
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CuII/CuI decorated N-doped carbonaceous electrocatalysts for the oxygen reduction reaction†
Janina
Kossmann
 ,
María Luz
Ortíz Sánchez-Manjavacas
,
Hannes
Zschiesche
,
Nadezda V.
Tarakina
,
Markus
Antonietti
,
Josep
Albero
* and
Nieves
López-Salas
*
,
María Luz
Ortíz Sánchez-Manjavacas
,
Hannes
Zschiesche
,
Nadezda V.
Tarakina
,
Markus
Antonietti
,
Josep
Albero
* and
Nieves
López-Salas
*
Colloid Chemistry Department, Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476, Potsdam, Germany. E-mail: nieves.lopezsalas@mpikg.mpg.es; josep.albero@mpikg.mpg.de
First published on 9th November 2021
Abstract
The oxygen reduction reaction (ORR) that for instance takes place at the cathode of fuel cells is one of the most examined model reactions of energy conversion. The ORR presents sluggish reaction kinetics, thus limiting the overall efficiency of these cells. Pt-based catalysts are still the widest choice though they exhibit important drawbacks such as long-term instability and intolerance to methanol crossover. In this context, engineering transition metals in the form of nano- and subnano-sites on carbonaceous supports has the potential of becoming an alternative to scarce noble metal-based catalysts. Herein, we describe a simple synthetic route towards CuII/CuI decorated N-doped carbonaceous ORR electrocatalysts. CuII/CuI nanosites are obtained by calcination in air of an ionic liquid derived noble carbonaceous support impregnated with copper(II) acetate. The strong interaction between the copper and the noble support foster the co-formation of CuII/CuI nanosites. Larger amounts of copper(II) acetate translate into larger amounts of CuI and lower Tafel slopes. The material with 4 wt% of copper catalyzes the selective reduction of oxygen through a 4-electron transfer pathway and exhibits a lower Tafel slope than commercial platinum, a minimal overpotential, and a higher limiting current density. Moreover, all materials show promising durability and high methanol stability, which makes them promising to replace noble metals for the ORR.
Introduction
The oxygen reduction reaction (ORR) is the cathodic reaction of several types of fuel cells (FCs) and it is well-known as the kinetic bottleneck of the overall process since, for instance, hydrogen oxidation at the anode is six times faster.1 Due to its importance for renewable energy technologies, the development of sustainable and efficient electrocatalysts towards the ORR is a key step in the further development of fuel cells.2 Pt-based electrocatalysts are widely used since this noble metal has demonstrated high efficiency and selectivity towards the reduction of oxygen through a 4-electron pathway mechanism.3–6 However, Pt-based materials show important drawbacks, including Pt scarcity and thus their high cost, poor stability towards methanol cross-over, and poor durability, among others, limiting their practical applications.2,7–9 Therefore, many research efforts are currently underway focusing on developing new catalysts containing abundant and affordable elements, which can outperform Pt-based materials and overcome their drawbacks, especially for simpler cell management designs.Transition metals supported on N-doped carbonaceous materials (M–NCs) such as Fe10–12 or Co13,14 have shown good ORR activity and stability and they have been broadly analysed as catalysts in cathodes.2,15–17 However, their practical applicability is limited due to their ability to participate in Fenton-like radical oxygen activation pathways, and hence, promoting the degradation of the polymeric membrane in polymer membrane fuel cells.1,18 Another abundant transition metal of great potential in the ORR is Cu. Cu is nature's choice for the reduction of oxygen in metalloenzymes such as laccase and amine oxidase.2,19,20 Lu and co-workers reported that the larger activity of Cu oxidases over Fe ones could be assigned to a more positive redox potential together with the higher electron density in the d-orbital of Cu, which results in weaker O–O bonds.21 Many studies, bioinspired by laccase and naturally occurring Cu oxidases, have investigated the influence of ligands in Cu complexes on their performance as ORR electrocatalysts. The reports point at multiatom centres with different Cu oxidation states fostering different steps of the ORR mechanism.22 Even more interestingly, similar results have been observed using heterogeneous catalysts consisting of Cu single sites stabilized in N-modified carbon supports using in situ EXAFS analysis.23 Moreover, CO2 and O2 activation have also been reported using Cu-modified ZSM-5 zeolites.24
On the other hand, the use of heteroatom doped carbonaceous materials as transition metal supports in electrocatalysis has been widely explored since these have multifarious roles. Among the properties that justify the interest of these materials as supports, the most relevant ones are their capability to anchor metal active sites, and the pore structure and surface area tunability, which are of utmost importance for the fast transport of different species to the active sites.25 Ionic liquids (ILs) are attractive precursors for conductive and N-doped carbonaceous materials due to their high amount of N and ionic character. The preparation of carbonaceous materials from ILs can be achieved in high yields without any pre-polymerization, for instance, if polymerizable moieties in the IL backbones (i.e. cyano group) can cross-link upon heat treatment.26 Moreover, combining ILs with salt melts offers the possibility to tune simultaneously the pore structure and surface area of these materials.27,28 Moreover, IL derived materials have been proposed as noble carbon precursors due to their stability upon oxidation. Their nobility, and that of other selected precursors, was hypothesized by Oschatz et al. to come from the thermodynamic inheritance of the original large stability towards elimination reactions that leads to the formation of very stable bonds upon carbonization.29
Herein, we describe the synthesis of a highly N-doped carbonaceous material derived from the IL 1-butyl-4-methylpyridinium tricyanomethanide (BTCN), and its use as a support for Cu nanoclusters. The as-prepared carbonaceous material stability upon oxidation allows Cu loading by a simple strategy of impregnation with copper(II) acetate plus calcination in air. The samples were subsequently fully analysed by energy-dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD), thermogravimetric analysis (TGA), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), and transmission electron microscopy (TEM). Their performance as catalysts for the ORR has been evaluated and correlated to their physico-chemical properties.
Methods
Materials
1-Butyl-4-methylpyridinium tricyanomethanide (>98%) was purchased from IOLITEC. Potassium chloride (99.5%) was obtained from PanReac AppliChem and zinc chloride (98.5%) from ACROS ORGANICS. Fuming hydrochloric acid (37%) was purchased from Roth and acetonitrile, potassium hydroxide, and methanol were from MERCK. Copper(II) acetate (98%) and Nafion (5 wt% in lower aliphatic alcohols and water, contains 15–20% water) were purchased from Sigma-Aldrich. Platinum on graphitized carbon (20 wt% loading) was purchased from Aldrich.Synthesis
Characterization
X-ray powder diffraction patterns (XRD) were recorded on a Bruker D8 Advance diffractometer with Cu-Kα radiation (λ = 1.5418 Å) in the 2θ range 5–70°, 1625 steps and 1 step per second. Thermogravimetric analysis (TGA) was performed on a NETZSCH TG 209 F1 device from 25 °C to 1000 °C in either a nitrogen or synthetic air atmosphere and at a heating rate of 10 °C min−1 in a Pt crucible. Inductively coupled plasma (ICP) spectroscopy is performed with a PerkinElmer ICP-OES Optima 8000. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo Fisher Scientific Escalab 250 Xi. Physisorption measurements were performed on a Quantachrome Quadrasorb SI apparatus with nitrogen measurements at 77 K, and carbon dioxide measurements at 298 K. The specific surface area of each material was obtained from the nitrogen adsorption data (P/P0 < 0.2) using the Brunauer–Emmett–Teller (BET) method and the total pore volume (VT) was calculated from the amount of nitrogen adsorbed at P/P0 = 0.995. The N2 pore size distribution was obtained by using a quenched solid density functional theory (QSDFT) model with a slit/cylindrical pore shape using nitrogen adsorption data. Scanning electron microscopy (SEM) and energy-disperse X-ray spectroscopy were performed on a LEO 1550 Gemini Zeiss microscope equipped with an Oxford Instruments EDX spectrometer after sputtering the samples with an 80% gold/20% palladium mixture. For transmission- and scanning transmission electron microscopy (STEM) observations, a suspension of the sample in ethanol was sonicated for 15 minutes and then drop-cast on a Ni grid with a lacey carbon support and dried. We carried out the transmission electron microscopy (TEM) study on a Zeiss 912 transmission electron microscope, operated at 120 kV. The STEM study was performed using a double Cs corrected JEOL JEM-ARM200F operated at 80 kV, equipped with a cold-field emission gun and a high-angle silicon drift energy dispersive X-ray (EDX) detector (solid angle up to 0.98 steradians with a detection area of 100 mm2). High-angle annular dark-field STEM (ADF-STEM) images were collected with an angular collection semi angle ranging from 33 mrad to 125 mrad. Bright field STEM (BF-STEM) images were acquired on a maximal semi acceptance angle of 20 mrad.Electrochemical characterization
Electrochemical tests were carried on an Interface 1000 (Gamry) with a standard three-electrode cell system with an Ag/AgCl reference electrode and a platinum counter electrode. As the working electrode, a rotating disk electrode (RDE) from PINE or a rotating ring disk electrode (RRDE) from PINE were deposited with 10 μL of the catalyst ink and slowly dried at room temperature. The ink was prepared by mixing 50 μL Nafion with 500 μL distilled water and 250 μL isopropyl alcohol and subsequent adding of 5 mg catalyst followed by 5 min sonication. Commercial Pt/C (platinum, 20% on carbon) was used as the reference catalyst.Cyclic voltammetry (CV) curves were recorded in a nitrogen saturated 0.1 M KOH solution with a scan rate of 10 mV s−1. The oxygen reduction reaction (ORR) activity was tested with CV curve measurements in oxygen saturated 0.1 M KOH solution and determined with linear sweep voltammetry (LSV) with an RDE at a scan rate of 10 mV s−1 and different rotation speeds (0 rpm to 1600 rpm). The electron transfer number n was calculated by using the Koutecký–Levich eqn (1) at various rotation speeds.
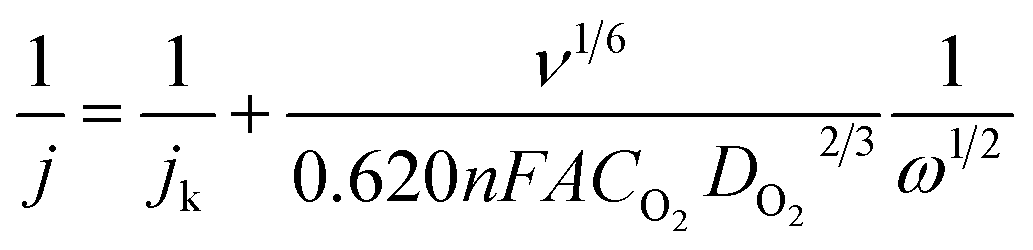 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), v is the kinetic viscosity (0.01004 cm2 s−1), CO is the oxygen concentration in the electrolyte (1.39 × 10−3 mol L−1) and DO is the diffusion coefficient of oxygen in the electrolyte (1.9 × 10−5 cm2 s−1).
485 C mol−1), v is the kinetic viscosity (0.01004 cm2 s−1), CO is the oxygen concentration in the electrolyte (1.39 × 10−3 mol L−1) and DO is the diffusion coefficient of oxygen in the electrolyte (1.9 × 10−5 cm2 s−1).
Additionally, the electron transfer number n and the hydrogen peroxide yield% H2O2 are calculated using eqn (2) and (3) from RRDE measurements in oxygen saturated 0.1 M KOH with a constant ring potential of 0.3 V (vs. Ag/AgCl),
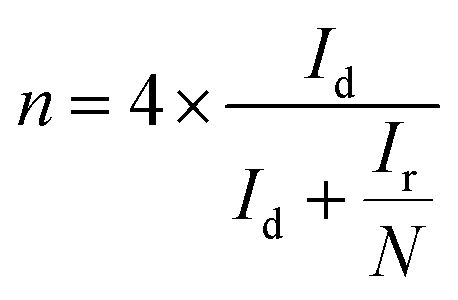 | (2) |
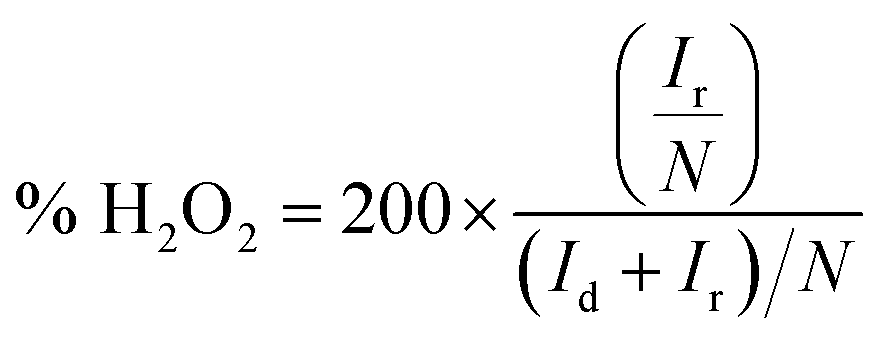 | (3) |
Stability tests are done with chronoamperometry measurements for 4 h in oxygen saturated 0.1 M KOH by applying a potential of −0.15 V vs. Ag/AgCl and MeOH stability tests are performed by chronoamperometry measurements and adding 0.5 mL MeOH after 1000 s to the electrolyte.
Results and discussion
Materials preparation and characterization
BTCN ionic liquid (IL) contains large amounts of N in its chemical structure. Moreover, the cyano-groups of the anionic counterpart are known to condense upon carbonization, rendering large yields of N-doped carbon, BTCN being a very convenient candidate as a N-doped carbonaceous material precursor.30,31 In the present case, a yield as large as 24% at 800 °C was calculated based on the experimental data obtained from the TGA of the IL precursor (Fig. S1†). The N-doped carbon support preparation procedure is graphically depicted in Fig. 1. In brief, N-doped carbon was obtained by mixing BTCN as the N-doped carbon precursor and KCl/ZnCl2 as solvent and porogen (BTCN:KCl:ZnCl2 weight ratio of 1:5:5), and subsequent thermal treatment at 800 °C in a N2 atmosphere. Samples obtained by BTCN condensation at 800 °C will be named hereafter BTCN–Cu0.
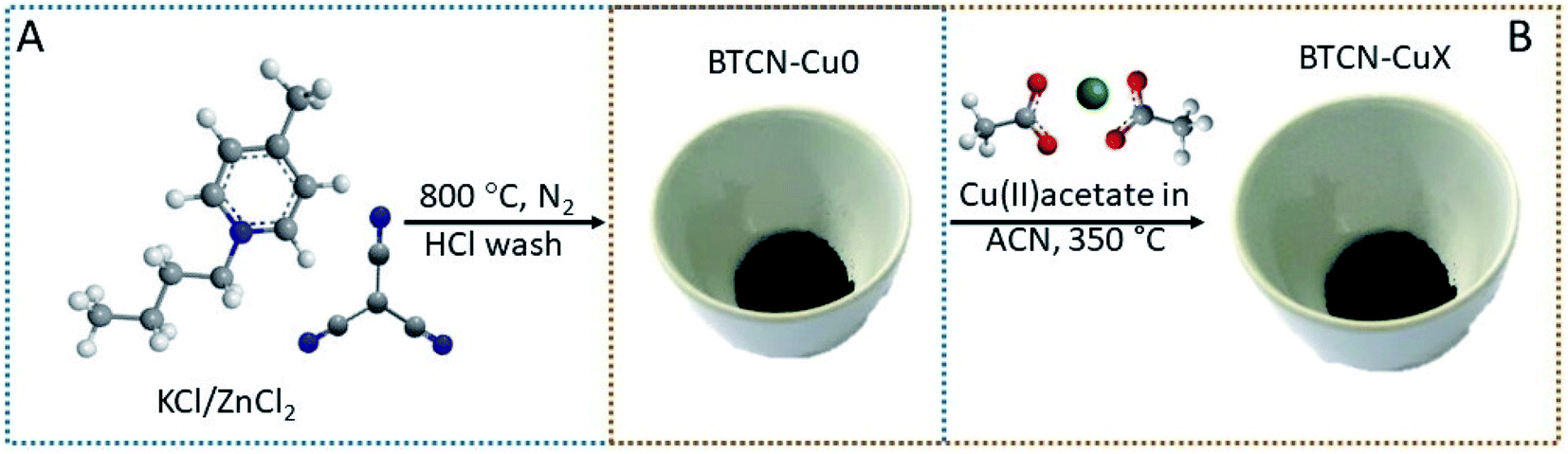 | ||
| Fig. 1 Schematic illustration of the synthetic procedure. (A) 1-Butyl-4-methylpyridinium tricyanomethanide (BTCN) and the salt mixture (KCl/ZnCl2) are mixed and heat treated at 800 °C under a N2 atmosphere and washed with HCl. (B) The product (BTCN–Cu0) is further used as a support for Cu loading: BTCN–Cu0 is added to a solution of Cu(II) acetate in acetonitrile and stirred for 2 h. After solvent removal, the Cu–BTCN–Cu0 mixture is heated to 350 °C in air. The obtained product is named BTCN–CuX, X being the Cu loading. | ||
BTCN–Cu0 was obtained with a yield of ca. 24 wt% and has a remarkably high N content of 20 at% according to SEM-EDX analysis (Table 1). Cu loading was carried out by stirring the BTCN–Cu0 powder in Cu(II) acetate solution in acetonitrile at different concentrations for 2 h. After removing the solvent, the impregnated BTCN–Cu0/Cu(II) acetate mixtures were heated at 350 °C for 2 h in an air atmosphere (Fig. 1B). Different copper loadings were achieved by simple adjustment of Cu(II) acetate solution concentrations (Table S1†). Samples are hereafter named BTCN–CuX, where X is ascribed to their Cu loading (i.e. BTCN–Cu1 was synthesized with 1 wt% copper salt loading). Inductively coupled plasma mass spectrometry (ICP-MS) measurements show that the samples BTCN–Cu1, BTCN–Cu2, and BTCN–Cu4 have Cu contents of 0.8 wt%, 2.2 wt%, and 3.7 wt%, respectively, which are in the range of the initially loaded amounts (Table 1) and point at the outstanding adsorption capacity of the carbonaceous support to stabilize even larger amounts of Cu. ICP-MS shows that zinc traces were present in the support even after intensive HCl washing.
| Sample | Yield | SEM-EDX (at%) | ICP-MS (wt%) | S BET | |||
|---|---|---|---|---|---|---|---|
| wt% | C | N | O | C/Nc | Cu | m2 g−1 | |
| a BTCN to BTCN–Cu0. b BTCN–Cu0 to Cu decorated material. c Propagation of error from the standard deviation of the determination of carbon and nitrogen content. d Obtained using the QSDFT method applied to N2 adsorption isotherms at 77 K. | |||||||
| BTCN–Cu0 | 24a | 74.0 (2) | 20.4 (2) | 5.0 (1) | 3.6 (0.4) | — | 1643 |
| BTCN–Cu1 | 90b | 72.3 (3) | 18.0 (2) | 9.7 (1) | 4.0 (0.5) | 0.8 | 1615 |
| BTCN–Cu2 | 88b | 68.1 (4) | 19.6 (3) | 12.3 (3) | 3.5 (0.6) | 2.2 | 1522 |
| BTCN–Cu4 | 85b | 72.1 (1) | 16.5 (1) | 11.4 (1) | 4.4 (0.3) | 3.7 | 1358 |
Further chemical analysis was carried out using SEM-EDX. The results are summarized in Table 1 and spectra are shown in Fig. S2.† Despite the changes in the absolute nitrogen (i.e. from 20.4 at% N for BTCN–Cu0 to 16.5 at% N for BTCN–Cu4) and carbon content (decreases from 74.0 at% for BTCN–Cu0 to 72.1 at% for BTCN–Cu4) of the samples, the C/N ratios of the obtained materials remain similar, pointing at the stability of the support. The O content increases from 5 at% to 9.7 at%, 12.3 at%, and 11.4 at% for BTCN–Cu0, BTCN–Cu1, BTCN–Cu2, and BTCN–Cu4, respectively. We ascribe this increase of the oxygen content to O functionalities introduced by the copper(II) acetate salt remaining on the surface of the support during the calcination process.
It is worth noticing that BTCN–Cu0 was also thermally treated at 350 °C in an air atmosphere to ensure that the increase of O is not just caused by oxidation during calcination. SEM-EDX analysis shows that the sample composition does not change during this thermal treatment under oxidative conditions (see Table S2†).
N2 adsorption/desorption isotherms at 77 K (Fig. S3A†) of the samples show type I curves for all samples, indicating that the materials are mainly microporous. All isotherms are very similar despite a slight decrease of N2 uptake with increasing Cu loading, which is also confirmed by the specific surface areas calculated using the BET method (Table 1). The similarity between all the isotherms further points at the support stability (i.e. the low pore volume change indicates that no pores are created because of the material decomposition or disappear due to shrinking). The slight decreases in specific surface area with increasing Cu content (from 1643 m2 g−1 for BTCN–Cu0 to 1358 m2 g−1 for BTCN–Cu4), make us hypothesize that the deposition of the Cu species causes a small percentage of pore blocking in the material.
The XRD patterns (Fig. S3B†) show no peaks related to Cu, CuO or Cu2O in any of the materials. The broad and low intense diffraction peak at ca. 27° has been assigned to graphitic stacking of the carbonaceous support. Fig. 2 shows the selected SEM and TEM images of BTCN–CuX. SEM images show a colloidal carbon structure as typically obtained with salt melt templating formed by primary carbon particles with a diameter of ca. 200 nm. After Cu loading, the samples do not suffer notable changes, which is in accordance with the physisorption analysis. TEM micrographs also show similar morphology before and after Cu loading. Furthermore, no Cu nanoparticles or big clusters can be identified on the surface of the samples, which is also in good agreement with the powder XRD results.
 | ||
| Fig. 2 Micrographs of BTCN–Cu0, BTCN–Cu1, BTCN–Cu2 and BTCN–Cu4. Top row: surface morphology visualized by SEM, using a secondary electron detector. Bottom row: overview TEM images. | ||
Higher resolution analysis of the samples was performed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 3). While the carbonaceous support appears greyish, brighter single atom or atom-cluster spots of a size of ca. 0.3 nm can also be identified, which must originate from elements with a higher atomic number. The resolution of the STEM does not allow us to clearly discern whether these bright spots correspond only to sub-nanometric clusters, but the presence of Cu single atoms together with nanoclusters containing different Cu atoms (i.e. Cu2, Cu3, Cu4, etc.) cannot be discarded. The number of bright spots per projected area increases qualitatively with the Cu loading (comparing BTCN–Cu0, BTCN–Cu1, and BTCN–Cu4), thus referring likely to a homogeneous distribution of Cu atoms or sub-nanometric clusters instead of the formation of nanoparticles or bigger clusters.
 | ||
| Fig. 3 HAADF-STEM images of BTCN–Cu0, BTCN–Cu1, and BTCN–Cu4. | ||
X-ray photoelectron spectroscopy (XPS) measurements were performed in order to evaluate the oxidation state of the Cu species in the prepared materials. The high-resolution Cu 2p spectra (Fig. 4) show the typical peaks centred at 954.6 and 934.4 eV, attributed to CuII, in all samples. Upon increasing the initial Cu(II) acetate loading, additional peaks at 951.8 and 931.9 eV are developed, which can be ascribed either to CuI or Cu0.32 In order to elucidate the actual Cu species oxidation state, the Auger Cu LMM spectra have been measured for BTCN–Cu1, BTCN–Cu2 and BTCN–Cu4 samples, and the results are presented in Fig. S4 in the ESI.† As can be seen there, the Cu LMM spectrum of BTCN–Cu2 shows a peak at 915.95 eV assigned to CuI, while BTCN–Cu4 presents a small peak at 919.32 eV together with a major peak at 915.95 eV, indicating the coexistence of CuI and Cu0, although the latter to a much lesser extent. Thus, we can conclude that the BTCN–CuX samples contain mainly the redox pair CuII/CuI, the existence of Cu0 being negligible. CuII/CuI ratios of 3.5, 2, and 1 were calculated for BTCN–Cu1, BTCN–Cu2, and BTCN–Cu4, respectively. The decreasing CuII/CuI ratio when increasing the copper(II) acetate loading points at a possible redox valence tautomerism of the Cu centres formed. Such redox valence tautomerism has been previously reported for N and O based ligands in molecular compounds.33–35 The carbonaceous support offers a high amount of N moieties where CuII/CuI centers can interact with the support. Such stabilization would imply electron withdrawal from N functionalities. Fig. S5† shows the high-resolution C 1s and N 1s spectra and the best deconvolution to their individual components for BTCN–Cu0, BTCN–Cu1, BTCN–Cu2, and BTCN–Cu4 samples. In this case, drastic changes can be seen upon Cu loading (i.e. from BTCN–Cu0 to BTCN–Cu1). The C 1s peak in BTCN–Cu0 shows three different components attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds (284.7 eV), C–N bonds (286.1 eV), and a third component at 288 eV, which can be ascribed to the electron poor conjugated C–N bonds. After Cu loading, the C–N to CC ratio increases (from BTCN–Cu0 to BTCN–Cu1) and remains approximately unaltered upon further Cu loading. However, the component at 288 eV becomes very well defined, clearly indicating the formation of electron deficient C–N bonds, presumably the locus of CuII coordination. On the other hand, all N 1s signals are the sum of four species centered at 398.5 eV, 400.0 eV, 401.2 eV, and 404 eV typically ascribed to pyridinic-N, pyrrolic-N, pyridonic-N, and in plane quaternary-N, respectively. Interestingly, upon Cu loading, the component centred at 401.2 eV increases, related to electron poor nitrogen, which also points to nitrogen electron withdrawal to stabilize Cu sites.
C bonds (284.7 eV), C–N bonds (286.1 eV), and a third component at 288 eV, which can be ascribed to the electron poor conjugated C–N bonds. After Cu loading, the C–N to CC ratio increases (from BTCN–Cu0 to BTCN–Cu1) and remains approximately unaltered upon further Cu loading. However, the component at 288 eV becomes very well defined, clearly indicating the formation of electron deficient C–N bonds, presumably the locus of CuII coordination. On the other hand, all N 1s signals are the sum of four species centered at 398.5 eV, 400.0 eV, 401.2 eV, and 404 eV typically ascribed to pyridinic-N, pyrrolic-N, pyridonic-N, and in plane quaternary-N, respectively. Interestingly, upon Cu loading, the component centred at 401.2 eV increases, related to electron poor nitrogen, which also points to nitrogen electron withdrawal to stabilize Cu sites.
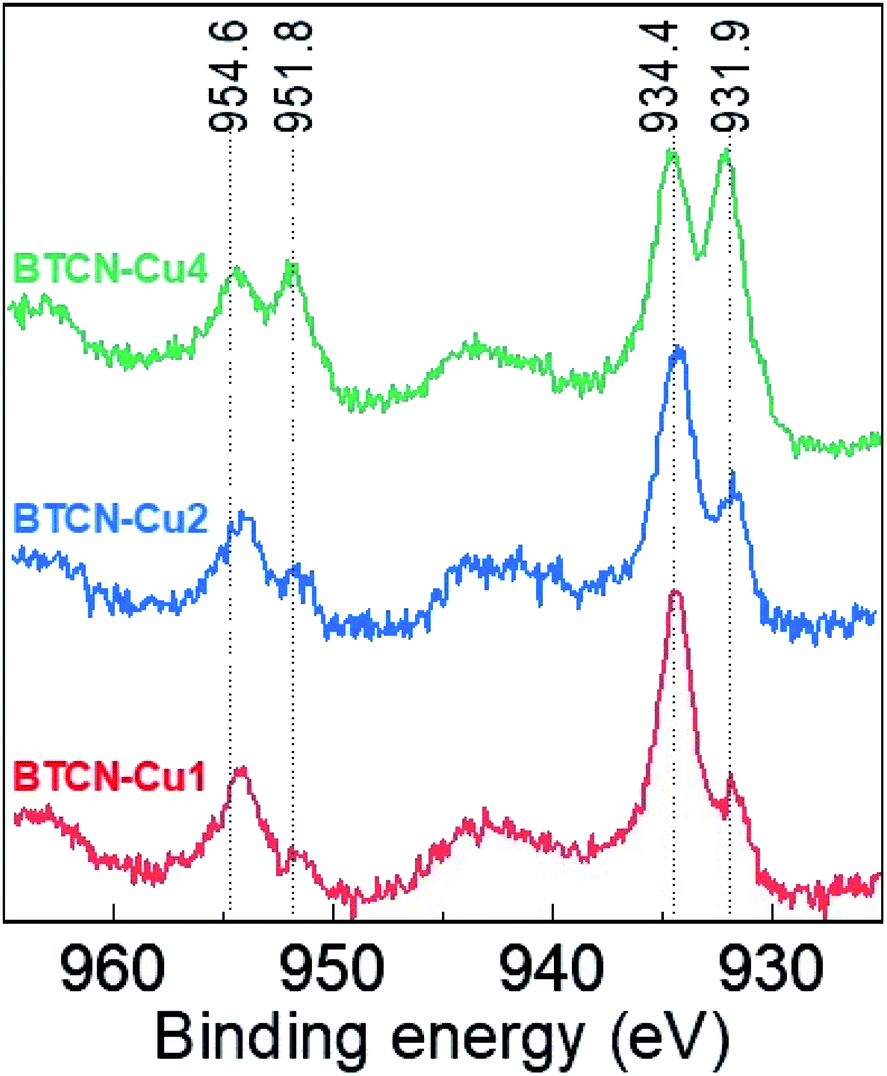 | ||
| Fig. 4 Cu 2p XPS spectra of BTCN–Cu1, BTCN–Cu2, and BTCN–Cu4. | ||
Electrocatalytic activity
The electrocatalytic activity of BTCN–Cu0, BTCN–Cu1, BTCN–Cu2, and BTCN–Cu4 was evaluated in a three-electrode setup. The three-electrode setup consisted of a Pt wire as the counter electrode, Ag/AgCl(sat. KCl) as the reference electrode, and a rotating disk electrode (RDE) with a glassy carbon tip coated with the as prepared catalyst as the working electrode. For comparison purposes, the benchmark catalysts based on Pt nanoparticles supported on active carbon (Pt/C) (carbon/Pt 20%) were also measured under similar conditions to the BTCN–CuX materials. Moreover, to disregard the effect of support oxidation during copper loading, a sample was prepared by subjecting the carbonaceous support to a similar air treatment to BTCN–CuX samples but in the presence of no copper. The sample was named “BTCN–Cu0 350 °C air”. Cyclic voltammetry (CV) curves (Fig. S6†) recorded in N2 saturated 0.1 M KOH solution showed a rectangular shape. Alternatively, a reduction peak can be observed for all samples in O2 saturated 0.1 M KOH solution, confirming the catalytic activity of these samples for the ORR. Linear sweep voltammetry (LSV) curves of the different samples and Pt/C in O2 saturated electrolyte were measured with a scan rate of 10 mV s−1 and a rotation rate of 1600 rpm (Fig. 5A). The LSV curves of all BTCN–CuX materials show higher onset potential than that of Pt/C although larger limiting current densities can be reached.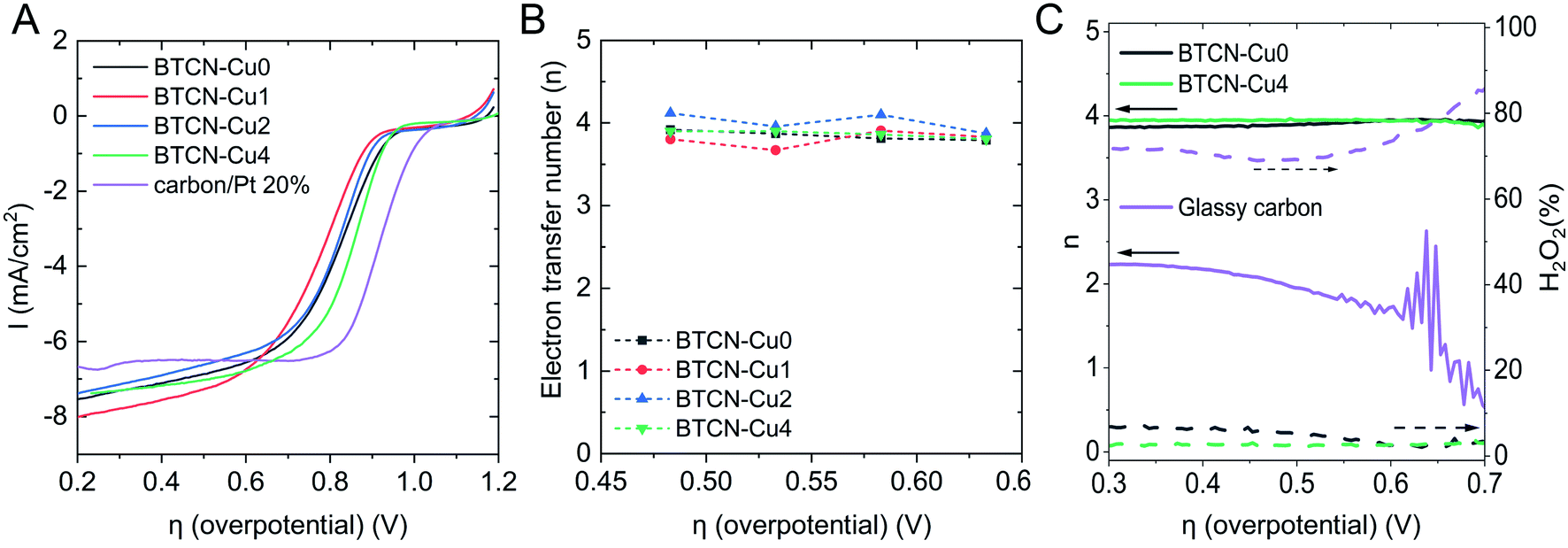 | ||
| Fig. 5 (A) LSV curves with a rotation speed of 1600 rpm and a step size of 10 mV in O2 saturated 0.1 M KOH solution. (B) Number of transferred electrons (n) obtained using the Koutecký–Levich method at different potentials. (C) RRDE BTCN–Cu0, BTCN–Cu4 and glassy carbon ORR electron transfer number (lines) and H2O2 yields (dashed lines) in O2 saturated 0.1 M KOH solution with a scan rate of 10 mV s−1 and at a stirring rate of 1000 rpm with a ring potential of 0.3 V vs. Ag/AgCl. | ||
Interestingly, the half-wave potential (E1/2) decreased from 0.83 V to 0.78 V with the initial Cu loading BTCN–Cu1, while this value increases up to 0.86 V for BTCN–Cu4, this value being very close to the benchmark Pt/C catalysts (0.92 V). The same trend can be seen for the onset potentials (see Table 2), decreasing in BTCN–Cu1 from 0.93 V to 0.81 V, while increasing to 0.91 V and 0.94 V for BTCN–Cu2 and BTCN–Cu4, respectively. Additional LSV curves were measured with different rotation rates (Fig. S7†) to calculate the electron transfer number (n) using the Koutecký–Levich equation. This gave an n of ca. 4 for all materials (Fig. 5B). Further evidence of the 4-electron transfer mechanism, without H2O2 production, was obtained by performing the ORR with a rotating ring disc electrode (RRDE). The obtained electron transfer numbers for BTCN–Cu0, BTCN–Cu1, BTCN–Cu2, BTCN–Cu4, and glassy carbon without the catalyst are shown together with the hydrogen peroxide yield obtained from the ring and disc currents in Fig. 5C and S8.† The results show almost no H2O2 evolution and confirmed the 4-electron transfer. In comparison, glassy carbon shows a H2O2 production of 70–90%, and an electron transfer number of 2. On the other hand, the comparison of the results of the BTCN–CuX series and BTCN–Cu0 350 °C air shows that the improved ORR performance is caused by the copper loading and not by oxidation of the material during heat treatment. For instance, even though BTCN–Cu0 350 °C is able to reduce oxygen through the 4-electron transfer mechanism, it shows lower Eonset, E1/2 and JL than the rest of the samples.
| Sample | η Onset | η 1/2 | J L | Tafel slope |
|---|---|---|---|---|
| V | V | mA cm−2 | mV dec−1 | |
| a Obtained at 0.5 V. | ||||
| BTCN–Cu0 | 0.93 | 0.83 | −7.0 | 131 |
| BTCN–Cu1 | 0.81 | 0.78 | −7.4 | 128 |
| BTCN–Cu2 | 0.91 | 0.83 | −5.1 | 99 |
| BTCN–Cu4 | 0.94 | 0.86 | −7.0 | 87 |
| Carbon/Pt 20% | 1.00 | 0.92 | −6.5 | 113 |
Moreover, the Tafel slope (Fig. 6A) decreases upon Cu loading from 131 mV dec−1 to 128 mV dec−1 for BTCN–Cu0 and BTCN–Cu1, respectively. With increasing Cu content, the Tafel slope further decreases from 99 mV dec−1 to 87 mV dec−1 for BTCN–Cu2 and BTCN–Cu4, respectively. This value is much lower than that obtained for the Pt-based benchmark catalyst used as the reference (i.e. 113 mV dec−1), indicating that the ORR occurs faster with BTCN–Cu4. According to XPS, sample BTCN–Cu4 shows the largest amount of CuI, pointing out CuI as the active sites for the ORR. Interestingly, Chidsey, Stack, and co-workers reported in 2007 that CuI centers, in Cu complexes using substituted 1,10-phenanthroline ligands, were responsible for the adsorption of O2 as well as the reduction from CuII to CuI in these complexes as the limiting step for the ORR.36 Later, in 2011, Langerman and Hetterscheid reported similar results using the Cu(II) tris(2-pyridylmethyl)amine complex where the rate limiting step was the coordination of O2 to CuI.37 Thus, we hypothesize here that the N-doped carbon stabilized CuI centres on the surface of the materials facilitate the adsorption of O2 and, thus, foster a faster ORR on BTCN–Cu4, which accounts for the highest CuI content. These CuI centres can, thus, be assigned as the catalytically active sites for this reaction under these conditions.
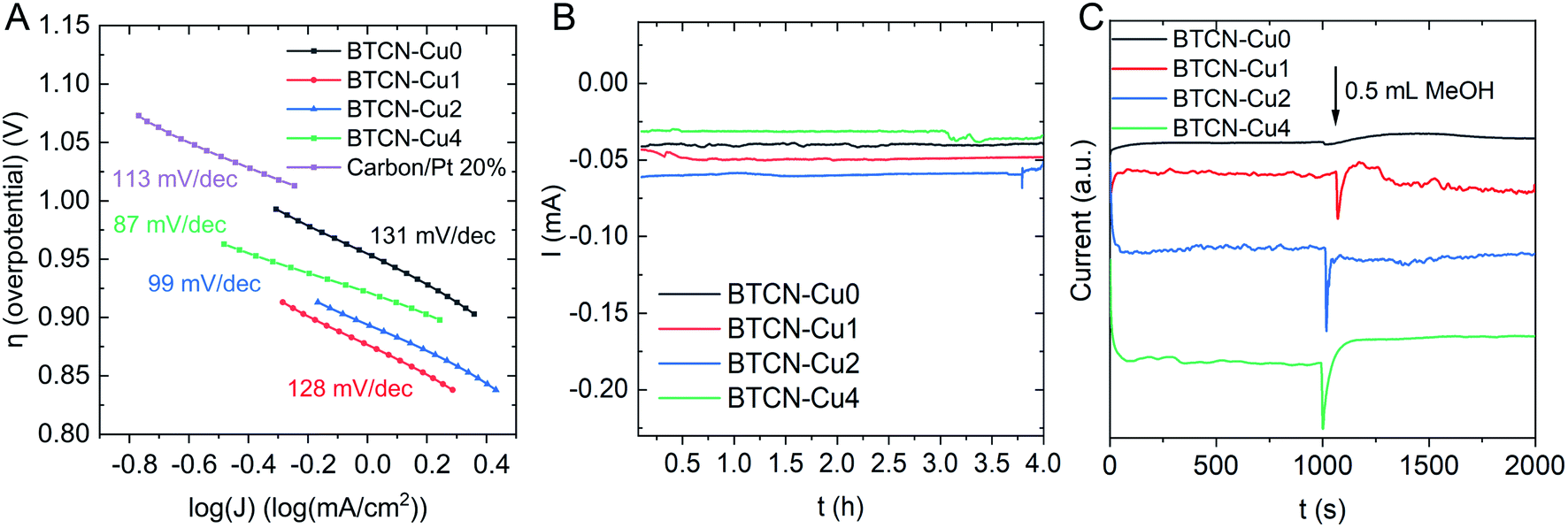 | ||
| Fig. 6 (A) Calculated Tafel slopes of BTCN–Cu0, BTCN–Cu1, BTCN–Cu2, BTCN–Cu4 and Pt/C, (B) chronoamperometry curves at 0.15 V vs. Ag/AgCl (sat. KCl) over 4 hours and (C) chronoamperometry curves at 0.15 V vs. Ag/AgCl (sat. KCl) after the injection of 0.5 mL MeOH. | ||
To evaluate the durability of the catalysts, chronoamperometric measurements over 4 hours were performed (Fig. 6B), showing a stable current for all samples when a potential of 0.15 V vs. Ag/AgCl was applied. ICP measurements showed Cu leaching below 0.03 wt% for all the samples. In addition, the stability of the materials towards methanol crossover was tested by injecting 0.5 mL MeOH after 1000 s in chronoamperometric measurements, applying a potential of 0.15 V. As can be seen in Fig. 6C, after the addition of MeOH (that provokes a peak due to electrolyte agitation), the reducing current was recovered ca. 50 s after MeOH addition for all materials, indicating their good stability towards MeOH crossover.
The significantly lower Tafel slope, large stability, and activity of BTCN–Cu4 compared to those of the Pt-based benchmark catalyst make it a very promising electrocatalyst for the ORR. In Table S3,† a comparison of the analysed parameters with other Cu-based materials reported in the literature can be found. As shown in the table, the values presented here are also among the best reported in the literature. Thus, we believe that the simple and cost-efficient synthesis of CuII/CuI decorated N-doped carbonaceous materials holds great potential for the new generation of ORR cathode electrocatalysts.
Conclusions
In this paper, a simple and scalable synthesis of an N-doped carbonaceous material decorated with CuII/CuI nanoclusters is described. Samples comprising 0, 1, 2, and 4 wt% Cu loading were analyzed, and their performance as ORR electrocatalysts was discussed according to their composition and morphology. HAADF-STEM images show that Cu is homogeneously distributed as sub-nanometric clusters or even single atoms. XPS shows that the composition of these sites changes from 3.5 to 1 CuII/CuI when the Cu loading increases from 1 wt% to 4 wt%. The ORR results show that BTCN–Cu4 exhibits a larger limiting current and faster reaction rate, according to the Tafel slopes, pointing at CuI species as the active sites, facilitating O2 binding during the ORR. Additionally, the material does not show current loss within 4 hours and a remarkable MeOH stability. Further studies are currently underway to investigate the suitability of these catalysts in fuel cells; however, we believe this work sets the precedent for mass-production of Cu site-heterogeneous catalysts matching the performance of Cu complexes, whose synthesis involves multiple synthetic and purification steps, and therefore, they are difficult to scale up.Author contributions
M. L. O. S.-M. prepared the catalysts. J. K. prepared the catalysts, characterized the materials, performed the electrochemical tests, and wrote the draft. H. Z. and N. T. performed the TEM analysis. The work was supervised by M. A., J. A. and N. L.-S. J. A. and N. L.-S. conceptualized the project. N. L.-S. managed it. The manuscript was revised by all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We want to gratefully thank Antje Voelkel for measuring TGA and Heike Runge and Bolortuya Badamdorj for the microscopy work and the Max Planck Society is gratefully acknowledged for financial support. Open Access funding provided by the Max Planck Society.Notes and references
- D. Wang, C. Ao, X. Liu, S. Fang, Y. Lin, W. Liu, W. Zhang, X. Zheng, L. Zhang and T. Yao, ACS Appl. Energy Mater., 2019, 2, 6497–6504 CrossRef CAS.
- L. Cui, L. Cui, Z. Li, J. Zhang, H. Wang, S. Lu and Y. Xiang, J. Mater. Chem. A, 2019, 7, 16690–16695 RSC.
- F. Li, G.-F. Han, H.-J. Noh, S.-J. Kim, Y. Lu, H. Y. Jeong, Z. Fu and J.-B. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- J. Greeley, I. Stephens, A. Bondarenko, T. P. Johansson, H. A. Hansen, T. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef CAS.
- S. Mondal and C. R. Raj, J. Phys. Chem. C, 2018, 122, 18468–18475 CrossRef CAS.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef CAS.
- H. Yang, Z. Li, S. Kou, G. Lu and Z. Liu, Appl. Catal., B, 2020, 119270 CrossRef CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang and D. Wang, Angew. Chem., 2017, 129, 7041–7045 CrossRef.
- G. Wan, C. Yang, W. Zhao, Q. Li, N. Wang, T. Li, H. Zhou, H. Chen and J. Shi, Adv. Mater., 2017, 29, 1703436 CrossRef.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. T. Goh, T. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- E. Chen, M. Bevilacqua, C. Tavagnacco, T. Montini, C.-M. Yang and P. Fornasiero, Catal. Today, 2020, 356, 132–140 CrossRef CAS.
- C. Zhu, Q. Shi, B. Z. Xu, S. Fu, G. Wan, C. Yang, S. Yao, J. Song, H. Zhou, D. Du, S. P. Beckman, D. Su and Y. Lin, Adv. Energy Mater., 2018, 8, 1801956 CrossRef.
- G. Han, Y. Zheng, X. Zhang, Z. Wang, Y. Gong, C. Du, M. N. Banis, Y.-M. Yiu, T.-K. Sham, L. Gu, Y. Sun, Y. Wang, J. Wang, Y. Gao, G. Yin and X. Sun, Nano Energy, 2019, 66, 104088 CrossRef CAS.
- K. Iwase, T. Yoshioka, S. Nakanishi, K. Hashimoto and K. Kamiya, Angew. Chem., Int. Ed., 2015, 54, 11068–11072 CrossRef CAS.
- H. Wu, H. Li, X. Zhao, Q. Liu, J. Wang, J. Xiao, S. Xie, R. Si, F. Yang, S. Miao, X. Guo, G. Wang and X. Bao, Energy Environ. Sci., 2016, 9, 3736–3745 RSC.
- A. Bhagi-Damodaran, M. A. Michael, Q. Zhu, J. Reed, B. A. Sandoval, E. N. Mirts, S. Chakraborty, P. Moënne-Loccoz, Y. Zhang and Y. Lu, Nat. Chem., 2017, 9, 257–263 CrossRef CAS.
- Y. M. Zhao, G. Q. Yu, F. F. Wang, P. J. Wei and J. G. Liu, Chemistry, 2019, 25, 3726–3739 CrossRef CAS.
- Z. Jiang, W. Sun, H. Shang, W. Chen, T. Sun, H. Li, J. Dong, J. Zhou, Z. Li, Y. Wang, R. Cao, R. Sarangi, Z. Yang, D. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2019, 12, 3508–3514 RSC.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS.
- W. He, C. Jiang, J. Wang and L. Lu, Angew. Chem., 2014, 126, 9657–9661 CrossRef.
- S. Zhang, K. Dokko and M. Watanabe, Mater. Horiz., 2015, 2, 168–197 RSC.
- X. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237–8265 RSC.
- N. Fechler, T. P. Fellinger and M. Antonietti, Adv. Mater., 2013, 25, 75–79 CrossRef CAS PubMed.
- M. Antonietti and M. Oschatz, Adv. Mater., 2018, 30, 1706836 CrossRef PubMed.
- J. Yuan, C. Giordano and M. Antonietti, Chem. Mater., 2010, 22, 5003–5012 CrossRef CAS.
- J. S. Lee, X. Wang, H. Luo, G. A. Baker and S. Dai, J. Am. Chem. Soc., 2009, 131, 4596–4597 CrossRef CAS.
- D. Li, G. Wang, L. Cheng, C. Wang and X. Mei, ACS Omega, 2018, 3, 14755–14765 CrossRef CAS PubMed.
- T. Ivanova, K. Maslakov, A. Sidorov, M. Kiskin, R. Linko, S. Savilov, V. Lunin and I. Eremenko, J. Electron Spectrosc. Relat. Phenom., 2020, 238, 146878 CrossRef CAS.
- G. Speier, Z. Tyeklar, P. Toth, E. Speier, S. Tisza, A. Rockenbauer, A. Whalen, N. Alkire and C. Pierpont, Inorg. Chem., 2001, 40, 5653–5659 CrossRef CAS.
- S. Haaf, E. Kaifer, H. Wadepohl and H.-J. Himmel, Chem.–Eur. J., 2021, 27, 959–970 CrossRef CAS.
- C. C. L. McCrory, X. Ottenwaelder, T. D. P. Stack and C. E. D. Chidsey, J. Phys. Chem. A, 2007, 111, 12641–12650 CrossRef CAS.
- M. Langerman and D. G. H. Hetterscheid, Angew. Chem., Int. Ed., 2019, 58, 12974–12978 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta09459a |
| This journal is © The Royal Society of Chemistry 2022 |