
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrospinning based on benign solvents: current definitions, implications and strategies
Joshua
Avossa†
 ,
Gordon
Herwig†
,
Claudio
Toncelli
,
Fabian
Itel
* and
René Michel
Rossi
*
,
Gordon
Herwig†
,
Claudio
Toncelli
,
Fabian
Itel
* and
René Michel
Rossi
*
Laboratory for Biomimetic Membranes and Textiles, Empa, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, CH-9014 St. Gallen, Switzerland. E-mail: Fabian.Itel@empa.ch; Rene.Rossi@empa.ch
First published on 7th March 2022
Abstract
Electrospinning evolved as a powerful technology to produce membranes consisting of nano- to micron-scaled fibers offering unique membrane properties due to the extremely high surface-to-volume ratio and high membrane porosity. However, this technology is often far removed from a green and environmentally-friendly process, as it employs solvents with issues such as high flammability, toxicity, difficult disposal, or energy-intensive synthesis. Indeed, the most commonly used solvents in the electrospinning field are halogenated (e.g. chloroform, trifluoroethanol) and toxic solvents (e.g. dimethylformamide), the use of which is now restricted by the Chemical Control Regulation in the European Union (REACH). This is especially important when considering the commercialization of electrospun products due to the more limited solvent choice. In order to render electrospinning more attractive as a commercial technique and further improve scalability, solvent alternatives and other green routes have to be established, which adhere to societal and legal restrictions especially in regard to the environmental and health impact. Therefore, this review provides the current ecological and economical context of this technology, and summarizes recent approaches towards green electrospinning. Since electrospinning from solutions and dispersions constitutes the predominantly used form of this technique both in research and industry, the focus of this work is set on the use of solvents or solvent mixtures classified exclusively by the term “green”. The approaches presented comprehensively cover the production of polymeric fibers via solution, emulsion, suspension, and in situ cross-linking electrospinning.
 Joshua Avossa | Joshua Avossa received his Bachelor and Master degree in Industrial Chemistry from the University of Naples Federico II in 2011 and 2014, respectively. Subsequently, he obtained his PhD from the Department of Chemical, Materials and Production Engineering, University of Naples Federico II, in 2018. In 2020, he joined Empa Laboratories as postdoc under supervision of Prof. Dr René Rossi in the Department of Biomimetic Membranes and Textiles working on the development of nanofiber membranes for transparent face masks using pilot-scale needleless electrospinning, biodegradable electrospun fibers using green solvents, and hydrogel-based composite materials for the development of skin models. |
 Gordon Herwig | Gordon Herwig conducted his doctoral research on 3D-printable biodegradable polymers with support by The Leverhulme Trust at the University of Warwick, UK. After receiving his PhD in 2019, he joined the Empa Laboratories in St. Gallen under supervision of Prof. Dr René M. Rossi, where he leads the research within industrial projects, employing electrospinning and coating strategies for functional textiles, controlled release and particle filtration applications. His research focus lies on functional nanofiber membranes produced by pilot-scale needleless electrospinning, using biocompatible materials, sustainable solvents and functional additives. |
 Fabian Itel | Fabian Itel studied Nanoscience at the University of Basel, Switzerland, and obtained his PhD in 2015. From 2016–2018, he did a postdoctoral stay at the Interdisciplinary Nanoscience Center (iNANO) at the University of Aarhus, Denmark, and then joined Empa Laboratories in 2019 with a Marie Skłodowska-Curie Cofund scholarship under supervision of Prof. Dr René Rossi. His current research interests include the generation of functional hybrid hydrogel/electrospun fiber scaffolds for applications in drug delivery, sensors, and tissue engineering. |
 René Michel Rossi | René M. Rossi studied applied physics at the University of Neuchâtel and obtained his PhD at ETH Zurich. Since 2003, he is leading the Laboratory for Biomimetic Membranes and Textiles at Empa, developing novel smart fibers, textiles and membranes for body monitoring, drug delivery and tissue engineering applications. A special focus is to develop physical and numerical skin/body models for the analysis of the interactions between materials and the skin. René Rossi is adjunct professor at the Department of Health Sciences and Technology at ETH Zurich and invited professor at the University of Haute-Alsace in Mulhouse/France. |
1. Introduction
Electrospinning (ES) has developed into a well-known, versatile technique for the preparation of polymeric non-woven fibers. The range of applications for electrospun fibers is broad: particle filtration from liquid or gas,1 drug-delivery,2 tissue engineering,3 functional wound dressings,4 light-weight battery membranes,5 customizable sensors,6 sound absorption,7,8etc. The success of the ES technique originates in the possibility to generate fibers from the micro- down to the lower nano-meter range by tailoring fiber diameter, surface area, porosity and other fiber surface properties, offering a very high surface-to-volume ratio superior to common textile fiber production techniques.9 Furthermore, the obtained nanofiber membranes possess very high air and liquid permeability, while commonly enabling diffusion of solutes and cells, which are requirements for the majority of their current applications. This versatility is further extended by the range of different materials that can be electrospun, including biomaterials and tailored synthetic polymers. In contrast, ES of polymer solutions is limited by the use of a solvent or solvent mixtures. Standard ES compositions often include halogenated compounds due to their high solvation power for hydrophobic polymers and low boiling points. These types of solvents have caused more and more concern from a scientific, public, and industrial point of view mainly due to their high toxicity and high environmental impact. Additionally, halogenated compounds recently became strongly regulated, for example by the Chemical Control Regulation in the European Union (REACH).10 Regarding the commercialization of products using the ES technique, many companies hesitate to implement this method into their production line since it implies large know-how on solvent handling and their proper disposal, which is also extremely cost-intensive. For industrial-scale production, ES involves the use of large volumes of solvents requiring recycling or waste management facilities. This is mainly due to relatively low polymer concentrations in the spinning solutions needed to obtain a defined viscosity of the commonly used high molecular weight polymers.Generally, solvent-free fiber manufacturing techniques, such as melt-ES,11,12 as well as the industrially well-established melt blowing13,14 and melt spinning,15 offer the greenest options to pursue. However, such techniques require high temperatures to melt the polymers, that can lead to chemical degradation and may not be suitable for the processing of bioactive molecules.16,17 Furthermore, solvent-free techniques result in fibers with usually larger diameters than their solution electrospun counterparts, but under certain circumstances, also nanofibers can be obtained.18 Melt ES in particular combines fiber diameters in the nanometer range and a solvent-free methodology, while also offering spatial precision for nano-patterning; unfortunately it is significantly less accessible.19 As this review focusses on recent accomplishments in improving the greenness of the classical solvent-based ES technique, the vast field of solvent-free ES falls beyond its scope.
Regarding classical solvent-based ES, many studies in literature have shown that it is possible to spin almost all commonly employed polymers with green solvents rather than the generally used toxic solvents. However, the significantly lower number of published articles using greener alternatives decreases their visibility dramatically. In order to emphasize greener alternatives of the ES technique with the goal to reduce the environmental impact, we highlight the implications and strategies arising from using more environmentally friendly solvents for the production of electrospun fibers.
First, we briefly highlight the latest methods and concepts behind the definition of “greenness” regarding solvent choice, followed by a comprehensive overview over recent reports of electrospun fibers generated by the most commonly used water-insoluble polymers, based entirely on green solvents. In the subsequent chapter, water-based ES techniques as the greenest options are presented, based on versatile dispersion ES methodologies to further extend the available range of polymers and combinations with active compounds in green ES. Lastly, reactive ES is introduced, which aims at enhancing the chemical inertness and mechanical stability of weak, water-soluble polymers by performing cross-linking steps during (in situ) or after (post) fiber generation for support structures such as used in tissue engineering. The final conclusion discusses limitations of green ES and offers a future perspective.
2. Electrospinning with green solvents
The solvent choice probably represents the most important parameter in ES, since only a limited range of solvents and compositions can dissolve the respective polymers and subsequently achieve fiber formation via ES. However, many polymers are generally difficult to dissolve and, therefore, require solvents with high solvation properties. These solvents are usually toxic (e.g. halogenated solvents), difficult to recycle and with high environmental impact (e.g. ionic liquids20). Consequently, the solvent choice has a tremendous effect on the environmental impact, safety and overall sustainability of the ES process.2.1 Definition of green solvents
The definition of green solvents expresses the goal to minimize the environmental impact resulting from the chemical production until the disposal of the solvents. The simple question “what is a green solvent?”21 cannot be answered so easily, as it requires many criteria to be considered. Fischer et al. addressed this question by assessing the substance-specific hazards by quantifying the emissions and resources used over the life cycle of the solvent. For each solvent, the EHS (environmental, health, and safety) indicator was calculated by adding the scores of water hazard, persistency, chronic toxicity, irritation, acute toxicity, reactivity, fire-, and explosion-hazard and release potential that characterize each compound. On the other hand, the life-cycle assessment (LCA) method quantifies the cumulative energy demand (CED) taking into account the production, usage, potential recycling, and disposal of the solvent. Taken together, CED and EHS indicators provide an estimation of how green a solvent is, where the lower the score, the greener or less problematic the solvent is (Fig. 1).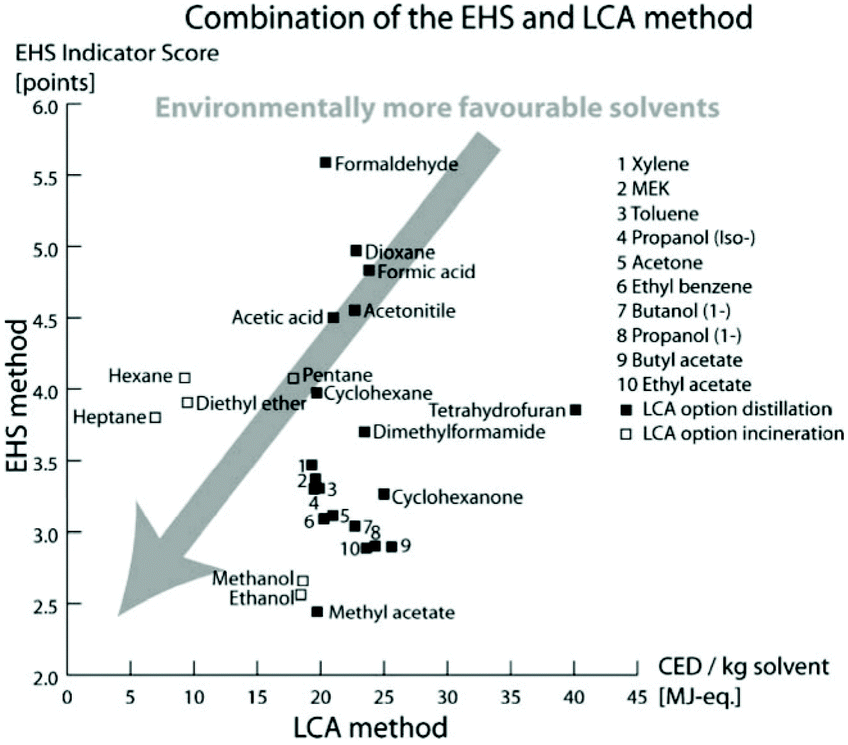 | ||
| Fig. 1 Classification of 26 solvents by EHS and LCA methods representing the greenest solvents with the lowest scores. Reproduced from ref. 21 with permission from the Royal Society of Chemistry. | ||
Nevertheless, both the EHS indicator and CED are only estimations and do not fully reflect objective values. Indeed, a solvent can be synthesized from different commodities or raw materials and can be disposed of or recycled in different ways. Therefore, the energy demand through these different steps can vary significantly, resulting in different CED values. Also, the EHS indicator can change if new insights into the EHS impact of the solvent are revealed. Indeed more recently, new regulations and restrictions entered into force: the Global Harmonized System (GHS) as applied by the European Classification, and the Labelling and Packaging (CLP) regulation.22,23 Therefore, this method requires the adjustment of EHS indicators in light of the introduction of the new regulations and restrictions.
Slater and Savelski from Rowan University developed a similar method for evaluating the greenness of a solvent.24 Compared to the EHS method, it takes into account occupational health and environmental hazards (threshold limit value, biodegradation, ingestion toxicity, carcinogenicity, half-time, ozone depletion, global warming potential, smog formation, acidification, soil adsorption coefficient, bio concentration factor), as well as the amount of solvent used in a process. However, peroxide formation, flash point, and CED of the solvent were not used as selection parameters. Since the definition of “green” depends on the method and its solvent score, the threshold value defining the solvent “greenness” is not always defined. For instance, ethanol (EtOH) is considered green for both EHS and LCA methods, with both having low scores. Whereas the approach developed by Slater and Savelski can make a distinction between the “greenness” of the hydrocarbons, the EHS method considers the hydrocarbons nearly all as green.23
In 2015, a public-private partnership European consortium comprised of pharmaceutical companies, universities, and small-medium enterprises, called the Innovative Medicines Initiative (IMI)-CHEM21, developed a ranking of the most common solvents to compare the “greenness” of processes or syntheses.25,26 Each solvent was classified according to benchmarks of existing guides, which were combined to limit their number to three safety, health and environmental criteria, each represented with scores from 1 to 10 and a color code (green for 1–3, yellow for 4–6, and red for 7–10). The final ranking of the solvent (recommended, problematic and hazardous) is usually derived from its least green characteristic, i.e. not from an average or summation of all properties, as performed in the EHS/LCA or Slater and Savelski methods. Moreover, bio-derived solvents (derived from sustainable and renewable resources) were evaluated accordingly, obtaining a final ranking. However, the environmental scoring of this method lacks the CED from LCA assessment or the carbon footprint that depends on the production process and usage of the solvent determined case by case by experts.
In the pharmaceutical sector, companies have focused on the safety and the health- and environmental-impact related to the practical use of the solvent. For instance, Pfizer developed a color-coded list of solvents using green, yellow, and red as flags for “Preferred”, “Usable” and “Undesirable”, respectively.27 GlaxoSmithKline (GSK)28,29 and Sanofi30 also used a visual “traffic light” code with tables displaying a list of solvents (Fig. 2). According to the solvent tables provided and in agreement with the EHS and Slater/Savelsky methods, the greenest solvents are water; C1–C4 alcohols, acids, ketones and esters; as well as 2-methyl tetrahydrofuran (2-MeTHF), dimethyl sulfoxide (DMSO), acetonitrile and dimethylpropylene urea. Conclusions can also be drawn regarding the least green solvents, considered as undesirable if not already banned by REACH: chloroform, 1,2-dichloroethane, carbon tetrachloride, N-methyl pyrrolidone, dimethylformamide (DMF), dimethylacetamide (DMAc), benzene, hexane, 1,4-dioxane, 1,2-dimethoxyethane, diethyl ether, and 2-methoxyethanol.31 In 2016, GSK developed an updated solvent guide version29 that retains the overall approach (color-code), but with additional 44 solvents since the last GSK guidance publication25 updating the data behind all scores. All the solvents in the guide are scored assigning a value from 1 (the worst) to 10 (the best) to the following categories: incineration, recycling, biotreatment, and volatile organic compound (VOC) emissions related to the “waste” area; environmental air impact and aqueous impact; potential of exposure and heath hazard; flammability & explosion potential, and reactivity & stability are related to the “safety” area; life cycle assessment. For each general area of assessment (waste, environment, health, and safety), an overall summary score (called composite score) is defined as the geometric mean of each of the relevant category scores:
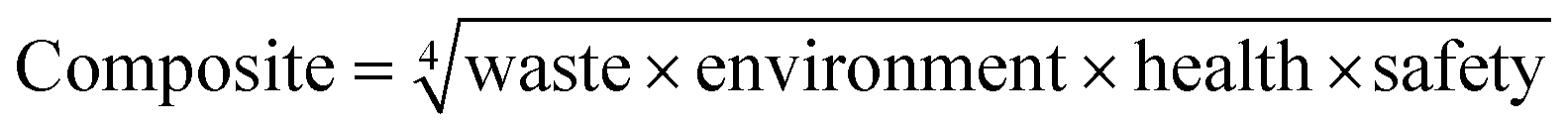
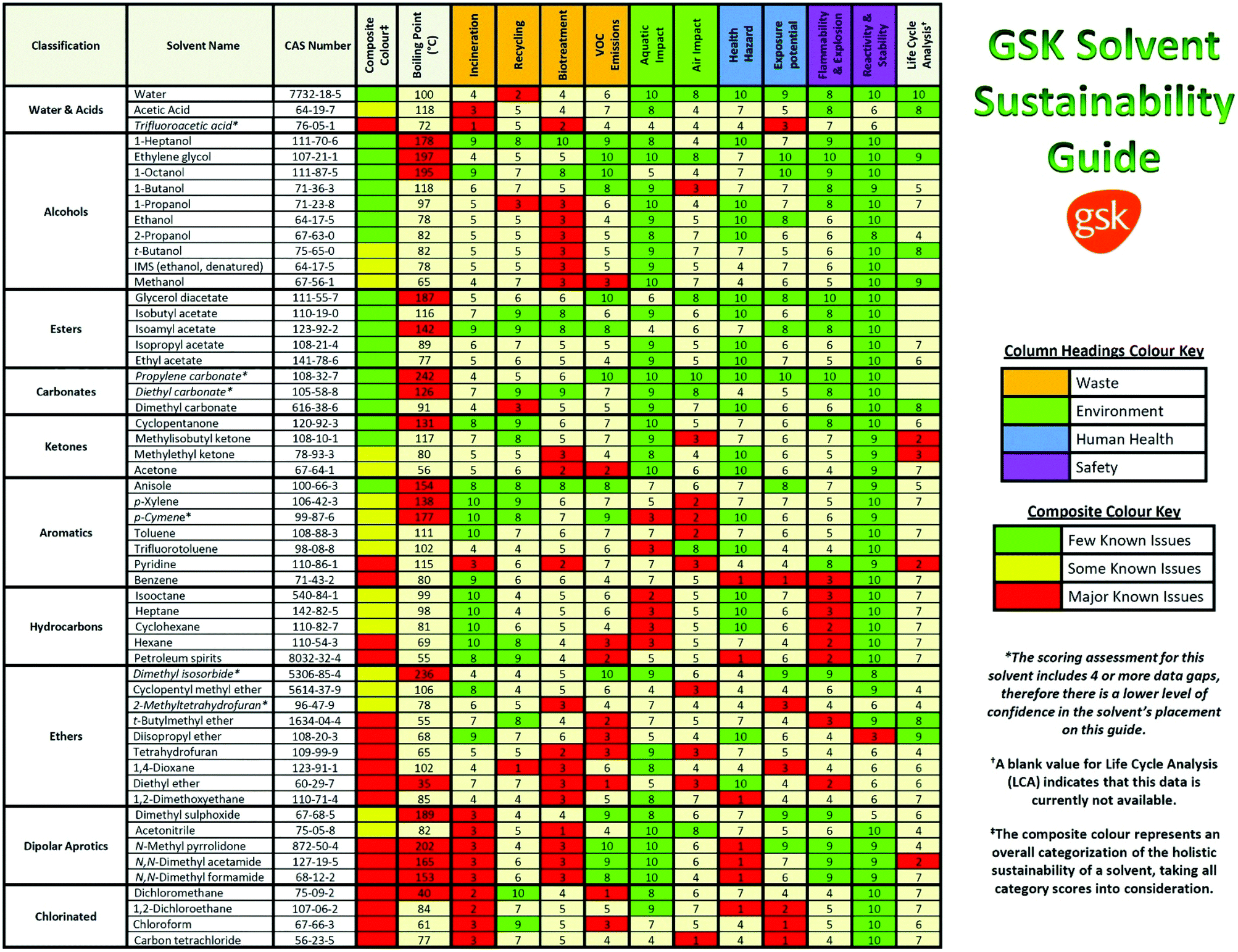 | ||
| Fig. 2 The GSK solvent sustainability selection guide showing the 55 most commonly used solvents scored and color-coded based on their greenness. In the ESI of ref. 29 is reported a chart classifying 154 solvents. Reproduced from ref. 29 with permission from the Royal Society of Chemistry. | ||
The color assignment of each category is based on the score: green (x > 7.5), amber (3.5 ≥ x < 7.5), and red (x < 3.5). Instead, the composite color assignment is made by taking into account possible data gaps in the classification and giving more importance to 4 out of 10 categories (i.e. VOC emissions, health hazard, flammability and explosion potential, and reactivity and stability) following a decision tree assignment diagram.29
According to the GSK classification (Fig. 2), anisole is the greenest aromatic solvent; DMSO and dimethylpropylene urea are the greenest among the dipolar aprotic solvents (with only some issues related to the “waste” category); formic acid (FA) and acetic acid (AcOH) can be considered green with some issues related to the fact that they are acids; ketones and alcohols can be more or less considered green; and all the halogenated compounds are considered not green.29
In 2018, Clarke et al. published a review focusing on ionic liquids, deep eutectic solvents, supercritical fluids, switchable solvents, liquid polymers, and renewable solvents.32 Plant biomass is considered a source of renewable solvents: crops such as corn and sugar cane, agricultural residues, and agroforestry products are all considered potential raw materials for the generation of fuels and solvents.33 However, the food versus fuel debate limits or hinders the use of potential food as raw material for chemical production.34 The reason why biofuel/chemical production from plants remains controversial lies on the competition between food and fuel production on limited resources (land, water, labor, and capital),35 since both the food and biofuel originate from crops.36 Instead, chemical production using agricultural residues, grasses, and agroforestry products (lignocellulosic biomass) or waste in general, provides a renewable resource without competing directly with food. However, processing of biomass waste requires usually more energy and cost. Glucose, for example, can be obtained from the hydrolysis of starch and cellulose as renewable sources that could be extracted from wood pulp (non-food source). Subsequently, transformations of glucose can provide bioderived solvents such as limonene, γ-valerolactone, and 2-methyl tetrahydrofuran (2-MeTHF). The latter is now being used as a renewable alternative to THF showing lower toxicity.37 Moreover, 2-MeTHF can be obtained from furfural, which may be derived from agricultural waste.38 Nevertheless, the CHEM21 methodology categorized this solvent as “problematic” because of its high flammability.25,26 Limonene (both enantiomers) is considered green with some issues (color code amber) according to the GSK chart29 due to the low score in the “environmental” category. Meanwhile, γ-valerolactone has better scores compared to both limonene and 2-MeTHF. Nevertheless, there is a lack of data, especially in the “environmental” and “health” categories, thus score values have been estimated via calculation or comparison to structurally similar solvents.
2.2 Polymers electrospun with green solvents
Many reports on the ES of polymers from single polymer solutions using solely green solvents are already available in literature, but are still a minority in the ES field. In the following section, the most common polymers used for ES with green solvents are summarized (see Table 1). ES of water-soluble polymers is not reported here, since such fibers are not stable over time because of air humidity. Therefore, a cross-linking process is needed, which is elaborated on in Section 4 “Reactive Electrospinning”.| Polymer | Solvent/solvent mixture | Electric field [kV cm−1] | Fiber morphology | Ref. |
|---|---|---|---|---|
| CA | AcOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 75:25 (w/w) :H2O 75:25 (w/w) |
1.2 | Fibers | 40 |
| AcOH:H2O 75:25 (w/w) |
1.7–2.0–2.5 | Beads on fibers | 41 | |
| Acetone | 1.7–2.0–2.5 | Cylindrical + ribbon-shaped | 41 | |
| DMSO:acetone 2:1 (v/v) |
0.73 | Fibers (650 ± 130 nm) | 43 | |
| PCL | AcOH | 0.75–1.0 | Microfibers | 48 |
| AcOH:H2O 90:10 (w/w) |
0.75–1.0 | Beads on fibers | 48 | |
| FA | 0.75–1.0 | Nanofibers (88 nm) | 48 | |
| AcOH:FA 3:1 (w/w) |
0.96 | Fibers (266 nm) | 49 | |
| Acetone | 0.36–1.0 | Fibers | 50 and 51 | |
| PLA | Acetone | 1.33 | Fibers (757 ± 275 nm) | 53 |
| PVDF | DMSO:acetone = 3: 2 (v/v) |
0.64 | Fibers (625 ± 113 nm) | 54 |
| PA6 | FA:water 85:15 (v/v) |
2.1 | Nanofibers | 55 |
| PA11 | FA:water 98:2 (v/v) |
1.5 up to 5.0 | Fibers and nanofibers | 56 |
| AcOH:FA 3:1 (v/v) |
3.3 | Fibers (124 nm) | 57 | |
| FA:Anisole 3:2 (v/v) |
4.2 | Nanofibers (53 ± 10 nm) | 58 | |
| Zein | EtOH:water 8:2 (v/v) |
1.7 | Fibers (146 ± 33 nm) with some beads | 60 |
| EtOH:water 8:2 (w/w) |
1.4 | Fibers (126 ± 31 nm) | 61 | |
| CS | AcOH:water 9:1 (w/w) |
1.1–4.0 | Fibers (140 ± 51; 130 nm) | 64 and 67 |
| PS | D-Limonene | 1 | Fibers (700 nm) | 68 |
| EtOAc | 1.0 to 3.0 | C-shaped and ribbon-like microfibers | 69 and 70 | |
| MEK | 1.0 to 3.0 | C-shaped and ribbon-like microfibers | 69 and 70 | |
| PMMA | EtOAc | 1.0–1.4–1.8 | Ribbon-like microfibers (∼6.5 μm) | 71 |
| 2-propanol:water 78:20 (w/w) |
0.59 | Fibers (770 ± 45) | 72 | |
| PAN | DMSO (at 80 °C) | 1.3 | Fibers (∼550 nm) | 73 |
| DMSO (needleless set-up) | 1.4–3.1 | Fibers | 74 and 75 |
Cellulose acetate (CA, acetyl content 39.8%, MW = 30000) was successfully electrospun using a mixture of AcOH and water by Han et al.40 Bead-free uniform nanofibers were obtained at concentrations higher than 17 wt% in an AcOH–water mixture 75:25 (w/w). Recently, Majumder et al. reported a formation of uniform fibers with some beads using the same type of CA and polymer concentration used in the work of Han.41 Majumder et al. used different types of solvent mixtures, the green ones being pure acetone, and AcOH and water (3:1 w/w) producing cylindrical/ribbon-shaped fibers and uniform fibers with some beads, respectively. The presence of beads using acetic acid and water can be ascribed by the fact that the RH during ES was likely higher than Han's work (Majumder and Han did not report RH); indeed high humidity can cause the formation of beads due to lower evaporation rates.42
Haas et al. dissolved cellulose acetate in a mixture of acetone:DMSO (2:1 v/v) and EtOH:DMSO (1:1 v/v). They showed that acetone allowed it to spin fibers without beads (18 wt% CA solution) thanks to the higher volatility compared to EtOH that caused the formation of beads. Their results on DMSO-based binary solvent mixtures revealed that the transition from spraying to spinning, starting from 12 wt%, can be shifted to lower concentrations (8–10 wt%) if EtOH is replaced by acetone.43
Reports on the electrospinning of other cellulose derivatives are beyond the scope of this review and the reader is referred to recent detailed review articles.44–47
:water, FA and FA:acetone (4:1) using different concentrations of PCL (Mw = 80 kDa).48 PCL microfibers without beads are formed using AcOH with a concentration of 15 wt%, and nanofibers with 88 ± 25 nm resulted when 20 wt% PCL solution was electrospun at 15 kV with a tip-to-collector distance of 20 cm. Using FA:acetone and 90% AcOH mixtures only beads-on-fibers were obtained.48 Schueren et al. successfully spun a 10 wt% PCL in AcOH:FA = 3:1 at a RH of 30% obtaining fibers with an average diameter of 266 ± 39 nm.49 Reneker et al. spun PCL (Mw = 80 kDa) solution of 15 wt% in acetone.50 Subsequently, Royen et al. produced fibers without beads from a solution of 15 wt% PCL (Mw = 40 kDa) in acetone with CTAB (15 mol%).51
Mosher et al. showed the production of a blend of PLGA/PCL with AcOH and non-green solvents (DMF/DCM) and compared the mechanical properties of the produced fibers between the green solvents and the traditional solvents for ES PLGA and PCL. The fibers were produced by applying a voltage of 0.67 kV cm−1 for an average fiber diameter of 1.27 ± 0.23 μm. Fibers produced with AcOH resulted in a 47% increase in ductility accompanied by a 29% reduction in elastic modulus compared with the traditional ES mixture (Fig. 3).52 Recently, PCL was also blended at different ratios together with a citric acid-based biodegradable elastomer, poly(1,8-octanediol-co-citrate) (POC), and electrospun from mixtures of AcOH and FA. They obtained defect-free nanofibers with diameters of 0.61–1.11 μm with wettability- and mechanical properties that were dependent on the PCL/POC ratio, post-crosslinking treatment, and fiber orientation.
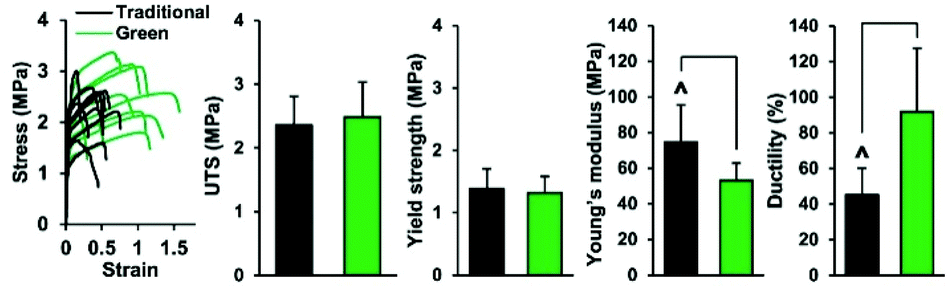 | ||
| Fig. 3 Comparison of the mechanical properties of membranes electrospun using green and traditional solvents. They performed similarly in ultimate tensile strength (UTS) and yield strength, while the elastic modulus was lower and the ductility higher for the green solvents. Reproduced from ref. 274 with permission from IOP Publishing. | ||
Finally, Casasola et al. were able to electrospin PLA in acetone (10% w/v). The PLA used was an amorphous polymer with an L-lactide content of around 88 wt%.53
:acetone mixtures. However, DMF can be replaced by DMSO, as demonstrated recently with PVDF at 10 wt% using DMSO and acetone (3:2 v/v) as a greener solvent mixture.54
:water (85:15 v/v)as solvent.55
A parametric study was also performed using 85 and 98 vol% FA as solvent. The investigated parameters were polymer grade, viscosity of the solution, salt content and solvent grade, voltage, distance to the collector, nozzle size, and feeding pressure of the solution.56
De Vrieze et al. performed a further parametric study by changing the ratio of AcOH and FA of different concentrations of PA6 of 10 kDa (from 12 up to 21 wt%). The study showed that a steady-state condition can be achieved using a solvent mixture from 33 to 50 vol% of AcOH. They also showed a reduction in the fiber diameter when the RH increased speculating that water acted as a plasticizer proven by the stable γ-crystals and a drop in the glass transition temperature.57
PA11 was successfully electrospun using a mixture of FA:anisole 60:40 (v/v) by Meireman et al. obtaining nanofibers with an average diameter of 53 ± 10 nm.58
While Zein can be spun both from EtOH or AcOH,59 8:2 EtOH:water (w/w) solutions are most commonly employed,60 as investigated comprehensively by Neo et al.61 The parameters studied were solution feed rate, applied voltage, tip-to-collector distance, and concentration. The authors reported that bead-less fibers were formed at 25, 30, and 35 wt% Zein solutions which are 2.1, 2.5, and 2.9 times the entanglement concentration of Zein, respectively. The results are in agreement with the observation of McKee et al.,62,63 who proposed that the entanglements between the polymers chains are enough to stabilize the jet and suppress the Raleigh instability, thus, promoting the formation of fibers rather than droplets during ES.
Geng et al. electrospun CS fibers with an average diameter of 130 nm from aqueous 90% AcOH solution containing 7 wt% of polymer. The authors reported that the acid content should be higher than 30%, a requirement for CS fiber formation, because more concentrated acetic acid decreased surface tension of the CS solution and increased charge density. However, an AcOH solution at ≥90% did not dissolve enough CS to reach a spinnable concentration. Instead, bead-free fibers were obtained from a molecular weight of 106 kDa, while lower or higher molecular weights (30 and 398 kDa) showed the formation of beads. Average fiber diameters and size distributions decreased with increasing electric field and more bead defects appeared at 5 kV cm−1 or more.67
Homayoni et al. used medium molecular weight CS of 1095 kDa and hydrolyzed it using NaOH with different treating times (from 0.75 to 48 h). The best result was obtained using 5wt% of the 48 h hydrolyzed CS (corresponding to a molecular weight of 294 kDa) in 90% AcOH. The hydrolyzed CS can also be spun in 80 and 70% AcOH producing bigger fibers.64 In comparison with the work of Geng,67 the applied electric field is much weaker (1.1 kV cm−1 instead of 4.0 kV cm−1) and this can explain why the fibers are bead-less even though the molecular weight is higher.
Jarusuwannapoom et al. used PS solutions (Mw = 299 kDa, Mn = 119 kDa) in MEK and EtOAc.69 EtOAc and MEK were both able to dissolve PS pellets within three and one day, respectively. Fibers and beads were observed for a 10% w/v solution of the polymer. SEM observations revealed that the fibers were c-shaped and ribbon-like. The authors stated that the occurrence of such appearance was a result of the low boiling point of the solvents (77.1 °C and 79.6 °C for EtOAc and MEK, respectively). Later, a similar paper was published by Manee-in et al.70 In both papers, the same PS was used with the same Mw and polydispersity. However, in this work, the average fiber diameter and standard deviation of the fibers obtained from 10, 20, and 30% w/v in EtOAc and MEK are reported in the function of the change of the applied voltage and tip-to-collector distance with only EtOAc and MEK. The diameters of the bead-less-fibers ranged from 3.5 to 24 μm.69,70
Chang et al. used 2-propanol:water 7.8:2 (w/w) mixture to spin PMMA with an average molecular weight of 400 kDa at a concentration of 1.5 wt%.72 Since RH was relatively high (45%), the formation of pores on the surface of the fibers was expected, but not documented due to the low SEM image resolution.
 | ||
| Fig. 4 SEM images of (a) PAN fibers electrospun at 24 °C, and (b–d) Acyclovir-loaded PAN fibers electrospun at 80 °C with different magnifications. Reproduced from ref. 73 with permission from Elsevier. | ||
 | ||
| Fig. 5 Droplets of solvent on fiber spun from PAN in DMSO solution showing phase separation of the stretched polymer (in white) and the solvent (transparent droplet). Reproduced from ref. 76 with permission from Wiley-VCH Verlag GmbH & Co. KGaA. | ||
3. Dispersion electrospinning
As biomolecules are most stable in aqueous environments, unfortunately, a variety of desirable active compounds is intolerant to even the majority of green solvents. Hence, a far safer and environmentally friendly approach can be found in choosing a predominantly aqueous ES medium; however, the selection of both water-soluble polymers and active compounds is limited. In order to bridge this gap, dispersion ES methodologies have been implemented in a compromise, which consists of either liquid-in-liquid (emulsion) or solid-in-liquid (suspension) approaches. While a comprehensive overview of the then-recent research into green ES was reported by Greiner and Agarwal in 2011,78 significant progress has been made in the last decade.3.1 Emulsion electrospinning
Emulsion ES is considered a scalable alternative to the classical blend and co-axial ES techniques, which aims at encapsulating hydrophilic and/or hydrophobic compounds within nanofibers to steer release rates (Fig. 6). Co-axial electrospinning with its channel-in-channel approach based on special needle architecture offers a stable homogeneous core–shell structure on a small scale, which is important for a range of applications as summarized elsewhere.79–81 In contrast, emulsion ES is in more demand as it provides scalability on both needle and needleless ES setups, and can be regarded as a greener alternative to encapsulate hydrophobic moieties since it allows to use cheaper hydrophilic biopolymers in water as a continuous phase while avoiding or reducing organic solvents.82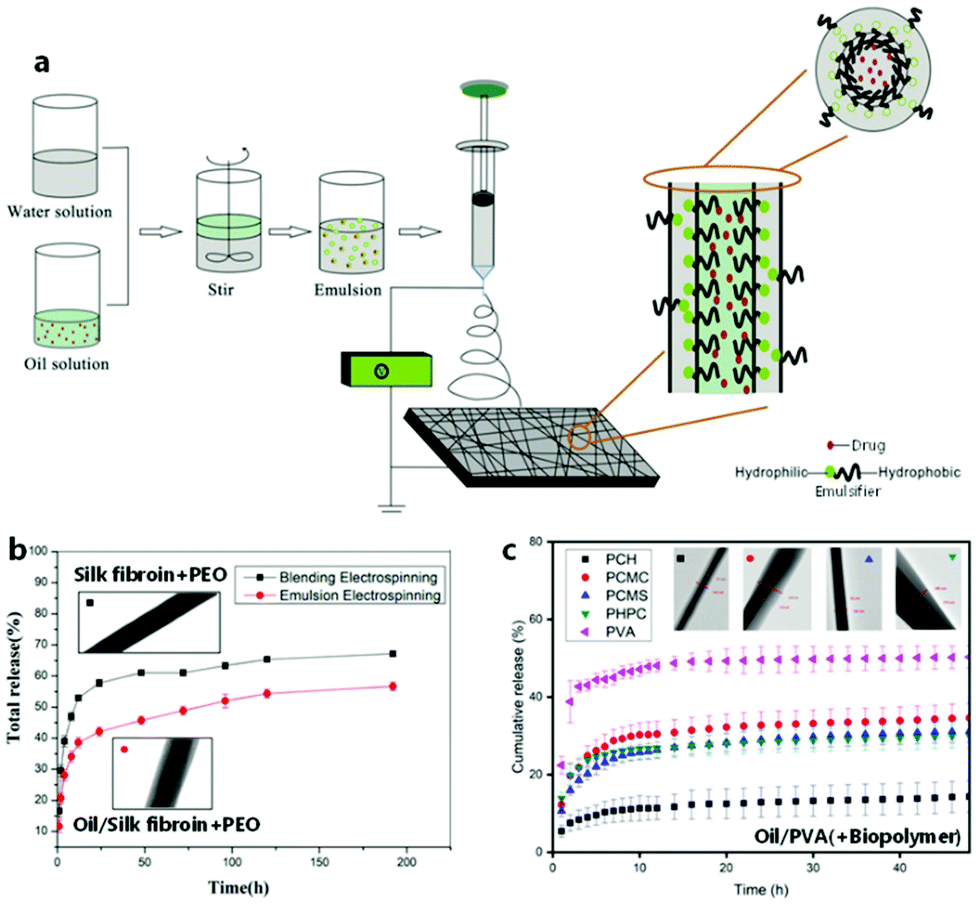 | ||
| Fig. 6 Principle and effect of electrospinning from emulsion for encapsulation purposes. (a) Schematic process of emulsion electrospinning. (b) Typical release profiles from emulsion-spun and blend-spun fibers including respective TEM images. (a and b) Adapted from ref. 85 with permission from Elsevier B.V. (c) Release profiles depending on different sheath materials including respective TEM images. Adapted from ref. 109 with permission from Informa UK Limited. | ||
An emulsion constitutes a dispersion of two immiscible liquids of which one forms the continuous phase and the other a dispersed phase. By definition, emulsions are described as water-in-oil (W/O) or oil-in-water (O/W) if the water phase is the dispersed or the continuous phase, respectively. The insoluble hydrophobic phase is commonly described as the oil phase, which includes solutions based on solvents that do not necessarily constitute a viscous “oil”. Specifically, “green” oil phases commonly consist predominantly of natural oily extracts, whereas occasionally green solvents are used to facilitate phase separation. Nevertheless, “green” O/W emulsions are by far the most commonly electrospun green compositions compared to W/O emulsions. One reason for this is the fact that only few polymeric scaffold materials are soluble in natural oils alone.
In order to prevent phase separation in W/O and O/W emulsions, emulsifiers can be used, which are typically found in surfactants (e.g. Tweens, Spans), particles (e.g. calcium carbonate, silica), biopolymers (e.g. gelatin, alginate), and globular biopolymers (e.g. soy proteins, whey protein).83 Therefore, the majority of modern approaches employs these naturally derived “green” biopolymers and minerals as stabilizers, which is additionally motivated by the desire to avoid potential mechanical weaknesses caused by the leaching of small-molecule surfactants.84 While finding the right type and amount of emulsifiers to stabilize the emulsion is often a challenge in the ES of emulsions, the 2-phase composition gives rise to advantageous fibrous encapsulation structures. During fiber generation, a rapid viscosity increase occurs at the outer layer of the jet due to the higher evaporation rate of the solvent from the continuous phase gradually towards the core, displacing the dispersed second phase and effectively pushing it towards the center. The concentrated dispersed phase can then condense in the core continuously or in the shape of local reservoirs, which results in a core–sheath or beaded structure, respectively (Fig. 7).80,82
 | ||
| Fig. 7 Morphology of emulsion-electrospun fibers depending on emulsion composition and spinning parameters.Top: TEM images of fibers spun from emulsions containing different total matrix and oil concentrations. Adapted from ref. 97 with permission from Elsevier B. V. Middle: SEM images of fibers spun from emulsions containing different ratios of matrix to oil. Adapted from ref. 86 with permission from the American Chemical Society. Bottom: SEM images of fibers spun from an emulsion at different humidity levels. Adapted from ref. 98 with permission from Springer Science + Business Media New York. | ||
Focusing on carbohydrates instead, Tampau et al. used casein to stabilize and spin an acidic aqueous emulsion of carvacrol in starch.88 Despite yielding only a small amount of well-defined fibers, which was attributed to insufficient chain entanglement, the authors achieved encapsulation efficiencies of up to 90%. In contrast, Krokida and coworkers observed smooth fibers from their emulsion-spinning of ulvan/pullulan with algae-extracts in coconut oil and Tween 20, while also encapsulating around 90% of the extract.89 CS was employed in combination with PEO in acidic aqueous solution by Schiffman and coworkers in the surfactant-free spinning to encapsulate cinnamaldehyde.90 The authors demonstrated the delayed release of the compound over several hours and improved antibacterial activity of the electrospun mats. In a follow-up study, the effect of molecular weight of CS, its degree of acetylation, and composition of the emulsion-components on the resulting fiber morphology was detailed.91 As expected, beaded or smooth fibers were obtained depending on the CS concentration and molecular weight, whereas increasing acetylation improved the incorporation of cinnamaldehyde while impairing the encapsulation efficiency of hydrocinnamyl alcohol. Similarly, Allafchian and coworkers conducted a comprehensive study on the composition and electro-spraying/spinning parameters of limonene and Tween 20 in Alyssum homolocarpum seed gum water solution.92 Originally focusing on spraying, their green approach also produced fibers as well as capsules with promising retention properties of limonene during storage over 90 days.
In contrast, Spano et al. demonstrated that the active ingredient does not always have to be incorporated into the core.93 In their study, a solution of polypropylene carbonate in EtOAc was emulsified without surfactant in an aqueous blend of PEO and sericin protein. The aqueous continuous phase and hence resulting outer hydrophilic layer incorporated an antigen, inorganic fluorescent nanoparticles or magnetic nanorods via dispersion, whereas the subsequent release of the dispersed species over 72 h was demonstrated to depend on the exact ratio of polymeric compounds used.
Despite their synthetic origin, many studies considered PVA for delayed release of oils for food and other storage applications, as a consequence of their controllable well-described properties and biocompatibility. Camerlo et al. demonstrated the potential of encapsulating limonene in PVA without surfactant.97,98 Fiber morphology shifted from beaded to uniform fibers from 5–12 wt% PVA concentration, whereas oil concentration only affected the number of beads. During storage, a slight loss of 20–35% of the volatile compound was recorded, unless conditions harsher than 25 °C and 60% RH were chosen. The effect of the humidity during the spinning was further investigated and revealed nanostructured beads at 7 wt% PVA concentration and a shift from smooth fibers to increasingly bigger beaded fibers with increasing moisture at 9 wt% PVA concentration. Similarly, Ciera et al. observed beaded structures as they incorporated different mosquito-repellant oils into an aqueous PVA solution and subsequently proved the repellent effect of the electrospun mat on insects.99 Cinnamon oil as the antifungal and acaricidal active component of choice was incorporated into an aqueous PVA solution with the help of Tween 80 by Lee and coworkers.100 In a comprehensive biological study, the authors established the inhibiting effect of the 250–500 nm thick and up to 30 wt% oil-containing fibers on microbes, bacteria, and fungi. Moreover, they used the analog system with phytoncide and palmarosa oil coupled with heat treatment at 170 °C for PVA layer stabilization in order to achieve both antimicrobial and water absorption capabilities, which promises potential wound treatment applications.101 Tampau et al. demonstrated the incorporation of carvacrol, both with and without Tween 85 as surfactant, into a beaded fiber structure with 75% encapsulation efficiency and higher.102 In fact, the authors suggested that the surfactant diminished the bonding between carvacrol and the PVA matrix by competitive micelle formation, which led to a fractional release of the compound before and after pyrolysis of the PVA occurred. Biological surfactants were used by García-Moreno et al. to stabilize an emulsion of fish oil in acidified aqueous PVA solution to obtain smooth or beaded fibers, depending on the polymer concentration.103
Considering the cost and efficiency of certain pharmaceuticals, carrier oils can also serve as a solvent in biological applications. Aiming for the oral delivery of the antimycotic clotrimazole, Opanasopit and coworkers employed a complex system of oleic acid as primary oil phase, as well as Tween 80 and co-surfactants, in an aqueous template of CS-EDTA (chitosan-ethylenediamine tetraacetic acid) and PVA.104 Sunflower oil and Phospholipon 90 H were used by Daniels and coworkers to dissolve birch extract, which was shown to achieve prolonged release and wound healing capabilities.105 Targeting antioxidant coatings instead, López-Rubio and coworkers dissolved carotene in soybean oil, which was emulsified with soy protein isolate in an aqueous PVA solution.106 Expectedly, the emulsifier facilitated the formation of more stable emulsions as well as beaded fiber structures after spinning, which were annealed to remove residual incorporated water and achieve delayed gradual release. The annealing methodology was also employed by Meera Moydeen et al. in the preparation of core–shell PVA/dextran nanofibers, which incorporated a commonly used antibiotic.107 Plant oil served as a carrier for this active substance and resisted the subsequent heat treatment at 120 °C, which was carried out to evaporate traces of water for increased intermolecular hydrogen-bonding between PVA/dextran molecules and hence improve mechanical stability of these fibers. Comparative release studies from coaxially and blend-spun nanofibers revealed a rapid release of a water-soluble ciprofloxacin salt, while the emulsion-spun antibiotic in oil exhibited efficient encapsulation with only minor release after 50 h. Subsequently, the authors further investigated the effect of the dextran content on the release kinetics using PVP as the main matrix polymer108 and other polysaccharides109 within an analogous system. Dextran content promoted intermolecular hydrogen bonding, thus affecting the viscosity and erosion of the polymer matrix.
Sesli Cetin and coworkers chose a system based on PVP for the encapsulation of antibiotic essential oils. Emulsions of 1–4 wt% cinnamon oil were stabilized with Cremophor RH 40 as a surfactant and successfully electrospun, although encapsulation efficiency decreased with increasing oil content.110 As expected, biological studies confirmed the antibacterial effect of the electrospun mat starting from concentrations of 2 wt% of essential oil after 24 h. In a follow-up study using thyme essential oil as active component and gelatin/PVP for improved durability, a similar spinning behavior was observed.111 Fiber morphology changed from beaded to smooth fibers with increasing oil content; however, a concentration of 5 wt% and more led to issues in spinnability and stability of the membranes. The optimal membrane of this study contained 3 wt% of the active component, showing antibacterial properties for up to 8 days.
Despite these promising properties, water-soluble sheath materials spun from an aqueous continuous phase can achieve only limited stability in hydrophilic media, which are used in biomedicine and the food industry. Furthermore, the application of O/W systems containing carrier oils requires that the application is able to tolerate the excess oily medium, which can be a considerable disadvantage.
:1 mixture of acetic and formic acid and adding an aqueous PVA solution, which incorporated strontium ranelate as an osteogenic agent.113 The spun fibers were subsequently coated with gelatin for enhanced hydrophilicity before characterization, which revealed that a higher content of strontium ranelate increased elasticity, water uptake, and stimulation of osteogenic growth.
Yet another alternative, a “water in water” emulsion, can be produced by dissolving two incompatible but both water-soluble components simultaneously, which subsequently separate into two or more phases. This approach was used by Fuenmayor and coworkers to spin an emulsion consisting of both limonene-loaded cyclodextrane and pullulan in water to achieve prolonged storability of the volatile compound.114
In contrast, a mostly water-free emulsion approach was chosen by De Clerck and coworkers to form gellable nanofibers, in which PCL and gelatin were dissolved in a mixture of acetic and formic acid.115 Interestingly, this composition resulted in an emulsion forming separate PCL- and gelatin-rich phases. Different fiber diameters of 140–550 nm were achieved via manipulation of the polymeric content, while optimizing the solvent ratio or addition of small quantities of water led to the formation of homogenous blend solutions instead. With an entirely different application in mind, Yu and coworkers employed a template system consisting of PVP in ethanol and acetic acid to emulsify a metalorganic titanium precursor.116 After spinning the sol emulsion and annealing at 500 °C, smooth TiO2 nanofibers with a diameter of roughly 300 nm were obtained, which were ground up to nanorods for incorporation into a solar cell assembly.
Despite such achievements using emulsion systems however, clearly the more direct and convenient route for the incorporation of solid active compounds into nanofibers leads via the direct spinning from suspension.
3.2 Suspension electrospinning
Solid-in-liquid dispersions, termed either “suspension” or “sol” depending on particle size and visual appearance, can be electrospun similarly to emulsions in a wide range of compositions. In most cases, water-soluble matrix polymers and surfactants are dissolved in an aqueous continuous phase, to which the powdered solid particles or pre-made dispersions are added. This technique is compatible with a range of different particle types from biology to catalysis and can simply be upscaled using wire ES (Fig. 8); in fact, unstable dispersions can often lead to clogging in needle-based setups. An exception to this general procedure is only found in rather specific techniques, such as the direct near-field electrospinning (NFES) of aqueous-dispersed bacteriophages without any template polymer by Sugimoto et al.117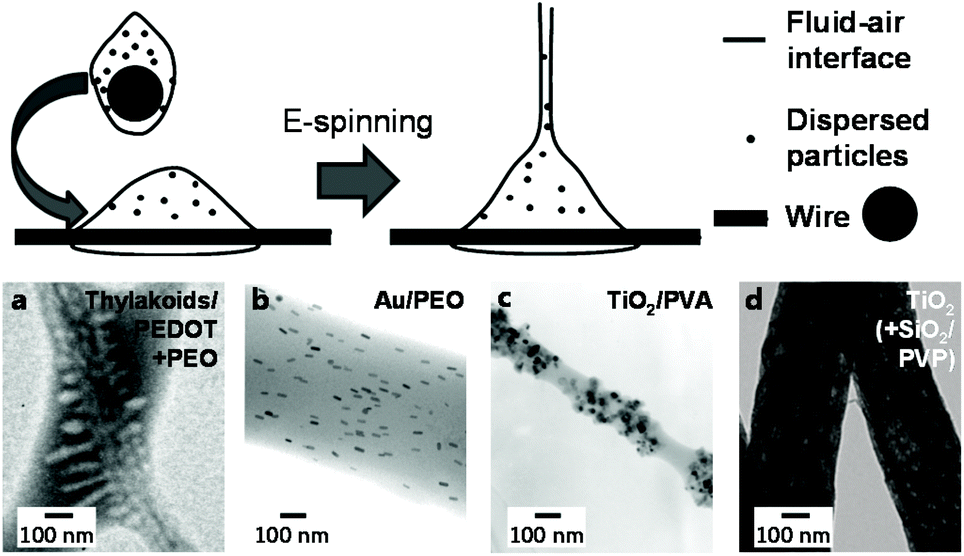 | ||
| Fig. 8 Principle and morphology of suspension-electrospun fibers depending on particle type. Top: schematic process of wire electrospinning using dispersions. Adapted from ref. 164 with permission from the American Chemical Society. Bottom: selected TEM images of fibers spun from suspensions incoporating different types of particles (a–c) and after their removal (d). (a) Adapted from ref. 149 with permission from the American Chemical Society. (b) Adapted from ref. 152 with permission from the American Chemical Society. (c) Adapted from ref. 134 with permission from Springer Science + Business Media New York. (d) Adapted from ref. 167 with permission from Elsevier B. V. | ||
 | ||
| Fig. 9 Morphological and mechanical effect of particle concentration and size in suspension-electrospun fibers. (a) SEM images of electrospun fibers obtained from dispersions with increasing concentration of spherical particles. (b) Typical strain-stress curves of electrospun fibrous mats obtained from suspensions containing increasing amounts of particles. (c and d) SEM images of typical electrospun fibers obtained from dispersions containing nanoparticles (c) or microparticles (d). (a) Adapted from ref. 124 with permission from the American Chemical Society. (b) Adapted from ref. 141 with permission from Elsevier B. V. (c) Adapted from ref. 125 with permission from Wiley-VCH Verlag GmbH & Co. KGaA. (d) Adaptedfrom ref. 144 with permission from Elsevier B. V. | ||
If, however, an even more water-soluble template polymer for suspension ES than PVA was required, PEO has frequently been chosen as an alternative. Colín-Orozco et al. incorporated water-insoluble whey protein isolate and rosemary extract into smooth PEO fibers for gradual complete release over just several hours.146 A two polyester-polyether block-copolymer was also reported on, which required no surfactants to stay dispersed and yielded slightly fused fibrous mats after spinning and re-dissolution of the PEO matrix.147 Cellulose nanofibers, a polythiophene and an epoxy-crosslinker were added by Latonen et al. to a PEO solution to produce conductive biocompatible fibers.148 A similar system was previously reported, which showcased successfully encapsulated intact thylakoid vesicles and correlated photo-induced currents and pH-changes.149 This approach required EtOH as co-solvent, which was also the case in the study concerning dispersion-spinning of thermos-responsive polyacrylamide(PAAm)/CS microgels by Borges and coworkers.150 Similarly, Mihindukulasuriya and Lim required this solvent system to incorporate TiO2 particles, methylene orange, and glycerol into PEO fibers for UV-activated oxygen sensors.151 Other structurally interesting compositions on the other hand did not require a co-solvent. For example, internal alignment of incorporated gold nanorods was achieved simply by influencing the spinning conditions,152 while silicate platelets were incorporated into a PEO polyacrylic acid (PAA) template in a well-dispersed manner despite the stark size difference.153 Interestingly, PAA represents another suitable matrix polymer by itself; however, it almost always requires an alcoholic co-solvent. Lei and coworkers spun a dispersion of boron nitride followed by thermal crosslinking to achieve stable thermally conductive mats for high-temperature thermoregulation.154 In contrast, catalytic applications were targeted with the surfactant-aided encapsulation of Pt by Sightler et al.155 and further improved by Kayarkatte et al.,156 who added carbon particles to improve conductivity and prevent catalyst erosion at the same time.
Finally, water-soluble PVP has also been widely employed for suspension ES. Rare uses of aqueous spinning media include a study concerning the incorporation of poly(methyl methacrylate)-polyethyleneimine (PMMA-PEI) capsules in PVP fibers,157 as well as the encapsulation of zeolites and subsequent calcination for hydrocracking catalysts.158 Similarly, fibrous alumina constructs were produced for high-temperature filtration or catalysis.159 Furthermore, in situ laser ablation enabled Uyar and coworkers to incorporate gold particles in a PVP matrix without the use of any harmful solvents whatsoever.160
Only a small number of studies investigated the dispersion spinning of any other polymers from aqueous solutions, such as the incorporation of live yeast from an aqueous EtOH PAAm solution by Fan et al.161 In contrast, most other potential matrix polymers, as well as further compositions involving the above-mentioned polymers, require non-aqueous media.
Catalytic systems based on metal oxides and others were also obtained from green ES of entirely water-insoluble systems. Augustine et al. described the non-inflammatory peroxide-mediated wound-healing properties of a ZnO/PCL mat, which was obtained using acetone as dispersion medium.172 Acetic acid served as a solvent in the ES of a PCL/gelatin/graphene oxide system, which showed antibacterial behavior combined with cytocompatibility, accelerated degradation, and gradual drug release capabilities.173
Other notable bioactive systems can be obtained using polyamides (PA, Nylon), for which FA is a green solvent. Ryu et al. prepared an antimicrobial and photocatalytic mat incorporating Ag-TiO2 nanoparticles; however, they found that a PA mat with sprayed-on particles exhibited superior properties.174 A similar composition was chosen by Chen and coworkers, who dispersed carbon nanotubes and generated Ag particles in situ to obtain conductive antibacterial mats.175 The effect of the carbon nanotube concentration on the conductivity of PA fibers was further investigated by Pan and coworkers from dispersions in FA.176 Meanwhile, Rahaman and coworkers combined acetic and formic acid as mixed medium and demonstrated the superior filtration and water-permeation capabilities of a PA membrane in contrast to common filter materials, whereby the PA membrane incorporated SiO2 nanoparticles and was coated with polyvinylacetate (PVAc) for increased hydrophobicity.177
Water-insoluble polymers such as PCL and PA significantly broaden the scope of potential applications for ES from dispersions. Nevertheless, even such water-resistant alternatives show degradation over time. Hence, even more durable formulations for more demanding purposes are highly desirable.
PTFE dispersions are commonly used in combination with a supporting template polymer for the ES of functional membranes directly, or further sintered into fibers above the PTFE melting point (approximately 330 °C). The particularly practical property of PTFE to maintain structural integrity under heat is based on its exceedingly high melt viscosity.178 Despite the simplicity of the system, particle concentration and sintering conditions can significantly influence the resulting membrane morphology (Fig. 10).
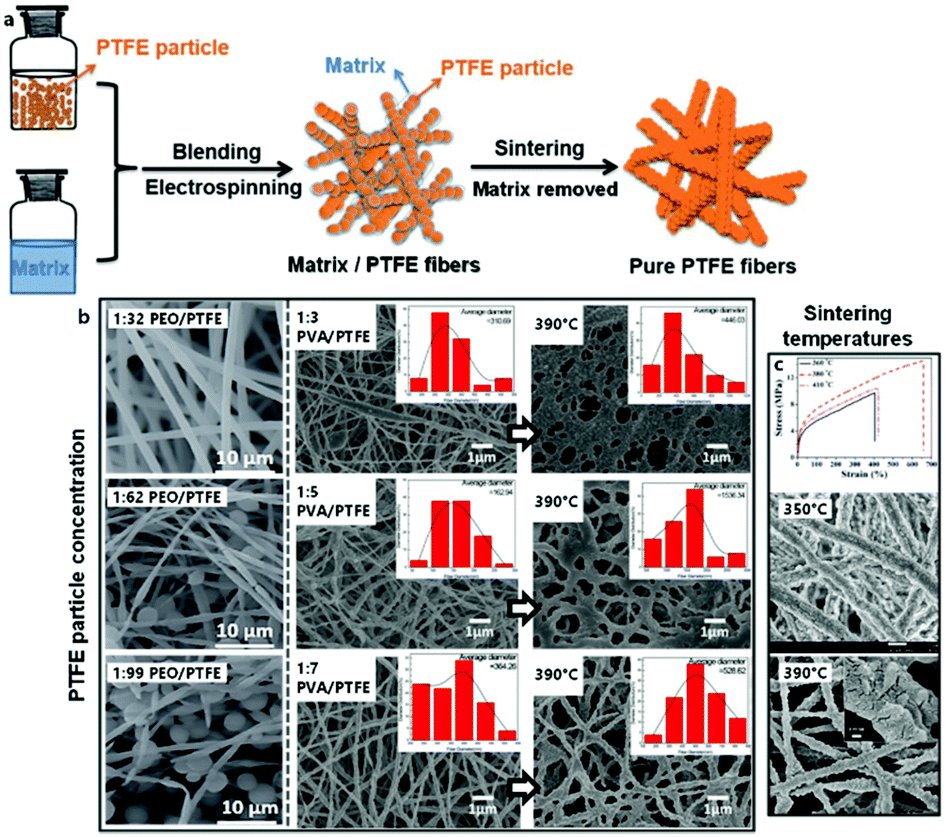 | ||
| Fig. 10 Principle and morphology of the electrospinning of blends based on PTFE suspensions. (a) Schematic process of electrospinning PTFE suspensions for pure PTFE fibers. (b) Left: SEM images of typical fibers obtained from electrospinning PTFE suspensions at different ratios of matrix to PTFE. (b) Right: SEM images of typcial fibers obtained from electrospinning PTFE suspensions at different ratios of matrix to PTFE and subsequent calcination. (c) Top: Typical strain-stress curves of electrospun PTFE-fiber membranes obtained after calcination at different temperatures. (c) Bottom: SEM images of typical electrospun PTFE-fiber membranes obtained after calcination at different temperatures. (a) and (b) Left, (c) top adapted from ref. 179 with permission from the Royal Society of Chemistry. (b) Right adapted from ref. 186 with permission from Springer Nature. (c) Bottom adapted from ref. 184 with permission from Elsevier B. V. | ||
PEO constitutes one of the more common matrix polymers for PTFE dispersion spinning. Feng et al. demonstrated that this system leads to remarkably acid- and base-resistant membranes after sintering, which was proven by unchanged tensile strength in such media.179 The addition of polyamide-imide (PAI) further increased the tensile strength while diminishing elasticity.180 Other important applications of PTFE fibers include triboelectric nano-generators,181 as well as membranes for oil/water separation.182
Alternatively, Kolesnik et al. reported a procedure for ES aqueous dispersions of PTFE using PVA as water-soluble sacrificial matrix polymer.183 The optimal sintering temperature and tensile strength of the resulting nanofibers from this system were detailed by Zhou et al.,184 while Zhu et al. determined that the aerosol filtration efficiencies of these membranes matched those of commercial PTFE filters.185 It was further demonstrated that the oil/water separation properties of an analogous PTFE mat could not be further improved by the incorporation of ZnO particles, however in contrast this study found that the addition of small amounts of boric acid lowered the concentration of PVA needed to form smooth fibers.186 Huang and coworkers subsequently employed the same system in near-field-ES (NFES) from dispersion to 3D-print micro-meshes for effective aqueous SiO2 particle filtration.187
While PTFE systems can never be considered truly “green” due to the environmental concerns of the persistent nature of fluoropolymers and their toxic pyrolysis products,188 the above-mentioned approaches can contribute to a greener manufacturing process of this still unrivalled material. Even further optimization in “greenness” is certainly under constant development, such as the focus on biopolymers as template materials. An example was recently demonstrated by Pang et al. with the ES and subsequent fabrication of hollow PTFE microfibers based on a pullulan template.189
4. Reactive electrospinning
One of the fields with the highest need for biocompatible materials concerns biomedical applications. Specifically, the formation of tissue produced by implanted cells is influenced greatly by the scaffold onto which they are seeded. It is often preferable to use a biodegradable material scaffold in order to naturally degrade the implanted materials and leaving only the generated tissue. Usually, the common materials used in this field are synthetic and natural biodegradable polymers. Among them, hydrogels are the most promising due to their ability to retain a great amount of water, good biocompatibility, low interfacial tension, and minimal mechanical and frictional irritation upon mechanical strains. Therefore, hydrogels and other soft composites more effectively promote cell expansion and tissue formation.190 The most logical option is the use of polymers that are already present in animal or human tissues, such as collagen191 and its hydrolyzed form gelatin,192 natural and modified polysaccharides,193 proteins,194 and glycosaminoglycans,195 as well as derivated polyaminoacids. Other synthetic polymers often used in this field are polyvinylalcohol (PVA), PEO and different types of acrylates. However, the mechanical properties of these hydrophilic polymers often quickly deteriorate once in contact with biological buffers. This creates a challenging obstacle for cell seeding, transfer from the cell culture, and in vivo implantation. Additionally, numerous findings in mechanobiology have strongly suggested that the closer the match between the elasticity modulus of the scaffold and the intended host tissue, the lower is the immunogenic and local inflammatory response during implantation.196,197In this context, reactive ES aims at enhancing the mechanical stability of weak, water-soluble polymers by performing an in situ cross-linking step during or after fiber generation. This procedure provides the balance between obtaining a soft hydrogel and enhancing the mechanical stability and water stability over time.
Different methods and chemistries to cross-link electrospun fibers involving water or other green solvents are presented in this section. However, only covalent cross-linking methods will be considered herein, whereas other non-covalent approaches, such as the use of inclusion complexes, hydrophobic interactions, or the formation of electrolyte complexes, can be found elsewhere.
4.1 Reactive electrospinning designs
Different designs of ES methodologies (co-axial, direct mixing, photo-curing, and co-spinning) with a focus on how this affects fiber morphology and cross-sectional composition are presented in Table 2.| Route | Reactive ES formulation | Polymer substrate | Polymer concentration [wt%] | Cross-linker (catalyst)/polymer [mol/mol] | Ref. | |
|---|---|---|---|---|---|---|
| The first column of the table is related to the routes of reactive ES, followed by the formulation, and which polymers are used at which concentration. The 5th column, “Cross-linker/polymer”, mentions the ratio between the cross-linker and the polymer. AA=acrylic acid, BAPO = bis-acylphosphinoxide, CQ=camphorquinone, EGDMA = ethylene glycol− dimethacrylate cross-linker, HA = Hyaluronic acid, MA = methacrylic acid, MCA = multi-carboxylic acids, MMA=methyl methacrylate, MPTMS = (3-mercaptopropyl)-trimethoxysilane, M-PVA = methacrylated polyvinyl alcohol, PAmA = Polyamic Acid, PEGDA = poly(ethylene glycol) diacrylate, PEtOx-EI = partially hydrolyzed PEtOx (poly(2-ethyl-2-oxazoline-)-co-ethylenimine, PI = Photoinitiator, PMC = Pentaerythritoltetrakis(2-mercaptoacetate), PO = phenyl-bis(2,4,6-trimethylbenzoyl)-phosphine oxide, POEGMA=poly(ethylene glycol) methacrylates, PAA-g-Az = Poly(acrylic acid-g-azidoaniline), PO-CMC = partially oxidized carboxymethylcellulose (bearing aldehydes groups), THA = Thiolated hyaluronic acid, TTT = 2,4,6,8-tetramethyl-2,4,6,8-tetravinylcyclotetrasiloxane. | ||||||
| 1 | Mixing | Glutaraldehyde | CS | 1.7–2.0 | 1:1 |
207 |
| Mixing | Carboxyethyl CS | 3.4 | 1:1 |
207 | ||
| Mixing | Gelatin | 10 | 0.07:1 |
209 | ||
| Mixing | PVA | 04/07/22 | 90–181:1 |
213 | ||
| Mixing | Pullulan | 22 | 0.056 (wt/wt) + 1% H2SO4 | 217 | ||
| Mixing | Glyoxal | Gelatin | 14 | 1.4–6.3:1 |
198 | |
| Mixing | Gelatin | 12/16/22 | 5.3–6:1 |
210 | ||
| Mixing | CS/PVA | 0.6 CS/6 PVA | 5 vol% Glyoxal | 218 | ||
| Mixing | Oxidized sugars | Carboxymethyl CS | 3 | 1.5 (wt/wt) | 208 | |
| Mixing | Gelatin | 20 | 5/30 (wt/wt) | 211 | ||
| Mixing | PO-CMC | Gelatin | 6.25–25 blend (80:20 wt%) |
1:0.25 |
212 | |
| 2 | Mixing | Collagen | 16 | 1.5–2.0:1 (NHS:EDC) |
222 | |
| Mixing | Recomb. Collagen Peptides/CS | 8 | 20 mM (only EDC) | 225 | ||
| Mixing | HA /PVA/CDs | 6 PVA/6 HA/0–40 CDs | 1:2 (wt/wt NHS:EDC) |
226 | ||
| 3 | Mixing | CS/PVA/MCA | 0.06–0.24 CS/6–9 PVA | 20–80 mM MCA | 227 | |
| Mixing | Alginate/PVA/Citric acid | 1 Alginate/5 PVA | 5 wt% Citric acid | 228 | ||
| Mixing | PAA/PEO | 5 to 12.5 PAA/5–10 PEO 25 | - | 230 | ||
| Mixing | Gelatin/Glucose | 2.5 CMC/2.5 PEO | up to 30 wt% Glucose | 231 | ||
| Mixing | CMC/PEO/MCA | 4.7 CS/0.25 PEO | 3–5–7–10 wt% MCA | 232 | ||
| Mixing | CS/PEO/MCA | 1 wt% MCA | 233 | |||
| 4 | Mixing | Di-epoxide | Gelatin | 15 | 2–6 wt% Di-epoxide | 235 |
| 5 | Co-axial | Gelatin | 30 | 0.18 wt% Genipin /polymer | 201 | |
| Mixing | Silk-fibroin | 12 | 0.12 wt% Genipin /polymer | 238 | ||
| Mixing | Gelatin | 20 | 0.01–0.05 wt% Genipin /polymer | 239 | ||
| 6 | Mixing | PVA/gelatin core/shell | 8 | 0.5 to 2 wt% enzyme/gelatin | 240 | |
| 7 | Mixing | TEOS | — | 1:0.01:2:2 TEOS:HCl:EtOH:H2O |
241 | |
| Mixing | TEOS | — | 1:1:2:2 wt% TEOS:HCl:EtOH:H2O |
242 and 243 | ||
| Mixing | PVA/TEOS | 1.4–5.6 | 0.04 wt% HCl/TEOS | 246 | ||
| Mixing | M-PVA | 10 | 5–10–20 wt% MPTMS/2 wt% PI | 247 | ||
| 8 | Mixing + UV | PHEMA | N/A | 318:1:1.33 (PHEMAc: EGDMA:PI) |
248 | |
| 9 | Dual-syringe + UV | THA/PEGDA/PEO | 2.5% w/v PEO/2.5% w/v THA | 9.0% w/v PEGDA | 250 | |
| Mixing + UV | PVP | 10 | 5 wt% PMC/5 wt% TTT/3 wt% PI | 251 | ||
| 10 | Mixing + UV | Gelatin/PAA-g-Az | 23% w/v – 10 Gelatin | 2.3% w/v - 1 wt% PAA-g-Az | 192 and 252 | |
| 11 | No mixing (2 syringes) | Aldehyde- & hydrazide-HA/PEO | 3.5 Aldehyde-HA/2.5 PEO | 3.5 wt% hydrazide-HA/2.5 wt% PEO | 205 | |
| Double-barrel | Aldehyde- & hydrazide-POEGMA | 7.5 | N/A | 253 | ||
| 12 | Mixing under Ar | Selenol-PEtOx-EI | 30 | 2–4–5–6 mol% Selenol (-SeH) | 256 | |
During the reactive ES process, if the fiber drawing and cross-linking are occurring at the same time, the performance of the two processes has to be especially coordinated to properly match with the targeted ES setup (e.g. single, multi-needle or needleless) and the available ES time window dictated by the kinetics of the cross-linking chemistry. Indeed, above a critical cross-linking degree depending on the type of polymer and the linker, the change in viscosity of the spinning solution alters the formation of a stable polymer jet and thus fiber formation. When a cross-linking reaction undergoes considerable changes in viscosity within the ES time window, as with using glyoxal and gelatin, a multi-layer scaffold with a gradient in fiber diameters can be generated.198 In order to overcome the problem of change in viscosity, cross-linking chemistries with extremely fast kinetic rates (i.e. aldehyde-hydrazine199 or blocked isocyanate-amine200) can be spun by separating the fluids before spinning with a double-barrel syringe (Fig. 11a). Alternatively, gelatin and genipin have also been mixed in situ with a customized co-axial ES setup, where the inner needle wall had 20 circular holes (∅ = 0.5 mm) at its tip.201
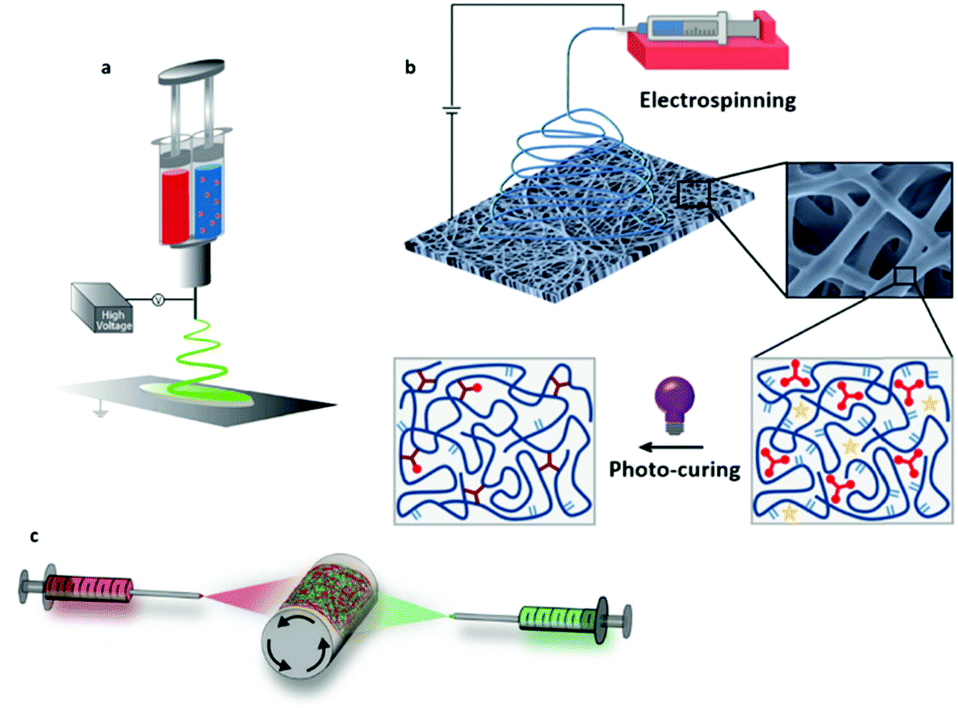 | ||
| Fig. 11 Different designs for in situ cross-linking strategies: (a) co-axial/double-barrel electrospinning; (b) photo-induced cross-linking; and (c) the development of multi-fiber hydrogel network. (a) Adapted from ref. 199 and (b) adapted from ref. 203 with permission from the American Chemical Society. (c) Adapted from ref. 205 with permission from Wiley-VCH Verlag GmbH & Co. KGaA. | ||
UV-induced ES covers a considerable number of studies regarding reactive ES, since its kinetic is generally fast and within the time window for ES (Fig. 11b).202,203 Since the cross-linking is triggered by UV exposure, the cross-linking degree during spinning has to be adjusted by optimizing the power amplitude of the UV lamp as well as the distance from the needle tip. Furthermore, a recent study has shown that the UV-spinning onto a plate collector submerged in a solvent for the un-cross-linked materials can lead to strikingly different mechanical properties compared to the more conventional “dry” UV ES.204 This aspect can also further contribute to expanding the range of mechanical properties achievable with the desired polymer network.
In 2020, an interesting combination of reactive chemistries has been proposed by integrating reactive units within functional block copolymers for in situ UV ES as well as complementarily reactive groups for covalent cross-linking.205 Two separate solutions of HA modified with norbornenes exclusively functionalized with hydrazides and aldehydes were oppositely spun at the same time on a rotating mandrel (Fig. 11c). Thanks to the presence of these reactive groups on the fiber surface, fiber-to-fiber cross-linking was promoted by mechanical loading, therewith endowing the scaffold with mechano-sensitive properties.
4.2 Cross-linking of amine and hydroxyl groups
This section and onwards will be focused on a description of already adopted cross-linking chemistries, including the reactants and eventual pre-functionalization of the polymer, experimental conditions, and residual reactive units.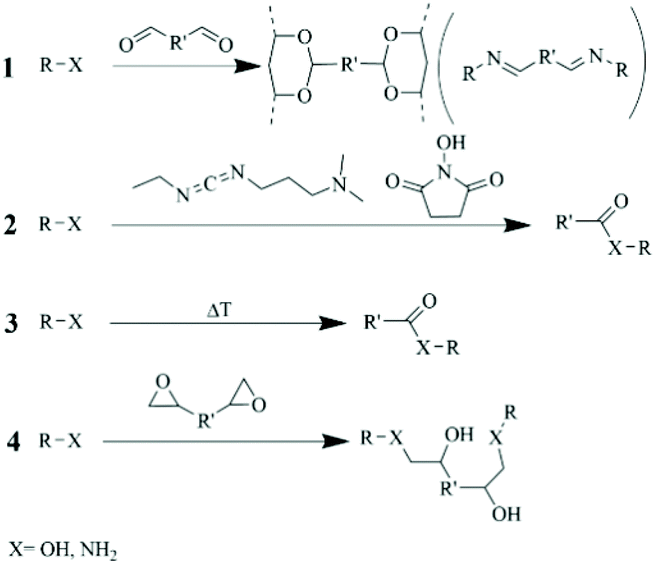 | ||
| Scheme 1 Cross-linking reactions occurring with polyelectrolytes bearing amine and hydroxyl groups. Route 1 yields acetal and imine groups whether the functional group was a hydroxyl groups (1,2-diols or 1,3-diols) or an amine group. Route 2 and 3 depict the reactions with R-X and carboxylic groups enabled by EDC/NHS and temperature, respectively. Route 4 shows the crosslinking behavior of epoxides. | ||
Glutaraldehyde has been one of the most investigated cross-linkers, probably because it is commercially available as a disinfectant and utilized for niche medical procedures, as with wart treatment.206 Imine cross-linking with CS or polyethyleneimine proceeds too fast and the solution cannot be spinnable unless its kinetic is quenched by using organic acids as solvents,207 mostly AcOH and formic acid. Another way to slow down the reaction kinetic is to use PEO. This polymer interpenetrates the network and ensures sufficient dilution of reactive groups to enable stable ES up to 5 h. The use of an ethyl carboxylated homolog of CS (CECS) further delays the cross-linking kinetic reaching up to 6 h of stable ES with the same experimental conditions. For both polymers, reactive cross-linking did not significantly alter the fiber diameter compared to the glutaraldehyde-free membrane homolog (∼60–140 nm for CS vs. ∼270 nm for CECS). More recently, partially oxidized saccharides displaying two aldehyde functionalities were synthesized with sodium periodate as oxidant.208 Alginate di-aldehyde delays the gelation point of carboxymethyl CS up to 12 min if the two solutions are mixed on-line prior to ES and they are diluted with PEO. The resulting scaffolds have shown better biocompatibility compared to glutaraldehyde-based ones. As well as improved retention of the fiber morphology.208
While CS possesses a high concentration of primary amino groups, as it is composed only of aminoglycosides, gelatin and collagen are proteins with only lysine as the primary amine containing amino acid unit (e.g. concentration of amino groups of 0.286 mmol g−1 in porcine skin type A vs. ∼4.0–5.0 mmol g−1 in CS). This feature, combined with the reactive ES with glutaraldehyde at relatively low cross-linker concentration (0.01:1 glutaraldehyde/gelatin by weight),209 delays the cross-linking process to yield relatively uniform fiber diameters of ∼600 nm. On the other hand, although the tensile strength increases up to the lower MPa range, the fibers are extremely fragile, with elongation at break of 3%.
The reaction velocity of the imine condensation between aldehydes and primary amines from lysine subunits in gelatin can be considerably reduced when a shorter di-aldehyde is utilized, such as glyoxal, achieving several hours of stability using standard ES. Glyoxal bears two aldehydes directly linked together and its reactivity is strikingly different from glutaraldehyde. After the reaction of glyoxal with an amine, the resulted imine further undergoes cyclization together with another mole of the imine (directly linked to aldehyde) group to yield imidazole group. Longer kinetic times for cross-linking can then be utilized for the development of gradient scaffolds with increasing fiber diameter (e.g. from 60 to 680 nm) from bottom to top.198 Despite the use of a cross-linker in excess of up to 6.7 times the amount of primary amines, the fibers showed an elongation at break at 180% and a lower Young's modulus of 880 kPa. Contrary to this, nanoindentation measurements of electrospun fibers from gelatin and glyoxal mixtures showed much lower elastic moduli down to 2.1, 3.2, and 10.9 kPa when the needle size was 2.1, 1.4, and 0.9 mm, respectively (e.g. compared to the 0.8 mm utilized by the previous work).210 Other cross-linking strategies for gelatin by using di-aldehydes include reactive ES with alginate211 and carboxymethylcellulose.212 When cross-linked with carboxymethylcellulose di-aldehyde, the fiber Young's moduli are between 10 and 15 MPa.
When di-aldehydes are reacting with 1,3-diols, as in the model compound PVA, a condensation reaction occurs to form an acetal bridge between the macromolecules without addition of catalyst. In contrast, the imine formation via amine-aldehyde reaction is too slow to proceed via reactive ES at room temperature without any catalyst. This issue has been tackled by the group of Khan,213 who added hydrochloric acid as catalyst to match the cross-linking within the ES window. They have shown that the formation of stable electrospun fibers upon soaking in water could be produced only above a glutaraldehyde/PVA molar ratio of 90:1. Moreover, in situ cross-linking enables stable ES of fibers at lower concentrations down to 4 wt%, which results in the formation of smaller fibers with an average diameter of 178 nm instead of 233 nm. This setup has been utilized also in other studies for producing conductive fibers when blended with semi-conductor ionomers,214 as a substrate for ultra-thin layer chromatography,215 and enzyme immobilization.216 Similarly, recent work has shown that this method is valid also for polysaccharides.217 Specifically, sulfuric acid was added to the formulation with glutaraldehyde to obtain in situ cross-linking of pullulan fibers. The cross-linking kinetic is even slower when glyoxal instead of glutaraldehyde is utilized as di-aldehyde cross-linker, similar to what was observed when reacting it with amines. In this case, CS added to the blend is the primary target for the initial cross-linking of amino groups, which increases the entanglements of the polymer blend favoring its early-stage mechanical integrity.218
The kinetics of this reaction is normally too fast to permit a stable ES process. A breakthrough was achieved by Meng et al., who used an excess of NHS/EDC to delay the coupling reaction to match the duration of the ES process.222 This concept was used to cross-link collagen in an EtOH/PBS buffer solution yielding fibers with diameters of 420 nm, which was roughly doubled when compared with un-cross-linked fibers with the same solvent system. Storage humidity of the membrane after ES was found to play a key role in the preservation of the fiber architecture. Unfortunately, fibers were stable in water only after production at relatively low humidity conditions (e.g. 43 and 53% after 3 and 1 day, respectively), with only the scaffold produced at 33% RH showing substantial retention of the fibrous structure. Indeed, water droplets condensing on the fiber, on one hand, increase the cross-linking conversion, but on the other hand, they provoke the fusion and merging of the fibers, therewith corrupting the fiber architecture and its porosity.
A similar strategy in terms of NHS/EDC amount was pursued to cross-link gelatin in combination with a thermo-sensitive polymer, poly(N-isopropylacrylamide) (PNIPAM), although utilizing a toxic solvent (e.g. 2,2,2-trifluoroethanol) for controlled drug release.223 Despite PNIPAM is not expected to contribute to cross-linking, several fiber-to-fiber coagulation areas are observed, which results in a loss of fibrous morphology. However, the gelatin-PNIPAM electrospun fibers could be potentially produced using water as a solvent since waterborne ES of PNIPAM has already been performed.224
A recent work, which combined CS and recombinant gelatin peptides, has shown complete retention of nanofibrous morphology when cross-linked with EDC in the absence of NHS as in situ cross-linking strategy.225 Contrary to conventional post-cross-linking by EDC, where the porosity decreases by one-third, the fibers retain a high surface-to-volume ratio with porosities above 60%, and consequently, their water uptake is much higher (1314% instead of 927%).
On the other hand, ester condensation does not occur fast enough to provoke cross-linked fibers during the ES process. Indeed, a polysaccharide bearing carboxylic functionalities, hyaluronic acid (HA), completes the cross-linking only after annealing it at 60 °C for 24 h, obtaining fibers with an average diameter around 200–300 nm and stable at all RH conditions.226
Different polymer blends bearing carboxylic and amine/hydroxyl units have been thermally cross-linked with this approach (120–170 °C from few minutes up to hours), namely PVA/CS,227 PVA/alginate,228 PVA/poly(acrylic acid) (PAA),229 polyamic acid/PEO,230 gelatin,231 carboxymethyl-cellulose,232 and CS.233 For most of these approaches, multi-carboxylic acids, namely citric acid,227,228,234 succinic acid,227 itaconic acid,233 1,2,3,4-butanetetracarboxylic acid,227,232 were added to the ES formulation to facilitate the diffusion of the reactive functional groups within a suitable distance for the thermal crosslinking to occur. A similar strategy was employed for this scope in other works, where the authors have explored the OH-rich cross-linkers glucose231 and cellulose nanocrystals.229 A systematic study of the respective morphologies of PVA/CS electrospun blends has clarified that the higher the carboxyl functionality of the multi-acid cross-linker, the higher its stability in water and retention of fibrous morphology without any substantial difference in the water uptake.227 This observation is in agreement with the observed decrease of its elongation at break from 43% with succinic acid compared to 20% with 1,2,3,4-butanetetracarboxylic acid.
000 times lower than conventional glutaraldehyde-based cross-linking methods. Genipin can be considered as a bi-functional cross-linker in the presence of polymers with pendant amino groups to form stable amine and amide linkages (route 5 in Scheme 2). Although the formation of a cross-linking network reduces its crystallinity,236 the scaffold is still relatively brittle with respective elongation at break of 4.5–5.0% similar to the un-cross-linked CS.237 Arginine and lysine residues in fibroin undergo cross-linking with genipin in FA after mixing for 2, 15, and 24 h before ES.238 The increase in conductivity at higher mixing times causes a reduction of the fiber diameter and, as expected the higher the cross-linking degree, the lower is the extent of fiber dilatation upon swelling in water. The same amino acids are also responsible for the cross-linking with genipin at 1–5 wt% with gelatin.239 ES of the precursors immediately aftermixing leads to fibers that still require storage in water vapor saturated conditions to complete the cross-linking reaction.
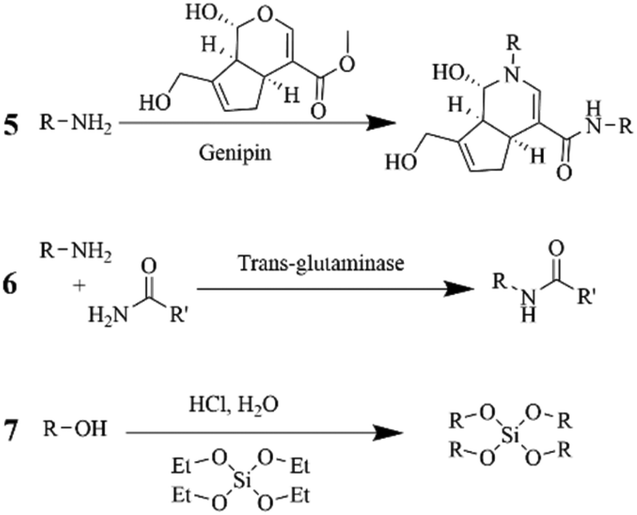 | ||
| Scheme 2 Cross-linking reactions specific for amino and hydroxyl groups using (5) Genipin, (6) Transglutaminase, and (7) TEOS. | ||
An innovative design for co-axial spinning of gelatin and genipin is described by Gualandi et al.201 In this case, through the inner needle that contained several holes, a genipin solution could diffuse out for thorough mixing with the sheath gelatin flow. Although the resulting fibers in a range of 320–350 nm were initially stable with minimal fused fiber-to-fiber intersections, the fibers still required a thermal and EtOH treatment to preserve their stability in water. Overall, despite its low toxicity and promising cell compatibility in combination with different polymeric substrates, the costs of genipin and the fiber generation process are still intolerable for most applications.
:7 TEOS/PVA. A twist to this strategy was combining the thiol–ene induced coupling of thiol-derived organosilane with methacrylated PVA during UV-assisted ES in DMSO.247 The product of this reaction was not a membrane scaffold, but rather an assembly of nanowires with an average length of 800 nm and diameters of 35–55 nm, which is rather peculiar among the different reactive ES processes.
4.3. In situ photo-induced cross-linking
The simultaneous UV-induced cross-linking of double bonds during the ES process is an appealing alternative to cross-link on demand the electrospun scaffolds and retain the high porosity features of this technique. The mechanism is mainly a radical-initiating polymerization of the terminal and substituted alkene or methacrylate groups.While substituted alkenes are generally formed by inserting functional monomers during the polymerization process, terminal alkenes and (meth)acrylates are formed by post-functionalization of the end groups or the pendent units of the macromolecule. Alternatively, even the direct polymerization of mono and di-functional (meth)acrylates, after thermal pre-conditioning can offer appealing versatility in terms of the nature of the functional groups incorporated into the polymer.
A twist to this approach has included in the past few years the use of thiol as co-reactant in these radical reactions, therewith remarkably expanding the window of polymer architectures achievable with this technology. Finally, other photo-induced cross-linking mechanisms, such as the recently explored nitrene chemistry will also be debated in this section.
 | ||
| Scheme 3 Overview of the different photo-induced cross-linking chemistries using (8) vinyl groups, (9) vinyl groups and thiols, and (10) polyacrylic acid and 4-azidoaniline forming nitrene intermediate under UV-light that can further react with amines or vinyl groups on polymers, thus acting as a cross-linker. | ||
 | ||
| Scheme 4 Chemical structure of molecules undergo photo-addition with thiols in order of reactivity: (a) norbornene, (b) vinyl silane, (c) vinyl ether, (d) fumarate, (e) allyl ether, (f) propene, (g) maleimide, and (h) methacrylate. | ||
Ji et al. used a double syringe setup containing aqueous solutions of hyaluronic acid functionalized with thiol groups and poly(ethylene oxide) in the first syringe and poly(ethylene glycol) di-acrylate (PEGDA) in the second syringe.250 The reactive ES process initially produced a uniform distribution of fibers, with a mean diameter of 90 ± 15 nm. Moreover, the thiol functionalized HA and the PEGDA underwent crosslinking reaction at RT in a time frame of 10 min without using UV light. The so prepared fiber network enabled fibroblasts to form 3D dendritic networks penetrating within the scaffold.
Not only macromonomers but also small multi-functional thiol–ene blocks can be easily translated into non-woven mats by reactive ES. Thiolated derivatives from pentaerythritol were utilized as tetra-functional blocks with cyclic siloxanes bearing four251 terminal alkene per molecule. When adding PVP to the tetra-functional polysiloxane/thiolated pentaerythritol formulation, phase separation occurred and core–shell morphologies were obtained.251 These fibers were generated in a mixture of EtOH and EtOAc.
:1 with these reactive units.192,252 Their Young's modulus of 5.5 ± 1.0 kPa were lower than gelatin cured with glutaraldehyde, which suggests a lower cross-linking degree.252 However, their cytotoxicity was lower than glutaraldehyde-based scaffolds, with better adhesion of fibroblast and good potential for cell scaffold in osteogenesis.192
4.4 Other cross-linking mechanisms
 | ||
| Scheme 5 Other cross-linking strategies involving hydrazone bond (11), and disulfide and diselenide formation (12). | ||
A follow-up study on the application of this type of cross-linking strategy for tissue engineering demonstrated the direct addition of biological cells to the ES solution in order to have cells inside or attached to the electrospun fibers, thus the cells were distributed in the whole scaffold already.199 Cell viability was ensured by using water as the solvent and by the good bio-inertness of this coupling chemistry. The generated fiber membranes not only showed higher cell viability and proliferation compared to a bulk cell-loaded hydrogel of the same material, but also maintained this high cell viability and proliferative capacity following a freeze/thaw cycle without requiring any cryoprotectant. Recently, the same group has also shown that, depending on the amount of PEO utilized as ES aid as well as on the weight ratio between hydrazide and aldehyde-based polymers, pure fibers to beaded fibers to bead network morphologies with tunable bead sizes can be fabricated.254 When utilizing ketone precursors, as with poly(acrylamide)-co-poly(diacetone acrylamide), the reaction proceeded slightly slower in the presence of a di-functional cross-linker, adipic dihydrazide, thus enabling the production of fibers with diameters of 200–220 nm by precursor mixing before usage.255
A new concept in the field of reactive ES for tissue engineering has been proposed by Davidson et al., where fiber-to-fiber cross-linking could be triggered by mechanical loading, therewith mimicking the force-responsive properties of the extra-cellular matrix.205 To achieve this novel mechano-sensitive hydrogel, they have utilized hyaluronic acid as a functional polymer for the selective attachment of aldehyde or hydrazide units. Aldehyde functionalities in polysaccharides can be easily obtained by oxidizing the glucoside ring with sodium periodate as previously discussed (see section 4.2.1), whereas hydrazine moieties were introduced by reacting to the carboxylic groups in hyaluronic acid with adipic dihydrazide. Additionally, both polymers contain norbornene units for UV-induced reactive ES according to the previously described thiol–ene chemistry (e.g. dithiothreitol is also added as a bi-functional thiol linker) (see section 4.3.3). The mixed fiber scaffold with both unreacted hydrazide and aldehyde fiber network has been achieved by spinning the two different polymers with separate needles onto a common rotating mandrel. The advantage of this chemistry also offers the possibility to reverse the cross-linking in acidic pH (acetate buffer, pH = 4.3), thus restoring the fiber architecture similar to the state before its plastic deformation induced by in situ fiber-to-fiber cross-linking.
Specifically, the reaction of the trimetaphosphate cyclic anion with 1,3-diols leads to ring-opening and cycle re-arrangement directly with the polymer backbone, for example with pullulan and dextran blends.257–259
In a grafting approach, (meth)acrylation of polymers bearing hydroxyl/amine moieties can be achieved with smaller building blocks such as glycidyl methacrylate,260–262 2-isocyanoethyl-methacrylate,263 methacrylic anhydride,264,265 and acryloyl chloride.266,267 Subsequently, the (meth)acrylate units can be crosslinked following the route 8 or 9 of Scheme 3.
Isocyanate coupling is another potentially transferrable strategy to cross-link polymers bearing amino and hydroxyl groups, which yields urea and urethane bridges by reacting 1,6-hexa-methylenediisocyanate (HMDI) in the presence of 1,4-diaza-bicyclo[2,2,2]octane (DABCO)200 or blocked isocyanates268,269 and hexamethylene-1,6-diaminocarboxysulfonate.270–272
Despite their potential, certainly the latter approaches and others based on quite toxic components (e.g. glutaraldehyde, epoxides, isocyanate, etc.) risk higher cytotoxicity in tissue engineering applications due to residual crosslinkers, impairing the scaffold transplantation. Therefore, selection of chemical routes involving less toxic crosslinker/catalyst (e.g. carboxylic acid, genipin, enzymes, etc.) is generally preferred.
5. Conclusions and future perspectives
Methods to achieve green ES, including exclusively sustainable and non-hazardous solvents, are under constant development and have been practically available for most common types of polymers. Available solvent chart tables that classify the solvents according to their greenness offer the possibility to easily select solvents based on their health and safety properties, environmental impact and disposal requirements. Since classical ES requires the use of copious volumes of solvents, the choice of solvent has a large impact on the sustainability of the production process. Especially the use of halogenated solvents should further be avoided due to their environmental and safety impact. Hence, greener solvent alternatives have been listed, among which the most common ones are water, EtOH, MeOH, AcOH, acetone and DMSO. By employing water-based spinning solutions, the fields of emulsion and suspension-based spinning offer vast potential and unrivalled opportunities for encapsulation and immobilization of active agents, by either completely avoiding or at least drastically minimizing the use of organic solvents. Another sophisticated strategy to avoid organic solvent completely is the suspension electrospinning, which allows the generation of fibers from undissolved polymer particles, and can be sintered after the ES process. To this end, various compositions and conditions to prepare not only carrier membranes for drug delivery and storage of volatiles, but also functional membranes for filtration and catalysis from dispersions have been summarized. In contrast, reactive ES was shown to be yet another promising technique to achieve green manufacturing conditions, whereas it often escalates in complexity compared to the more traditional ES approaches. Nevertheless, it offers the formation of particularly stable 3D fiber networks using water-soluble polymers and green solvents. And practically, the only necessary steps to achieve cross-linking reactions are the addition of a suitable cross-linking agents and curing conditions, according to the type of functionalities of the polymer.Despite these options, the final selection of solvents relies on the assessment by the user. Limiting the solvent choice to green solvents inherently reduces the possibilities to optimize the ES parameters of the spinning process for desired fiber and membrane production. Also, green solvents usually have higher boiling points, which renders them safer to work with, but can cause a higher energy demand, such as through higher required voltages, more time consuming and complex manufacturing processes, or higher temperature and/or lower air humidity. Furthermore, the polymer solubility sometimes depends on other polymer properties, such as conformation, molecular weight and polydispersity. For example, not all aliphatic polyesters and even just PLAs are equally well soluble in acetone, although they are synthesized from the same monomers.273 Another limitation of using green solvents in ES is the limitation in mixing different types of polymers in one spinning solution (polymer blend), particularly without the solvation capabilities of halogenated solvents.
As the environmental and ecological awareness of consumers is increasing from year to year, also the production and material selection of products that were not in focus yet, will have to adapt for a more green manufacturing practices. Members of the ES industry might be hesitant to introduce unfamiliar green solvent alternatives, therefore we hope that placing more emphasis on reactive and dispersion electrospinning could offer sufficient incentive for implementing these alternative green pathways. Apart from the obvious sustainability and customer interest benefits, this allows for an extension of the choice of materials for blends which are otherwise impossible to find a good co-solvent for, e.g. blends of hydrophilic and hydrophobic polymers or incorporation of additives and active compounds.
As restrictions for the use of certain solvents are increasing, solvent alternatives, especially to replace halogenated solvents, are increasingly investigated in the electrospinning field. While most reports still use classical solvents, literature has shown that the most common polymers can be electrospun with green solvents to obtain membranes exhibiting largely the same properties. In the end, it can and hopefully will be just a matter of habit to first select green solvents. After all, the drive towards safe, environmentally conscious and sustainable ES-manufacturing is only increasing.
Abbreviations
| AcOH | Acetic acid |
| CA | Cellulose acetate |
| CED | Cumulative energy demand |
| CS | Chitosan |
| CMC | Carboxymethylcellulose |
| DMAc | Dimethylacetamide |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EHS | Environmental, health, and safety indicator |
| ES | Electrospinning |
| EtOAc | Ethyl acetate |
| EtOH | Ethanol |
| FA | Formic acid |
| LCA | Life-cycle assessment |
| MEK | Methyl ethyl ketone |
| 2-MeTHF | 2-Methyl tetrahydrofuran |
| NHS | N-Hydroxylsuccinimide |
| Nylon-6 | see PA6 |
| Nylon-11 | see PA11 |
| O/W | Oil-in-water |
| PA6 | Polyamide 6 (Nylon-6) |
| PA11 | Polyamide 11 (Nylon-11) |
| PAA | Polyacrylic acid |
| PAAm | Polyacrylamide |
| PAN | Polyacrylonitrile |
| PCL | Polycaprolactone |
| PEO | Polyethylene oxide |
| PHEMA | Poly(hydroxyethyl)methacrylate |
| PLA | Poly-lactic acid |
| PLGA | Poly(lactic-co-glycolic acid) |
| PMMA | Polymethylmethacrylate |
| PS | Polystyrene |
| PTFE | Polytetrafluoroethylene |
| PVA | Polyvinylalcohol |
| PVDF | Poly(vinylidene fluoride) |
| PVP | Polyvinylpyrrolidone |
| REACH | Chemical Control Regulation in the European Union |
| RH | Relative humidity |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| TEOS | Tetraethylorthosilicate |
| THF | Tetrahydrofuran |
| UV | Ultraviolet light |
| W/O | Water-in-oil |
Author contributions
J. A., G. H. and C. T. conceptualized, compiled and drafted the review. J. A., G. H. and F. I. edited the manuscript, supported by C. T. F. I. and R. M. R. supervised the work. All authors checked and modified the manuscript, and agreed to the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was initiated as a contribution to the Subitex 2020-2025 project. F. I. would like to acknowledge the EMPAPOSTDOCS–II program, which received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska–Curie grant agreement no. 754364. The work is dedicated to our late colleague and supervisor Giuseppino Fortunato. Jean Schoeller and Mohammadreza Norouzi are acknowledged for their scientific input and discussions.References
- X. Qin and S. Subianto, in Electrospun Nanofibers, Elsevier, 2017, pp. 449–466 Search PubMed.
- A. Luraghi, F. Peri and L. Moroni, J. Controlled Release, 2021, 334, 463–484 CrossRef CAS PubMed.
- M. Rahmati, D. K. Mills, A. M. Urbanska, M. R. Saeb, J. R. Venugopal, S. Ramakrishna and M. Mozafari, Prog. Mater. Sci., 2021, 117, 100721 CrossRef CAS.
- Y. Liu, T. Li, Y. Han, F. Li and Y. Liu, Curr. Opin. Biomed. Eng., 2021, 17, 100247 CrossRef CAS.
- Y. Li, Q. Li and Z. Tan, J. Power Sources, 2019, 443, 227262 CrossRef CAS.
- Y. Li, M. A. Abedalwafa, L. Tang, D. Li and L. Wang, in Electrospinning: Nanofabrication and Applications, Elsevier, 2019, pp. 571–601 Search PubMed.
- T. Ulrich and J. P. Arenas, Sustainability, 2020, 12, 2361 CrossRef CAS.
- F. Salehi, M. Jamshidi Avanaki and M. Nouri, J. Text. Inst., 2021, 1–10 CAS.
- K. Ohmura, Nanofiber nonwoven fabrics by electrospinning and meltblown nonwovens, https://www.nonwovens-industry.com/issues/2018-11/view_far-east-report/nanofiber-nonwoven-fabrics-by-electrospinning-and-meltblown-nonwovens/.
- ECHA, Substances restricted under REACH, https://echa.europa.eu/substances-restricted-under-reach.
- N. Horzum, M. M. Demir, R. Muñoz-Espí and D. Crespy, Green Electrospinning, De Gruyter, 2019 Search PubMed.
- A. Bachs-Herrera, O. Yousefzade, L. J. del Valle and J. Puiggali, Appl. Sci., 2021, 11, 1808 CrossRef CAS.
- R. Gahan and G. C. Zguris, in Fifteenth Annual Battery Conference on Applications and Advances (Cat. No. 00TH8490), IEEE, 2000, vol. 2000-Janua, pp. 145–149.
- Y. Kara and K. Molnár, J. Ind. Text., 2021 DOI:10.1177/15280837211019488.
- R. Hufenus, Y. Yan, M. Dauner and T. Kikutani, Materials, 2020, 13, 4298 CrossRef CAS PubMed.
- T. D. Brown, P. D. Dalton and D. W. Hutmacher, Prog. Polym. Sci., 2016, 56, 116–166 CrossRef CAS.
- Z. Wei, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 452, 022002 Search PubMed.
- D. W. Hutmacher and P. D. Dalton, Chem. – Asian J., 2011, 6, 44–56 CrossRef CAS PubMed.
- M. M. Bubakir, H. Li, A. Barhoum and W. Yang, in Handbook of Nanofibers, Springer International Publishing, Cham, 2019, pp. 125–156 Search PubMed.
- V. G. Maciel, D. J. Wales, M. Seferin, C. M. L. Ugaya and V. Sans, J. Cleaner Prod., 2019, 217, 844–858 CrossRef CAS.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- The European Parliament and the Council of the European Union, REGULATION (EC) No 1272/2008 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 December 2008 on classification, labelling and packaging of substances and mixtures, amending and repealing Directives 67/548/EEC and 1999/45/EC, and amending Regulation (EC), https://eur-lex.Europa.eu/legal-content/EN/TXT/?uri=celex%3A32008R1272.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 1–24 CrossRef.
- C. S. Slater and M. Savelski, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2007, 42, 1595–1605 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 17, 4848–4848 RSC.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- D. Prat, O. Pardigon, H. W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- European Chemicals Agency (ECHA), Candidate List of substances of very high concern for Authorisation, https://echa.europa.eu/candidate-list-table.
- C. J. Clarke, W. C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- G. Berndes, M. Hoogwijk and R. van den Broek, Biomass Bioenergy, 2003, 25, 1–28 CrossRef.
- F. Cherubini and S. Ulgiati, Appl. Energy, 2010, 87, 47–57 CrossRef CAS.
- M. C. Rulli, D. Bellomi, A. Cazzoli, G. De Carolis and P. D'Odorico, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- A. Muscat, E. M. de Olde, I. J. M. de Boer and R. Ripoll-Bosch, Global Food Secur., 2020, 25, 100330 CrossRef.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez De María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- H. H. Khoo, L. L. Wong, J. Tan, V. Isoni and P. Sharratt, Resour., Conserv. Recycl., 2015, 95, 174–182 CrossRef.
- T. Heinze and T. Liebert, Prog. Polym. Sci., 2001, 26, 1689–1762 CrossRef CAS.
- S. O. Han, J. H. Youk, K. D. Min, Y. O. Kang and W. H. Park, Mater. Lett., 2008, 62, 759–762 CrossRef CAS.
- S. Majumder, M. A. Matin, A. Sharif and M. T. Arafat, Bull. Mater. Sci., 2019, 42, 1–9 CrossRef CAS.
- J. Pelipenko, J. Kristl, B. Janković, S. Baumgartner and P. Kocbek, Int. J. Pharm., 2013, 456, 125–134 CrossRef CAS PubMed.
- D. Haas, S. Heinrich and P. Greil, J. Mater. Sci., 2010, 45, 1299–1306 CrossRef CAS.
- M. W. Frey, Polym. Rev., 2008, 48, 378–391 CrossRef CAS.
- K. Ohkawa, Molecules, 2015, 20, 9139–9154 CrossRef CAS PubMed.
- M. Crabbe-Mann, D. Tsaoulidis, M. Parhizkar and M. Edirisinghe, Cellulose, 2018, 25, 1687–1703 CrossRef CAS.
- Y. Zhang, C. Zhang and Y. Wang, Nanoscale Adv., 2021, 3, 6040–6047 RSC.
- A. Gholipour-Kanani and S. H. Bahrami, J. Nanomater., 2011, 2011, 1–10 CrossRef.
- L. Van Der Schueren, B. De Schoenmaker, Ö. I. Kalaoglu and K. De Clerck, Eur. Polym. J., 2011, 47, 1256–1263 CrossRef CAS.
- D. H. Reneker, W. Kataphinan, A. Theron, E. Zussman and A. L. Yarin, Polymer, 2002, 43, 6785–6794 CrossRef CAS.
- P. Van Royen, E. Schacht, L. Ruys and L. Van Vaeck, Rapid Commun. Mass Spectrom., 2006, 20, 346–352 CrossRef CAS PubMed.
- C. Z. Mosher, P. A. P. Brudnicki, Z. Gong, H. R. Childs, S. W. Lee, R. M. Antrobus, E. C. Fang, T. N. Schiros and H. H. Lu, Biofabrication, 2021, 13, 035049 CrossRef CAS PubMed.
- R. Casasola, N. L. Thomas, A. Trybala and S. Georgiadou, Polymer, 2014, 55, 4728–4737 CrossRef CAS.
- S. Gee, B. Johnson and A. L. Smith, J. Membr. Sci., 2018, 563, 804–812 CrossRef CAS.
- P. Supaphol, C. Mit-Uppatham and M. Nithitanakul, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3699–3712 CrossRef CAS.
- P. Heikkilä and A. Harlin, Eur. Polym. J., 2008, 44, 3067–3079 CrossRef.
- S. De Vrieze, B. De Schoenmaker, Ö. Ceylan, J. Depuydt, L. Van Landuyt, H. Rahier, G. Van Assche and K. De Clerck, J. Appl. Polym. Sci., 2011, 119, 2984–2990 CrossRef CAS.
- T. Meireman, L. Daelemans, S. Rijckaert, H. Rahier, W. Van Paepegem and K. De Clerck, Compos. Sci. Technol., 2020, 193, 108126 CrossRef CAS.
- L. Vogt, L. Liverani, J. Roether and A. Boccaccini, Nanomaterials, 2018, 8, 150 CrossRef PubMed.
- C. L. S. De Oliveira Mori, N. A. Dos Passos, J. E. Oliveira, L. H. C. Mattoso, F. A. Mori, A. G. Carvalho, A. De Souza Fonseca and G. H. D. Tonoli, Ind. Crops Prod., 2014, 52, 298–304 CrossRef CAS.
- Y. P. Neo, S. Ray, A. J. Easteal, M. G. Nikolaidis and S. Y. Quek, J. Food Eng., 2012, 109, 645–651 CrossRef CAS.
- M. G. McKee, G. L. Wilkes, R. H. Colby and T. E. Long, Macromolecules, 2004, 37, 1760–1767 CrossRef CAS.
- M. G. McKee, J. M. Layman, M. P. Cashion and T. E. Long, Science, 2006, 311, 353–355 CrossRef CAS PubMed.
- H. Homayoni, S. A. H. Ravandi and M. Valizadeh, Carbohydr. Polym., 2009, 77, 656–661 CrossRef CAS.
- A. Keirouz, N. Radacsi, Q. Ren, A. Dommann, G. Beldi, K. Maniura-Weber, R. M. Rossi and G. Fortunato, J. Nanobiotechnol., 2020, 18, 51 CrossRef CAS PubMed.
- E. Antaby, K. Klinkhammer and L. Sabantina, Appl. Sci., 2021, 11, 11937 CrossRef CAS.
- X. Geng, O. H. Kwon and J. Jang, Biomaterials, 2005, 26, 5427–5432 CrossRef CAS PubMed.
- C. Shin and G. G. Chase, Polym. Bull., 2005, 55, 209–215 CrossRef CAS.
- T. Jarusuwannapoom, W. Hongrojjanawiwat, S. Jitjaicham, L. Wannatong, M. Nithitanakul, C. Pattamaprom, P. Koombhongse, R. Rangkupan and P. Supaphol, Eur. Polym. J., 2005, 41, 409–421 CrossRef CAS.
- J. Manee-in, M. Nithitanakul and P. Supaphol, Iran. Polym. J., 2006, 15, 341–354 CAS.
- L. Li, Z. Jiang, M. Li, R. Li and T. Fang, RSC Adv., 2014, 4, 52973–52985 RSC.
- H.-Y. Chang, C.-C. Chang and L.-P. Cheng, MATEC Web Conf., 2019, 264, 03004 CrossRef CAS.
- H. M. Chen and D. G. Yu, J. Mater. Process. Technol., 2010, 210, 1551–1555 CrossRef CAS.
- T. Grothe, T. Böhm, D. Wehlage and A. Remche, Tekstilec, 2017, 60, 290–295 CrossRef.
- T. Grothe, J. L. Storck, M. Dotter and A. Ehrmann, Tekstilec, 2020, 63, 225–232 CrossRef CAS.
- S. Kotomin, A. Malkin and I. Skvortsov, Macromol. Symp., 2020, 389, 3–5 CrossRef.
- A. L. Kalabin and E. A. Pakshver, Fibre Chem., 2000, 32, 274–278 CAS.
- S. Agarwal and A. Greiner, Polym. Adv. Technol., 2011, 22, 372–378 CrossRef CAS.
- P. McClellan and W. J. Landis, BioRes. Open Access, 2016, 5, 212–227 CrossRef CAS PubMed.
- M. Buzgo, A. Mickova, M. Rampichova and M. Doupnik, in Core-Shell Nanostructures for Drug Delivery and Theranostics, eds. M. L. Focarete and A. Tampieri, Elsevier, 2018, pp. 325–347 Search PubMed.
- B. Pant, M. Park and S.-J. Park, Pharmaceutics, 2019, 11, 305 CrossRef CAS PubMed.
- N. Nikmaram, S. Roohinejad, S. Hashemi, M. Koubaa, F. J. Barba, A. Abbaspourrad and R. Greiner, RSC Adv., 2017, 7, 28951–28964 RSC.
- C. Zhang, F. Feng and H. Zhang, Trends Food Sci. Technol., 2018, 80, 175–186 CrossRef CAS.
- G. Rosace, V. Migani, C. Colleoni, M. R. Massafra and E. Sancaktaroglu, Nanotechnology in Textiles, Jenny Stanford Publishing, 2020 Search PubMed.
- W. Chen, D. Li, A. EI-Shanshory, M. El-Newehy, H. A. EI-Hamshary, S. S. Al-Deyab, C. He and X. Mo, Colloids Surf., B, 2015, 126, 561–568 CrossRef CAS PubMed.
- C. Zhang and H. Zhang, J. Agric. Food Chem., 2018, 66, 11681–11690 CrossRef CAS PubMed.
- C. Zhang, Y. Li, P. Wang, A. Zhang, F. Feng and H. Zhang, Food Hydrocolloids, 2019, 94, 38–47 CrossRef CAS.
- A. Tampau, C. González-Martinez and A. Chiralt, J. Food Eng., 2017, 214, 245–256 CrossRef CAS.
- S. Papadaki, K. Kyriakopoulou and M. Krokida, IOSR J. Environ. Sci., Toxicol. Food Technol., 2017, 10, 53–58 Search PubMed.
- K. A. Rieger and J. D. Schiffman, Carbohydr. Polym., 2014, 113, 561–568 CrossRef CAS PubMed.
- K. A. Rieger, N. P. Birch and J. D. Schiffman, Carbohydr. Polym., 2016, 139, 131–138 CrossRef CAS PubMed.
- K. Khoshakhlagh, M. Mohebbi, A. Koocheki and A. Allafchian, Bioact. Carbohydr. Diet. Fibre, 2018, 16, 43–52 CrossRef CAS.
- F. Spano, A. Quarta, C. Martelli, L. Ottobrini, R. M. Rossi, G. Gigli and L. Blasi, Nanoscale, 2016, 8, 9293–9303 RSC.
- E. Zdraveva, J. Fang, B. Mijovic and T. Lin, Ind. Eng. Chem. Res., 2015, 54, 8706–8712 CrossRef CAS.
- L. Zhou, F. Shi, G. Liu, J. Ye, P. Han and G. Zhang, Sol. Energy, 2021, 213, 339–349 CrossRef CAS.
- C. Srikamut, K. Thongchaivetcharat, T. Phakkeeree and D. Crespy, J. Colloid Interface Sci., 2020, 577, 199–206 CrossRef CAS PubMed.
- A. Camerlo, C. Vebert-Nardin, R. M. Rossi and A. M. Popa, Eur. Polym. J., 2013, 49, 3806–3813 CrossRef CAS.
- A. Camerlo, A.-M. Bühlmann-Popa, C. Vebert-Nardin, R. M. Rossi and G. Fortunato, J. Mater. Sci., 2014, 49, 8154–8162 CrossRef CAS.
- L. Ciera, L. Beladjal, L. Van Landuyt, D. Menger, M. Holdinga, J. Mertens, L. Van Langenhove, K. De Clerck and T. Gheysens, R. Soc. Open Sci., 2019, 6, 182139 CrossRef CAS PubMed.
- Y. Jung, H. Yang, I.-Y. Lee, T.-S. Yong and S. Lee, Polymers, 2020, 12, 243 CrossRef CAS PubMed.
- K. Lee and S. Lee, Fibers Polym., 2020, 21, 999–1012 CrossRef CAS.
- A. Tampau, C. González-Martínez and A. Chiralt, React. Funct. Polym., 2020, 153, 104603 CrossRef CAS.
- P. J. García-Moreno, K. Stephansen, J. van der Kruijs, A. Guadix, E. M. Guadix, I. S. Chronakis and C. Jacobsen, J. Food Eng., 2016, 183, 39–49 CrossRef.
- P. Tonglairoum, T. Ngawhirunpat, T. Rojanarata, R. Kaomongkolgit and P. Opanasopit, Colloids Surf., B, 2015, 126, 18–25 CrossRef CAS PubMed.
- F. K. Mwiiri, J. M. Brandner and R. Daniels, Pharmaceutics, 2020, 12, 770 CrossRef CAS PubMed.
- A. López-Rubio, E. Sanchez, S. Wilkanowicz, Y. Sanz and J. M. Lagaron, Food Hydrocolloids, 2012, 28, 159–167 CrossRef.
- A. M. Moydeen, M. S. A. Padusha, E. F. Aboelfetoh, S. S. Al-Deyab and M. H. El-Newehy, Int. J. Biol. Macromol., 2018, 116, 1250–1259 CrossRef CAS PubMed.
- A. M. Moydeen, M. S. A. Padusha, B. M. Thamer, N. A. Ahamed, A. M. Al-Enizi, H. El-Hamshary and M. H. El-Newehy, Fibers Polym., 2019, 20, 2078–2089 CrossRef CAS.
- M. M. Abdul Hameed, S. A. P. Mohamed Khan, B. M. Thamer, A. Al-Enizi, A. Aldalbahi, H. El-Hamshary and M. H. El-Newehy, J. Macromol. Sci., Part A: Pure Appl.Chem., 2020, 58, 130–144 CrossRef.
- H. Kesici Güler, F. Cengiz Çallıoğlu and E. Sesli Çetin, J. Text. Inst., 2019, 110, 302–310 CrossRef.
- F. Cengiz Çallıoğlu, H. Kesici Güler and E. Sesli Çetin, Mater. Res. Express, 2019, 6, 125013 CrossRef.
- X. Wang, Y. Yuan, X. Huang and T. Yue, J. Appl. Polym. Sci., 2015, 132, 41811 Search PubMed.
- S. B. Abdollahi Boraei, J. Nourmohammadi, B. Bakhshandeh, M. M. Dehghan, H. Gholami, Z. Gonzalez, A. J. Sanchez-Herencia and B. Ferrari, Biomed. Mater., 2021, 16, 025009 CrossRef PubMed.
- C. A. Fuenmayora, E. Mascheronia, M. S. Cosioa, L. Piergiovannia, S. Benedettia, M. Ortenzic, A. Schiraldia and S. Mannino, Chem. Eng. Trans., 2013, 32, 1771–1776 Search PubMed.
- L. Daelemans, I. Steyaert, E. Schoolaert, C. Goudenhooft, H. Rahier and K. De Clerck, Nanomaterials, 2018, 8, 551 CrossRef PubMed.
- G. Wang, W. Xiao and J. Yu, Energy, 2015, 86, 196–203 CrossRef CAS.
- R. Sugimoto, J. H. Lee, J.-H. Lee, H.-E. Jin, S. Y. Yoo and S.-W. Lee, RSC Adv., 2019, 9, 39111–39118 RSC.
- K. Jalaja, V. S. Sreehari, P. R. A. Kumar and R. J. Nirmala, Mater. Sci. Eng., C, 2016, 64, 11–19 CrossRef CAS PubMed.
- L. Deng, Y. Li, A. Zhang and H. Zhang, Food Hydrocolloids, 2020, 103, 105640 CrossRef CAS.
- I. Kutzli, D. Beljo, M. Gibis, S. K. Baier and J. Weiss, Food Biophys., 2019, 15, 206–215 CrossRef.
- H. Sehaqui, S. Morimune, T. Nishino and L. A. Berglund, Biomacromolecules, 2012, 13, 3661–3667 CrossRef CAS PubMed.
- N. V. Herrera, A. P. Mathew, L. Y. Wang and K. Oksman, Plast., Rubber Compos., 2013, 40, 57–64 CrossRef.
- Y. Li, H. Yang, Y. Hong, Y. Yang, Y. Cheng and H. Chen, J. Therm. Anal. Calorim., 2017, 130, 1063–1068 CrossRef CAS.
- W. Yuan and K.-Q. Zhang, Langmuir, 2012, 28, 15418–15424 CrossRef CAS PubMed.
- K. Friedemann, T. Corrales, M. Kappl, K. Landfester and D. Crespy, Small, 2012, 8, 144–153 CrossRef CAS PubMed.
- E. Gonzalez, A. Barquero, B. Muñoz-Sanchez, M. Paulis and J. R. Leiza, Nanomaterials, 2021, 11, 706 CrossRef CAS PubMed.
- Z. Zhang, Y. Wu, Z. Wang, X. Zou, Y. Zhao and L. Sun, Mater. Sci. Eng., C, 2016, 69, 462–469 CrossRef CAS PubMed.
- R. Augustine, A. Hasan, V. K. Yadu Nath, J. Thomas, A. Augustine, N. Kalarikkal, A.-E. Al Moustafa and S. Thomas, J. Mater. Sci. Mater. Med., 2018, 29, 163 CrossRef PubMed.
- L. Sethuram, J. Thomas, A. Mukherjee and N. Chandrasekaran, Mater. Adv., 2021, 2, 2971–2988 RSC.
- S. Yao, Y. Li, Z. Zhou and H. Yan, RSC Adv., 2015, 5, 91878–91887 RSC.
- X. H. Zhang, H. G. Dai and Y. Cao, Adv. Mater. Res., 2013, 796, 311–316 Search PubMed.
- B. Wang, Z. Chen, J. Zhang, J. Cao, S. Wang, Q. Tian, M. Gao and Q. Xu, Colloids Surf., A, 2014, 457, 318–325 CrossRef CAS.
- P. Zhang, X. Zhao, Y. Ji, Z. Ouyang, X. Wen, J. Li, Z. Su and G. Wei, J. Mater. Chem. B, 2015, 3, 2487–2496 RSC.
- N. T. B. Linh, K.-H. Lee and B.-T. Lee, J. Mater. Sci., 2011, 46, 5615–5620 CrossRef.
- A. Naeimi and M.-S. Ekrami-Kakhki, Appl. Organomet. Chem., 2020, 34, e5453 CrossRef CAS.
- Y. Mizuno, E. Hosono, T. Saito, M. Okubo, D. Nishio-Hamane, K. Oh-ishi, T. Kudo and H. Zhou, J. Phys. Chem. C, 2012, 116, 10774–10780 CrossRef CAS.
- C. Y. Su, A. C. Yang, J. S. Jiang, Z. H. Yang, Y. S. Huang, D. Y. Kang and C. C. Hua, J. Phys. Chem. B, 2018, 122, 380–391 CrossRef CAS PubMed.
- A. Fuad, N. Fatriani, C. I. Yogihati, A. Taufiq and E. Latifah, J. Phys.: Conf. Ser., 2018, 1011, 012013 CrossRef.
- X. Yang, Y. Guo, Y. Han, Y. Li, T. Ma, M. Chen, J. Kong, J. Zhu and J. Gu, Composites, Part B, 2019, 175, 107070 CrossRef.
- A. Sutka, S. Kukle, J. Gravitis, R. Milašius and J. Malašauskienė, Adv. Mater. Sci. Eng., 2013, 2013, 1–6 CrossRef.
- S. Huan, L. Bai, W. Cheng and G. Han, Polymer, 2016, 92, 25–35 CrossRef CAS.
- H. Sosiati, W. Nur Fatihah, Y. Yusmaniar and M. B. Nur Rahman, Key Eng. Mater., 2018, 792, 74–79 Search PubMed.
- J. Lamarra, M. N. Calienni, S. Rivero and A. Pinotti, Int. J. Biol. Macromol., 2020, 160, 307–318 CrossRef CAS PubMed.
- W. Zhang, P. Yang, X. Li, Z. Zhu, M. Chen and X. Zhou, Int. J. Biol. Macromol., 2019, 141, 747–755 CrossRef CAS PubMed.
- E. Giebel, C. Mattheis, S. Agarwal and A. Greiner, Adv. Funct. Mater., 2013, 23, 3156–3163 CrossRef CAS.
- J. Colín-Orozco, M. G. Zapata-Torres, G. Rodríguez-Gattorno and R. Pedroza-Islas, Food Biophys., 2014, 10, 134–144 CrossRef.
- K. Bubel, Y. Zhang, Y. Assem, S. Agarwal and A. Greiner, Macromolecules, 2013, 46, 7034–7042 CrossRef CAS.
- R.-M. Latonen, J. A. W. Cabrera, S. Lund, S. Kosourov, S. Vajravel, Z. Boeva, X. Wang, C. Xu and Y. Allahverdiyeva, ACS Appl. Bio Mater., 2020, 4, 483–493 CrossRef PubMed.
- N. M. Bedford, G. D. Winget, S. Punnamaraju and A. J. Steckl, Biomacromolecules, 2011, 12, 778–784 CrossRef CAS PubMed.
- S. C. S. Marques, P. I. P. Soares, C. Echeverria, M. H. Godinho and J. P. Borges, RSC Adv., 2016, 6, 76370–76380 RSC.
- S. D. F. Mihindukulasuriya and L.-T. Lim, J. Mater. Sci., 2013, 48, 5489–5498 CrossRef CAS.
- K. E. Roskov, K. A. Kozek, W.-C. Wu, R. K. Chhetri, A. L. Oldenburg, R. J. Spontak and J. B. Tracy, Langmuir, 2011, 27, 13965–13969 CrossRef CAS PubMed.
- Z. Shami, N. Sharifi-Sanjani, B. Khanyghma, S. Farjpour and A. Fotouhi, RSC Adv., 2014, 4, 40892–40897 RSC.
- J. Wang, Q. Li, D. Liu, C. Chen, Z. Chen, J. Hao, Y. Li, J. Zhang, M. Naebe and W. Lei, Nanoscale, 2018, 10, 16868–16872 RSC.
- J. J. Sightler, E. McPherson, W. A. Rigdon and X. Huang, ECS Trans., 2013, 50, 1445–1451 CrossRef.
- M. K. Kayarkatte, Ö. Delikaya and C. Roth, Mater. Today Commun., 2018, 16, 8–13 CrossRef CAS.
- T. Vongsetskul, P. Kongjumnean, P. Sunintaboon, R. Rangkupan and P. Tangboriboonrat, Polym. Bull., 2012, 69, 1115–1123 CrossRef CAS.
- S. F. Anis, G. Singaravel and R. Hashaikeh, RSC Adv., 2018, 8, 16703–16715 RSC.
- Z. Zhao, X. Shen, H. Yao, J. Wang, J. Chen and Z. Li, J. Sol-Gel Sci. Technol., 2014, 70, 72–80 CrossRef CAS.
- A. E. Deniz, H. A. Vural, B. Ortaç and T. Uyar, Mater. Lett., 2011, 65, 2941–2943 CrossRef CAS.
- Y. Fan, X. Tian, L. Zheng, X. Jin, Q. Zhang, S. Xu, P. Liu, N. Yang, H. Bai and H. Wang, Mater. Sci. Eng., C, 2021, 120, 111747 CrossRef CAS PubMed.
- J. Faria, C. Echeverria, J. P. Borges, M. H. Godinho and P. I. P. Soares, RSC Adv., 2017, 7, 48972–48979 RSC.
- B. K. Brettmann, K. Cheng, A. S. Myerson and B. L. Trout, Pharm. Res., 2013, 30, 238–246 CrossRef CAS PubMed.
- B. K. Brettmann, S. Tsang, K. M. Forward, G. C. Rutledge, A. S. Myerson and B. L. Trout, Langmuir, 2012, 28, 9714–9721 CrossRef CAS PubMed.
- A. Guo, J. Liu, Y. Wang and H. Xu, Mater. Lett., 2012, 74, 107–110 CrossRef CAS.
- A. Guo, J. Liu, X. Dong and M. Liu, Mater. Lett., 2013, 95, 74–77 CrossRef CAS.
- K. Choi, W. Lee, S.-H. Lee and C. Lim, Mater. Lett., 2015, 158, 36–39 CrossRef CAS.
- Y. Wei, Y. Shi, X. Zhang, Z. Jiang, Y. Zhang, L. Zhang, J. Zhang and C. Gong, J. Mater. Sci.: Mater. Electron., 2019, 30, 14519–14527 CrossRef CAS.
- T. Tański, W. Matysiak, Ł. Krzemiński, P. Jarka and K. Gołombek, Appl. Surf. Sci., 2017, 424, 184–189 CrossRef.
- W. Matysiak, T. Tański and M. Zaborowska, in IOP Conference Series: Materials Science and Engineering, 2018, vol. 461, p. 012050.
- J. Passaro, P. Russo, A. Bifulco, M. T. De Martino, V. Granata, B. Vitolo, G. Iannace, A. Vecchione, F. Marulo and F. Branda, Polymers, 2019, 11, 4–6 CrossRef PubMed.
- R. Augustine, E. A. Dominic, I. Reju, B. Kaimal, N. Kalarikkal and S. Thomas, RSC Adv., 2014, 4, 24777 RSC.
- M. Heidari, S. H. Bahrami, M. Ranjbar-Mohammadi and P. B. Milan, Mater. Sci. Eng., C, 2019, 103, 109768 CrossRef CAS PubMed.
- S.-Y. Ryu, J. W. Chung and S.-Y. Kwak, Compos. Sci. Technol., 2015, 117, 9–17 CrossRef CAS.
- D. Huang, Q. Lin, M. Yin, Y. Wei, J. Du, Y. Hu, L. Zhao, X. Lian and W. Chen, J. Biomater. Sci., Polym. Ed., 2019, 30, 1744–1755 CrossRef CAS PubMed.
- Y. Liu, J. Li and Z. J. Pan, Adv. Mater. Res., 2011, 295–297, 1993–1997 CAS.
- M. S. Islam, J. R. McCutcheon and M. S. Rahaman, J. Membr. Sci., 2017, 537, 297–309 CrossRef CAS.
- L. C. Case, J. Appl. Polym. Sci., 1960, 3, 254–254 CrossRef CAS.
- Y. Feng, T. Xiong, S. Jiang, S. Liu and H. Hou, RSC Adv., 2016, 6, 24250–24256 RSC.
- Y. Feng, T. Xiong, H. Xu, C. Li and H. Hou, Mater. Lett., 2016, 182, 59–62 CrossRef CAS.
- P. Zhao, N. Soin, K. Prashanthi, J. Chen, S. Dong, E. Zhou, Z. Zhu, A. A. Narasimulu, C. D. Montemagno, L. Yu and J. Luo, ACS Appl. Mater. Interfaces, 2018, 10, 5880–5891 CrossRef CAS PubMed.
- S. J. Son, S. K. Hong and G. Lim, J. Sens. Sci. Technol., 2020, 29, 89–92 CrossRef.
- I. M. Kolesnik, E. N. Bolbasov, V. M. Buznik and S. I. Tverdokhlebov, in IOP Conference Series: Materials Science and Engineering, 2020, vol. 731, p. 012021.
- T. Zhou, Y. Yao, R. Xiang and Y. Wu, J. Membr. Sci., 2014, 453, 402–408 CrossRef CAS.
- S. J. Zhu, Y. Y. Zhou, O. Takashi and G. Wu, Mater. Sci. Forum, 2011, 675–677, 827–830 CAS.
- W. Kang, H. Zhao, J. Ju, Z. Shi, C. Qiao and B. Cheng, Fibers Polym., 2016, 17, 1403–1413 CrossRef CAS.
- J. Cheng, Q. Huang, Y. Huang, W. Luo, Q. Hu and C. Xiao, J. Membr. Sci., 2020, 603, 118014 CrossRef CAS.
- H. Arito and R. Soda, Ann. Occup. Hyg., 1977, 20, 247–255 CAS.
- H. Pang, K. Tian, Y. Li, C. Su, F. Duan and Y. Xu, Sep. Purif. Technol., 2021, 274, 118186 CrossRef CAS.
- A. Asti and L. Gioglio, Int. J. Artif. Organs, 2014, 37, 187–205 CrossRef PubMed.
- S. Yoshii and M. Oka, J. Biomed. Mater. Res., 2001, 56, 400–405 CrossRef CAS PubMed.
- W.-H. Lin, J. Yu, G. Chen and W.-B. Tsai, Colloids Surf., B, 2016, 138, 26–31 CrossRef CAS PubMed.
- S. Tiwari, R. Patil and P. Bahadur, Polymers, 2018, 11, 1 CrossRef PubMed.
- W. Qiu, Y. Huang, W. Teng, C. M. Cohn, J. Cappello and X. Wu, Biomacromolecules, 2010, 11, 3219–3227 CrossRef CAS PubMed.
- R. Menezes, S. Hashemi, R. Vincent, G. Collins, J. Meyer, M. Foston and T. L. Arinzeh, Acta Biomater., 2019, 90, 169–178 CrossRef CAS PubMed.
- Z. Zhuang, Y. Zhang, S. Sun, Q. Li, K. Chen, C. An, L. Wang, J. J. J. P. van den Beucken and H. Wang, ACS Biomater. Sci. Eng., 2020, 6, 3091–3102 CrossRef CAS PubMed.
- M. P. Nikolova and M. S. Chavali, Bioact. Mater., 2019, 4, 271–292 CrossRef PubMed.
- M. Angarano, S. Schulz, M. Fabritius, R. Vogt, T. Steinberg, P. Tomakidi, C. Friedrich and R. Mülhaupt, Adv. Funct. Mater., 2013, 23, 3277–3285 CrossRef CAS.
- F. Xu, M. Dodd, H. Sheardown and T. Hoare, Biomacromolecules, 2018, 19, 4182–4192 CrossRef CAS PubMed.
- A. P. Kishan, R. M. Nezarati, C. M. Radzicki, A. L. Renfro, J. L. Robinson, M. E. Whitely and E. M. Cosgriff-Hernandez, J. Mater. Chem. B, 2015, 3, 7930–7938 RSC.
- C. Gualandi, P. Torricelli, S. Panzavolta, S. Pagani, M. L. Focarete and A. Bigi, Biomed. Mater., 2016, 11, 025007 CrossRef PubMed.
- J. P. Chan, K. G. Battiston and J. P. Santerre, Acta Biomater., 2019, 96, 161–174 CrossRef CAS PubMed.
- A. Vitale, G. Massaglia, A. Chiodoni, R. Bongiovanni, C. F. Pirri and M. Quaglio, ACS Appl. Mater. Interfaces, 2019, 11, 24544–24551 CrossRef CAS PubMed.
- E. Shamirzaei Jeshvaghani, L. Ghasemi-Mobarakeh, R. Mansurnezhad, F. Ajalloueian, M. Kharaziha, M. Dinari, M. Sami Jokandan and I. S. Chronakis, J. Biomed. Mater. Res., Part B, 2018, 106, 2371–2383 CrossRef CAS PubMed.
- M. D. Davidson, E. Ban, A. C. M. Schoonen, M. H. Lee, M. D'Este, V. B. Shenoy and J. A. Burdick, Adv. Mater., 2020, 32, 1–8 Search PubMed.
- K. Pal, A. T. Paulson and D. Rousseau, in Handbook of Biopolymers and Biodegradable Plastics, ed. S. B. T.-H. of B. and B. P. Ebnesajjad, Elsevier, Boston, 2013, pp. 329–363 Search PubMed.
- H. Penchev, D. Paneva, N. Manolova and I. Rashkov, Macromol. Biosci., 2009, 9, 884–894 CrossRef CAS PubMed.
- X. Zhao, S. Chen, Z. Lin and C. Du, Carbohydr. Polym., 2016, 148, 98–106 CrossRef CAS PubMed.
- J. Zhan, Y. Morsi, H. Ei-Hamshary, S. S. Al-Deyab and X. Mo, J. Biomater. Sci., Polym. Ed., 2016, 27, 385–402 CrossRef CAS PubMed.
- N. Jedrusik, C. Meyen, G. Finkenzeller, G. B. Stark, S. Meskath, S. D. Schulz, T. Steinberg, P. Eberwein, S. Strassburg and P. Tomakidi, Adv. Healthcare Mater., 2018, 7, 1–15 Search PubMed.
- J. Chen, Z. Liu, M. Chen, H. Zhang and X. Li, Macromol. Biosci., 2016, 16, 1368–1380 CrossRef CAS PubMed.
- J. Joy, J. Pereira, R. Aid-Launais, G. Pavon-Djavid, A. R. Ray, D. Letourneur, A. Meddahi-Pellé and B. Gupta, Int. J. Biol. Macromol., 2018, 107, 1922–1935 CrossRef CAS PubMed.
- C. Tang, C. D. Saquing, J. R. Harding and S. A. Khan, Macromolecules, 2010, 43, 630–637 CrossRef CAS.
- D. Cho, N. Hoepker and M. W. Frey, Mater. Lett., 2012, 68, 293–295 CrossRef CAS.
- T. Lu and S. V. Olesik, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 912, 98–104 CrossRef CAS PubMed.
- C. Tang, C. D. Saquing, P. K. Sarin, R. M. Kelly and S. A. Khan, J. Membr. Sci., 2015, 472, 251–260 CrossRef.
- Z. Li, T. Pan, Y. Wu, W. Kang and Y. Liu, Adsorpt. Sci. Technol., 2019, 37, 401–411 CrossRef CAS.
- M. Koosha, M. Raoufi and H. Moravvej, Colloids Surf., B, 2019, 179, 270–279 CrossRef CAS PubMed.
- N. Nakajima and Y. Ikada, Bioconjugate Chem., 1995, 6, 123–130 CrossRef CAS PubMed.
- J. M. Lee, H. H. L. Edwards, C. A. Pereira and S. I. Samii, J. Mater. Sci. Mater. Med., 1996, 7, 531–541 CrossRef CAS.
- G. Beckers, R. J. Berzborn and H. Strotmann, Biochim. Biophys. Acta - Bioenerg., 1992, 1101, 97–104 CrossRef CAS.
- L. Meng, O. Arnoult, M. Smith and G. E. Wnek, J. Mater. Chem., 2012, 22, 19412–19417 RSC.
- P. Slemming-Adamsen, J. Song, M. Dong, F. Besenbacher and M. Chen, Macromol. Mater. Eng., 2015, 300, 1226–1231 CrossRef CAS.
- E. Schoolaert, P. Ryckx, J. Geltmeyer, S. Maji, P. H. M. Van Steenberge, D. R. D'Hooge, R. Hoogenboom and K. De Clerck, ACS Appl. Mater. Interfaces, 2017, 9, 24100–24110 CrossRef CAS PubMed.
- A. Deng, Y. Yang, S. Du and S. Yang, Biomater. Sci., 2018, 6, 2197–2208 RSC.
- M. Séon-Lutz, A. C. Couffin, S. Vignoud, G. Schlatter and A. Hébraud, Carbohydr. Polym., 2019, 207, 276–287 CrossRef PubMed.
- A. Pangon, S. Saesoo, N. Saengkrit, U. Ruktanonchai and V. Intasanta, Carbohydr. Polym., 2016, 138, 156–165 CrossRef CAS PubMed.
- S. A. Stone, P. Gosavi, T. J. Athauda and R. R. Ozer, Mater. Lett., 2013, 112, 32–35 CrossRef CAS.
- J. Han, S. Wang, S. Zhu, C. Huang, Y. Yue, C. Mei, X. Xu and C. Xia, ACS Appl. Mater. Interfaces, 2019, 11, 44624–44635 CrossRef CAS PubMed.
- S. Jiang, H. Hou, S. Agarwal and A. Greiner, ACS Sustainable Chem. Eng., 2016, 4, 4797–4804 CrossRef CAS.
- K. Siimon, P. Reemann, A. Põder, M. Pook, T. Kangur, K. Kingo, V. Jaks, U. Mäeorg and M. Järvekülg, Mater. Sci. Eng., C, 2014, 42, 538–545 CrossRef CAS PubMed.
- M. Kurečič, T. Mohan, N. Virant, U. Maver, J. Stergar, L. Gradišnik, K. S. Kleinschek and S. Hribernik, RSC Adv., 2019, 9, 21288–21301 RSC.
- L. Liang, L. Nie, M. Jiang, F. Bie, L. Shao, C. Qi, X. M. Zhang and X. Liu, New J. Chem., 2018, 42, 11023–11030 RSC.
- J. Gao, H. Guo, L. Zhao, X. Zhao and L. Wang, Fibers Polym., 2017, 18, 1496–1503 CrossRef CAS.
- J. R. Dias, S. Baptista-Silva, C. M. T. d. Oliveira, A. Sousa, A. L. Oliveira, P. J. Bártolo and P. L. Granja, Eur. Polym. J., 2017, 95, 161–173 CrossRef CAS.
- P. A. Norowski, S. Mishra, P. C. Adatrow, W. O. Haggard and J. D. Bumgardner, J. Biomed. Mater. Res., Part A, 2012, 100 A, 2890–2896 CrossRef PubMed.
- P. A. Norowski, T. Fujiwara, W. C. Clem, P. C. Adatrow, E. C. Eckstein, W. O. Haggard and J. D. Bumgardner, J. Tissue Eng. Regener. Med., 2015, 9, 577–583 CrossRef CAS PubMed.
- S. S. Silva, D. Maniglio, A. Motta, J. F. Mano, R. L. Reis and C. Migliaresi, Macromol. Biosci., 2008, 8, 766–774 CrossRef CAS PubMed.
- L. Li, W. Zhang, M. Huang, J. Li, J. Chen, M. Zhou and J. He, J. Biomater. Sci., Polym. Ed., 2018, 29, 2154–2167 CrossRef CAS PubMed.
- M. Sengor, A. Ozgun, O. Gunduz and S. Altintas, Mater. Lett., 2020, 263, 127233 CrossRef CAS.
- J. Geltmeyer, L. Van Der Schueren, F. Goethals, K. De Buysser and K. De Clerck, J. Sol-Gel Sci. Technol., 2013, 67, 188–195 CrossRef CAS.
- J. Geltmeyer, J. De Roo, F. Van den Broeck, J. C. Martins, K. De Buysser and K. De Clerck, J. Sol-Gel Sci. Technol., 2016, 77, 453–462 CrossRef CAS.
- E. Loccufier, J. Geltmeyer, L. Daelemans, D. R. D′hooge, K. De Buysser and K. De Clerck, Adv. Funct. Mater., 2018, 28, 1–10 Search PubMed.
- Y. B. Kim, D. Cho and W. H. Park, J. Appl. Polym. Sci., 2009, 114, 3870–3874 CrossRef CAS.
- A. Merlettini, S. Pandini, S. Agnelli, C. Gualandi, K. Paderni, M. Messori, M. Toselli and M. L. Focarete, RSC Adv., 2016, 6, 43964–43974 RSC.
- T. Pirzada, S. A. Arvidson, C. D. Saquing, S. S. Shah and S. A. Khan, Langmuir, 2012, 28, 5834–5844 CrossRef CAS PubMed.
- E. Baştürk, B. Oktay and M. V. Kahraman, J. Polym. Res., 2015, 22, 1–7 CrossRef.
- S. H. Kim, S. H. Kim, S. Nair and E. Moore, Macromolecules, 2005, 38, 3719–3723 CrossRef CAS.
- B. H. Northrop and R. N. Coffey, J. Am. Chem. Soc., 2012, 134, 13804–13817 CrossRef CAS PubMed.
- Y. Ji, K. Ghosh, B. Li, J. C. Sokolov, R. A. F. Clark and M. H. Rafailovich, Macromol. Biosci., 2006, 6, 811–817 CrossRef CAS PubMed.
- Q. Niu, L. Zeng, X. Mu, J. Nie and G. Ma, J. Ind. Eng. Chem., 2016, 34, 337–343 CrossRef CAS.
- W.-H. Lin and W.-B. Tsai, Biofabrication, 2013, 5, 035008 CrossRef CAS PubMed.
- F. Xu, H. Sheardown and T. Hoare, Chem. Commun., 2016, 52, 1451–1454 RSC.
- F. Xu, I. Gough, J. Dorogin, H. Sheardown and T. Hoare, Acta Biomater., 2020, 104, 135–146 CrossRef CAS PubMed.
- Y. Ido, A. L. B. Maçon, M. Iguchi, Y. Ozeki, S. Koeda, A. Obata, T. Kasuga and T. Mizuno, Polymer, 2017, 132, 342–352 CrossRef CAS.
- Y. Li, M. Vergaelen, X. Pan, F. E. Du Prez, R. Hoogenboom and K. De Clerck, Macromolecules, 2018, 51, 6149–6156 CrossRef CAS.
- X. Jiang, M. H. Nai, C. T. Lim, C. Le Visage, J. K. Y. Chan and S. Y. Chew, J. Biomed. Mater. Res., Part A, 2015, 103, 959–968 CrossRef PubMed.
- P. O. Rujitanaroj, R. Aid-Launais, S. Y. Chew and C. Le Visage, Biomater. Sci., 2014, 2, 843–852 RSC.
- L. Shi, C. Le Visage and S. Y. Chew, J. Biomater. Sci., Polym. Ed., 2011, 22, 1459–1472 CrossRef CAS PubMed.
- X. Xu, J. F. Zhang and Y. Fan, Biomacromolecules, 2010, 11, 2283–2289 CrossRef CAS PubMed.
- F. J. C. Baratéla, O. Z. Higa, E. D. dos Passos and A. A. A. de Queiroz, Mater. Sci. Eng., C, 2017, 73, 72–79 CrossRef PubMed.
- B. Zeytuncu, M. Ürper, İ. Koyuncu and V. V. Tarabara, Sep. Purif. Technol., 2018, 197, 432–438 CrossRef CAS.
- A. Kishan, T. Walker, N. Sears, T. Wilems and E. Cosgriff-Hernandez, J. Biomed. Mater. Res., Part A, 2018, 106, 1155–1164 CrossRef CAS PubMed.
- R. Wu, J.-F. Zhang, Y. Fan, D. Stoute, T. Lallier and X. Xu, Biomed. Mater., 2011, 6, 035004 CrossRef PubMed.
- J.-F. Zhang, Y. Wang, M. L. Lam, R. J. McKinnnie, W. C. Claycomb and X. Xu, Mater. Today Commun., 2017, 13, 306–316 CrossRef CAS.
- I. Stefani and J. J. Cooper-White, Acta Biomater., 2016, 36, 231–240 CrossRef CAS PubMed.
- H. M. Ismail, S. Zamani, M. A. Elrayess, W. Kafienah and H. M. Younes, Polymers, 2018, 10, 1–18 CrossRef PubMed.
- J. Sheng, M. Zhang, W. Luo, J. Yu and B. Ding, RSC Adv., 2016, 6, 29629–29637 RSC.
- J. H. Lee, U. S. Lee, K. U. Jeong, Y. A. Seo, S. J. Park and H. Y. Kim, Polym. Int., 2010, 59, 1683–1689 CrossRef CAS.
- M. S. Austero, A. E. Donius, U. G. K. Wegst and C. L. Schauer, J. R. Soc., Interface, 2012, 9, 2551–2562 CrossRef CAS PubMed.
- M. A. Kiechel, L. T. Beringer, A. E. Donius, Y. Komiya, R. Habas, U. G. K. Wegst and C. L. Schauer, J. Biomed. Mater. Res., Part A, 2015, 103, 3201–3211 CrossRef CAS PubMed.
- L. T. Beringer, M. A. Kiechel, Y. Komiya, A. E. Donius, R. Habas, U. G. K. Wegst and C. L. Schauer, J. Biomed. Mater. Res., Part A, 2015, 103, 3026–3033 CrossRef CAS PubMed.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed.
- C. Z. Mosher, P. A. P. Brudnicki, Z. Gong, H. R. Childs, S. W. Lee, R. M. Antrobus, E. C. Fang, T. N. Schiros and H. H. Lu, Biofabrication, 2021, 13, 035049 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |