM6 CF, BTSCN-IDT:PM6 CB, and BTSCN-IDT:PM6 CF blends was much lower than the hole mobility, with values of 1.51 × 10−6, 4.36 × 10−6, 1.99 × 10−7, 2.32 × 10−7 cm2 V−1 s−1, respectively. The general trend agrees with the JSC parameter of the device performance. The FBTSCN-IDT:PM6 CF device delivered slightly higher JSC than FBTSCN-IDT:PM6 CB and the fluorinated acceptor had higher JSC than the non-fluorinated. However the relatively modest overall device performance can be partly explained by the low electron mobility.58 The imbalance in hole and electron mobility can also result in increased recombination, reducing fill factor and photocurrent.59
The recombination mechanism has been studied by measuring the light-intensity dependence of VOC and JSC, of the optimized devices of the FBTSCN-IDT and BTSCN-IDT blend system. The data was fitted according to linear law of VOC ∝ nkT/q![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnPlight and formula JSC ∝ Pαlight, for calculating the device ideality factor (n) and recombination parameters (α), respectively.60Fig. 4c shows the linear correlation between VOC and logarithm of the light intensity. The calculated slope for PM6:FBTSCN-IDT CB and PM6:BTSCN-IDT CB devices were 1.9 and 2.6kt/q−1, respectively. The lower slope of PM6:FBTSCN-IDT CB than PM6:BTSCN-IDT CB, indicates that trap-assisted recombination is suppressed at open-circuit of FBTSCN-IDT molecules-based devices.61Fig. 4d shows the JSCvs. light intensity relationship, plotted on log–log scale, of PM6:FBTSCN-IDT CB and PM6:BTSCN-IDT CB devices. The exponent α value close to unity indicates the weak bimolecular recombination.62 The PM6:FBTSCN-IDT CB (α = 0.967) device has lower bimolecular recombination than PM6:BTSCN-IDT CB (α = 0.935) system. The device with PM6:FBTSCN-IDT shows lower bimolecular recombination, which agrees well with the higher FF (61.5%) than PM6:BTSCN-IDT (34.95%).60
lnPlight and formula JSC ∝ Pαlight, for calculating the device ideality factor (n) and recombination parameters (α), respectively.60Fig. 4c shows the linear correlation between VOC and logarithm of the light intensity. The calculated slope for PM6:FBTSCN-IDT CB and PM6:BTSCN-IDT CB devices were 1.9 and 2.6kt/q−1, respectively. The lower slope of PM6:FBTSCN-IDT CB than PM6:BTSCN-IDT CB, indicates that trap-assisted recombination is suppressed at open-circuit of FBTSCN-IDT molecules-based devices.61Fig. 4d shows the JSCvs. light intensity relationship, plotted on log–log scale, of PM6:FBTSCN-IDT CB and PM6:BTSCN-IDT CB devices. The exponent α value close to unity indicates the weak bimolecular recombination.62 The PM6:FBTSCN-IDT CB (α = 0.967) device has lower bimolecular recombination than PM6:BTSCN-IDT CB (α = 0.935) system. The device with PM6:FBTSCN-IDT shows lower bimolecular recombination, which agrees well with the higher FF (61.5%) than PM6:BTSCN-IDT (34.95%).60
The overall performance of both acceptors under AM1.5 was moderate compared to the current state of the art for medium band gap materials. Nevertheless, the relatively wide gap of both acceptor materials is well matched to indoor light harvesting, which requires absorption in the 380–780 nm wavelength range. Therefore the devices were measured under fluorescent lamp (at 1000 Lux, see ESI†) and the J–V characteristics and device characteristics are shown in Fig. 5 and Table 2, respectively. As expected, similar trends to the AM1.5 devices are observed, with PM6:BTSCN-IDT exhibiting a PCE of 1.80% with a JSC of 24.12 μA cm−2, VOC of 0.72 V, and FF of 36.08%, Whereas FBTSCN-IDT shows a much better performance, with the best device exhibiting a PCE of 13.69% with a JSC of 73.9 μA cm−2, VOC of 0.77 V, and FF of 66.15% under similar conditions. Although comparison to other systems is somewhat complicated by the lack of standardised testing conditions for indoor lighting, the power output is reasonable compared to other NFA systems such as ITIC,63 although clearly limited by the low FF. Overall, the results highlight that annulated BT is a weak acceptor group, leading to medium band gap materials which function as electron acceptors in solar cell blends. The moderate device performance is largely associated with the low electron mobility of the acceptor, which could potentially be improved by replacement of IDT core with a less electron rich aromatic.
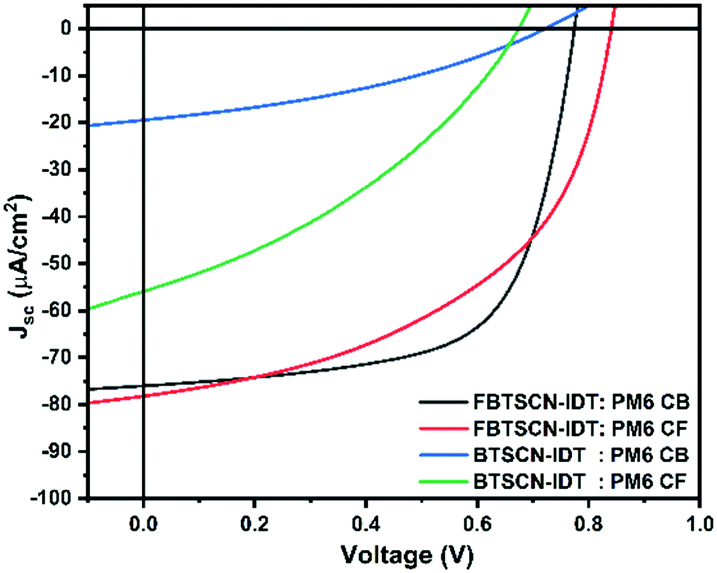 | ||
| Fig. 5 J–V characteristics of FBTSCN-IDT/BTSCN-IDT:PM6 blend system devices processed using chlorobenzene or chloroform under fluorescent lamp. | ||
| Active layer | Thermal annealing (°C) | V OC (V) | J SC (μA cm−2) | FF (%) | PCE (%) | Pout (μw cm−2) |
|---|---|---|---|---|---|---|
| a OSCs performance obtained from the best devices. b OSCs performance averaged of 4 devices with their standard deviations. | ||||||
| PM6:FBTSCN-IDT, chlorobenzene | As-cast | 0.70b ± 0.069(0.77)a | 73.8b ± 0.005(73.9)a | 64.6b ± 1.51(66.15)a | 11.9b ± 1.39(13.69)a | 34.9b ± 3.3(38.2)a |
| PM6:FBTSCN-IDT, chloroform | 140 | 0.83b ± 0.008(0.84)a | 78.1b ± 0.001(78.2)a | 47.7b ± 2.09(49.86)a | 10.9b ± 0.86(11.76)a | 30.6b ± 2.30(32.9)a |
| PM6:BTSCN-IDT, chlorobenzene | As-cast | 0.60b ± 0.112(0.72)a | 24.1b ± 0.002(24.12)a | 33.3b ± 2.71(36.08)a | 1.4b ± 0.35(1.80)a | 3.9b ± 1.15(5.07)a |
| PM6:BTSCN-IDT, chloroform | 140 | 0.64b ± 0.052(0.70)a | 59.6b ± 0.005(59.7)a | 34.2b ± 1.48(35.7)a | 4.4b ± 0.41(4.83)a | 12.4b ± 1.01(13.4)a |
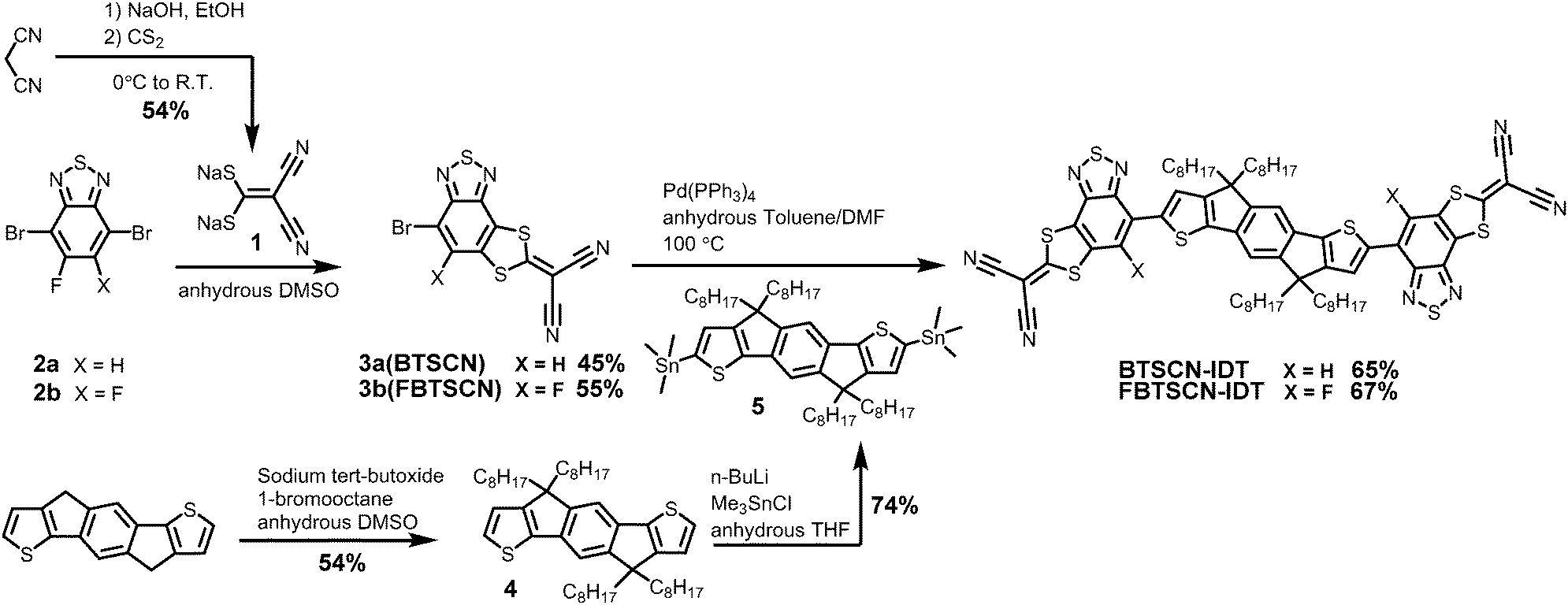 | ||
| Scheme 1 Synthesis route towards BTSCN-IDT, FBTSCN-IDT. | ||
Conclusions
In summary, we report a facile approach to annulate 2,1,3-benzothiadiazole with 2-(1,3-dithiol-2-ylidene)malonitrile in the 4,5-positions via a nucleophilic aromatic substitution reaction of a fluorinated BT precursor. Two annulated BT derivatives were prepared, differing only by the presence of a fluoro substituent in the 6-position. Both derivatives were coupled to an alkylated indacenodithiophene core in good yield. The resulting NFA materials exhibited identical absorption spectra in the solution state, but significant differences were observed in the solid state, with the fluorinated material exhibiting a more redshifted absorption maxima and smaller band gap, likely due to enhanced inter and intramolecular interactions. Cyclic voltammetry measurements suggested both materials were moderate electron acceptors, with LUMO levels from −3.7 to −3.8 eV. Both materials were investigated as the electron accepting component in bulk-heterojunction blends with a variety of donor polymers. The fluorinated material outperformed the non-fluorinated, with devices exhibiting high open circuit voltage over 1 V under 1 sun illumination. Due to the good match of their absorption spectra with the output of a fluorescent lamp, devices were also investigated under indoor lighting conditions. Reasonable performance was observed, with a PCE of 13.7% in the best device. Coupling of such annulated acceptors with different donor cores may be a route to further improve the performance for such low light applications.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We would like to thank the Engineering and Physics Science Research Council (EPSRC) (EP/N020863/1, EP/V048686/1 and EP/T028513/1) and the Royal Society and Wolfson Foundation for financial support.Notes and references
- M. Moser, A. Wadsworth, N. Gasparini and I. McCulloch, Adv. Energy Mater., 2021, 11, 2100056 CrossRef CAS.
- X. Guo, L. Han and X. Hou, Mater. Chem. Front., 2021, 5, 6760–6778 RSC.
- L. Zhang, X. Zhu, D. Deng, Z. Wang, Z. Zhang, Y. Li, J. Zhang, K. Lv, L. Liu, X. Zhang, H. Zhou, H. Ade and Z. Wei, Adv. Mater., 2022, 34, 2106316 CrossRef CAS PubMed.
- Z. Luo, R. Ma, Y. Xiao, T. Liu, H. Sun, M. Su, Q. Guo, G. Li, W. Gao, Y. Chen, Y. Zou, X. Guo, M. Zhang, X. Lu, H. Yan and C. Yang, Small, 2020, 16, 2001942 CrossRef CAS PubMed.
- X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC.
- A. Armin, W. Li, O. J. Sandberg, Z. Xiao, L. Ding, J. Nelson, D. Neher, K. Vandewal, S. Shoaee, T. Wang, H. Ade, T. Heumüller, C. Brabec and P. Meredith, Adv. Energy Mater., 2021, 2003570 CrossRef CAS.
- P. Kafourou, B. Park, J. Luke, L. Tan, J. Panidi, F. Glöcklhofer, J. Kim, T. D. Anthopoulos, J.-S. Kim, K. Lee, S. Kwon and M. Heeney, Angew. Chem., Int. Ed., 2021, 60, 5970–5977 CrossRef CAS PubMed.
- H. Nakanotani, Y. Tsuchiya and C. Adachi, Chem. Lett., 2021, 50, 938–948 CrossRef CAS.
- M. Kim, S. U. Ryu, S. A. Park, Y.-J. Pu and T. Park, Chem. Sci., 2021, 12, 14004–14023 RSC.
- M. Ans, J. Iqbal, B. Eliasson, M. J. Saif, H. M. A. Javed and K. Ayub, J. Mol. Model., 2019, 25, 129 CrossRef PubMed.
- T. C. Parker, D. G. Patel, K. Moudgil, S. Barlow, C. Risko, J.-L. Brédas, J. R. Reynolds and S. R. Marder, Mater. Horiz., 2015, 2, 22–36 RSC.
- C. Tang, X. Ma, J.-Y. Wang, X. Zhang, R. Liao, Y. Ma, P. Wang, P. Wang, T. Wang, F. Zhang and Q. Zheng, Angew. Chem., Int. Ed., 2021, 60, 19314–19323 CrossRef CAS.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200–6214 RSC.
- Q. Nie, A. Tang, Q. Guo and E. Zhou, Nano Energy, 2021, 87, 106174 CrossRef CAS.
- F. Liu, Z. Du, X. Yuan, X. Bao, Y. Wang, J. Zhang, M. Sun and R. Yang, Polymer, 2019, 168, 1–7 CrossRef CAS.
- X. Wang, P. Jiang, Y. Chen, H. Luo, Z. Zhang, H. Wang, X. Li, G. Yu and Y. Li, Macromolecules, 2013, 46, 4805–4812 CrossRef CAS.
- Y. Nian, F. Pan, S. Li, H. Jiang, S. Feng, L. Zhang, Y. Cao and J. Chen, Asian J. Org. Chem., 2018, 7, 2285–2293 CrossRef CAS.
- A. Balan, D. Baran and L. Toppare, Polym. Chem., 2011, 2, 1029–1043 RSC.
- A. C. Stuart, J. R. Tumbleston, H. Zhou, W. Li, S. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef CAS PubMed.
- H. Yu, Z. Qi, J. Yu, Y. Xiao, R. Sun, Z. Luo, A. M. H. Cheung, J. Zhang, H. Sun, W. Zhou, S. Chen, X. Guo, X. Lu, F. Gao, J. Min and H. Yan, Adv. Energy Mater., 2021, 11, 2003171 CrossRef CAS.
- A. Casey, S. D. Dimitrov, P. Shakya-Tuladhar, Z. Fei, M. Nguyen, Y. Han, T. D. Anthopoulos, J. R. Durrant and M. Heeney, Chem. Mater., 2016, 28, 5110–5120 CrossRef CAS.
- A. Creamer, A. Casey, A. V. Marsh, M. Shahid, M. Gao and M. Heeney, Macromolecules, 2017, 50, 2736–2746 CrossRef CAS.
- A. E. London, H. Chen, M. A. Sabuj, J. Tropp, M. Saghayezhian, N. Eedugurala, B. A. Zhang, Y. Liu, X. Gu, B. M. Wong, N. Rai, M. K. Bowman and J. D. Azoulay, Sci. Adv., 2019, 5, eaav2336 CrossRef CAS PubMed.
- J. D. Yuen, M. Wang, J. Fan, D. Sheberla, M. Kemei, N. Banerji, M. Scarongella, S. Valouch, T. Pho, R. Kumar, E. C. Chesnut, M. Bendikov and F. Wudl, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 287–293 CrossRef CAS.
- J. D. Yuen, R. Kumar, D. Zakhidov, J. Seifter, B. Lim, A. J. Heeger and F. Wudl, Adv. Mater., 2011, 23, 3780–3785 CAS.
- T. T. Steckler, X. Zhang, J. Hwang, R. Honeyager, S. Ohira, X.-H. Zhang, A. Grant, S. Ellinger, S. A. Odom, D. Sweat, D. B. Tanner, A. G. Rinzler, S. Barlow, J.-L. Brédas, B. Kippelen, S. R. Marder and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 2824–2826 CrossRef CAS PubMed.
- T. L. Dexter Tam, T. Salim, H. Li, F. Zhou, S. G. Mhaisalkar, H. Su, Y. M. Lam and A. C. Grimsdale, J. Mater. Chem., 2012, 22, 18528–18534 RSC.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- S. Holliday, R. S. Ashraf, C. B. Nielsen, M. Kirkus, J. A. Röhr, C.-H. Tan, E. Collado-Fregoso, A.-C. Knall, J. R. Durrant, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 898–904 CrossRef CAS PubMed.
- M. U. Saeed, J. Iqbal, R. F. Mehmood, S. J. Akram, Y. A. El-Badry, S. Noor and R. A. Khera, Surf. Interfaces, 2022, 30, 101875 CrossRef.
- M. Hagimori, S. Matsui, N. Mizuyama, K. Yokota, J. Nagaoka and Y. Tominaga, Eur. J. Org. Chem., 2009, 5847–5853 CrossRef CAS.
- F. Zhang, Y. Hu, T. Schuettfort, C.-A. Di, X. Gao, C. R. McNeill, L. Thomsen, S. C. B. Mannsfeld, W. Yuan, H. Sirringhaus and D. Zhu, J. Am. Chem. Soc., 2013, 135, 2338–2349 CrossRef CAS PubMed.
- Z. Zhao, F. Zhang, Y. Hu, Z. Wang, B. Leng, X. Gao, C.-A. Di and D. Zhu, ACS Macro Lett., 2014, 3, 1174–1177 CrossRef CAS PubMed.
- H. Luo, D. He, Y. Zhang, S. Wang, H. Gao, J. Yan, Y. Cao, Z. Cai, L. Tan, S. Wu, L. Wang and Z. Liu, Org. Lett., 2019, 21, 9734–9737 CrossRef CAS PubMed.
- M. A. Kolaczkowski, A. Garzón-Ruiz, A. Patel, Z. Zhao, Y. Guo, A. Navarro and Y. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 53328–53341 CrossRef CAS PubMed.
- H. S. Ryu, S. Y. Park, T. H. Lee, J. Y. Kim and H. Y. Woo, Nanoscale, 2020, 12, 5792–5804 RSC.
- A. Creamer, C. S. Wood, P. D. Howes, A. Casey, S. Cong, A. V. Marsh, R. Godin, J. Panidi, T. D. Anthopoulos, C. H. Burgess, T. Wu, Z. Fei, I. Hamilton, M. A. McLachlan, M. M. Stevens and M. Heeney, Nat. Commun., 2018, 9, 3237 CrossRef PubMed.
- S. Cong, A. Creamer, Z. Fei, S. A. J. Hillman, C. Rapley, J. Nelson and M. Heeney, Macromol. Biosci., 2020, 20, 2000087 CrossRef CAS PubMed.
- J. F. Bunnett and R. E. Zahler, Chem. Rev., 1951, 49, 273–412 CrossRef CAS.
- G. M. Brooke, J. Fluorine Chem., 1989, 43, 393–403 CrossRef CAS.
- G. M. Brooke and J. M. Meara, J. Fluorine Chem., 1990, 50, 229–242 CrossRef CAS.
- R. D. Chambers, D. Close and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1980, 778–780, 10.1039/P29800000778.
- Q. He, M. Shahid, J. Wu, X. Jiao, F. D. Eisner, T. Hodsden, Z. Fei, T. D. Anthopoulos, C. R. McNeill, J. R. Durrant and M. Heeney, Adv. Funct. Mater., 2019, 29, 1904956 CrossRef.
- S. J. Akram, J. Iqbal, M. Ans, Y. A. El-Badry, R. F. Mehmood and R. A. Khera, Sol. Energy, 2022, 237, 108–121 CrossRef CAS.
- U. Yaqoob, A. Raza Ayub, S. Rafiq, M. Khalid, Y. A. El-Badry, Z. M. El-Bahy and J. Iqbal, J. Mol. Liq., 2021, 341, 117428 CrossRef CAS.
- Q. He, M. Shahid, X. Jiao, E. Gann, F. D. Eisner, T. Wu, Z. Fei, T. D. Anthopoulos, C. R. McNeill and M. Heeney, ACS Appl. Mater. Interfaces, 2020, 12, 9555–9562 CrossRef CAS PubMed.
- Q. Zhang, M. A. Kelly, N. Bauer and W. You, Acc. Chem. Res., 2017, 50, 2401–2409 CrossRef CAS PubMed.
- T. Hodsden, K. J. Thorley, J. Panidi, A. Basu, A. V. Marsh, H. Dai, A. J. P. White, C. Wang, W. Mitchell, F. Glöcklhofer, T. D. Anthopoulos and M. Heeney, Adv. Funct. Mater., 2020, 30, 2000325 CrossRef CAS.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS PubMed.
- K. J. Thorley and I. McCulloch, J. Mater. Chem. C, 2018, 6, 12413–12421 RSC.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z.-G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef PubMed.
- L. Ye, X. Jiao, M. Zhou, S. Zhang, H. Yao, W. Zhao, A. Xia, H. Ade and J. Hou, Adv. Mater., 2015, 27, 6046–6054 CrossRef CAS PubMed.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655–4660 CrossRef CAS PubMed.
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef PubMed.
- W. Xu, X. Li, S. Y. Jeong, J. H. Son, Z. Zhou, Q. Jiang, H. Y. Woo, Q. Wu, X. Zhu, X. Ma and F. Zhang, J. Mater. Chem. C, 2022, 10, 5489–5496 RSC.
- N. Yao, J. Wang, Z. Chen, Q. Bian, Y. Xia, R. Zhang, J. Zhang, L. Qin, H. Zhu, Y. Zhang and F. Zhang, J. Phys. Chem. Lett., 2021, 12, 5039–5044 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- M. M. Mandoc, L. J. A. Koster and P. W. M. Blom, Appl. Phys. Lett., 2007, 90, 133504 CrossRef.
- S. Zhang, X. Ma, C. Xu, W. Xu, S. Y. Jeong, H. Y. Woo, Z. Zhou, X. Zhang and F. Zhang, Macromol. Rapid Commun., 2022, 2200345 CrossRef PubMed.
- A. Tang, W. Song, B. Xiao, J. Guo, J. Min, Z. Ge, J. Zhang, Z. Wei and E. Zhou, Chem. Mater., 2019, 31, 3941–3947 CrossRef CAS.
- C. Liu, J. Tu, X. Hu, Z. Huang, X. Meng, J. Yang, X. Duan, L. Tan, Z. Li and Y. Chen, Adv. Funct. Mater., 2019, 29, 1808059 CrossRef.
- A. K. K. Kyaw, D. H. Wang, D. Wynands, J. Zhang, T.-Q. Nguyen, G. C. Bazan and A. J. Heeger, Nano Lett., 2013, 13, 3796–3801 CrossRef CAS PubMed.
- Z. Ding, R. Zhao, Y. Yu and J. Liu, J. Mater. Chem. A, 2019, 7, 26533–26539 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2164513 and 2164514. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc01433e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |