
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe design, synthesis, and in vitro trypanocidal and leishmanicidal activities of 1,3-thiazole and 4-thiazolidinone ester derivatives
Muhammad Haroona,
Mabilly Cox Holanda de Barros Diasb,
Aline Caroline da Silva Santosc,
Valéria Rêgo Alves Pereirac,
Luiz Alberto Barros Freitasb,
Rodolfo Bento Balbinot d,
Vanessa Kaplumd,
Celso Vataru Nakamurad,
Luiz Carlos Alvesef,
Fábio André Brayneref,
Ana Cristina Lima Leite*b and
Tashfeen Akhtar*a
d,
Vanessa Kaplumd,
Celso Vataru Nakamurad,
Luiz Carlos Alvesef,
Fábio André Brayneref,
Ana Cristina Lima Leite*b and
Tashfeen Akhtar*a
aDepartment of Chemistry, Mirpur University of Science and Technology (MUST), Mirpur, Allama Iqbal Road, 10250-Mirpur, AJK, Pakistan. E-mail: tashfeenchem@must.edu.pk
bLaboratório de Planejamento em química medicinal, Department of Pharmaceutical Sciences, Health Sciences Centre, Federal University of Pernambuco, 50740-520, Recife, PE, Brazil. E-mail: acllb2003@yahoo.com.br
cAggeu Magalhães Research Center, Fundação Oswaldo Cruz, 50670-420, Recife, PE, Brazil
dLaboratório de Inovação Tecnológica no Desenvolvimento de Fármacos e Cosméticos, State University of Maringá, Paraná, Brazil
eLaboratório de Imunopatologia Keizo Asami (LIKA), Campus UFPE, 50670-901, Recife, PE, Brazil
fInstituto Aggeu Magalhães, Fundação Oswaldo Cruz, 50670-420, Recife, PE, Brazil
First published on 11th January 2021
Abstract
Chagas and leishmaniasis are both neglected tropical diseases, whose inefficient therapies have made them remain the cause for millions of deaths worldwide. Given this, we synthesized 27 novel 1,3-thiazoles and 4-thiazolidinones using bioisosteric and esterification strategies to develop improved and safer drug candidates. After an easy, rapid and low-cost synthesis with satisfactory yields, compounds were structurally characterized. Then, in vitro assays were performed, against Leishmania infantum and Leishmania amazonensis promastigotes, Trypanosoma cruzi trypomastigotes and amastigotes, for selected compounds to determine IC50 and SI, with cytotoxicity on LLC-MK2 cell lines. Overall, 1,3-thiazoles exhibited better trypanocidal activity than 4-thiazolidinones. The compound 1f, an ortho-bromobenzylidene-substituted 1,3-thiazole (IC50 = 0.83 μM), is the most potent of them all. In addition, compounds had negligible cytotoxicity in mammalian cells (CC50 values > 50 μM). Also noteworthy is the examination of the cell death mechanism of T. cruzi, which showed that compound 1f induced necrosis and apoptosis in the parasite. Scanning electron microscopy analysis demonstrated that the treatment of Trypanosoma cruzi trypomastigote cells with the compound 1f at different IC50 concentrations promoted alterations in the shape, flagella and body surface, inducing parasite death. Together, our data revealed a novel series of 1,3-thiazole structure-based compounds with promising activity against Trypanosoma cruzi and Leishmania spp., broadening ways for scaffold optimization.
Introduction
Leishmaniasis consists of a group of diseases caused by the Leishmania protozoan, a component of the Trypanosomatidae family, which also includes Trypanosoma cruzi, the etiological agent of chagas disease. Beyond other characteristics, they have the presence of differentiated mitochondria called kinetoplastid in common.1 They are so-called Neglected Tropical Diseases (NTDs) due to various characteristics regarding their occurrence, to be noted poor housing conditions, lack of an adequate sanitary system, treatment, control scheme, malnutrition and scarce resources for investment in research.2The current therapy of chagas disease is mainly based on two compounds, Nifurtimox (NFX) and Benznidazole (BZD), which were developed about 40 years ago,3 and are associated with long-term treatments, severe side effects and low access worldwide.4,5 In fact, NFX and BZD can eliminate latent parasitemia and reduce serological titters in acute and early chronic infections.6,7 However, they have low efficacy in prolonged chronic infections.8
The treatment of choice for leishmaniasis has been based on pentavalent antimonials.9 The second line treatment of chagas disease includes drugs, such as Amphotericin B, Pentamidine and Miltefosine.10 Most treatments are inadequate, owing to factors like a low therapeutic index, which leads to high toxicity and side effects, the emergence of resistant parasites, high costs beyond the means of the affected countries and others. These disadvantages, together with the lack of an effective vaccine, indicate the need to investigate new drugs for NTDs.10
Drug discovery approaches, for NTDs such as the aforementioned ones, have been a hot topic in medicinal chemistry in the last years since their last annual occurrence was of more than 1 billion people with significant mortality of 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000,11 calling for urgent attention to this topic.
000,11 calling for urgent attention to this topic.
Considering the lack of effective, affordable, or easy-to-use drug treatments for NTDs, the adoption of the Privileged Structures strategy has been advantageous to provide new chemical entities with good ‘drug-like’ properties. Privileged motifs are recurring in a wide range of biologically active compounds that reach different pharmaceutical targets and pathways. The drug-like properties of privileged structures and substructures might produce many drug-like compound libraries and leads.12,13
For example, 1,3-thiazole and 4-thiazolidinone nuclei are privileged structures that are found in several natural products, and have also been used to develop synthetic drugs and drug-like molecules with a variety of pharmacological effects.14–22
Taking account of their biological diversity and potential, together with the easy and rapid synthesis, 4-thiazolidinones and 1,3-thiazoles are outlined scaffolds in medicinal chemistry. A good strategy for optimizing bioactive compounds is the bioisosterism. This approach has been considered efficient through thiosemicarbazones cyclization to 1,3-thiazoles/4-thiazolidinones, taking to anti-T. cruzi and anti-Leishmania spp. activity enhancement.23–26
Indeed, we have used the bioisosteric strategy on previously synthesized compounds by our group. The thiosemicarbazones cyclization to 4-thiazolidinones obtained 23 different bioactive 4-oxo-thiazolidinones-5-acetic-acids. As a common structural feature, this series of compounds presented an acetic acid in the 5th position of the 4-oxo-thiazolidinones ring system, and a varied ring core at the hydrazone moiety. Some of these reported compounds showed potent trypanocidal activity and low toxicity to mammalian cells.27
Herein, we present two novel series, exploring the bioisosteric strategy on thiosemicarbazones cyclization to 1,3-thiazoles/4-thiazolidinones rings and developing a novel derivatives from the carboxylic acids produced by our group.27 The hypothesis was based on the concept of whether the classical medicinal chemistry using esterification could be used to simultaneously improve the solubility and permeability to obtain acetates. In two different series, we used a simple two-step synthesis approach to obtain novel 1,3-thiazoles (1a–k) and 4-thiazolidinones (2a–p) from the thiosemicarbazones nucleus, as summarized in Fig. 1. Following the synthesis, we characterized these molecules and evaluated their trypanocidal, leishmanicidal and cytotoxic activities. The utmost promising molecule was chosen to assess necrosis development on trypomastigotes by flow cytometry assay and evaluation of activity against the intracellular forms of T. cruzi, as well as evaluation of its ultrastructural impacts on T. cruzi trypomastigotes, seen by scanning electron microscopy (SEM). All data were collected to establish the structure–activity relationships (SAR), and provide a comparison with the previous results of their bioisosteric analogues27 and reference drugs. Hence, our work demonstrates that 1,3-thiazoles and 4-thiazolidinone acetates are a versatile building block for lead generation, easily yielding access to diverse derivatives for drug optimization.
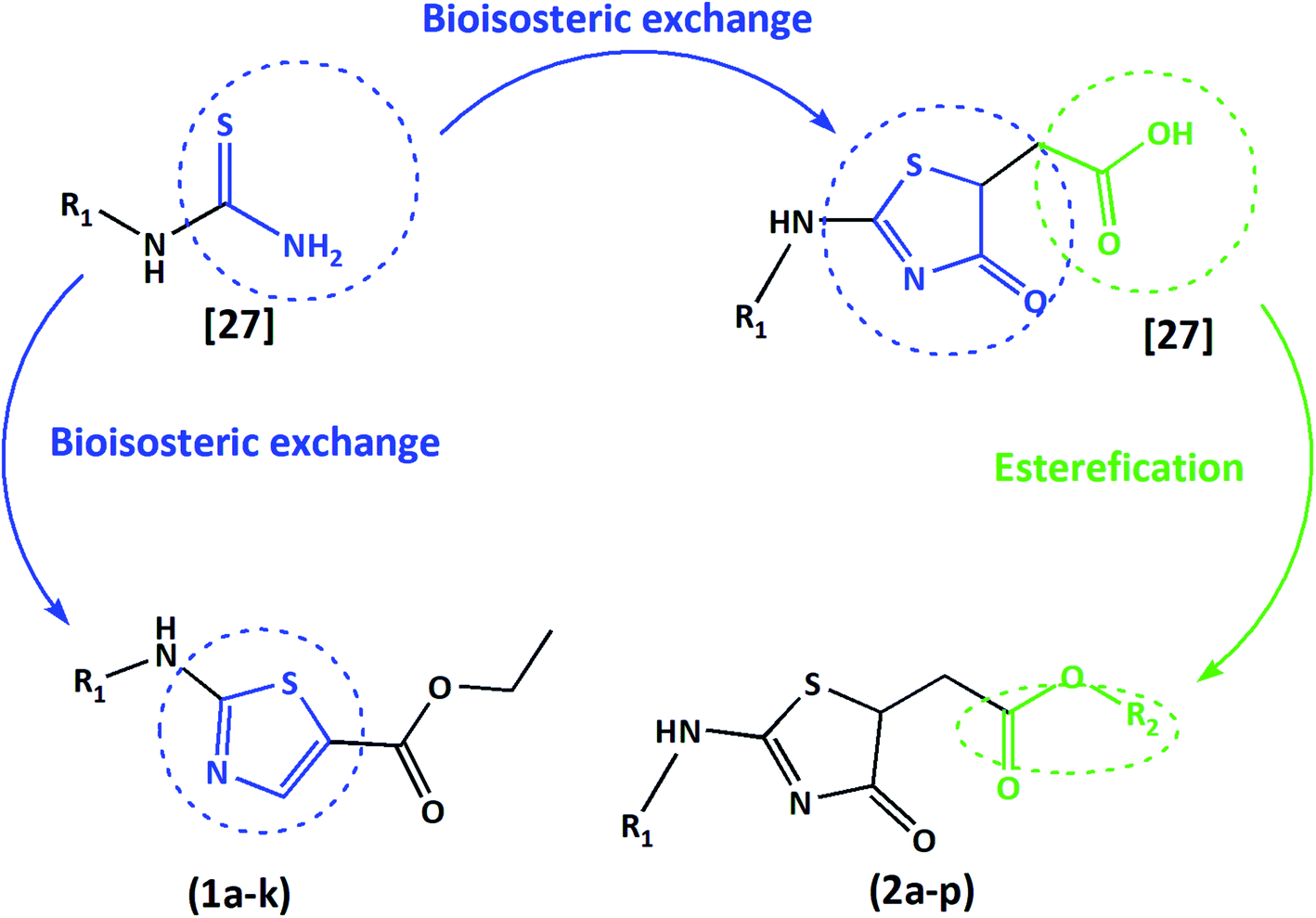 | ||
| Fig. 1 Summary of the structural planning of the novel 1,3-thiazoles and 4-thiazolidinones reported in this work, based on previous results.27 | ||
Results and discussion
Chemistry
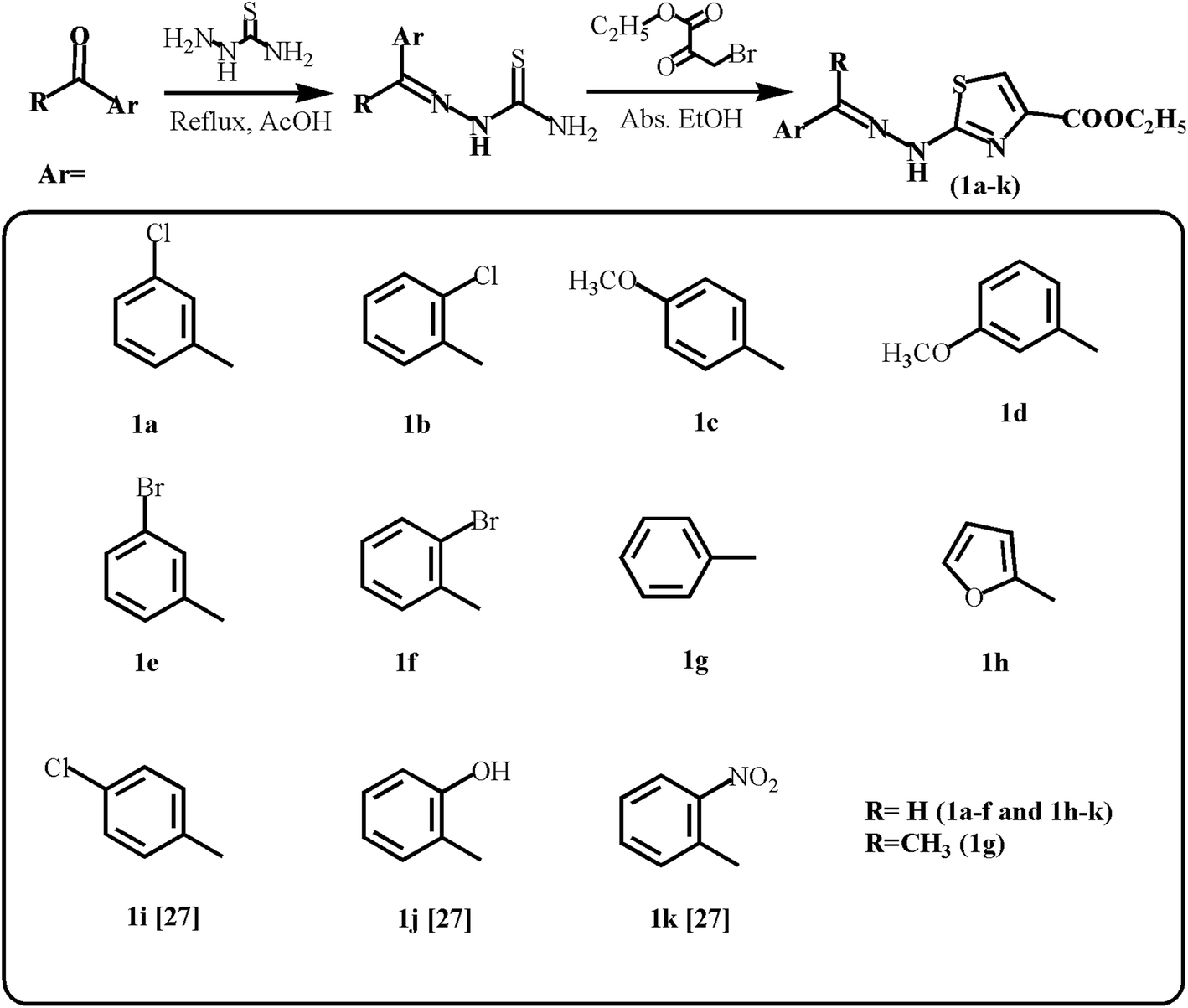 | ||
| Scheme 1 Synthetic route for ethyl 2-(2-(arylidene)hydrazinyl)thiazole-4-carboxylates (1a–h; 1i–k27). | ||
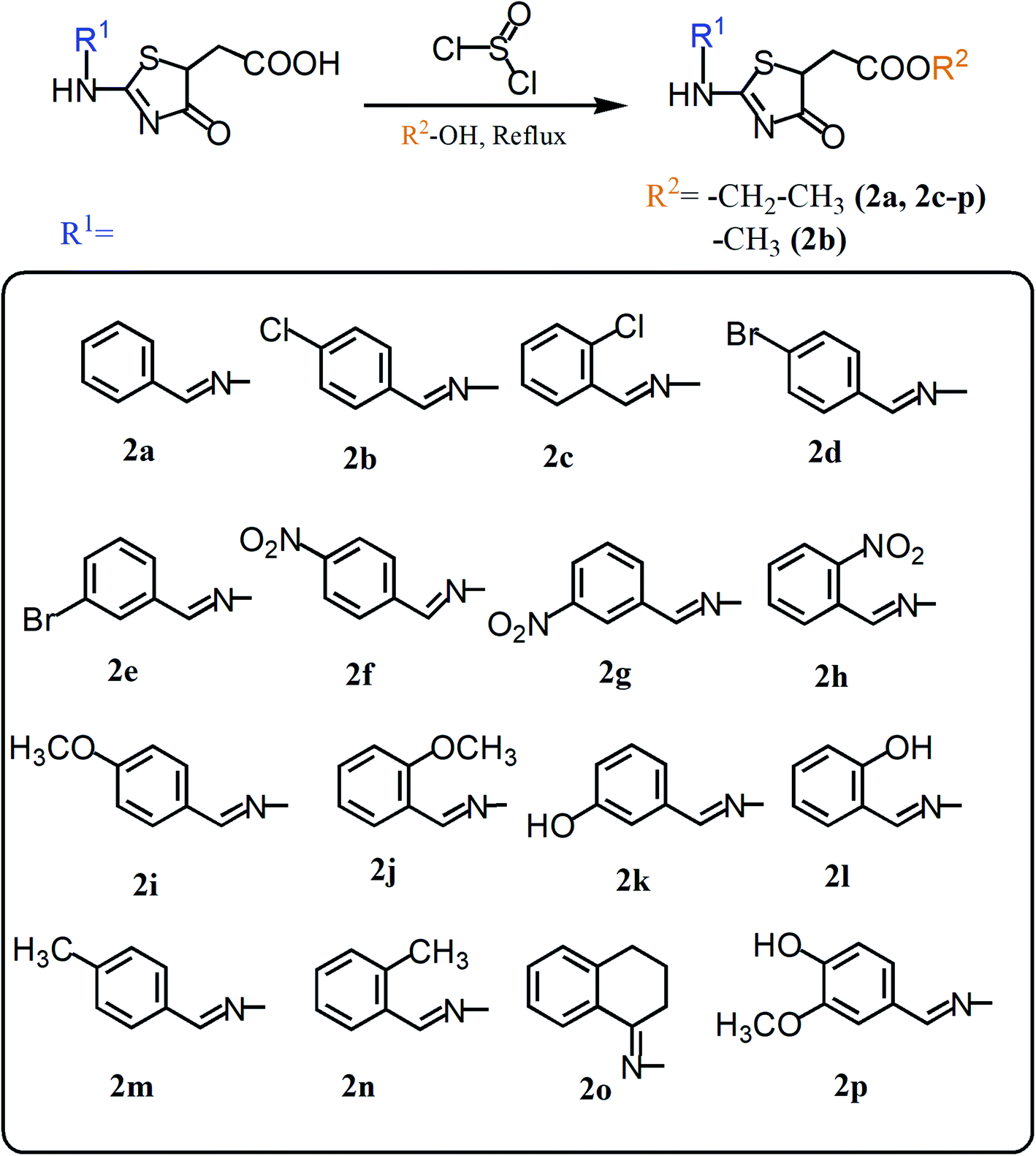 | ||
| Scheme 2 Synthetic route for alkyl 2-((2-(arylidene)hydrazinyl)-4-oxo-4,5-dihydrothiazol-5-yl)acetate (2a–p). | ||
Cytotoxic studies
The potential cytotoxic profile of novel compounds against mammalian cells was investigated by MTT assay28 using LLC-MK2 cells. Compounds 1a–k and 2a–p were added in increasing concentrations (10 to 1000 μM), and incubated for 96 hours for the determination of the 50% cytotoxic concentration (CC50). As we can see in Table 1, no compounds were considered cytotoxic to mammalian cells since all of the CC50 values were higher than 50 μM. However, compounds 1d, 1j, 1k, 2b and 2n exhibited higher CC50 values than the reference BZD with highlights to the o-methyl substituted 2n (CC50 = 770.37; SITrypo = 61) and the p-methyl substituted 2b (CC50 = 659.08; SIL.inf = 19.8) being less cytotoxic.| Compound code | LLC-MK2, CC50a (μM) | Trypomastigotes | Promastigotes | ||||
|---|---|---|---|---|---|---|---|
| EC50b (μM) | SIc | IC50b (μM) | SIc | ||||
| IC50L.Ama | IC50L.Inf | SIL.Ama | SIL.Inf | ||||
| a CC50 = cytotoxic concentration for 50% of the cells.b EC50 = effective concentration for 50% of the parasites.c SI = selectivity index (CC50/IC50).d NA = non-applicable; ND = no difference. | |||||||
| 1a | 538.34 | 4.44 | 121.2 | 189.27 | 164.32 | 2.8 | 3.3 |
| 1b | 258.4 | 12.50 | 20.7 | 27.89 | 14.39 | 9.3 | 18.0 |
| 1c | >1000 | 20.51 | >48.8 | 391.68 | 332.08 | 2.6 | 3.0 |
| 1d | 640.79 | 12.91 | 15.5 | 159.19 | 103.85 | 4.0 | 6.2 |
| 1e | 600.46 | 46.73 | 12.8 | 245.55 | 170.28 | 2.4 | 3.5 |
| 1f | 251.66 | 0.83 | 303.2 | 24.34 | 23.46 | 10.3 | 10.7 |
| 1g | 292.4 | 2.83 | 103.3 | 42.96 | 37.43 | 6.8 | 7.8 |
| 1h | 422.4 | 2.75 | 153.6 | 451.2 | 374.16 | 0.9 | 1.1 |
| 1i | 598.56 | 24.87 | 24.1 | 145.07 | 108.11 | 4.1 | 5.5 |
| 1j | 926.07 | ND | NA | 484.67 | 289.74 | 1.9 | 3.2 |
| 1k | 661.54 | ND | NA | 359.02 | 158.9 | 1.8 | 4.2 |
| 2a | 471.36 | 33.83 | 13.9 | 366.79 | 365.48 | 1.3 | 1.3 |
| 2b | 659.08 | ND | NA | 91.35 | 33.24 | 7.2 | 19.8 |
| 2c | 229.11 | ND | NA | 113.86 | 128.72 | 2.0 | 1.8 |
| 2d | 397.45 | 8.45 | 47.0 | 14.63 | 14.49 | 27.2 | 27.4 |
| 2e | 445.76 | 18.24 | 24.4 | 119.76 | 115.55 | 3.7 | 3.9 |
| 2f | 722.89 | ND | NA | 57.5 | 324.99 | 12.6 | 2.2 |
| 2g | 430.47 | ND | NA | 308.54 | 329.38 | 1.4 | 1.3 |
| 2h | 577.05 | ND | NA | 17.01 | 295.13 | 33.9 | 2.0 |
| 2i | 399.41 | 12.72 | 31.4 | 169.44 | 146.52 | 2.4 | 2.7 |
| 2j | 538.62 | 24.15 | 22.3 | 185.58 | 236.03 | 2.9 | 2.3 |
| 2k | 613.59 | ND | NA | 363.77 | 370.31 | 1.7 | 1.7 |
| 2l | 53.45 | 19.76 | 2.7 | 84.3 | 67.27 | 0.6 | 0.8 |
| 2m | 609.45 | 29.26 | 20.8 | 13.35 | 18.82 | 45.7 | 32.4 |
| 2n | 770.37 | 12.62 | 61.0 | 267.54 | 167.85 | 2.9 | 4.6 |
| 2o | 279.57 | 27.87 | 10.0 | 179.75 | 176.27 | 1.6 | 1.6 |
| 2p | 544.65 | 35.49 | 15.3 | 232.76 | 320.16 | 2.3 | 1.7 |
| BZD10 | 614.7 | 34.5 ± 7.6 | 17.8 | NA | NA | NA | NA |
| Miltefosine29 | 12.27 | NA | NA | 36.31 | 53.71 | 0.3 | 0.2 |
Trypanocidal activity evaluated against trypomastigote form
The compounds 1a–k and 2a–p were evaluated in vitro against T. cruzi trypomastigote (strain Y). As shown in Table 1, most of the tested compounds showed a promising IC50 value and selectivity in comparison to the reference drug, BZD (IC50 = 34.5 μM and SI = 17.8). The compounds that had outstanding performance with minimal inhibitory concentration for 50% of the parasite cells (less than 10 μM) were 1f, 1h, 1g, 1a and 2d (0.83 μM, 2.75 μM, 2.83 μM, 4.44 μM and 8.45 μM, respectively). The synthesized compounds 1h (a furan moiety derivative), 1g (benzylidene) and 1a (m-Cl-benzylidene) were over a hundred times more selective to T. cruzi than to the mammalian cells (SI = 153.6, 103.3 and 121.2, respectively), outlining 1f (o-Br-benzylidene) that performed a selectivity of 303.2 in comparison with 17.8 of BZD. Concerning the 4-thiazolidinones derivatives, it was verified that among the 2a–p series, compound 2d exhibited the best IC50 value (8.45 μM). Regarding the selectivity index, we can highlight that compound 2d (p-Br-benzylidene, SI = 47), 2e (m-Br-benzylidene, SI = 24.2), 2i (p-CH3O-benzylidene, SI = 31.4), 2j (o-CH3O-benzylidene, SI = 22.3), 2m (p-CH3-benzylidene, SI = 20.8), and 2n (o-CH3-benzylidene, SI = 61), with 2p (m-methoxy and p-hydroxy benzylidene, SI = 15.3) performed better than BZD. The methyl substitution on the aromatic ring was evidenced as a good change, resulting in satisfactory IC50 values in both synthesized positions (ortho and para), as well as the methoxy substitution. In addition, the two leading compounds were ortho-substituted in terms of the trypanocidal activity of both series, evidencing the importance of this position and corroborating our previous results with the non-cyclized 1g* (IC50 = 5.65 μM),27 which presents an o-chlorine substitution.In general, the 1,3-thiazole derivatives (1a–k) were more effective than the 4-thiazolidinones (2a–p) ones. The compounds 1f and 1h can be highlighted as the hit compounds, given their enhanced trypanocidal activity with high selectivity to parasite cells, and the low concentrations needed to kill half of the parasite population, with 1f at the nanomolar scale (830 nM).
Trypanocidal activity evaluated against the amastigote forms
The compounds 1a, 1g, 1h and 1f were tested in six different concentrations (0.625, 1.25, 2.5, 5, 10 and 20 μg mL−1) against T. cruzi amastigotes (Tulahuen strain). The analysis of the obtained results, displayed in Table 2, indicate that compound 1a was the best among the tested compounds, with an IC50 = 16.85 μM and SI = 31.9. Nevertheless, it was weaker than the standard drug BZD (IC50 = 5.65 μM, SI = 108.8).| Compound code | LLC-MK2, CC50a (μM) | Amastigote, IC50b (μM) | Amastigote, SIc |
|---|---|---|---|
| a CC50 = cytotoxic concentration for 50% of cells.b IC50 = inhibitory concentration for 50% of cells.c SI = selectivity index (CC50/IC50). | |||
| 1a | 538.34 | 16.85 | 31.9 |
| 1g | 292.4 | 40.71 | 7.2 |
| 1h | 422.4 | 75.39 | 5.6 |
| 1f | 251.66 | 56.46 | 4.5 |
| BZD | 614.7 | 5.65 | 108.8 |
Cell death assessment
After the outstanding trypanocidal activity, the cell death mechanism rates of the nanomolar-active compound 1f was investigated using BZD as a standard. This was assessed through conventional staining with Propidium Iodide (PI) and Annexin-V in the flow cytometry method. First, PI is a stain that passes within the cell membrane whenever it is damaged, reaching and binding directly to the DNA. On the other hand, Annexin V binds to the phospholipid phosphatidylserine that is found in the inner side of the cell membrane. There, it stains whenever this phospholipid is exposed, which happens when the cells go through the apoptosis process. Moreover, when the apoptotic process is late or involves necrosis, there is PI staining on DNA.31As shown in Fig. 2, compound 1f (at 1.66 μM and 3.32 μM) was able to induce significant labeling, which is consistent with necrosis in trypomastigotes. Similar results were found in parasites treated with BZD (34.5 μM, 70.8 μM, and 141.6 μM), the reference drug, and the positive control used in this assay (Fig. 2A). Furthermore, at 3.32 μM concentration of 1f, an equivalent necrosis was observed (17.58% double-stained) as that for the 70.8 μM of BZD (19.56% double-stained), reinforcing the potency of the 1,3-thiazole derivatives in comparison with the standard drug (Fig. 2B).
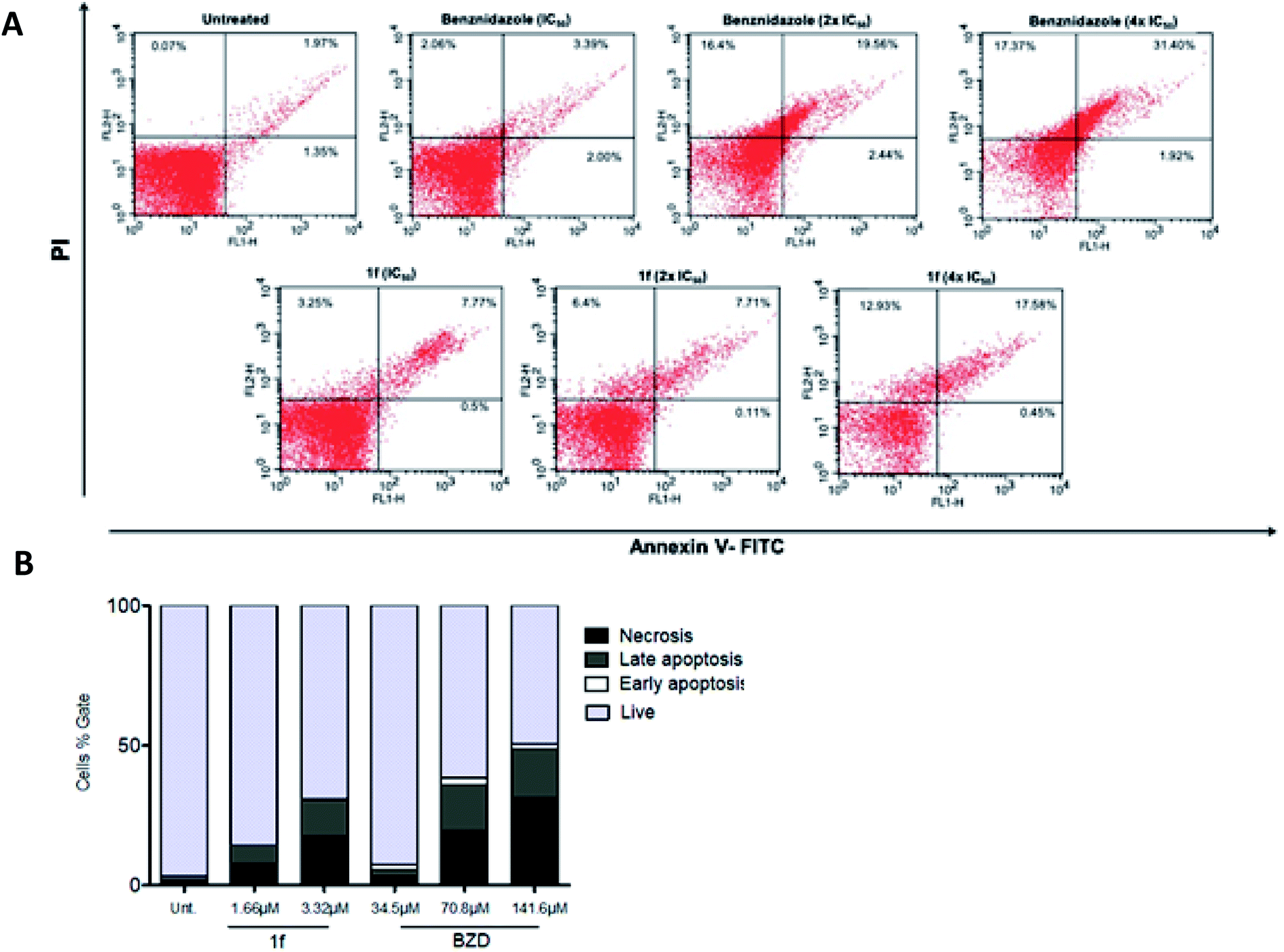 | ||
| Fig. 2 Flow cytometry results on T. cruzi trypomastigotes untreated and treated with 1f at 2 × IC50 (1.66 μM) and 4 × IC50 (3.32 μM); and treated with BZD at 1 × IC50 (34.5 μM), 2 × IC50 (70.8 μM), and 4 × IC50 (141.6 μM), following staining with PI and Annexin-V. (A) Plotted flow cytometry graphs with FITC on the X-axis and PI on the Y-axis. (B) Bar chart illustrating the proportions of each cell death mechanism among all concentrations of 1f and BZD tested, and comparison with the untreated group. The percentage of populations can be separated on double-negative (live) cells, Annexin-V positive (early apoptotic cells), PI-positive (late apoptotic), and double-positive (necroptotic) cells. All p values of the shown concentrations were below 0.05. The graphs were plotted with the GraphPad Prism ® software. | ||
Ultrastructural studies for T. cruzi
To evaluate the effects of 1,3-thiazoles on the parasite morphology, the most active compound of this work (1f) was selected. Scanning electron microscopy analysis (Fig. 3) showed that T. cruzi trypomastigotes in the control group had preserved morphologies in the body and flagella (Fig. 3A), while cells treated with BZD had their bodies contorted with the extravasation of cytoplasmic content (Fig. 3B). In contrast, cells treated with 1f at IC50, 2 × IC50, and 4 × IC50 (0.8 μM, 1.66 μM and 3.32 μM) concentrations, respectively, promoted dose-dependent alterations in the shape, flagella and surface of the parasite's body (Fig. 3C–E) when compared to the untreated control group. Furthermore, the compound 1f promoted body writhing with a swollen and rounded appearance, shortening of the flagellum, plus a disruption of the membrane with extravasation of the cytoplasmic content. The activity of compound 1f can be observed through ultrastructural alterations caused in T. cruzi, which certainly makes the cell unfeasible.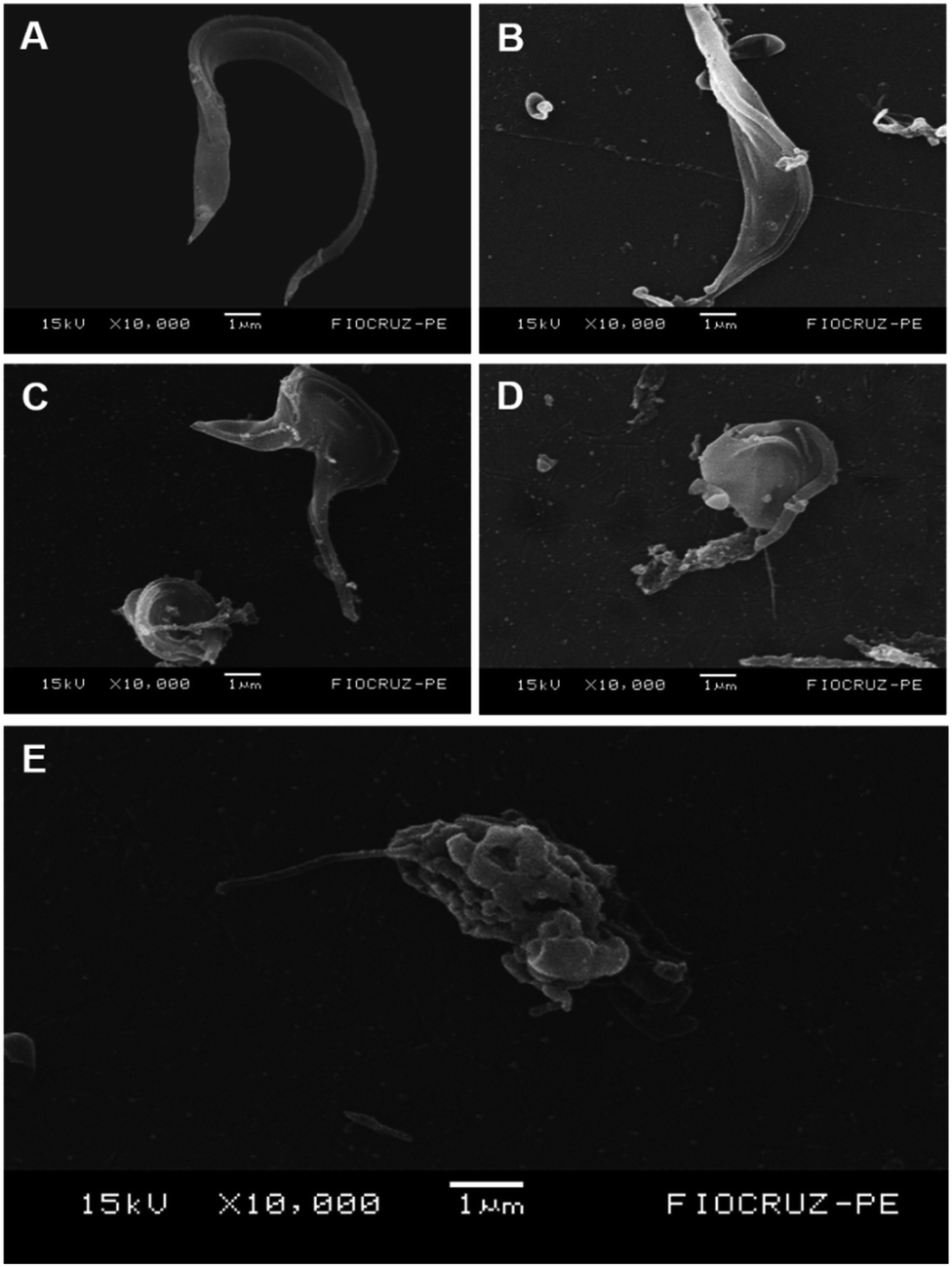 | ||
| Fig. 3 Ultrastructural alterations in trypomastigote forms of T. cruzi treated with BZD and compound 1f by SEM. (A) Control untreated trypomastigotes showing the typical elongated body with the slick cell surface and preserved flagellum. (B) Treated trypomastigotes with BZD (IC50 = 34.5 μM) showing a contorted and swollen body, with disruption displaying internal cell content. (C) Treated trypomastigotes with 1f (IC50 = 0.8 μM) showing body writhed with appearance swollen and rounded, beyond flagellum shortening. (D) Treated trypomastigotes with 1f (2 × IC50 = 1.66 μM), showing a rounded body, rupture of the cytoplasmic membrane, and exposure of internal cellular content. (E) Treated trypomastigotes with 1f (4 × IC50 = 3.32 μM) showing alterations in the flagella form and cell body, with total membrane destruction and extravasation of the cytoplasmic content. | ||
Leishmanicidal activity evaluated against promastigote forms
The ability of the new derivatives (1a–k and 2a–p) to inhibit the growth of the promastigote forms of L. amazonensis and L. infantum was investigated using Miltefosine as a standard.30 It was verified that all of the synthesized compounds were less toxic and more selective than the standard drug Miltefosine, as shown in Table 1.The compounds 1b, 1f, 2d and 2m exhibited better IC50 values for both L. amazonensis (27.89 μM, 23.34 μM, 14.63 μM and 13.35 μM) and L. infantum (14.39 μM, 23.46 μM, 14.49 μM and 18.81 μM), in comparison to the standard Miltefosine (36.31 μM and 53.71 μM). They also displayed good selectivity, with a focus on 2d (around 27 times, both) and 2m (SI = 45.7 and SI = 32.4) that had high SI for both Leishmania spp. tested. In addition, 2m performed the highest selectivity value for L. amazonensis of all tested compounds, being 46 times more selective to the parasite than to the LLC-MK2 cells (and 32.4 to L. infantum). Among the 1,3-thiazoles, the compound 1f was once again evidenced for its bioactivity (L. amazonensis – IC50 = 24.34 μM, SI = 10.3; L. infantum – IC50 = 23.46 μM, SI = 10.7), together with 1g (L. infantum – IC50 = 37.43 μM, SI = 7.8) and 1b (L. amazonensis – IC50 = 27.89 μM, SI = 9.3; L. infantum – IC50 = 14.39 μM, SI = 18). Furthermore, the ortho-substitution with a halogen group on the benzylidene moiety was demonstrated to be beneficial for the bioactivity of the substituted 1,3-thiazole-4-carboxylates, whereas the 4-thiazolidinone-5-acetates displayed enhanced leishmanicidal activity when para-substituted (2m, 2d, 2b). In addition, the compound 2d (p-Br-benzylidene) is outlined as a promising structure with satisfactory activity against both T. cruzi (SI = 47) and Leishmania sp. (SIL.ama = 27.2; SIL.inf = 27.4), having the same atom allocated on the benzylidene ring as 1f (o-bromine). Moreover, our strategy of esterification was proven to be efficient since the outstanding carboxylic acids synthesized by our group had their activity greatly improved, to enumerate: 1b* (IC50L.ama = 34.48 μM, IC50L.inf = 59.52 μM) p-bromine substituted that was esterified to 2d, and 1c* (IC50L.inf = 85.30 μM), p-chlorine that originated from 2b.27
The summarized SAR evaluation for both 1,3-thiazoles and 4-thiazolidinones can be seen in Fig. 4, while the trypanocidal and leishmanicidal activities regarding the SI values are compiled in Fig. 5.
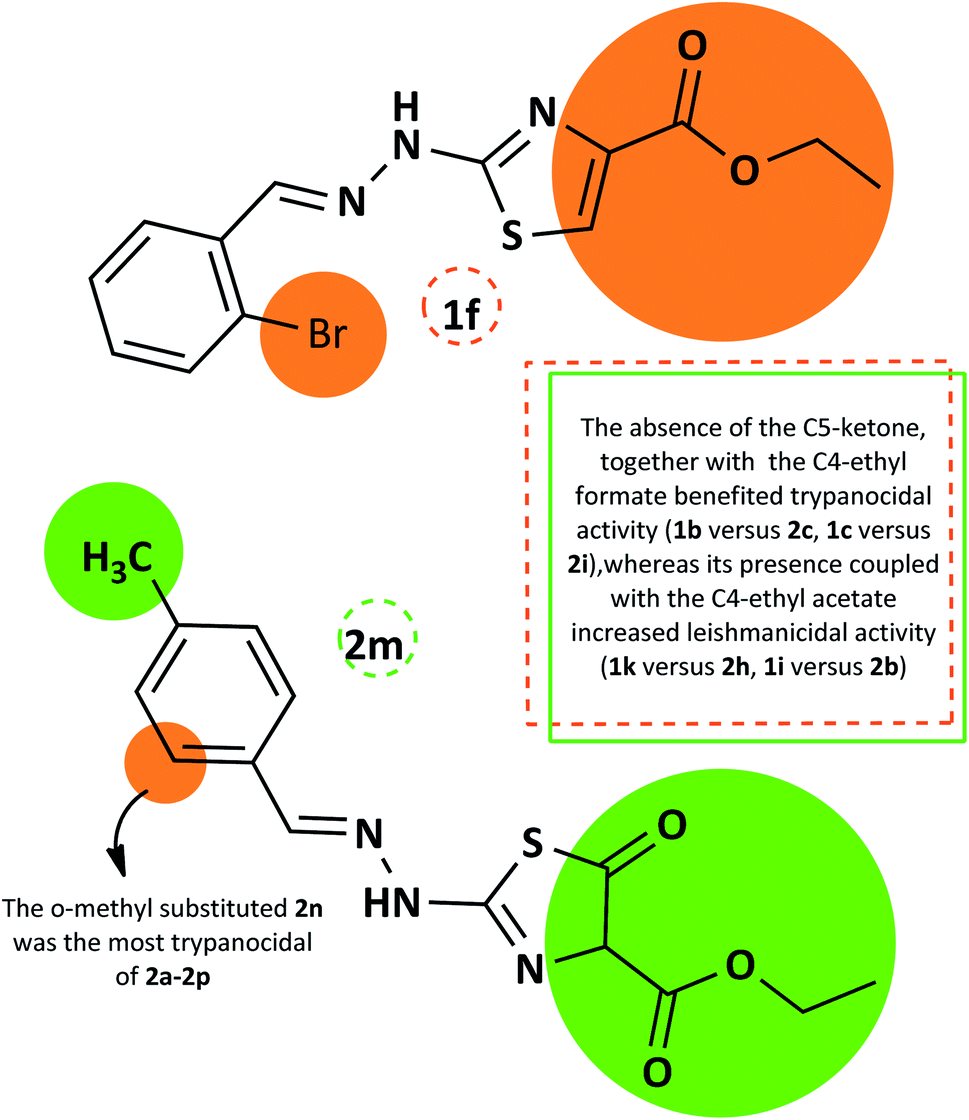 | ||
| Fig. 4 Summary of the SAR evaluation between the top compounds of each 1,3-thiazole series (1f) and 4-thiazolidinone series (2m), regarding the trypanocidal and leishmanicidal activity. | ||
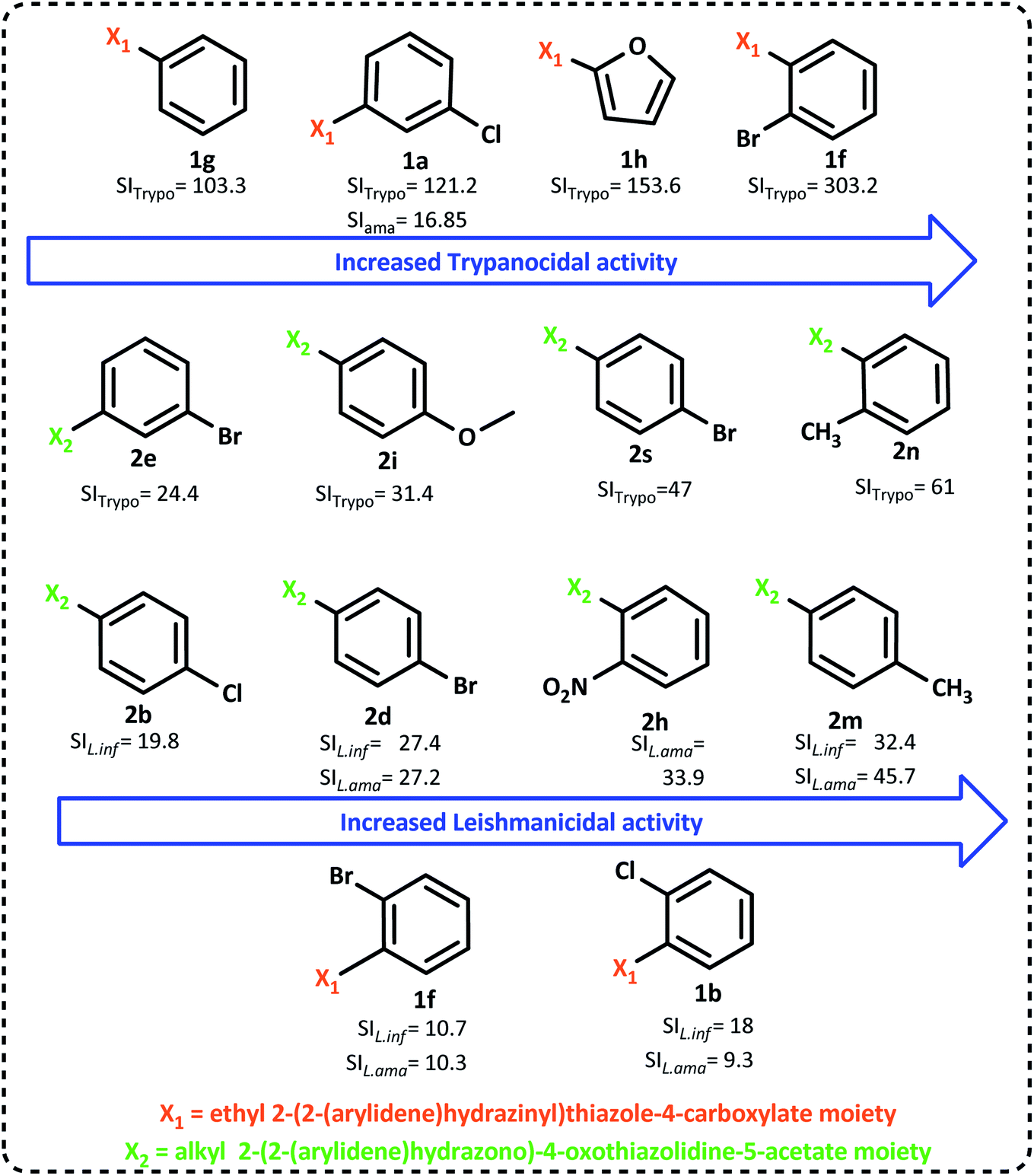 | ||
| Fig. 5 Improvement in the SI (selectivity index) values was observed by the change in the radicals of 1,3-thiazoles and 4-thiazolidinones. | ||
Experimental
Synthesis and characterization of novel 1,3-thiazole and thiazolidin-4-one analogues
High-purity grade reagents and solvents were used for the synthesis of ethyl 2-(2-(arylidene)hydrazinyl)thiazole-4-carboxylates (1a–k) and alkyl 2-((2-(arylidene)hydrazinyl)-4-oxo-4,5-dihydrothiazol-5-yl)acetates (2a–p). All synthesized esters of 1,3-thiazoles and thiazolidin-4-ones were initially characterized by their physical parameters, like the change in color, melting points, and the Rf (retardation factor) values. Spectroscopic techniques, like FT-IR, NMR and high-resolution mass spectrometry, were used for the confirmation of the synthesized compounds. To evaluate the purity of all synthesized compounds, the Rf values were calculated by thin-layer chromatography using silica gel 60 HF254 pre-coated aluminum sheets (Merck). Melting points were recorded on DMP-300 A&E Lab UK, apparatus. Characteristic functional groups were determined by the FT-IR spectrophotometer using ATR. Bruker 300 MHz, Varian VNMRS 400 MHz, and 500 MHz spectrometers were used to check protons and carbon signals. HRMS was recorded using a MALDI-TOF mass spectrometer, Bruker. The purity of the selected compound was calculated by qualitative HNMR method using qHNMR normalization (100%) method, established by Pauli et al. The spectra were recorded using this optimized protocol.32Synthesis and characterization of ethyl 2-(2-(arylidene)hydrazinyl)thiazole-4-carboxylates (1a–k)
Different aryl-substituted thiosemicarbazones (1.0 mmol) and ethyl bromopyruvate (1:1 equivalent) were refluxed in absolute ethanol for 4 to 6 hours. Thin-layer chromatography (TLC, acetone:n-hexane; 1:2) was used to check the completion and purity of the reaction. The reaction mixture solidified when put on ice. The solid was filtered and washed with plenty of water to obtain a pure product, and dried at room temperature.
:2); IR (ATR, cm−1): 1093 (C–O stretching, ester), 1446 (C–H bending, aliphatic), 1573 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C ring stretching), 1716 (CO stretching), 2987 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.28 (3H, t, –CH3 ester, J = 7.20 Hz), 4.25 (2H, q, –CH2–CH3 ester, J = 7.20 Hz), 7.44 (2H, m, Ar-H), 7.61 (1H, m, Ar-H), 7.70 (1H, m, Ar-H), 7.78 (1H, s, 1,3-thiazole ring C-5), 7.90 (1H, s, –CHN- azomethine), 12.49 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 60.9 (–OCH2– ester), 119.7 (1,3-thiazole ring C-5), 125.5, 126.1, 129.5, 131.2, 134.2, 136.9 (Ar-C), 140.6 (–CHN– azomethine), 143.6 (1,3-thiazole ring C-4), 161.4 (CO), 168.4 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C13H12ClN3NaO2S (M + Na) = 332.02364, found: 332.01055.:2); IR (ATR, cm−1): 1091 (C–O stretching, ester), 1423 (C–H bending, aliphatic), 1585 (CC ring stretching), 1687 (CN stretching), 1724 (CO stretching), 2985 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.27 (3H, t, –CH3 ester, J = 7.20 Hz), 4.28 (2H, q, –CH2–CH3 ester, J = 7.20 Hz), 7.39 (2H, m, Ar-H), 7.47 (1H, m, Ar-H), 7.79 (1H, s, 1,3-thiazole ring C-5), 7.90 (1H, m, Ar-H), 8.34 (1H, s, –CHN– azomethine), 12.48 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 60.9 (–OCH2– ester), 119.8 (1,3-thiazole ring C-5), 126.7, 128.1, 130.4, 131.3, 131.8, 132.7 (Ar-C), 138.0 (–CHN– azomethine), 143.3 (1,3-thiazole ring C-4), 161.4 (CO), 168.3 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C13H12ClN3NaO2S (M + Na) = 332.02364, found: 331.99641.:2); IR (ATR, cm−1): 1091 (C–O stretching, ester), 1423 (C–H bending, aliphatic), 1585 (CC ring stretching), 1687 (CN stretching), 1724 (CO stretching), 2985 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.28 (3H, t, –CH3 ester, J = 7.20 Hz), 3.79 (3H, s, –OCH3), 4.23 (2H, q, –CH2–CH3 ester, J = 7.20 Hz), 6.99 (2H, d, Ar-H, J = 8.70 Hz), 7.59 (2H, d, Ar-H, J = 8.70 Hz), 7.73 (1H, s, 1,3-thiazole ring C-5), 7.95 (1H, s, –CHN– azomethine), 12.14 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 55.7 (–OCH3), 60.8 (–OCH2– ester), 119.1 (1,3-thiazole ring C-5), 114.8, 127.2, 128.4, 160.8 (Ar-C), 142.4 (-CHN- azomethine), 143.3 (1,3-thiazole ring C-4), 161.5 (CO), 168.7 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C14H15N3NaO3S (M + Na) = 328.0732, found: 327.97609.:2); IR (ATR, cm−1): 1071 (C–O stretching, ester), 1456 (C–H bending, aliphatic), 1573 (CC ring stretching), 1617 (CN stretching), 1708 (CO stretching), 2980 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.28 (3H, t, –CH3 ester, J = 7.20 Hz), 3.79 (3H, s, –OCH3), 4.24 (2H, q, –CH2–CH3 ester, J = 6.90 Hz), 6.95 (1H, m, Ar-H), 7.22 (2H, m, Ar-H), 7.33 (1H, t, Ar-H, J = 7.80 Hz), 7.76 (1,3-thiazole ring C-5), 7.97 (1H, s, –CHN– azomethine), 12.34 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 55.5 (–OCH3), 60.9 (–OCH2– ester), 119.4 (1,3-thiazole ring C-5), 111.6, 115.8, 119.4, 130.4, 136.0, 159.9 (Ar-C), 142.1 (–CHN– azomethine), 143.3 (1,3-thiazole ring C-4), 161.5 (CO), 168.6 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C14H15N3NaO3S (M + Na) = 328.0732, found: 327.96025.:2); IR (ATR, cm−1): 1095 (C–O stretching, ester), 1446 (C–H bending, aliphatic), 1579 (CC ring stretching), 1687 (CN stretching), 1724 (CO stretching), 2999 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.28 (3H, t, –CH3 ester, J = 7.20 Hz), 4.24 (2H, q, –CH2–CH3 ester, J = 7.20 Hz), 7.38 (1H, t, Ar-H, J = 7.80 Hz), 7.56 (1H, d, Ar-H, J = 8.70 Hz), 7.65 (1H, d, Ar-H, J = 7.80 Hz), 7.78 (1H, s, 1,3-thiazole ring C-5), 7.83 (1H, s, Ar-H), 7.96 (1H, s, –CHN– azomethine), 12.44 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 60.9 (–OCH2– ester), 119.7 (1,3-thiazole ring C-5), 122.7, 125.8, 128.9, 131.5, 132.4, 137.1 (Ar-C), 140.5 (–CHN– azomethine), 143.3 (1,3-thiazole ring C-4), 161.41 (CO), 168.4 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C13H12BrN3NaO2S (M + Na) = 375.97313, found: 376.16224.:2); IR (ATR, cm−1): 1103 (C–O stretching, ester), 1431 (C–H bending, aliphatic), 1575 (CC ring stretching), 1687 (CN stretching), 1699 (CO stretching), 3045 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.27 (3H, t, –CH3 ester, J = 7.09 Hz), 4.23 (2H, q, –CH2–CH3 ester, J = 7.10 Hz), 7.30 (1H, ddd, Ar-H, J = 1.72 Hz, J = 7.23 Hz, J = 8.16 Hz), 7.44 (1H, m, Ar-H), 7.65 (1H, dd, Ar-H, J = 1.17 Hz, J = 8.05 Hz), 7.78 (1H, s, 1,3-thiazole ring C-5), 7.87 (1H, dd, Ar-H, J = 1.69 Hz, J = 7.91 Hz), 8.30 (1H, s, –CHN– azomethine), 12.50 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 60.9 (–OCH2– ester), 119.8 (1,3-thiazole ring C-5), 123.1, 127.1, 128.6, 131.5, 133.3, 133.6 (Ar-C), 140.4 (–CHN– azomethine), 143.3 (1,3-thiazole ring C-4), 161.4 (CO), 168.3 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C13H12BrN3NaO2S (M + Na) = 375.97313, found: 376.12838.:2); IR (ATR, cm−1): 1087 (C–O stretching, ester), 1442 (C–H bending, aliphatic), 1573 (CC ring stretching), 1699 (CN stretching), 1712 (CO stretching), 2978 (C–H stretching, aliphatic); 1H-NMR (500 MHz): δ 1.27 (3H, t, –CH3 ester, J = 7.14 Hz), 2.29 (3H, s, –CH3), 4.23 (2H, q, –CH2–CH3 ester, J = 7.08 Hz), 7.39 (3H, m, Ar-H), 7.75 (2H, m, Ar-H + 1,3-thiazole ring C-5), 7.91 (1H, s, –CHN– azomethine), 12.31 (1H, s, –N–NH–C–); 13C-NMR (126 MHz): δ 14.2 (–CH3 ester), 14.9 (–CH3), 60.8 (–OCH2– ester), 119.7 (1,3-thiazole ring C-5), 126.2, 128.7, 128.9, 129.3 (Ar-C), 138.2 (–CHN– azomethine), 147.4 (1,3-thiazole ring C-4), 161.6 (CO), 170.1 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C14H15N3NaO2S (M + Na) = 312.0783, found: 311.86282.:2); IR (ATR, cm−1): 1091 (C–O stretching, ester), 1431 (C–H bending, aliphatic), 1573 (CC ring stretching), 1622 (CN stretching), 1720 (CO stretching), 2976 (C–H stretching, aliphatic); 1H-NMR (500 MHz): δ 1.26 (3H, t, –CH3 ester, J = 7.12 Hz), 4.22 (2H, q, –CH2–CH3 ester, J = 7.05 Hz), 6.59 (1H, m, Ar-H), 6.81 (1H, d, Ar-H, J = 3.41 Hz), 7.73 (1H, s, 1,3-thiazole ring C-5), 7.78 (1H, m, Ar-H), 7.86 (1H, s, –CHN– azomethine), 12.54 (1H, s, –N–NH–C–); 13C-NMR (126 MHz): δ 14.6 (–CH3 ester), 60.8 (–OCH2– ester), 119.7 (1,3-thiazole ring C-5), 112.5, 113.1, 145.1, 149.5 (Ar-C), 132.6 (–CHN– azomethine), 143.2 (1,3-thiazole ring C-4), 161.4 (CO), 168.3 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C11H11N3NaO3S (M + Na) = 288.0419, found: 287.63438.
C ring stretching), 1716 (CO stretching), 2987 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.28 (3H, t, –CH3 ester, J = 7.20 Hz), 4.25 (2H, q, –CH2–CH3 ester, J = 7.20 Hz), 7.44 (2H, m, Ar-H), 7.61 (1H, m, Ar-H), 7.70 (1H, m, Ar-H), 7.78 (1H, s, 1,3-thiazole ring C-5), 7.90 (1H, s, –CHN- azomethine), 12.49 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 60.9 (–OCH2– ester), 119.7 (1,3-thiazole ring C-5), 125.5, 126.1, 129.5, 131.2, 134.2, 136.9 (Ar-C), 140.6 (–CHN– azomethine), 143.6 (1,3-thiazole ring C-4), 161.4 (CO), 168.4 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C13H12ClN3NaO2S (M + Na) = 332.02364, found: 332.01055.:2); IR (ATR, cm−1): 1091 (C–O stretching, ester), 1423 (C–H bending, aliphatic), 1585 (CC ring stretching), 1687 (CN stretching), 1724 (CO stretching), 2985 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.27 (3H, t, –CH3 ester, J = 7.20 Hz), 4.28 (2H, q, –CH2–CH3 ester, J = 7.20 Hz), 7.39 (2H, m, Ar-H), 7.47 (1H, m, Ar-H), 7.79 (1H, s, 1,3-thiazole ring C-5), 7.90 (1H, m, Ar-H), 8.34 (1H, s, –CHN– azomethine), 12.48 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 60.9 (–OCH2– ester), 119.8 (1,3-thiazole ring C-5), 126.7, 128.1, 130.4, 131.3, 131.8, 132.7 (Ar-C), 138.0 (–CHN– azomethine), 143.3 (1,3-thiazole ring C-4), 161.4 (CO), 168.3 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C13H12ClN3NaO2S (M + Na) = 332.02364, found: 331.99641.:2); IR (ATR, cm−1): 1091 (C–O stretching, ester), 1423 (C–H bending, aliphatic), 1585 (CC ring stretching), 1687 (CN stretching), 1724 (CO stretching), 2985 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.28 (3H, t, –CH3 ester, J = 7.20 Hz), 3.79 (3H, s, –OCH3), 4.23 (2H, q, –CH2–CH3 ester, J = 7.20 Hz), 6.99 (2H, d, Ar-H, J = 8.70 Hz), 7.59 (2H, d, Ar-H, J = 8.70 Hz), 7.73 (1H, s, 1,3-thiazole ring C-5), 7.95 (1H, s, –CHN– azomethine), 12.14 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 55.7 (–OCH3), 60.8 (–OCH2– ester), 119.1 (1,3-thiazole ring C-5), 114.8, 127.2, 128.4, 160.8 (Ar-C), 142.4 (-CHN- azomethine), 143.3 (1,3-thiazole ring C-4), 161.5 (CO), 168.7 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C14H15N3NaO3S (M + Na) = 328.0732, found: 327.97609.:2); IR (ATR, cm−1): 1071 (C–O stretching, ester), 1456 (C–H bending, aliphatic), 1573 (CC ring stretching), 1617 (CN stretching), 1708 (CO stretching), 2980 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.28 (3H, t, –CH3 ester, J = 7.20 Hz), 3.79 (3H, s, –OCH3), 4.24 (2H, q, –CH2–CH3 ester, J = 6.90 Hz), 6.95 (1H, m, Ar-H), 7.22 (2H, m, Ar-H), 7.33 (1H, t, Ar-H, J = 7.80 Hz), 7.76 (1,3-thiazole ring C-5), 7.97 (1H, s, –CHN– azomethine), 12.34 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 55.5 (–OCH3), 60.9 (–OCH2– ester), 119.4 (1,3-thiazole ring C-5), 111.6, 115.8, 119.4, 130.4, 136.0, 159.9 (Ar-C), 142.1 (–CHN– azomethine), 143.3 (1,3-thiazole ring C-4), 161.5 (CO), 168.6 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C14H15N3NaO3S (M + Na) = 328.0732, found: 327.96025.:2); IR (ATR, cm−1): 1095 (C–O stretching, ester), 1446 (C–H bending, aliphatic), 1579 (CC ring stretching), 1687 (CN stretching), 1724 (CO stretching), 2999 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.28 (3H, t, –CH3 ester, J = 7.20 Hz), 4.24 (2H, q, –CH2–CH3 ester, J = 7.20 Hz), 7.38 (1H, t, Ar-H, J = 7.80 Hz), 7.56 (1H, d, Ar-H, J = 8.70 Hz), 7.65 (1H, d, Ar-H, J = 7.80 Hz), 7.78 (1H, s, 1,3-thiazole ring C-5), 7.83 (1H, s, Ar-H), 7.96 (1H, s, –CHN– azomethine), 12.44 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 60.9 (–OCH2– ester), 119.7 (1,3-thiazole ring C-5), 122.7, 125.8, 128.9, 131.5, 132.4, 137.1 (Ar-C), 140.5 (–CHN– azomethine), 143.3 (1,3-thiazole ring C-4), 161.41 (CO), 168.4 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C13H12BrN3NaO2S (M + Na) = 375.97313, found: 376.16224.:2); IR (ATR, cm−1): 1103 (C–O stretching, ester), 1431 (C–H bending, aliphatic), 1575 (CC ring stretching), 1687 (CN stretching), 1699 (CO stretching), 3045 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.27 (3H, t, –CH3 ester, J = 7.09 Hz), 4.23 (2H, q, –CH2–CH3 ester, J = 7.10 Hz), 7.30 (1H, ddd, Ar-H, J = 1.72 Hz, J = 7.23 Hz, J = 8.16 Hz), 7.44 (1H, m, Ar-H), 7.65 (1H, dd, Ar-H, J = 1.17 Hz, J = 8.05 Hz), 7.78 (1H, s, 1,3-thiazole ring C-5), 7.87 (1H, dd, Ar-H, J = 1.69 Hz, J = 7.91 Hz), 8.30 (1H, s, –CHN– azomethine), 12.50 (1H, s, –N–NH–C–); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 60.9 (–OCH2– ester), 119.8 (1,3-thiazole ring C-5), 123.1, 127.1, 128.6, 131.5, 133.3, 133.6 (Ar-C), 140.4 (–CHN– azomethine), 143.3 (1,3-thiazole ring C-4), 161.4 (CO), 168.3 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C13H12BrN3NaO2S (M + Na) = 375.97313, found: 376.12838.:2); IR (ATR, cm−1): 1087 (C–O stretching, ester), 1442 (C–H bending, aliphatic), 1573 (CC ring stretching), 1699 (CN stretching), 1712 (CO stretching), 2978 (C–H stretching, aliphatic); 1H-NMR (500 MHz): δ 1.27 (3H, t, –CH3 ester, J = 7.14 Hz), 2.29 (3H, s, –CH3), 4.23 (2H, q, –CH2–CH3 ester, J = 7.08 Hz), 7.39 (3H, m, Ar-H), 7.75 (2H, m, Ar-H + 1,3-thiazole ring C-5), 7.91 (1H, s, –CHN– azomethine), 12.31 (1H, s, –N–NH–C–); 13C-NMR (126 MHz): δ 14.2 (–CH3 ester), 14.9 (–CH3), 60.8 (–OCH2– ester), 119.7 (1,3-thiazole ring C-5), 126.2, 128.7, 128.9, 129.3 (Ar-C), 138.2 (–CHN– azomethine), 147.4 (1,3-thiazole ring C-4), 161.6 (CO), 170.1 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C14H15N3NaO2S (M + Na) = 312.0783, found: 311.86282.:2); IR (ATR, cm−1): 1091 (C–O stretching, ester), 1431 (C–H bending, aliphatic), 1573 (CC ring stretching), 1622 (CN stretching), 1720 (CO stretching), 2976 (C–H stretching, aliphatic); 1H-NMR (500 MHz): δ 1.26 (3H, t, –CH3 ester, J = 7.12 Hz), 4.22 (2H, q, –CH2–CH3 ester, J = 7.05 Hz), 6.59 (1H, m, Ar-H), 6.81 (1H, d, Ar-H, J = 3.41 Hz), 7.73 (1H, s, 1,3-thiazole ring C-5), 7.78 (1H, m, Ar-H), 7.86 (1H, s, –CHN– azomethine), 12.54 (1H, s, –N–NH–C–); 13C-NMR (126 MHz): δ 14.6 (–CH3 ester), 60.8 (–OCH2– ester), 119.7 (1,3-thiazole ring C-5), 112.5, 113.1, 145.1, 149.5 (Ar-C), 132.6 (–CHN– azomethine), 143.2 (1,3-thiazole ring C-4), 161.4 (CO), 168.3 (1,3-thiazole ring C-2); HRMS (MALDI-TOF): calcd C11H11N3NaO3S (M + Na) = 288.0419, found: 287.63438.Synthesis of alkyl 2-(2-(arylidene)hydrazono)-4-oxothiazolidine-5-acetates (2a–p)
Thionyl chloride (2.0 equivalent) and the respective alcohol (20 mL) were initially stirred together at 0 °C, followed by the addition of 2-(2-(arylidene)hydrazono)-4-oxothiazolidine-5-acetic acids (1.0 mmol). The reaction mixture was further stirred at room temperature for 30 to 50 minutes. The reaction mixture was then refluxed for 30 to 50 minutes. Conversion of acids into respective esters was observed by taking TLC (acetone:n-hexane; 1:3). The solid appeared when the reaction mixture was put on ice, which was filtered and washed with an excess of water. The esters were dried at room temperature.
:3); IR (ATR, cm−1): 1018 (C–O stretching, ester), 1338 (C–H bending, aliphatic), 1516 (CC ring stretching), 1643 (CN stretching), 1708 (CO stretching), 2993 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.19 (3H, t, –CH3, ester, J = 7.05 Hz), 2.68 (1H, dd, –CH2–, J = 9.70 Hz, J = 16.95 Hz), 3.08 (1H, dd, –CH2-, J = 3.75 Hz, J = 16.95 Hz), 4.10 (2H, q, –CH2–CH3 ester, J = 6.45 Hz), 4.50 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.50 Hz, J = 7.70 Hz), 7.42 (3H, m, Ar-H), 7.70 (2H, m, Ar-H), 8.29 (1H, s, –CHN– azomethine); 13C-NMR (75 MHz): δ 14.5 (–CH3 ester), 38.6 (–CH2–), 45.8 (1,3-thiazolidin-4-one ring C-5), 60.9 (–OCH2– ester), 127.5, 127.8, 129.2, 130.1 (Ar-C), 135.7 (–CHN– azomethine), 152.6 (1,3-thiazolidin-4-one ring C-2), 171.5 (CO), 181.9 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C14H15N3NaO3S (M + Na) = 328.07318, found: 329.35526.:3); IR (ATR, cm−1): 1087 (C–O stretching, ester), 1442 (C–H bending, aliphatic), 1600 (CC ring stretching), 1643 (CN stretching), 1712 (CO stretching), 1735 (CO stretching), 2985 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 3.01 (1H, dd, –CH2–, J = 8.40 Hz, J = 17.70 Hz), 3.11 (1H, dd, –CH2-, J = 4.30 Hz, J = 17.30 Hz), 3.64 (3H, s, –OCH3), 4.42 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.20 Hz, J = 8.30 Hz), 7.56 (2H, m, Ar-H), 7.77 (2H, d, Ar-H, J = 8.40 Hz), 8.41 (1H, s, –CHN– azomethine), 12.08 (1H, s, –NH); 13C-NMR (75 MHz): δ 36.6 (-CH2-), 43.8 (1,3-thiazolidin-4-one ring C-5), 52.4 (–OCH3), 129.4, 130.5, 133.1, 136.5 (Ar-C), 155.5 (–CHN– azomethine), 165.3 (1,3-thiazolidin-4-one ring C-2), 171.3 (CO), 175.8 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C12H13ClN3NaO3S (M + Na) = 348.0186, found: 348.04657.:3); IR (ATR, cm−1): 1086 (C–O stretching, ester), 1445 (C–H bending, aliphatic), 1601 (CC ring stretching), 1647 (CN stretching), 1710 (CO stretching), 1731 (CO stretching), 2985 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.17 (3H, t, –CH3, ester, J = 7.03 Hz), 2.67 (1H, dd, –CH2-, J = 9.69 Hz, J = 16.95 Hz), 3.08 (1H, dd, –CH2–, J = 3.70 Hz, J = 16.95 Hz), 4.08 (2H, q, –CH2–CH3 ester, J = 6.45 Hz), 4.42 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.20 Hz, J = 8.30 Hz), 7.56 (2H, m, Ar-H), 7.77 (2H, d, Ar-H, J = 8.40 Hz), 8.41 (1H, s, –CHN– azomethine), 12.02 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.5 (–CH3 ester), 38.7 (–CH2–), 45.7 (1,3-thiazolidin-4-one ring C-5), 60.8 (–OCH2– ester), 127.5, 127.8, 131.2, 132.1 (Ar-C), 136.7 (-CHN- azomethine), 153.6 (1,3-thiazolidin-4-one ring C-2), 173.5 (CO), 181.9 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C14H14ClN3NaO3S (M + Na) = 362.03421, found: 362.12087.:3); IR (ATR, cm−1): 1010 (C–O stretching, ester), 1334 (C–H bending, aliphatic), 1512 (CC ring stretching), 1643 (CN stretching), 1712 (CO stretching), 2981 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.18 (3H, t, –CH3, J = 6.90 Hz), 2.98 (1H, dd, –CH2–, J = 7.80 Hz, J = 17.40 Hz), 3.08 (1H, dd, –CH2–, J = 4.50 Hz, J = 17.85 Hz), 4.09 (2H, q, –CH2–CH3 ester, J = 6.60 Hz), 4.41 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.50 Hz, J = 7.80 Hz), 7.69 (4H, m, Ar-H), 8.40 (1H, s, –CHN– azomethine), 12.09 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.4 (–CH3), 36.8 (–CH2–), 43.8 (1,3-thiazolidin-4-one ring C-5), 61.1 (–OCH2– ester), 124.5, 129.7, 129.9, 130.7, 132.1, 132.3 (Ar-C), 132.4 (–CHN– azomethine), 133.9 (1,3-thiazolidin-4-one ring C-2), 155.7 (CO), 170.6 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C14H14BrN3NaO3S (M + Na) = 405.98369, found: 406.43054.:3); IR (ATR, cm−1): 1021 (C–O stretching, ester), 1341 (C–H bending, aliphatic), 1600 (CC ring stretching), 1647 (CN stretching), 1710 (CO stretching), 2981 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.19 (3H, t, –CH3, J = 6.89 Hz), 2.99 (1H, dd, –CH2–, J = 7.80 Hz, J = 17.40 Hz), 3.08 (1H, dd, –CH2–, J = 4.50 Hz, J = 17.85 Hz), 4.10 (2H, q, –CH2–CH3 ester, J = 6.65 Hz), 4.42 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.52 Hz, J = 7.80 Hz), 7.78 (4H, m, Ar-H), 8.42 (1H, s, –CHN– azomethine), 11.51 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.4 (–CH3), 38.7 (–CH2–), 43.9 (1,3-thiazolidin-4-one ring C-5), 61.2 (–OCH2– ester), 124.5, 129.7, 129.9, 130.7, 132.1, 132.4 (Ar-C), 133.5 (–CHN– azomethine), 135.9 (1,3-thiazolidin-4-one ring C-2), 159.7 (CO), 172.6 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C14H14BrN3NaO3S (M + Na) = 405.98369, found: 406.89014.:3); IR (ATR, cm−1): 1033 (C–O stretching, ester), 1342 (C–H bending, aliphatic), 1518 (CC ring stretching), 1643 (CN stretching), 1728 (CO stretching), 2985 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.19 (3H, t, –CH3, ester, J = 6.90 Hz), 2.97 (1H, dd, –CH2–, J = 6.90 Hz, J = 16.95 Hz) 3.09 (1H, dd, –CH2–, J = 3.30 Hz, J = 17.70 Hz), 4.10 (2H, q, –CH2–CH3 ester, J = 6.60 Hz), 4.40 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.10 Hz, J = 7.70 Hz), 8.00 (2H, d, Ar-H, J = 7.80 Hz), 8.30 (2H, d, Ar-H, J = 8.10 Hz), 8.53 (1H, s, –CHN– azomethine), 12.00 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.4 (–CH3, ester), 37.0 (–CH2–), 44.2 (1,3-thiazolidin-4-one ring C-5), 61.1 (–OCH2– ester), 124.6, 128.9, 141.0, 148.5 (Ar-C), 154.0 (–CHN– azomethine), 168.7 (1,3-thiazolidin-4-one ring C-2), 170.8 (CO), 176.9 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C14H14N4NaO5S (M + Na) = 373.05826, found: 373.37363.:3); IR (ATR, cm−1): 1080 (C–O stretching, ester), 1354 (C–H bending, aliphatic), 1523 (CC ring stretching), 1627 (CN stretching), 1732 (CO stretching), 3093 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.19 (3H, t, –CH3 ester, J = 7.00 Hz), 3.03 (1H, dd, –CH2-, J = 7.50 Hz, J = 17.40 Hz), 3.12 (1H, dd, –CH2–, J = 4.80 Hz, J = 9.00 Hz), 4.10 (2H, q, –CH2–CH3 ester, J = 6.60 Hz), 4.45 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.80 Hz, J = 7.50 Hz), 7.76 (1H, t, Ar-H, J = 8.00 Hz), 8.1 (1H, d, Ar-H, J = 7.80 Hz), 8.29 (1H, dd, Ar-H, J = 1.65 Hz, J = 8.25 Hz), 8.50 (1H, s, Ar-H), 8.58 (1H, s, –CHN– azomethine), 12.17 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.4 (–CH3 ester), 36.6 (–CH2–), 43.9 (1,3-thiazolidin-4-one ring C-5), 61.1 (–OCH2– ester), 122.3, 125.3, 130.9, 133.9, 136.4, 148.6 (Ar-C), 154.9 (–CHN– azomethine), 166.3 (1,3-thiazolidin-4-one ring C-2), 170.7 (CO), 175.8 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C14H14N4NaO5S (M + Na) = 373.05826, found: 373.27241.:3); IR (ATR, cm−1): 1086 (C–O stretching, ester), 1387 (C–H bending, aliphatic), 1589 (CC ring stretching), 1643 (CN stretching), 1730 (CO stretching), 3089 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.19 (3H, t, –CH3 ester, J = 7.01 Hz), 3.09 (1H, dd, –CH2-, J = 7.55 Hz, J = 17.45 Hz), 3.13 (1H, dd, –CH2–, J = 4.90 Hz, J = 9.00 Hz), 4.12 (2H, q, –CH2–CH3 ester, J = 6.65 Hz), 4.42 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.85 Hz, J = 7.55 Hz), 7.78 (4H, m, Ar-H), 8.60 (1H, s, –CHN– azomethine); 13C-NMR (75 MHz): δ 14.4 (–CH3 ester), 36.8 (–CH2–), 42.9 (1,3-thiazolidin-4-one ring C-5), 61.2 (–OCH2– ester), 123.4, 126.3, 131.9, 133.9, 136.5, 149.6 (Ar-C), 155.9 (–CHN– azomethine), 165.3 (1,3-thiazolidin-4-one ring C-2), 171.7 (CO), 176.9 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C14H14N4NaO5S (M + Na) = 373.05826, found: 373.27236.:3); IR (ATR, cm−1): 1033 (C–O stretching, ester), 1311 (C–H bending, aliphatic), 1512 (CC ring stretching), 1651 (CN stretching), 1720 (CO stretching), 2989 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.18 (3H, t, –CH3 ester, J = 7.20 Hz), 2.97 (1H, dd, –CH2–, J = 8.10 Hz, J = 17.40 Hz), 3.07 (1H, dd, –CH2-, J = 4.35 Hz, J = 17.55 Hz), 3.81 (3H, s, –OCH3), 4.09 (2H, q, –CH2–CH3 ester, J = 6.30 Hz), 4.38 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.50 Hz, J = 7.80 Hz), 7.03 (2H, d, Ar-H, J = 9.00 Hz), 7.69 (2H, d, Ar-H, J = 8.70 Hz), 8.33 (1H, s, –CHN– azomethine), 12.03 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.4 (–CH3 ester), 36.9 (–CH2–), 43.8 (1,3-thiazolidin-4-one ring C-5), 55.8 (–OCH3), 61.1 (–OCH2– ester), 114.8, 127.3, 129.9, 161.7 (Ar-C), 156.2 (-CHN- azomethine), 170.7 (1,3-thiazolidin-4-one ring C-2), 175.9 (CO), 189.2 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C15H17N3NaO4S (M + Na) = 358.08375, found: 359.64858.:3); IR (ATR, cm−1): 1018 (C–O stretching, ester), 1334 (C–H bending, aliphatic), 1521 (CC ring stretching), 1639 (CN stretching), 1720 (CO stretching), 2985 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.18 (3H, t, –CH3, ester, J = 7.20 Hz), 2.97 (1H, dd, –CH2–, J = 8.10 Hz, J = 17.40 Hz), 3.07 (1H, dd, –CH2–, J = 4.50 Hz, J = 17.40 Hz), 3.88 (3H, s, –OCH3), 4.03 (2H, q, –CH2–CH3 ester, J = 6.00 Hz), 4.38 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.50 Hz, J = 7.80 Hz), 7.01 (1H, t, Ar, J = 7.50 Hz), 7.10 (1H, d, Ar, J = 8.40 Hz), 7.45 (1H, t, Ar-H, J = 8.40 Hz), 7.82 (1H, d, Ar-H, J = 7.80 Hz), 8.60 (1H, s, –CHN– azomethine), 12.07 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.5 (-CH3, ester), 36.9 (–CH2–), 43.9 (1,3-thiazolidin-4-one ring C-5), 56.2 (–OCH3), 61.1 (–OCH2– ester), 112.4, 121.2, 122.5, 126.5, 132.7, 151.5 (Ar-C), 158.5 (–CHN– azomethine), 170.7 (1,3-thiazolidin-4-one ring C-2), 176.2 (CO), 189.2 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C15H17N3NaO4S (M + Na) = 358.08375, found: 358.19232.:3); IR (ATR, cm−1): 1022 (C–O stretching, ester), 1330 (C–H bending, aliphatic), 1600 (CC ring stretching), 1647 (CN stretching), 1732 (CO stretching), 2993 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.18 (3H, t, –CH3 ester, J = 7.05 Hz), 2.99 (1H, dd, –CH2-, J = 8.10 Hz, J = 17.70 Hz), 3.08 (1H, dd, –CH2–, J = 4.50 Hz, J = 17.70 Hz), 4.17 (2H, q, –CH2–CH3 ester, J = 6.90 Hz), 4.40 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.50 Hz, J = 7.80 Hz), 6.87 (1H, m, Ar-H), 7.19 (3H, m, Ar-H), 8.30 (1H, s, –CHN– azomethine), 9.74 (1H, s, –OH), 12.04 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.5 (–CH3 ester), 36.9 (–CH2–), 43.8 (1,3-thiazolidin-4-one ring C-5), 61.1 (–OCH2– ester), 113.7, 118.5, 119.8, 130.3, 135.8, 158.1 (Ar-C), 156.9 (–CHN– azomethine), 170.7 (1,3-thiazolidin-4-one ring C-2), 175.8 (CO), 189.2 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C14H15N3NaO4S (M + Na) = 344.0681, found: 344.16745.:3); IR (ATR, cm−1): 1014 (C–O stretching, ester), 1338 (C–H bending, aliphatic), 1525 (CC ring stretching), 1639 (CN stretching), 1716 (CO stretching), 2989 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.18 (3H, t, –CH3 ester, J = 6.75 Hz), 3.03 (1H, dd, –CH2–, J = 8.10 Hz, J = 17.40 Hz), 3.09 (1H, dd, –CH2-, J = 4.20 Hz, J = 7.10 Hz), 4.10 (2H, q, –CH2–CH3 ester, J = 6.45 Hz), 4.50 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.20 Hz, J = 6.30 Hz), 6.95 (2H, m, Ar-H), 7.35 (1H, m, Ar-H), 7.65 (1H, m, Ar-H), 8.64 (1H, s, –CHN– azomethine), 10.87 (1H, s, –OH), 12.16 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.4 (-CH3, ester), 36.6 (–CH2–), 44.2 (1,3-thiazolidin-4-one ring C-5), 61.2 (–OCH2– ester), 116.8, 118.9, 119.9, 130.8, 132.7, 159.1 (Ar-C), 158.5 (–CHN– azomethine), 170.6 (1,3-thiazolidin-4-one ring C-2), 175.5 (CO), 189.2 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C14H15N3NaO4S (M + Na) = 344.0681, found: 344.11239.:3); IR (ATR, cm−1): 1014 (C–O stretching, ester), 1336 (C–H bending, aliphatic), 1618 (CC ring stretching), 1647 (CN stretching), 1712, 1726 (CO stretching), 2987 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.19 (3H, t, –CH3 ester, J = 7.00 Hz), 2.34 (3H, s, –CH3), 2.99 (1H, dd, –CH2–, J = 8.10 Hz, J = 17.70 Hz), 3.08 (1H, dd, –CH2-, J = 4.50 Hz, J = 17.40 Hz), 4.10 (2H, q, –CH2–CH3 ester, J = 6.30 Hz), 4.41 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.50 Hz, J = 7.50 Hz), 7.27 (2H, d, Ar-H, J = 8.10 Hz), 7.65 (2H, d, Ar-H, J = 8.10 Hz), 8.36 (1H, s, –CHN– azomethine), 12.07 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.5 (–CH3 ester), 21.6 (–CH3), 36.8 (–CH2–), 43.7 (1,3-thiazolidin-4-one ring C-5), 61.1 (–OCH2–, ester), 128.1, 129.9, 131.9, 141.1 (Ar-C), 156.7 (–CHN– azomethine), 163.9 (1,3-thiazolidin-4-one ring C-2), 170.7 (CO), 175.8 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C15H18N3NaO3S (M + Na) = 342.08883, found: 342.11165.:3); IR (ATR, cm−1): 1022 (C–O stretching, ester), 1338 (C–H bending, aliphatic), 1523 (CC ring stretching), 1643 (CN stretching), 1724 (CO stretching), 2993 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.18 (3H, t, –CH3, ester, J = 7.20 Hz), 2.49 (3H, s, –CH3), 2.98 (1H, dd, –CH2–, J = 7.95 Hz, J = 17.55 Hz), 4.09 (2H, q, –CH2–CH3 ester, J = 7.20 Hz), 4.39 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.50 Hz, J = 8.10 Hz), 7.31 (3H, m, Ar-H), 7.76 (1H, m, Ar-H), 8.92 (1H, s, –CHN– azomethine), 12.09 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.5 (–CH3, ester), 20.4 (–CH3), 36.9 (–CH2-), 43.9 (1,3-thiazolidin-4-one ring C-5), 61.1 (–OCH2–, ester), 126.6, 128.4, 130.6, 131.6, 132.6, 137.9 (Ar-C), 155.8 (–CHN– azomethine), 170.7 (1,3-thiazolidin-4-one ring C-2), 176.3 (CO), 189.2 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C15H18N3NaO3S (M + Na) = 342.08883, found: 342.07265.:3); IR (ATR, cm−1): 1031 (C–O stretching, ester), 1346 (C–H bending, aliphatic), 1616 (CC ring stretching), 1716 (CO stretching), 2909 (C–H stretching, aliphatic); 1H-NMR (300 MHz): δ 1.18 (3H, t, –CH3 ester, J = 7.20 Hz), 1.81 (2H, m, –CH2–, tetralone), 2.79 (4H, m, tetralone), 2.98 (1H, dd, –CH2–, J = 7.80 Hz, J = 17.40 Hz), 3.07 (1H, dd, –CH2–, J = 4.50 Hz, J = 17.40 Hz), 4.09 (2H, q, –CH2–CH3 ester, J = 6.90 Hz), 4.37 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.50 Hz, J = 7.80 Hz), 7.26 (3H, m, tetralone), 8.06 (1H, m, tetralone), 12.01 (1H, s, –NH); 13C-NMR (75 MHz): δ 14.6 (–CH3 ester), 22.3, 27.4, 29.6 (3-CH2–, tetralone), 36.9 (–CH2–), 43.4 (1,3-thiazolidin-4-one ring C-5), 61.1 (–OCH2–, ester), 125.0, 126.6, 129.3, 130.2, 132.6, 140.9 (Ar-C), 160.3 (–CN–), 162.9 (1,3-thiazolidin-4-one ring C-2), 170.7 (CO), 175.6 (1,3-thiazolidin-4-one ring C-4); HRMS (MALDI-TOF): calcd C17H19N3NaO5S (M + Na) = 368.01448, found: 368.27513.:3); IR (ATR, cm−1): 1026 (C–O stretching, ester), 1338 (C–H bending, aliphatic), 1595 (CC ring stretching), 1637 (CN stretching), 1724 (CO stretching), 2990 (C–H stretching, aliphatic), 3375 (–OH, stretching); 1H-NMR (300 MHz): δ 1.18 (3H, t, –CH3, ester, J = 7.20 Hz), 3.04 (1H, dd, –CH2–, J = 4.80 Hz, J = 13.20 Hz), 3.01 (1H, dd, –CH2–, J = 4.80 Hz, J = 13.20 Hz), 3.80 (3H, s, –OCH3), 4.09 (2H, q, –CH2–CH3 ester, J = 7.20 Hz), 4.38 (1H, dd, 1,3-thiazolidin-4-one ring C-5, J = 4.50 Hz, J = 7.50 Hz), 6.84 (1H, d, Ar-H, J = 8.10 Hz), 7.18 (1H, dd, Ar-H, J = 1.50 Hz, J = 8.40 Hz), 7.31 (1H, d, Ar-H, J = 1.50 Hz), 8.26 (1H, s, –CHN– azomethine), 9.65 (1H, s, –OH), 11.94 (1H, s, –NH); 13C-NMR (75 MHz): δ solubility issue. HRMS (MALDI-TOF): calcd C15H17N3NaO5S (M + Na) = 374.07866, found: 374.31122.In vitro evaluation and mechanism of action assays
For this purpose, LLC-MK2 cells (2.5 × 105 cells per mL) in DMEM medium supplemented with 10% SFB were cultured in 96-well plates, and maintained at 37 °C and 5% CO2 for 24 h. The substances were then added at increasing concentrations (10, 50, 100, 500, and 1000 μM), and the plate incubated for 96 hours. After treatment, cells were washed with PBS (pH 7.2) and incubated in the presence of MTT (2 mg mL−1) for 4 hours. The supernatant was removed, the Formazan crystals were solubilized in DMSO, and the absorbance reading was taken at 570 nm on a plate spectrophotometer (Power Wave XS-BioTek). The percentage of viable cells was calculated concerning the control. The CC50 values (50% cytotoxic concentration) were determined by nonlinear regression analysis.
Cell death assessment through flow cytometry
After confirmation of trypanocidal activity, Annexin-FITC/propidium iodide labeling was used to characterize cell death modalities induced by incubation with compounds. Metacyclic trypomastigotes were collected from the supernatant of infected L929 cells, and then seeded at 4 × 105 cells per well in RPMI-1640 medium. Compound 1f was dissolved in DMSO, and added to wells at IC50, 2 × IC50, and 4 × IC50 concentrations. Benznidazole (IC50, 2 × IC50, and 4 × IC50) and the culture medium were used as a positive and negative control, respectively. Plates were incubated at the same conditions used for anti-trypomastigote activity (37 °C, 24 hours). Briefly, after treatment, parasites were washed with PBS and resuspended in binding buffer (Annexin V Binding Buffer- BD Pharmingen™, USA). For labeling, 10 μL of propidium iodide (50 μg mL−1) and 5 μL of Annexin-FITC (BD Pharmingen™, USA) were added for 15 min at room temperature in the dark. Flow cytometry was conducted in a FACS Caliber (Becton & Dickinson, USA). For each sample, we acquired 20000 events and the data were analyzed using Cell Quest software (Becton & Dickinson, USA). Assays were conducted in triplicate. For significance analysis, ANOVA and Dunnett's test was used, taking into account p < 0.05.
Conclusions
Eleven 1,3-thiazole-4-carboxylates and sixteen 4-thiazolidinone-acetates were obtained with reasonable yields using a simple methodology by bioisosteric and esterification strategies. These molecules were evaluated for their trypanocidal, leishmanicidal and cytotoxicity. Some of them exhibited very good trypanocidal activity, especially compounds 1f, 1h, 1g, 1a, and 2d. From the tested scaffolds, the 1,3-thiazoles exhibited better trypanocidal activity than 4-thiazolidinones. The flow cytometry study showed that compound 1f (IC50 = 0.83 μM) kills the parasite via necrosis and apoptosis. However, the activity was not observed in the amastigote form. Scanning electron microscopy analysis showed that compound 1f at IC50 concentrations promoted alterations in the shape, flagella and surface of the body, inducing parasite death. Besides, compounds 1b, 1f, 2d and 2m exhibited better IC50 values for both L. amazonensis and L. infantum in comparison to standard Miltefosine. Our work corroborated the bioisosteric and esterification approaches for drug optimization and discovery with regards to chagas disease and leishmaniasis through 1,3-thiazole and 4-thiazolidinone scaffolds.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Ana Cristina Lima Leite is receiving a CNPq senior fellowship. The authors are thankful for the Centro de Tecnologias Estratégicas do Nordeste (CETENE) for recording LCMS of all compounds. All authors declare no competing financial interest. The author Muhammad Haroon is grateful to Prof. Dr Alan S. Goldman. Department of chemistry and chemical biology, Rutgurs University New Brunswick 610 Taylor Road, Piscataway, NJ 08854-8087 USA for providing NMR facility. The corresponding author is thankful to Prof. Dr Shahid Hameed, Department of Chemistry, QAU, Islamabad-45320, Pakistan for providing NMR facility.References
- Laboratory of immunomodulation, Department of Protozoology/IOC – FIOCRUZ, The Leishmaniasis, 1997, accessed on 28th August 2019, http://www.dbbm.fiocruz.br/tropical/leishman/leishext/html/morfologia.html Search PubMed.
- Pan American Health Organization, Leishmaniasis: Epidemiological Report in the Americas, Pan American Health Organization, Washington, 2019, http://www.paho.org/leishmaniasis Search PubMed.
- J. A. Urbina, J. Eukaryotic Microbiol., 2015, 62, 149–156 CrossRef CAS.
- M. J. Pinazo, L. Guerrero, E. Posada, E. Rodríguez, D. Soy and J. Gascon, Antimicrob. Agents Chemother., 2013, 57, 390e395 CrossRef.
- D. H. Molyneux, L. Savioli and D. Engels, Lancet, 2017, 389, 312–325 CrossRef.
- J. A. Urbina and R. Docampo, Trends Parasitol., 2003, 19, 495–501 CrossRef CAS.
- J. Urbina, Chemotherapy of chagas disease, Curr. Pharm. Des., 2002, 8, 287–295 CrossRef CAS.
- A. L. Ribeiro, M. P. Nunes, M. M. Teixeira and M. O. C. Rocha, Nat. Rev. Cardiol., 2012, 9, 576–589 CrossRef CAS.
- C. Marín, I. Ramírez-Macías, M. J. Rosales, B. Muro, F. Reviriego, P. Navarro, V. J. Arán and M. Sánchez-Moreno, Acta Trop., 2015, 148, 170–178 CrossRef.
- S. Espuelas, D. Plano, P. Nguewa, M. Font, J. A. Palop, J. M. Irache and C. Sanmartín, Curr. Med. Chem., 2012, 19, 4259–4288 CrossRef CAS.
- World Health Organization, Defeating Neglected Tropical Diseases: Progress, Challenges and Opportunities, WHO/CDS/NTD/2019.01, 2019, access in 8th September 2019, https://apps.who.int/iris/handle/10665/325755 Search PubMed.
- R. W. DeSimone, K. S. Currie, S. A. Mitchell, J. W. Darrow and D. A. Pippin, Comb. Chem. High Throughput Screening, 2004, 7(5), 473–494 CrossRef CAS.
- J. Polanski, A. Kurczyk, A. Bak and R. Musiol, Curr. Med. Chem., 2012, 19(13), 1921–1945 CrossRef CAS.
- D. González Cabrera, F. Douelle, T.-S. Feng, A. T. Nchinda, Y. Younis, K. L. White, Q. Wu, E. Ryan, J. N. Burrows, D. Waterson, M. J. Witty, S. Wittlin, S. A. Charman and K. J. Chibale, Med. Chem., 2011, 54(21), 7713–7719 CrossRef.
- P. A. de Moraes Gomes, M. de Oliveira Barbosa, E. Farias Santiago, M. V. de Oliveira Cardoso, N. T. Capistrano Costa, M. Z. Hernandes, D. R. Moreira, A. C. da Silva, T. A. Dos Santos, V. R. Pereira, F. A. Brayner Dos Santos, G. A. do Nascimento Pereira, R. S. Ferreira and A. C. Leite, Eur. J. Med. Chem., 2016, 121, 387–398 CrossRef.
- G. C. Moraski, N. Seeger, P. A. Miller, A. G. Oliver, H. I. Boshoff, S. Cho, S. Mulugeta, J. R. Anderson, S. G. Franzblau and M. J. Mille, ACS Infect. Dis., 2016, 2, 393–398 CrossRef CAS.
- M. V. Cardoso, L. R. de Siqueira, E. B. da Silva, L. B. Costa, M. Z. Hernandes, M. M. Rabello, R. S. Ferreira, L. F. da Cruz, D. R. Moreira, V. R. Pereira, M. C. de Castro, P. V. Bernhardt and A. C. Leite, Eur. J. Med. Chem., 2014, 86, 48–59 CrossRef CAS.
- H. M. Ashour, I. M. El-Ashmawy and A. E. Bayad, Monatsh. Chem., 2016, 147, 605–618 CrossRef CAS.
- G. Cihan-Üstündağ, E. Gürsoy, L. Naesens, N. Ulusoy Güzeldemirci and G. Çapan, Bioorg. Med. Chem., 2016, 24(2), 240–246 CrossRef.
- D. Secci, S. Carradori, B. Bizzarri, P. Chimenti, C. De Monte, A. Mollica, D. Rivanera, A. Zicari, E. Mari, G. Zengin and A. Aktumsek, Eur. J. Med. Chem., 2016, 117, 144–156 CrossRef CAS.
- D. R. Moreira, A. C. Leite, M. V. Cardoso, R. M. Srivastava, M. Z. Hernandes, M. M. Rabello, L. F. da Cruz, R. S. Ferreira, d. C. A. Simone, C. S. Meira, E. T. Guimaraes, A. C. da Silva, T. A. dos Santos, V. R. Pereira and M. B. Soares, ChemMedChem, 2014, 9(1), 177–188 CrossRef CAS.
- G. B. de Oliveira Filho, M. V. de Oliveira Cardoso, J. W. Espíndola, L. F. Ferreira, C. A. de Simone, R. S. Ferreira, P. L. Coelho, C. S. Meira, M. D. R. Moreira, M. B. Soares and A. C. Lima Leite, Bioorg. Med. Chem., 2015, 23(23), 7478–7486 CrossRef CAS.
- G. B. de Oliveira Filho, M. V. d. O. Cardoso, J. W. P. Espíndola, D. A. Oliveira e Silva, R. S. Ferreira, P. L. Coelho, P. S. dos Anjos, E. d. S. Santos, C. S. Meira, D. R. M. Moreira, M. B. P. Soares and A. C. L. Leite, Eur. J. Med. Chem., 2017, 141, 346–361 CrossRef CAS.
- M. V. O. Cardoso, G. B. Oliveira Filho, L. R. P. Siqueira, J. W. P. Espíndola, E. B. D. Silva, A. P. O. Mendes, V. R. A. Pereira, M. C. A. B. Castro, R. S. Ferreira, F. S. Villela, F. M. R. D. Costa, C. S. Meira, D. R. M. Moreira, M. B. P. Soares and A. C. L. Leite, Eur. J. Med. Chem., 2019, 180, 191–203 CrossRef CAS.
- A. S. dos Santos Aliança, A. R. Oliveira, A. P. S. Feitosa, K. R. C. Ribeiro, M. C. A. B. de Castro, A. C. L. Leite and F. A. Brayner, Eur. J. Pharm. Sci., 2017, 105, 1–10 CrossRef.
- C. M. Queiroz, G. B. de Oliveira Filho, J. W. P. Espíndola, A. V. do Nascimento, A. S. dos Santos Aliança, V. M. B. de Lorena, A. P. S. Feitosa, P. R. da Silva, L. C. Alves, A. C. L. Leite and F. A. Brayner, Med. Chem. Res., 2020, 29, 2050–2206 CrossRef.
- M. Haroon, T. Akhtar, A. C. d. S. Santos, V. R. A. Pereira, L. F. G. R. Ferreira, M. Z. Hernandes, R. E. O. Rocha, R. S. Ferreira, P. A. T. d. M. Gomes, F. A. de Sousa, M. C. H. d. B. Dias, M. N. Tahir, S. Hameed and A. C. L. Leite, ChemistrySelect, 2019, 4, 13163–13172 CrossRef CAS.
- C. da Silva, T. A. R. Dos Santos, I. V. B. da Silva, M. V. G. de Oliveira, D. R. M. Moreira, A. C. L. Leite and V. R. A. Pereira, Exp. Parasitol., 2017, 177, 57–65 CrossRef.
- E. Izumi, T. V. J. V. F. Ueda-Nakamura, A. C. Pinto and C. V. Nakamura, J. Med. Chem., 2012, 55, 2994–3001 CrossRef CAS.
- L. P. Borba-Santos, T. Gagini, K. Ishida, W. de Souza and S. Rozental, J. Med. Microbiol., 2015, 64(4), 415–422 CrossRef CAS.
- S. Pietkiewicz, J. H. Schmidt and I. N. Lavrik, J. Immunol. Methods, 2015, 423, 99–103 CrossRef CAS.
- G. F. Pauli, S.-N. Chen, C. Simmler, D. C. Lankin, T. Gödecke, B. U. Jaki, J. B. Friesen, J. B. McAlpine and J. G. Napolitano, J. Med. Chem., 2014, 57, 9220–9231 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |