
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Encapsulation of polyprodrugs enables an efficient and controlled release of dexamethasone†
Mengyi
Li
a,
Shuai
Jiang
 *ab,
Adelina
Haller
c,
Sebastian
Wirsching
d,
Michael
Fichter
d,
Johanna
Simon
ac,
Manfred
Wagner
a,
Volker
Mailänder
ac,
Stephan
Gehring
d,
Daniel
Crespy
e and
Katharina
Landfester
*a
*ab,
Adelina
Haller
c,
Sebastian
Wirsching
d,
Michael
Fichter
d,
Johanna
Simon
ac,
Manfred
Wagner
a,
Volker
Mailänder
ac,
Stephan
Gehring
d,
Daniel
Crespy
e and
Katharina
Landfester
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, Mainz 55128, Germany. E-mail: jiangs@mpip-mainz.mpg.de; landfester@mpip-mainz.mpg.de
bKey Laboratory of Marine Drugs, Chinese Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, China
cDepartment of Dermatology, University Medical Center, Johannes-Gutenberg University Mainz, Langenbeckstr. 1, Mainz 55131, Germany
dChildren's Hospital, University Medical Center, Johannes-Gutenberg University Mainz, Germany
eDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong 21210, Thailand
First published on 24th July 2021
Abstract
Water-soluble low molecular weight drugs, such as the synthetic glucocorticoid dexamethasone (DXM), can easily leak out of nanocarriers after encapsulation due to their hydrophilic nature and small size. This can lead to a reduced therapeutic efficacy and therefore to unwanted adverse effects on healthy tissue. Targeting DXM to inflammatory cells of the liver like Kupffer cells or macrophages is a promising approach to minimize typical side effects. Therefore, a controlled transport to the cells of interest and selective on-site release is crucial. Aim of this study was the development of a DXM-phosphate-based polyprodrug and the encapsulation in silica nanocontainers (SiO2 NCs) for the reduction of inflammatory responses in liver cells. DXM was copolymerized with a linker molecule introducing pH-cleavable hydrazone bonds in the backbone and obtaining polyprodrugs (PDXM). Encapsulation of PDXMs into SiO2 NCs provided a stable confinement avoiding uncontrolled leakage. PDXMs were degraded under acidic conditions and subsequently released out of SiO2 NCs. Biological studies showed significantly enhanced anti-inflammatory capacity of the polyprodrug nanoformulations over non-encapsulated DXM or soluble polyprodrugs. These results demonstrate the advantage of combining the polyprodrug strategy with nanocarrier-mediated delivery for enhanced control of the delivery of water-soluble low molecular weight drugs.
New conceptsApplication of water-soluble low molecular weight drugs faces important limitations including a rapid blood and renal clearance, nonspecific targeting and poor bioavailability at diseased sites. Water-soluble drugs cannot be effectively entrapped in nanocarriers, therefore drug leakage occurs before reaching the targeting tissues. Although a variety of polymer–prodrug conjugates and self-assembled nanoformulations were developed for the delivery of hydrophobic drugs, efficient encapsulation and controlled release of water-soluble drugs have been challenging. We develop here a nanocarrier-mediated polyprodrug delivery strategy for selectively delivering water-soluble drugs. The new polyprodrugs are synthesized by click chemistry and are efficiently encapsulated in porous silica nanocapsules, so that the prodrug polymer can be selectively decomposed in mildly acidic conditions to release water-soluble drug. This synthetic strategy allows a pH-controlled and sustained release of the drug so that drug concentration is below toxicity level. This is particularly important given the toxicity of drugs like dexamethasone and their detrimental side effects. Our results demonstrate the advantage of combining polyprodrug with nanocarrier-mediated delivery for enhanced control over the delivery of water-soluble drugs. This strategy paves the way for a selective delivery of other water-soluble drugs that are highly toxic for healthy cells. |
Introduction
Applicability of water-soluble low molecular weight drugs faces several limitations including the rapid blood/renal clearance, nonspecific targeting, and poor bioavailability at diseased sites. These limitations lead to low therapeutic efficacies and adverse effects on healthy tissues. Nanocarrier-mediated delivery has shown increased efficacy while reducing side effects due to an increased bioavailability of drugs in diseased tissues.3 However, due to the hydrophilic nature and small size, water-soluble low molecular weight drugs cannot be efficiently entrapped in the nanocontainers leading to a low encapsulation efficiency and premature drug leakage. Therefore, an efficient delivery of water-soluble drugs to the targeted tissue and controlled release in the cells of interest while preventing leakage during blood circulation is required.4–6 Although, a variety of polymer–prodrug conjugates and self-assembled nanoformulations have been developed for delivering hydrophobic drugs,4,7,8 efficient encapsulation and controlled release of water-soluble drugs have been challenging.9–11Herein, we developed a nanocontainer-mediated polyprodrug delivery strategy for controlled delivery and selective release of water-soluble drugs. In this approach, we first synthesized polyprodrug chains by copolymerizing the drug molecule with a linker molecule in order to form prodrug polymers connected by stimuli-cleavable linkages in the backbone. Secondly, we encapsulated the polyprodrugs in core–shell silica nanocontainers. The advantage of using nanocontainers is to provide a stable confinement for the polyprodrugs against premature leakage. We designed semipermeable nanocapsules to allow the entering of trigger molecules and subsequent release of depolymerized drug monomers upon triggering. As a proof-of-concept, we chose dexamethasone phosphate (DXM) as a model water-soluble low molecular weight drug. DXM is a synthetic glucocorticoid with vigorous anti-inflammatory properties representing standard care of treatment of different inflammatory liver diseases, such as autoimmune hepatitis or biliary atresia.12 However, prolonged systemic administration of DXM causes severe side effects such as hyperglycemia, hypertension, and intestinal bleeding. Therefore, controlled delivery of DXM without leakage is required.13–16
In this study, we synthesized pH-cleavable polydexamethasone (PDXM) prodrugs with defined molecular weights through a polycondensation reaction of DXM with adipic acid dihydrazide as the linker molecule. High molecular weight PDXM prodrugs were efficiently encapsulated in the nanocontainers without leakage, showing a selective release of DXM under acidic conditions. Biodistribution studies demonstrated an accumulation of nanocontainers in the liver and their preferential uptake by liver macrophages playing the major role for inducing inflammatory responses by secreting inflammatory cytokines.17 The nanoencapsulated PDXM showed an efficient modulation of the inflammatory response of liver macrophages over free DXM and non-encapsulated polyprodrugs. Our results demonstrated the advantage of combining the polyprodrug strategy with nanocontainer-mediated delivery for the selective delivery of water-soluble low molecular weight drugs.
Results and discussion
Synthesis of polydexamethasone (PDXM)
The concept of polyprodrug nanoencapsulation is illustrated in Fig. 1. First, PDXM polyprodrugs were synthesized from dexamethasone phosphate (DXM) via a condensation reaction between the ketone groups of DXM and acyl hydrazide groups of linker molecule adipic acid dihydrazide (ADH) (Fig. 2a). The DXM molecule has two ketone groups that can react with hydrazide moieties to form polymers with pH-responsive hydrazone linkages. Such chemistry was advantageously employed for conjugating DXM to poly(N-(2-hydroxypropyl)methacrylamide) copolymers18 and to peptide nanofiber gels,19 pluronic P123 micelles,20 and avidin.21 The molecular structure of PDXM was characterized by 1H NMR spectroscopy (Fig. 2b). In the NMR spectrum of ADH, the peak at δ = 1.53 ppm represents the internal methylene (H2, H3) protons, and the peak at δ = 2.17 ppm represents the H1 and H4 methylene protons. Upon condensation of ADH with DXM, splitting of the above two peaks was observed. The H1 and H4 methylene protons of ADH became distinguishable and shifted to low field as a bundle from 2.27 to 2.37 ppm. Because the reaction of hydrazide with carbonyl groups creates cis and trans isomers in the copolymer,22,23 the closer protons like H15, H14, H6, H20, H1, H2, H3 and H4 were changed in the absorption and their peaks became broader (more signals) due to the differences in the structure. New signals appeared at 3.2 ppm and 6.3–7.0 ppm after polymerization (will be discussed later). The total concentration of DXM in the PDXM polymer is 74 wt%.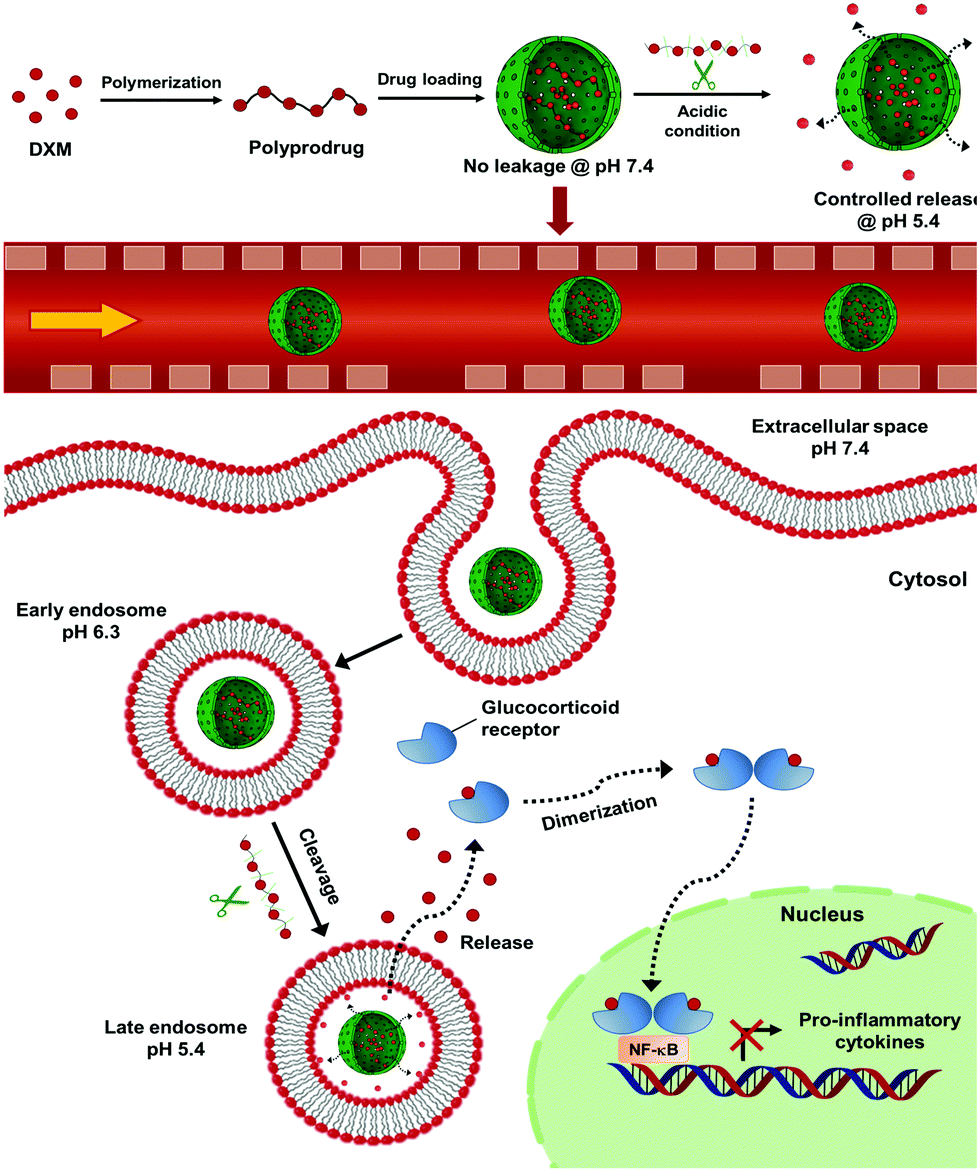 | ||
| Fig. 1 Scheme of pH-responsive delivery of dexamethasone phosphate via nanoencapsulation of polyprodrug for liver anti-inflammatory therapy. NF-κB: nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells. | ||
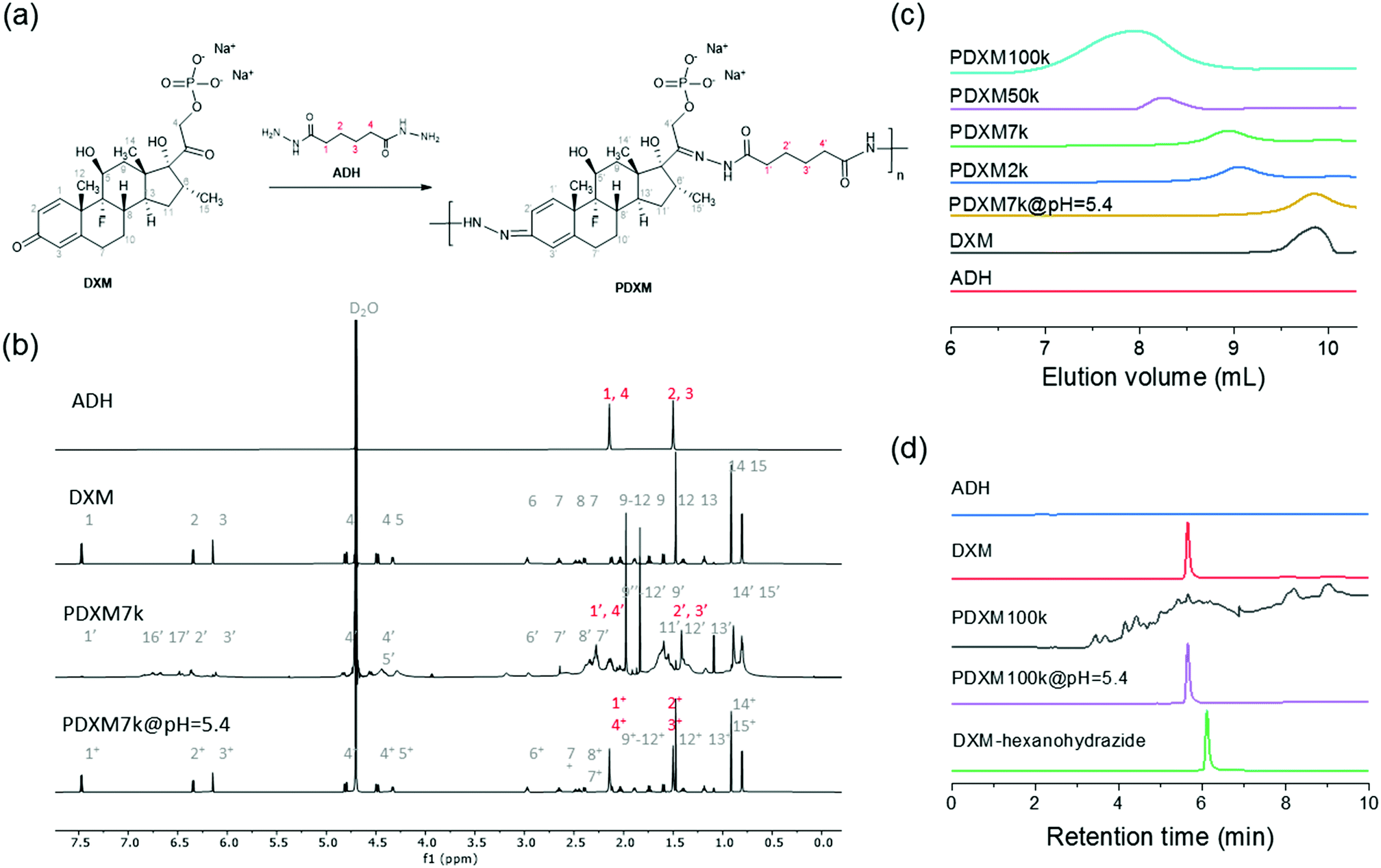 | ||
| Fig. 2 (a) The route for the synthesis of PDXM prodrugs. (b) 1H NMR (850 MHz at 298 K) spectra of ADH, DXM, PDXM7k, and PDXM7k@pH5.4. (c) GPC chromatograms of ADH, DXM, and PDXM with different Mn, as well as degradation products of PDXM7k at pH 5.4. (d) HPLC chromatograms of ADH, DXM, PDXM100k, degradation products of PDXM100k at pH 5.4, and the DXM-hexanohydrazide conjugate. | ||
PDXM polymers with acid-labile acyl hydrazone bonds in the main chain and with different molecular weights (Mn) were obtained upon a systematic variation of reaction parameters. The apparent Mn of PDXM was determined in water by gel permeation chromatography (GPC). Reaction time, temperature, and concentration of catalyst were varied to obtain different Mn of PDXM (Table S1, ESI†). First, we extended the reaction time from 12 h to 96 h (temperature at 50 °C, catalyst concentration 1.59 mmol mL−1), the Mn of PDXM increased from 2000 g mol−1 to 133![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1 (Fig. S1a, ESI†). As shown in the GPC chromatograms of Fig. 2c, the retention time of PDXM gradually decreased with increasing reaction time. The sizes of obtained PDXM polymers were measured by multi-angle DLS. The hydrodynamic radius of PDXM7k, PDXM50k and PDXM100k was 3.0, 4.9 and 6.2 nm, respectively (Fig. S2, ESI†). PDXM with Mn of ∼ 2000 g mol−1 could not be detected by DLS. Moreover, the Mn of PDXM increased from 101100 g mol−1 to 122100 g mol−1 when the temperature was increased from 50 °C to 70 °C (reaction time of 72 h, catalyst concentration 1.59 mmol mL−1) (Fig. S1b, ESI†). Increasing the concentration of catalyst from 0.43 mmol mL−1 to 0.82 and 1.59 mmol mL−1 led to an increase of Mn from 10300 g mol−1 to 36400 and 101100 g mol−1, respectively, with temperature kept at 50 °C and reaction time of 72 h (Fig. S1c, ESI†).
400 g mol−1 (Fig. S1a, ESI†). As shown in the GPC chromatograms of Fig. 2c, the retention time of PDXM gradually decreased with increasing reaction time. The sizes of obtained PDXM polymers were measured by multi-angle DLS. The hydrodynamic radius of PDXM7k, PDXM50k and PDXM100k was 3.0, 4.9 and 6.2 nm, respectively (Fig. S2, ESI†). PDXM with Mn of ∼ 2000 g mol−1 could not be detected by DLS. Moreover, the Mn of PDXM increased from 101100 g mol−1 to 122100 g mol−1 when the temperature was increased from 50 °C to 70 °C (reaction time of 72 h, catalyst concentration 1.59 mmol mL−1) (Fig. S1b, ESI†). Increasing the concentration of catalyst from 0.43 mmol mL−1 to 0.82 and 1.59 mmol mL−1 led to an increase of Mn from 10300 g mol−1 to 36400 and 101100 g mol−1, respectively, with temperature kept at 50 °C and reaction time of 72 h (Fig. S1c, ESI†).
The polyprodrug PDXM7k with a maximum peak at the elution volume of 8.9 mL (Mn = 7100 g mol−1) was studied for the degradation of PDXM under acidic conditions. As shown in Fig. 2c, the maximum peak of PDXM7k shifted to an elution volume of 9.3 mL after incubation at pH 5.4 for 24 h. The peak of degradation products is identical to the elution volume of pure DXM monomer, which indicated the degradation of PDXM into DXM monomer at pH 5.4. To confirm the complete degradation of PDXM rather than the formation of oligomers, we studied the degradation profile of PDXM by using HPLC. The degraded products (PDXM@pH = 5.4) displayed the same retention time as pure DXM monomer in HPLC chromatograms (Fig. 2d), showing the complete degradation of PDXM to the monomers. As comparison, an oligomer, prepared by reacting DXM with hexanohydrazide to produce DXM conjugates with mono or di-hexanohydrazide, clearly showed a delayed retention time compared to DXM monomer and degraded products of PDXM.
pH-responsive degradation of PDXM
As mentioned earlier (Fig. 2b), new signals appeared at 3.2 ppm and 6.3–7.0 ppm in the NMR spectra after polymerization. These signals then disappeared after the degradation of the polymer at pH 5.4 (indicated by green arrows in Fig. 3b). This result is attributed to the isomer signals from different cis and trans configurations in the copolymer structure, which falls back to monomers with an identical configuration. From NMR DOSY spectra in Fig. 3b, these protons present the same diffusion coefficient as the polymer PDXM7k, therefore confirming these protons belong to the polymer. We monitored the degradation process of PDXM7k under acidic conditions through a 1H NMR kinetic study. The characteristic peaks of DXM at 6.15, 6.35, and 7.45 ppm increased over 10 h (Fig. 3a), showing the degradation of PDXM polymer into DXM monomers. After degradation, the monomers DXM and ADH were discriminated from PDXM by showing different diffusion coefficients (Fig. 3b). After 20 h degradation, DXM and ADH were found in the solution present individually (Fig. 3c). The NMR COSY spectrum of degradation products corresponded well with pure DXM (Fig. 3d), indicating the recovery of molecular structure of the drug after the depolymerization process.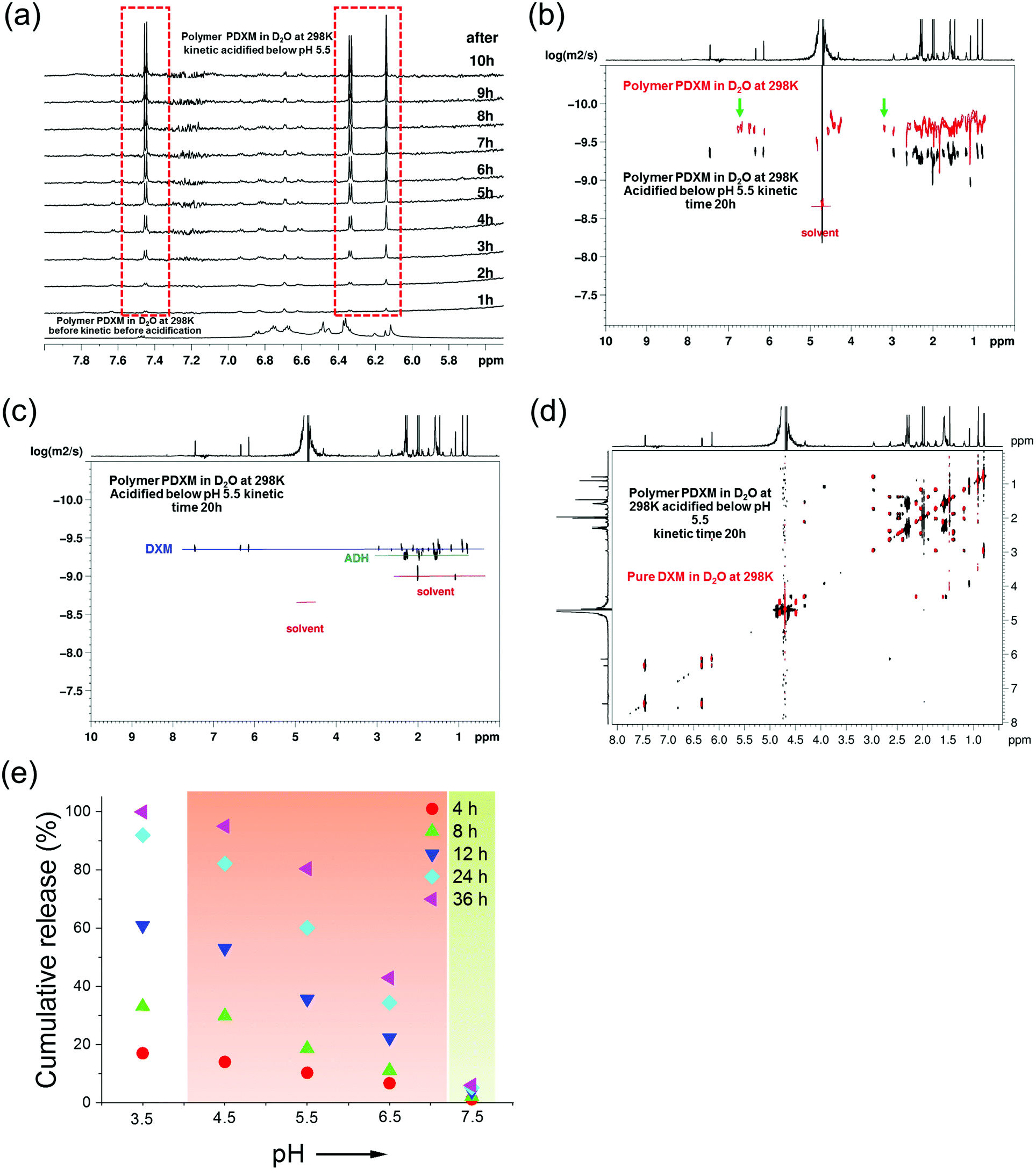 | ||
| Fig. 3 (a) 1H NMR (850 MHz at 298 K) in situ kinetic study of the degradation of PDXM in acidic condition. (b) Overlay of NMR DOSY spectra of PDXM7k polymer (red) and the degradation products in D2O after acidifying at kinetic time of 20 h (black). Green arrows point out the new peaks appearing after polymerization and disappearing after degradation. (c) NMR DOSY spectra of the degradation products of PDXM7k at kinetic time of 20 h. (d) Overlay of NMR COSY spectrum of the degradation products of PDXM7k at kinetic time of 20 h (black) with the spectrum of pure DXM monomer (red). (e) pH-dependency of the degradation and release of PDXM7k. The red background covers the range of pH values in different acidic extra- and intracellular environments reported in the literature.1,2 The green background represents physiological pH values (around 7.4). | ||
The release of DXM from degradation of PDXM was studied at different pH conditions for 36 h. PDXM7k powder, obtained by lyophilization, was first dissolved in PBS at pH 7.4 to avoid non-controlled degradation of polymer. The PDXM7k was stable in aqueous environment for more than 36 h at pH 7.4. In acidic conditions, as shown in Fig. 3e, the cleavage of PDXM7k was both pH- and time-dependent. The release of cleaved DXM started slowly at pH 6.5, which corresponds to the pH of early endosomes.24 Degradation of PDXM gradually accelerated as pH decreased to 3.5. The acid-sensitivity of PDXM7k around pH 4.5–5.5 was notable since late endosomes and lysosomes in cells bear pH values of about 5.0.24 Cumulative release of cleaved DXM reached 100% at pH 3.5 in 36 h, hence confirming the full conversion of PDXM into monomers.
Encapsulation and controlled release of PDXM by silica nanocapsules (SiO2 NCs)
Core–shell SiO2 NCs were synthesized via an interfacial confined sol–gel process in an inverse (water-in-oil) miniemulsion (see Fig. 4a). The inverse miniemulsion process was used since PDXM was highly water soluble (>40 mg mL−1). DXM or PDXM was first dissolved in the aqueous phase, forming the nanodroplets after emulsification. CTAC was used as a templating agent for confining the condensation of negatively charged silica species at water–oil interface.4,25 The core–shell morphology of the SiO2 NCs was visualized by TEM (Fig. 4b). Average size of the SiO2 NCs containing DXM or PDXM with different Mn was ca. 300–350 nm determined by dynamic light scattering (see Table S2, ESI†). As shown in Fig. 4c, the N2 adsorption–desorption isotherm of SiO2 NCs exhibited a type-IV isotherm with a BET surface area of 139 m2 g−1, pore volume of 0.55 cm3 g−1, and pore size of 4.4 nm. The H3-type hysteresis loops at relative pressure (P/P0) above 0.5 suggested the adsorption of N2 molecules in the hollow voids. The porous shell allowed the diffusion of protons into the capsule inducing the cleavage of acyl hydrazone bonds in the polymer, while the depolymerized DXM monomers presumably released into the environment (Fig. 4d). Compared with the pore size of 4.4 nm in the shell, the hydrodynamic diameters of PDXM7k, PDXM50k and PDXM100k polymers were measured as 6.0 nm, 9.8 nm, and 12.4 nm, respectively (Fig. S2, ESI†). Therefore, the polyprodrugs could be efficiently entrapped in the NCs. The encapsulation efficiency of PDXM increased with increasing of its Mn (Fig. S3, ESI†).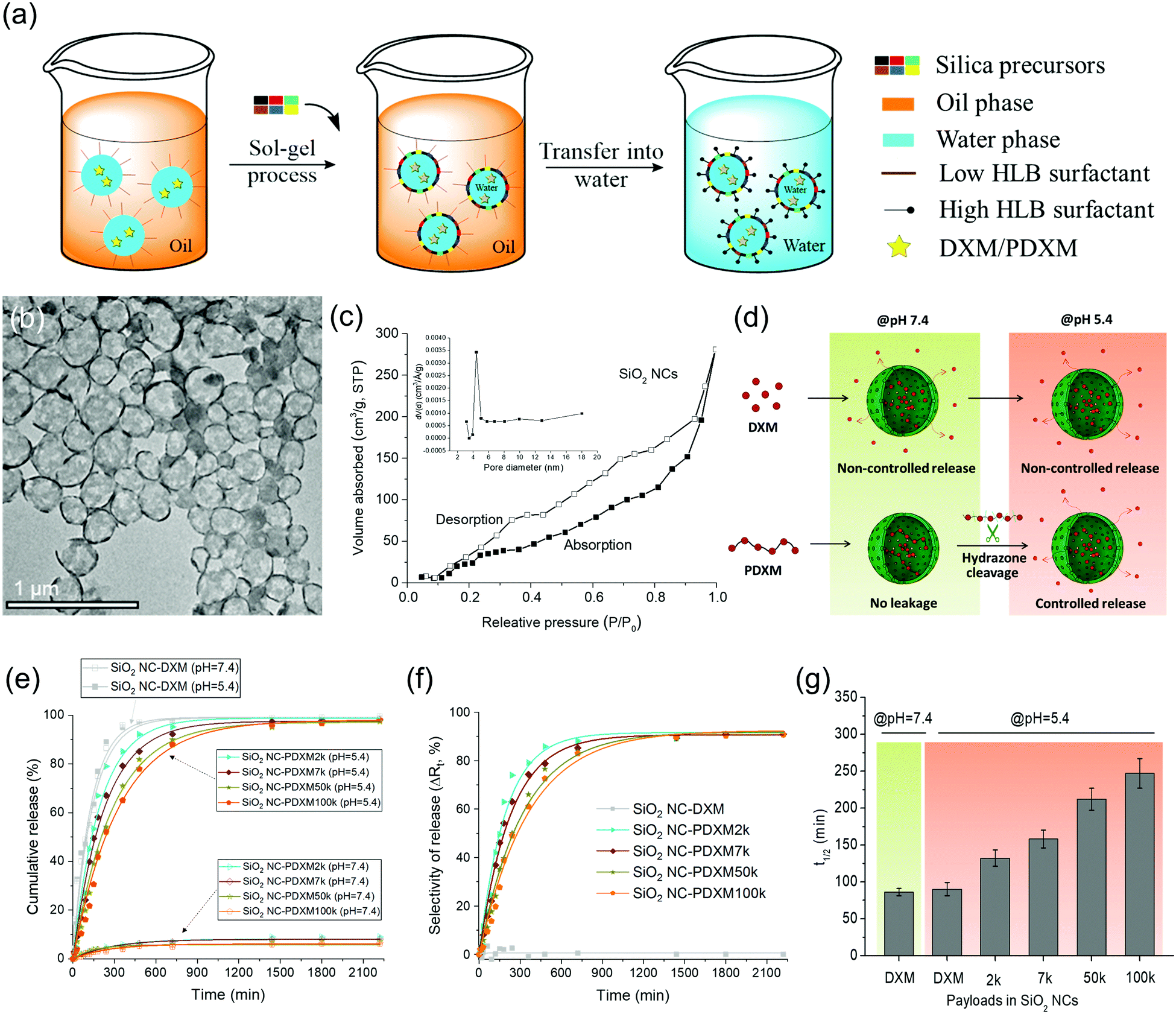 | ||
| Fig. 4 (a) Schematic illustration of the synthesis of SiO2 NCs in miniemulsion. (b) TEM micrograph of SiO2 NC-PDXM100k. (c) Nitrogen adsorption–desorption spectrum of SiO2 NCs. (d) Schematic illustration of the encapsulation and pH-responsive release mechanism. (e) pH-responsive release profiles (at 37 °C, mean ± SD, n = 3). (f) Selectivity ΔRt defined as difference between cumulative release of DXM from NCs at pH 5.4 and 7.4. (g) Half-time t1/2 of the pH-responsive release of DXM from SiO2 NC-DXM and SiO2 NC-PDXM. | ||
pH-responsive release of DXM from SiO2 NC-DXM/PDXM was studied at pH 7.4 and 5.4 to mimic the physiological pH conditions and acidic microenvironments in biological systems.26Fig. 4d shows the scheme of the release mechanism. For SiO2 NCs containing DXM monomer, 100% DXM was released after 6 h (Fig. 4e). No significant difference could be observed between pH 7.4 and 5.4. In this case, the release of DXM was mainly dominated by the diffusion of entrapped DXM across the silica shell. In contrast, at pH 7.4, less than 10% of DXM was released from SiO2 NC-PDXM after 36 h for all Mn (Fig. 4e). This is due to the fact that larger PDXM molecules were entrapped in the silica NCs and cannot diffuse through the shell (Fig. S3, ESI†). The low amount of released DXM is probably due to the low Mn fraction of the polymers as well as of the very slow hydrolysis of PDXM at neutral pH value. When the pH was lowered to 5.4, the release from SiO2 NC-PDXM was dramatically enhanced due to the cleavage of acyl hydrazone bonds in the polymer under acidic condition. The release profile of SiO2 NC-PDXM at pH 5.4 was slower as the Mn of PDXM increased. The release kinetics fit best with the one-order kinetics model with a correlation coefficient R2adjusted close to 1 (Table S3, ESI†). The release rate of DXM reduced linearly with decreasing the concentration of the remaining drug in the nanocapsules (Fig. S4, ESI†). Comparing the release profiles of PDXM and SiO2 NC-PDXM under same conditions showed that the pH-responsiveness of encapsulated PDXM was still addressable but the release process was delayed due to the diffusion across the silica shell (Fig. S5, ESI†).
A major advantage of the encapsulation of polyprodrugs over encapsulation of DXM monomer is the increased selectivity for the control of release (see Fig. 4f). Here we defined the selectivity ΔRt as the difference of release of DXM from nanocapsules at pH 5.4 and 7.4. This value therefore gives a relative quantity reflecting the selectivity of the release system–the part of the performance in the release that is only dependent on the pH-responsive property of the system. The selectivity of the SiO2 NCs-PDXM with different Mw was ∼ 90%. In contrast, there was almost no selectivity for the release of DXM from the sample SiO2 NC-DXM in response to pH change. The half-life t1/2 was calculated from the equation t1/2 = (ln2)/k. Increasing the Mw of PDXM resulted in a longer half-time and therefore a prolonged release of DXM (Fig. 4g). In summary, the polymerization of DXM dramatically increased the encapsulation efficiency of the drug and enabled the selective and sustained release of DXM in response to change of pH values.
Stability of SiO2 NCs-PDXM in blood plasma and influence of protein adsorption on the release profile of PDXM
The SiO2 NC-PDXM samples were incubated with human plasma, to further study the stability and drug release behavior of the nanocapsules closer to an in vivo situation. The stability of SiO2 NC-PDXM100k in blood plasma was studied by using multiangle DLS. Fig. S6 (ESI†) displays the autocorrelation function for the plasma/SiO2 NC mixture at different angles. The results showed that no objects with sizes larger than the largest size of either plasma or SiO2 NCs were formed in the mixture,27 indicating the stability (no aggregation) of the SiO2 NCs in blood plasma. Composition of the protein corona on SiO2 NCs-PDXM was analyzed by LC-MS and presented in Fig. S7 (ESI†). The release of DXM from the SiO2 NCs incubated with human plasma was compared with nanocapsules incubated in PBS as control group (Fig. S8, ESI†). The encapsulated PDXM molecules were still addressable by the protons after protein corona formation. For both, SiO2 NC-PDXM7k and SiO2 NC-PDXM100k, the release profiles were prolonged due to the increased diffusion barrier generated by the protein corona, as previously observed with other nanocarriers.28In vivo biodistribution and cellular uptake of SiO2 NCs
Since the strategy of this system is that the nanocapsules primarily reach the tissue and cells where the loaded substance is supposed to act, we have investigated the SiO2 NCs in vivo. The aim of this study was to deliver PDXM to the non-parenchymal cells (NPCs) of the liver to demonstrate a potential model for treating liver inflammation. The SiO2 NCs were covalently labelled with a fluorescent probe (Cy5) via NHS ester-amine chemistry (Cy5-SiO2 NC, Dh = 370 ± 70 nm, complete characterization of NCs shown in Table S4, ESI†). In vivo imaging using the IVIS SpectrumCT revealed a strong Cy5 fluorescence signal in the liver 5 min following intravenous injection of the NCs (Fig. 5a). The fluorescence intensity slightly declined within the subsequent 2 h probably due to intracellular degradation and clearance mechanisms. After sacrificing the animals 2 h post injection, relevant organs were isolated and imaged. The IVIS SpectrumCT imaging showed a slight accumulation of Cy5-SiO2 NCs in the lungs while the nanocapsules primarily accumulated in the livers (Fig. 5b and c). The NC-treated animals did not show any clinical signs of toxicity like lethargy or changes in posture. In a next step, the biodistribution of Cy5-SiO2 NCs was analyzed on a cell level. Therefore, NPCs of the liver were analyzed by flow cytometry. This heterogeneous group of cells consists of endothelial cells, stellate cells, Kupffer cells (KC), monocytes (Mono), and macrophages (MO) among others.29 The Kupffer cells in particular, representing the resident macrophages of the liver, play a major role in the pathogenesis of hepatic inflammations.17 CD68 is a pan marker for macrophages and is also expressed by Kupffer cells.30 Immunofluorescent staining (Fig. 5d) indicated that CD68-expressing cells are associated with an accumulation of Cy5-SiO2 NCs (Fig. S9, ESI†). Flow cytometric analyses were performed to measure the uptake of Cy5-SiO2 NC into specific NPC populations applying a set of fluorophore-conjugated antibodies and using the Cy5 intensity as a readout (see Fig. 5e and Fig. S10, S11, ESI†). Kupffer cells and other macrophages showed the strongest frequency of NC+ cells with 87% for both populations after injection in vivo followed by monocytes with 44% of NC+ cells. Kupffer cells and monocytes exhibited a very high median fluorescence intensity compared to all other cell populations investigated emphasizing a high uptake rate of Cy5-SiO2 NCs (Fig. 5f). Efficient delivery of DXM to these key player cell types bears the potential for robust suppression of intrahepatic inflammation.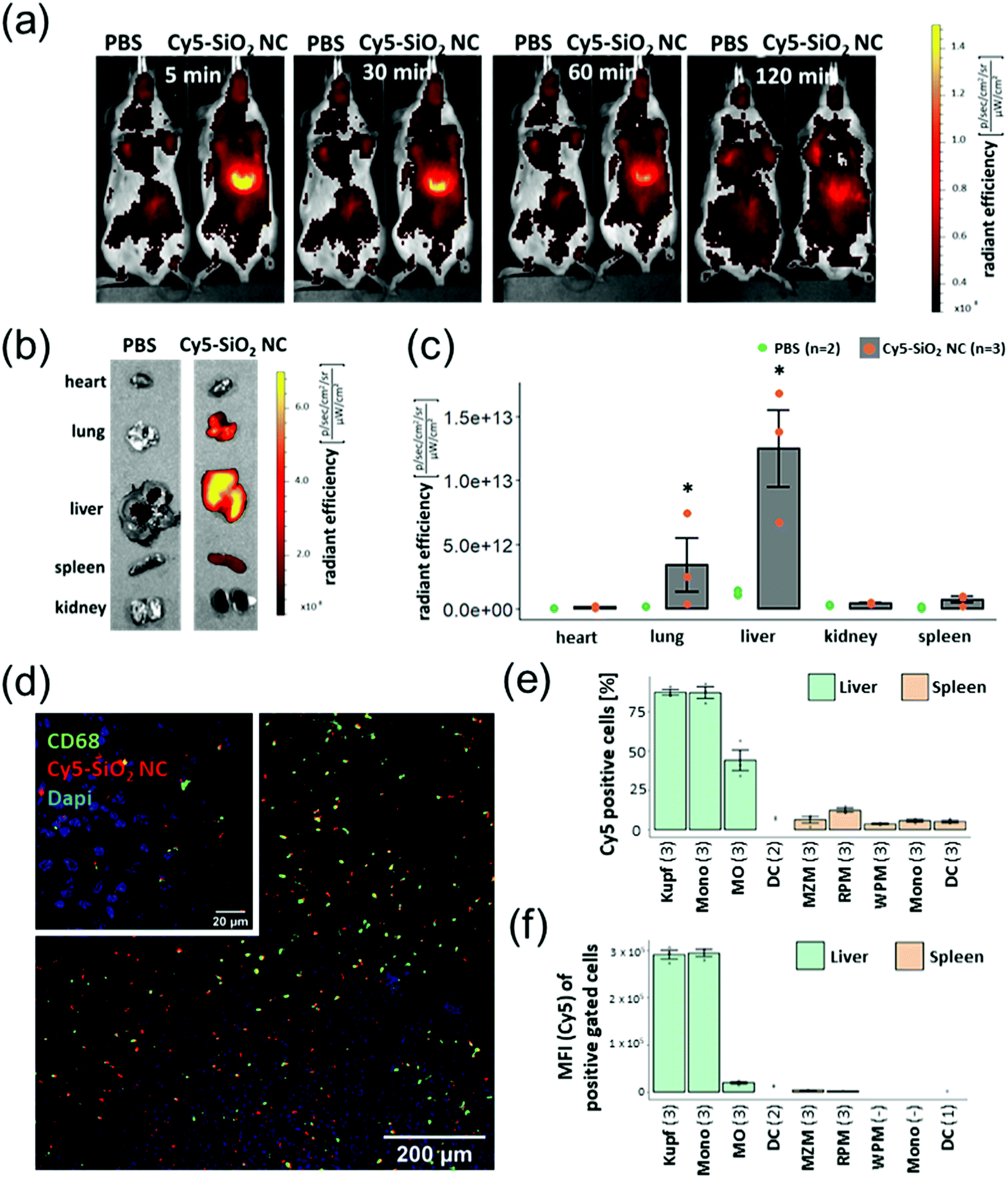 | ||
| Fig. 5 (a) In vivo imaging (IVIS) acquisitions of Cy5-SiO2 NCs-treated mice at different time points. (b–f) In vivo imaging, fluorescence microscopy and flow cytometric analyses were performed 2 h after NC administration. (b) Fluorescent imaging of Cy5-SiO2 NCs in organs 2 h after NC injection. (c) The graph shows the means ± SD of radiant efficiency for three animals investigated (grey columns and red points). The green points represent the background fluorescence intensity of two animals injected with PBS as a control. P < 0.05 (*) (Kruskall–Wallis test). (d) CLSM images of Cy5-SiO2 NC and CD68-based fluorescence on liver sections. The large image was acquired using the 10× objective, the insert (upper left) was taken using the 63× oil immersion objective at a separate spot. (e) Percentage of Cy5-SiO2-NC positive cells within defined populations. The graph shows the means ± SD of Cy5 positive cells of each population as described in Table S2 (ESI†). If the population included less than 50 cells, the value was excluded from the analysis. The number of animals investigated are indicated in brackets. (f) Median fluorescence intensity (MFI) of Cy5 positive cells. | ||
The uptake into splenic phagocyte populations was significantly lower (Fig. 5e, f and Fig. S12, ESI†) compared to NPCs. This finding is consistent with the in vivo imaging data showing a significantly reduced accumulation of Cy5-labelled SiO2 nanocapsules in the spleen in comparison to the liver. Injection of an equivalent amount of Cy5-NHS ester (free dye) showed a substantially lower fluorescence signal in the liver as detected by in vivo imaging and immunohistochemistry (Fig. S13, ESI†).
Functional analysis of PDXM and SiO2 NC-PDXM
In addition to the preferential uptake of NCs by NPCs, the potential of various polyprodrug formulations to decrease lipopolysaccharide (LPS)-induced secretion of the pro-inflammatory cytokine interleukin-6 (IL-6) was evaluated.31 As shown in Fig. 6a, PDXM polyprodrugs significantly suppressed the secretion of IL-6 by NPCs in a dose-dependent manner regardless of the PDXM chain length, while cytotoxicity could not be observed for any of the applied concentrations and formulations (Fig. S15, ESI†). The PDXM showed a similar activity compared with the monomer DXM applied as a positive control. When PDXM was formulated into SiO2 NCs, an even more pronounced IL-6 reduction was achieved (Fig. 6b). Additionally, the anti-inflammatory effects of either free PDXM or PDXM encapsulated in SiO2 NCs were compared by applying equimolar amounts of the polyprodrug (Fig. 6c). Encapsulation of PDXM led to a substantial reduction of IL-6 secretion. To achieve a reduction of IL-6 secretion by 80%, only 10 ng mL−1 of PDXM was required when formulated in NCs. In comparison, 10 μg mL−1 of free DXM or PDXM was required to achieve the same effect representing a dose-sparing effect of about 1000-fold. Empty SiO2 NCs served as a negative control and did not induce suppression of IL-6 secretion in LPS-treated cells (Fig. 6b). The efficient downregulation of IL-6 by the SiO2 NC-PDXM can be attributed to the preferential uptake by Kupffer cells as the major key players for LPS-induced intrahepatic inflammation. The acidic pH conditions in endosomes and lysosomes could trigger the degradation of PDXM to DXM (Fig. 1). DXM subsequently diffuses through the endosomal and lysosomal membranes into the cytoplasm where it binds to glucocorticoid receptors leading to a transcriptional blockade of pro-inflammatory cytokines including IL-6 in the nucleus.32–34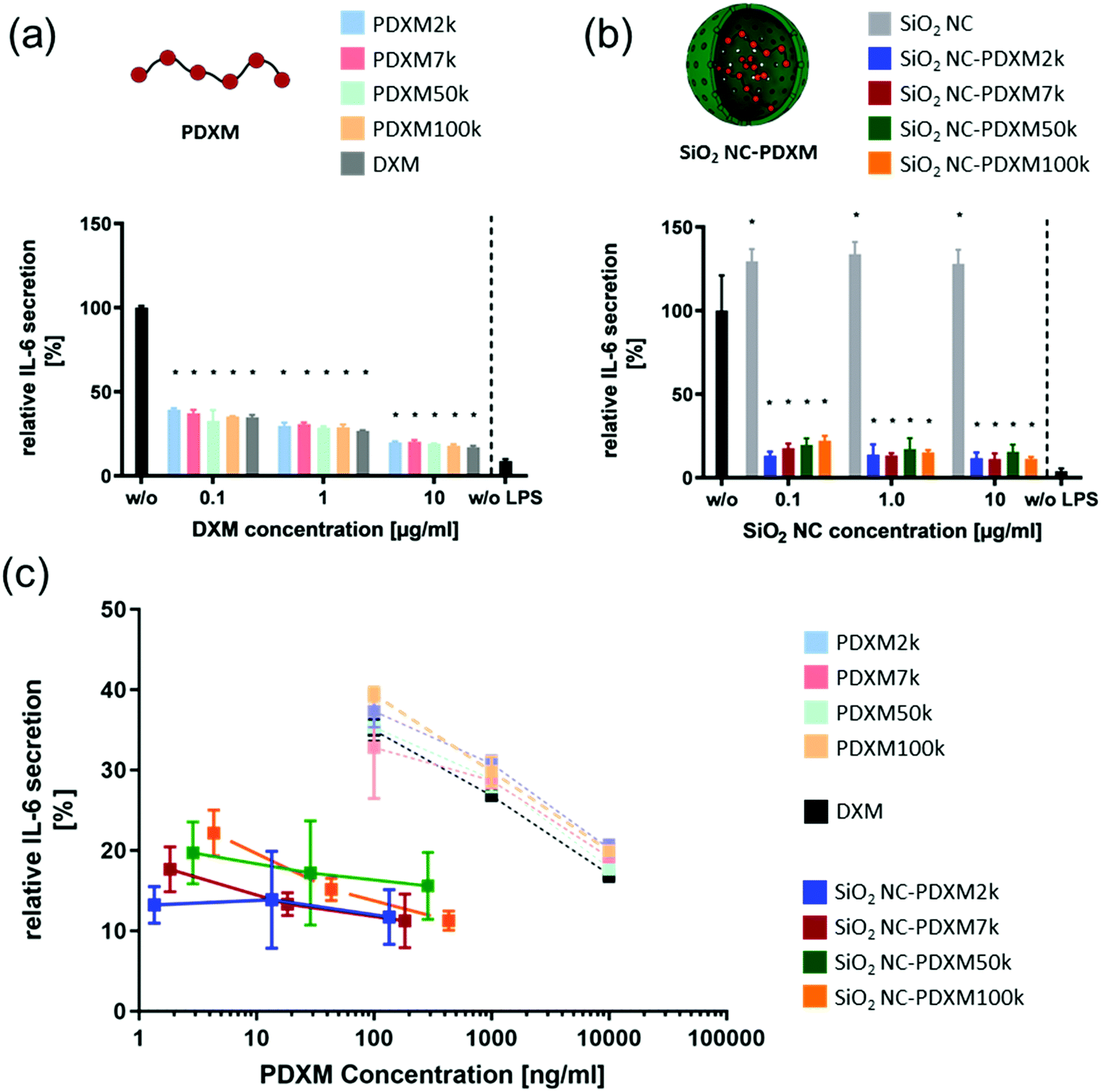 | ||
| Fig. 6 (a) Relative IL-6 secretion of LPS-stimulated (2.5 μg ml−1) NPCs treated with PDXM formulations for 24 h at different concentrations. Given are the means ± SD of 3 replicates each. The IL-6 expression was normalized to the untreated condition (black). P < 0.001 (*) (one-way ANOVA). (b) Relative IL-6 secretion of LPS-stimulated (2.5 μg ml−1) NPCs treated with PDXM-SiO2 NC formulations for 24 h at different concentrations. The IL-6 expression was normalized on the untreated condition. Given are the means ± SD of 3 replicates each. P < 0.001 (*) (one-way ANOVA). (c) The relative IL-6 expression from Fig. 6a and b plotted against the applied final concentration of PDXM in the cell culture medium. | ||
Conclusion
We developed a nanocontainer-based polyprodrug delivery strategy for selective and efficient delivery of water-soluble low molecular weight drugs. In the present proof-of-concept study, we generated PDXM-loaded SiO2 NC, addressing KC in the liver and reducing inflammatory responses by intracellular release of DXM out of the nanocapsule. We copolymerized DXM phosphate with adipic acid dihydrazide as a linker molecule in order to form prodrug polymers containing pH-cleavable hydrazone linkages in the backbone. In a next step, PDXM polyprodrugs were encapsulated in mesoporous silica nanocapsules, allowing protons at acidic pH (e.g. in the lysosome) to permeate the capsule shell and entering the PDXM-loaded core. Subsequently, this leads to cleavage of PDXM into DXM monomers and the release into the cytosol. In comparison to non-encapsulated polyprodrug, the herein presented SiO2-PDXM NCs required an approx. 1000-Fold lower drug concentration to induce a comparable anti-inflammatory effect in vitro. The finding that intravenously injected PDXM-loaded SiO2 NCs accumulated in KCs in the liver demonstrates the potential for this nanocarrier system to be applied in future studies for the treatment of chronic or acute intrahepatic inflammatory diseases.Author contributions
S. J., D. C. and K. L. designed the project; M. L. did the synthesis and characterization of polyprodrugs and nanocapsules; A. H. did the in vivo biodistribution and cellular uptake study of nanocapsules with the guidance from V. M.; S. W. and M. F. did the functional analysis of polyprodrugs and nanocapsules with the guidance from S. G.; J. S. did the protein corona study with the guidance from V. M.; M. W. did the NMR study for the polyprodrug synthesis and degradation; M. L. and S. J. drafted the manuscript with inputs from all the authors. S. J., V. M., S. G., D. C., and K. L. corrected the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge Stefan Schuhmacher for advices on scheme design. D. C. acknowledges the support of the Office of Higher Education Commission of Thailand (OHEC). Open Access funding provided by the Max Planck Society.References
- Y. Bae, W.-D. Jang, N. Nishiyama, S. Fukushima and K. Kataoka, Mol. BioSyst., 2005, 1, 242–250 RSC.
- L. Quan, Y. Zhang, B. J. Crielaard, A. Dusad, S. M. Lele, C. J. Rijcken, J. M. Metselaar, H. Kostková, T. S. Etrych and K. Ulbrich, ACS Nano, 2013, 8, 458–466 CrossRef PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef CAS PubMed.
- D. Maiolo, C. Pigliacelli, P. Sánchez Moreno, M. B. Violatto, L. Talamini, I. Tirotta, R. Piccirillo, M. Zucchetti, L. Morosi and R. Frapolli, ACS Nano, 2017, 11, 9413–9423 CrossRef CAS PubMed.
- F. Li, J. Lu, J. Liu, C. Liang, M. Wang, L. Wang, D. Li, H. Yao, Q. Zhang and J. Wen, Nat. Commun., 2017, 8, 1390 CrossRef PubMed.
- B. Demirdirek, J. J. Faig, R. Guliyev and K. E. Uhrich, Polymerized Drugs –- A Novel Approach to Controlled Release Systems: Synthesis, Characterization, and Applications, Polymers for Biomedicine, 2017 Search PubMed.
- L. Jing, Y. Fei, C. Yi and O. David, J. Controlled Release, 2015, 219, 369–382 CrossRef PubMed.
- Y. Zhu and J. Shi, Microporous Mesoporous Mater., 2007, 103, 243–249 CrossRef CAS.
- F. Liu, L.-X. Wen, Z.-Z. Li, W. Yu, H.-Y. Sun and J.-F. Chen, Mater. Res. Bull., 2006, 41, 2268–2275 CrossRef CAS.
- L. Maggi, L. Segale, M. Torre, E. O. Machiste and U. Conte, Biomaterials, 2002, 23, 1113–1119 CrossRef CAS PubMed.
- A. D. Yeoman, R. H. Westbrook, Y. Zen, W. Bernal, T. Al-Chalabi, J. A. Wendon, J. G. O’Grady and M. A. Heneghan, J. Hepatol., 2014, 61, 876–882 CrossRef CAS PubMed.
- A. Gauthier, A. Fisch, K. Seuwen, B. Baumgarten, H. Ruffner, A. Aebi, M. Rausch, F. Kiessling, M. Bartneck and R. Weiskirchen, Biomaterials, 2018, 178, 481–495 CrossRef CAS PubMed.
- L. W. Doyle, R. A. Ehrenkranz and H. L. Halliday, Neonatology, 2010, 98, 217–224 CrossRef CAS PubMed.
- U. Soiberman, S. P. Kambhampati, T. Wu, M. K. Mishra, Y. Oh, R. Sharma, J. Wang, A. E. Al Towerki, S. Yiu and W. J. Stark, Biomaterials, 2017, 125, 38–53 CrossRef CAS PubMed.
- M. C. C. Ferrer, V. V. Shuvaev, B. J. Zern, R. J. Composto, V. R. Muzykantov and D. M. Eckmann, PLoS One, 2014, 9, e102329 CrossRef PubMed.
- A. Boltjes, D. Movita, A. Boonstra and A. M. Woltman, J. Hepatol., 2014, 61, 660–671 CrossRef CAS PubMed.
- X.-M. Liu, L.-D. Quan, J. Tian, Y. Alnouti, K. Fu, G. M. Thiele and D. Wang, Pharm. Res., 2008, 25, 2910–2919 CrossRef CAS PubMed.
- M. J. Webber, J. B. Matson, V. K. Tamboli and S. I. Stupp, Biomaterials, 2012, 33, 6823–6832 CrossRef CAS PubMed.
- H. Wang, Y. Li, H. Bai, J. Shen, X. Chen, Y. Ping and G. Tang, Adv. Funct. Mater., 2017, 27, 1700339 CrossRef.
- A. G. Bajpayee, M. A. Quadir, P. T. Hammond and A. J. Grodzinsky, Osteoarthrotic Cartilage, 2016, 24, 71–81 CrossRef CAS PubMed.
- P. Kumar, K. Kadyan, M. Duhan, J. Sindhu, V. Singh and B. S. Saharan, Chem. Cent. J., 2017, 11, 115 CrossRef PubMed.
- M. Zhang, D. Wang, B. Bai, H. Wang, L. He and M. Li, Chin. Chem. Lett., 2018, 29, 497–500 CrossRef CAS.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS.
- M. A. Hood, U. Paiphansiri, D. Schaeffel, K. Koynov, M. Kappl, K. Landfester and R. Munoz-Espi, Chem. Mater., 2015, 27, 4311–4318 CrossRef CAS.
- Y. Wang, C. Wang, Y. Li, G. Huang, T. Zhao, X. Ma, Z. Wang, B. D. Sumer, M. A. White and J. Gao, Adv. Mater., 2017, 29, 1603794 CrossRef PubMed.
- K. Rausch, A. Reuter, K. Fischer and M. Schmidt, Biomacromolecules, 2010, 11, 2836–2839 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, R. Sakhtianchi, B. Müller, K. Landfester, D. Crespy and M. Mahmoudi, Colloids Surf., B, 2014, 123, 143–149 CrossRef CAS PubMed.
- D. L. Laskin, Biological Reactive Intermediates IV, Springer, 1991, pp. 499–505 Search PubMed.
- E. Zigmond, S. Samia-Grinberg, M. Pasmanik-Chor, E. Brazowski, O. Shibolet, Z. Halpern and C. Varol, J. Immunol., 2014, 193, 344–353 CrossRef CAS.
- R. Taub, Nat. Rev. Mol. Cell Biol., 2004, 5, 836–847 CrossRef CAS PubMed.
- K. Scheschowitsch, J. A. Leite and J. Assreuy, Front. Endocrinol., 2017, 8, 16 Search PubMed.
- M. Kadmiel and J. A. Cidlowski, Trends Pharmacol. Sci., 2013, 34, 518–530 CrossRef CAS PubMed.
- M. Zen, M. Canova, C. Campana, S. Bettio, L. Nalotto, M. Rampudda, R. Ramonda, L. Iaccarino and A. Doria, Autoimmun. Rev., 2011, 10, 305–310 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nh00266j |
| This journal is © The Royal Society of Chemistry 2021 |