
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
What is special about silicon in functionalised organic semiconductors?†
Karl J.
Thorley
 *ab,
Micai
Benford
c,
Yang
Song
c,
Sean R.
Parkin
a,
Chad
Risko
ab and
John E.
Anthony
ab
*ab,
Micai
Benford
c,
Yang
Song
c,
Sean R.
Parkin
a,
Chad
Risko
ab and
John E.
Anthony
ab
aUniversity of Kentucky, Department of Chemistry, Lexington, KY 40508, USA. E-mail: karl.thorley@uky.edu
bUniversity of Kentucky, Center for Applied Energy Research, Lexington, KY 40511, USA
cCentre College, Department of Chemistry, Danville, KY 40422, USA
First published on 13th July 2021
Abstract
A carbon side-chain analogue to the high-performance organic semiconductor triethylsilylethynyl difluoroanthradithiophene has been synthesised and characterized. Atomic substitution of carbon for silicon results in subtle changes to opto-electronic properties, which are rationalised by density functional theory and balance of electron donating and withdrawing effects. Larger differences are observed in photostability and solid-state packing of the new material in comparison to known silicon and germanium derivatives. Comparison of the group 14 elements teaches us about the newly synthesised system, but also how the silylethynyl substituents used for the last two decades contribute to successful employment of functionalised polycyclic aromatic hydrocarbons as organic semiconductors.
Introduction
Since 2001,1 silylethynyl substituents have been a mainstay in the functionalisation of organic semiconductors to impart solubility and stability.2–4 Control of the size of the silyl group has allowed for the engineering of solid-state packing favourable for multi-dimensional charge transport as well as a multitude of optical effects.5 While originally considered as a space filling effect, further research has looked at the stabilising non-covalent interactions between side chains as a means to rationalise the crystal packing in such substituted systems.6 Substitution of silicon by its downstairs neighbour germanium on the same chromophore results in polymorphism which is closely linked to the electron density in the solubilising groups.7 The missing group 14 element is of course carbon, prompting us to investigate the synthesis of isostructural side chains built only of carbon and hydrogen atoms to determine the specific contribution of silicon (and germanium) in these systems.Results and discussion
Synthesis
Alkynyl sidechains containing only carbon and hydrogen atoms are known in the literature, and many are even commercially available, but are mostly limited to linear alkyl chains.8 Introduction of a tertiary substituent9 to the alkynyl group is expected to give sufficient bulk for the functionalized materials to adopt desirable π stacking interactions in the solid state. In particular, triethylsilyl ethynyl (TES) and triethylgermyl ethynyl (TEG) groups have been attached to the difluoro anthradithiophene (FADT, Fig. 1) acene core to yield 2D brickwork packing and high p-type charge mobility.10 Therefore, we set out to synthesise the triethyl all-carbon equivalent side chain, which was achieved in three steps that should be applicable to the synthesis of a wide range of alkynes containing a quaternary carbon centre (Scheme 1). Deprotonation of acetonitrile with lithium diisopropylamide (LDA) and reaction with ethyl bromide introduced the tertiary carbon group in nitrile 1 in a one-pot reaction. The nitrile group then underwent a single addition of methyllithium, resulting in an imine intermediate that could be hydrolysed to the corresponding ketone 2.11 This hydrolysis was quite sensitive to acid concentration, with potential for carbocation rearrangement and further reactions of the ketone.12 A more consistent hydrolysis was achieved with saturated ammonium chloride, albeit at a much increased reaction time and lower yield. Unreacted imine could be easily recovered after each hydrolysis attempt and the process repeated. Methyl ketone 2 then underwent a one-pot elimination through the action of LDA and diethyl chlorophosphate.13 Although the yields are somewhat low, influenced perhaps by the volatility imparted by the short alkyl substituents, this represents a general synthetic scheme to the preparation of branched alkyl acetylenes with further substrates under investigation for detailed structural evaluations. Following literature procedures,10 alkyne 314 was deprotonated with butyllithium and FADT quinone added, with aromatisation by tin(II) chloride yielding the final TEC-FADT product as a mixture of syn and anti isomers of the thiophene ring orientation.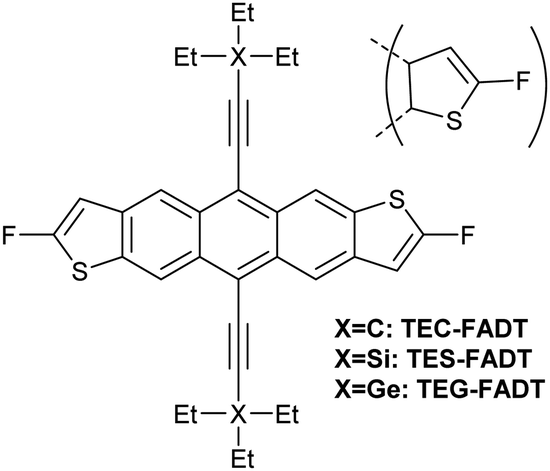 | ||
| Fig. 1 Chemical structure of FADT molecules bearing carbon, silicon or germanium side chains. Anti isomer is shown in full, but materials discussed here are a mixture of anti and syn isomers. | ||
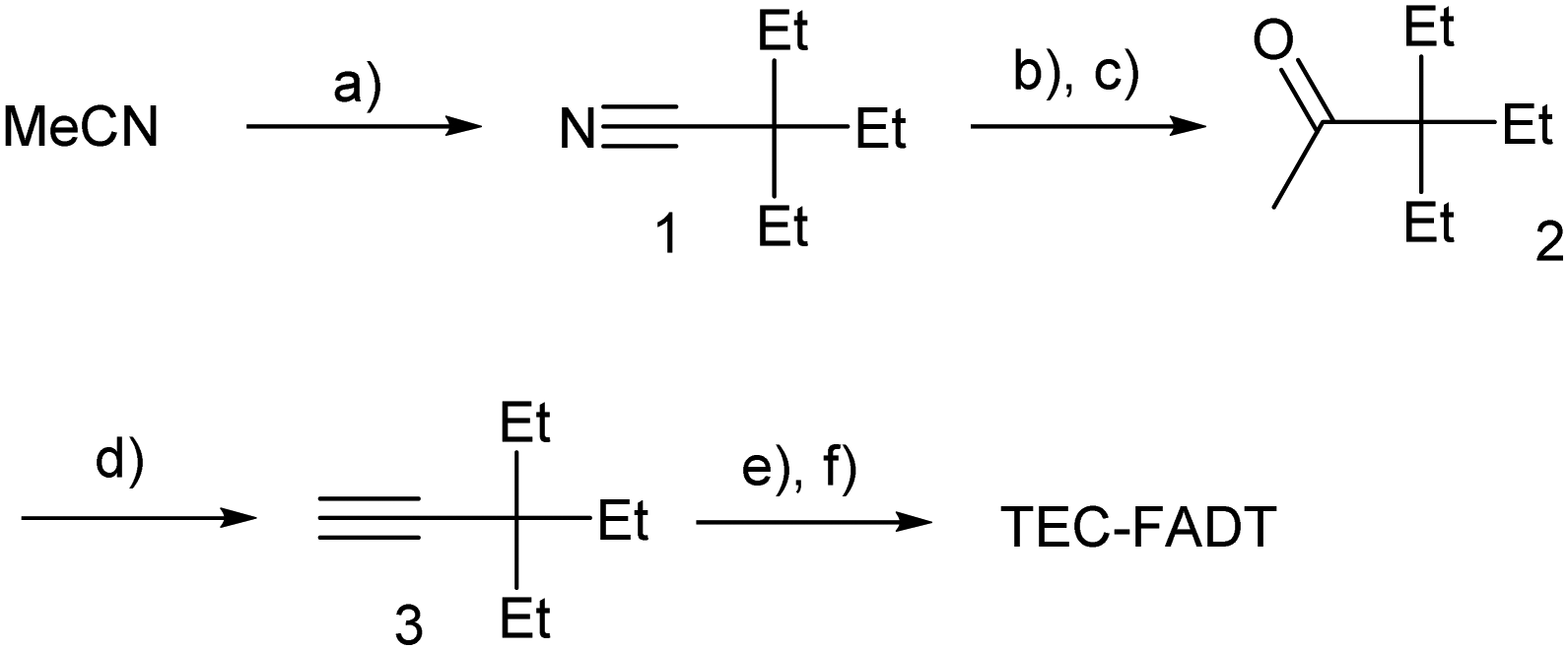 | ||
| Scheme 1 Synthesis of TEC-FADT. (a) 3.5 eq. iPr2NLi, 3.2 eq. EtBr, THF 0 °C, 95%; (b) MeLi, Et2O, 0 °C; (c) sat. NH4Cl (aq), THF, 41%; (d) 1.1 eq. iPr2NLi, 1.12 eq. (EtO)2P(O)Cl, 2 eq. iPr2NLi, THF, −78 °C to rt, 75%; (e) nBuLi, THF, 0 °C then FADT quinone; (f) SnCl2·2H2O, HCl, acetone, MeOH, 40%. | ||
Molecular properties
Upon isolating the carbon side-chained FADT, the colour was noticeably more yellow than the TES equivalent. Solution absorption spectra revealed an 11 nm (0.04 eV) blue-shift by introducing carbon in place of silicon, as well as a change in the relative intensities of the vibronic peaks. These spectral features were well reproduced by time-dependent density functional theory (TD-DFT, see Fig. S17, ESI†), suggesting a change in electronic structure rather than any kind of aggregation effects. TD-DFT indicated that the lowest energy transitions were indeed HOMO to LUMO excitations. The changes in absorption spectra were mirrored in the emission spectra, where both a shift in peak position (by 11 nm or 0.05 eV) and a change in band shape were observed (Fig. S15, ESI†). The vibronic pattern is more defined in the case of TEC-FADT, whereas broadening is observed for emission spectra of TES-FADT. A larger Stokes shift in TES-FADT (by 53 cm−1) suggests a larger reorganisation energy with the heavier group 14 element. Perhaps surprisingly, TEG-FADT exhibited optical properties intermediate between TES-FADT and TEC-FADT despite residing further down the group. These properties more closely resembled TES-FADT than TEC-FADT.The redox properties of the FADT derivatives were compared using cyclic voltammetry in solution, all showing reversible oxidation and reduction peaks within the solvent window (Table 1). TES-FADT exhibits redox potentials around 0.1 eV more positive than TEC-FADT, with estimated HOMO and LUMO energies being pushed to lower energy. As with the optical properties, TEG-FADT appears to have oxidation and reduction potentials in between those of the other two FADTs, and closer to TES-FADT. Trends in the DFT calculated ionisation potentials and electron affinities agree well with these experimental values.
| λ absmax /nm (eV)a | λ emmax /nm (eV)a | Stokes shifta cm−1 | E ox V | E red V | Estimated EHOMOc eV | Estimated ELUMOc eV | IP(calc)d eV | EA(calc)d eV | |
|---|---|---|---|---|---|---|---|---|---|
| a Measured in CH2Cl2 at 298 K at a concentration of 10−6 M. b Determined by cyclic voltammetry half-potentials in 0.1 M nBu4PF6 in CH2Cl2 relative to ferrocene oxidation. c Estimated from cyclic voltammetry redox potentials, assuming EHOMO for ferrocene = −4.8 eV. d Obtained using tuned ωB97XD/6-31G* in CHCl3 PCM. | |||||||||
| TEC-FADT | 517 (2.40) | 530 (2.34) | 474 | 0.45 | −1.88 | −5.25 | −2.92 | 5.09 | 2.16 |
| TES-FADT | 526 (2.36) | 541 (2.29) | 527 | 0.55 | −1.73 | −5.35 | −3.07 | 5.24 | 2.37 |
| TEG-FADT | 524 (2.37) | 538 (2.30) | 497 | 0.51 | −1.77 | −5.31 | −3.02 | 5.17 | 2.31 |
The trend in molecular electronic properties can be explained by considering the electron donating and withdrawing effects of the group 14 elements. Silicon and germanium are more electropositive than carbon and thus the Cδ−–Xδ+ bonds are polarised and provide a small inductive electron donating effect, which is about the same for both elements. This is seen in Hirshfeld atomic charges of the DFT optimised structures of the FADT series (Table 2). However, this effect would be expected to raise the energy of the frontier orbitals, and so the effect is considered to be small.
| qX a.u. |
|
|
|
Electron donation | |
|---|---|---|---|---|---|
| TEC-FADT | 0.03 | 15.48 | 9.99 | 1.02 | 4.47 |
| TES-FADT | 0.37 | 7.99 | 10.76 | 1.6 | −4.37 |
| TEG-FADT | 0.41 | 8.95 | 10.1 | 0.15 | −1.3 |
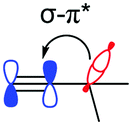
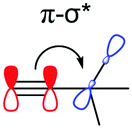
Previous studies have highlighted the ability for both silicon and germanium to accept electrons by resonance due to the presence of empty d-orbitals.15,16 This effect is expected to be weaker for germanium due to an elongated C–Ge bond and a mismatch in size and energy of the available d-orbitals with the alkynyl orbitals. Natural bond orbital (NBO) calculations hint at these resonance effects, made up of multiple small energetic stabilisations into numerous Rydberg states, which possess mostly d-orbital character (Table 2, π–d). Surprisingly, these are also present and comparable in energy in TEC-FADT, where the Rydberg states are made of contributions from both p and d vacant atomic orbitals based on carbon. While this effect is strongest for silicon, it does not fully explain the differences in molecular properties of the FADT system.
The alkyl chains attached directly to the group 14 element can also participate by hyperconjugation; their bonding orbitals can donate electron density into the two π* orbitals of the alkyne, while the alkyl σ* antibonding orbitals can accept electron density from the alkyne π orbitals. For carbon, the electron donating hyperconjugation heavily outweighs the electron withdrawing effects, while for silicon and germanium the electron withdrawing effects are stronger. This may be related to the bond polarisation of the Cδ−–Xδ+ bonds at the start of the alkyl chains, where Si or Ge can more readily accept electron density from the alkyne (π–σ*) due to the partial positive charge at these atoms. In contrast, the electron density in the alkyl C–X bonds is skewed towards the carbon atom and further away from the alkyne, so electron donation (σ–π*) is weaker for Si and Ge. Notably, these trends are not confined to the FADT (or acene) systems explored here, but simple model substituted benzenes show the same trends in molecular properties and NBO delocalisation (see Table S5, ESI†). An NBO comparison of tert-butyl benzene, trimethylsilyl benzene and trimethylgermyl benzene reveals that the silyl group is the most electron withdrawing for the same reasons as discussed here. The result of this is that silicon can be considered the strongest electron withdrawing group of the series, while germanium is weakly electron withdrawing and carbon is electron donating (Table 2). The electron withdrawing effects are further corroborated by the downfield shift of aromatic protons in the 1H NMR spectrum of TES-FADT relative to either TEC- or TEG-substituted systems.
Photostability
Acene materials are known to be sensitive to light, undergoing reaction with photogenerated oxygen species or photocycloadditions to give dimer species. The alkynyl groups are known to help stabilise the acene core against these reactions by adding steric bulk, lowering LUMO energies, which reduces the amount of reactive oxygen species generated, and allowing reversibility of these reactions by directing oxidation to only the centremost ring.17 Since the molecular optoelectronic properties are changed upon substitution of the group 14 element, we investigated whether the photostability is also influenced. Acene photostability is affected by choice of solvent and concentration,18 so all samples were made up to comparable absorbance at λmax, and then illuminated at close range with a high powered 365 nm LED. All samples were photostable over several hours in hexanes, but chromophore bleaching was observed in toluene (Fig. 2). Photodecomposition was fastest for TEC-FADT, then TEG-FADT, with TES-FADT being the most stable.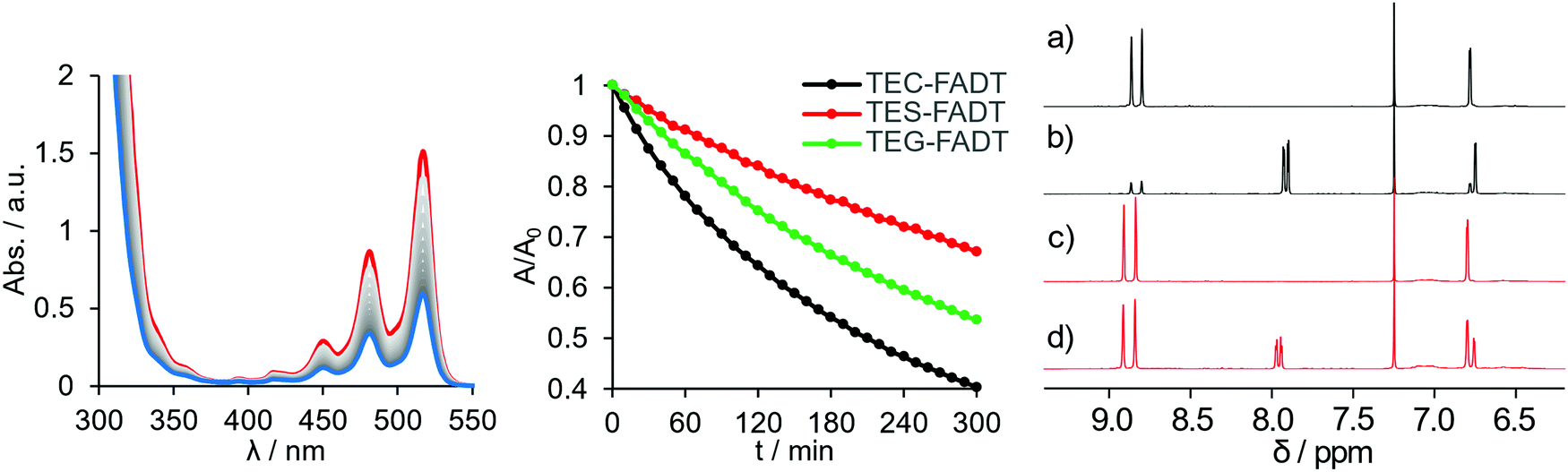 | ||
| Fig. 2 Photodecomposition of FADT molecules. (Left) UV-vis absorption spectra of TEC-FADT over 5 hours. Initial spectrum in red, final spectrum in blue, measured in PhMe at 298 K. (Middle) Normalised absorption at λmax over time for TEC-, TES- and TEG-FADT. (Right) Aromatic region of 1H NMR spectra of TEC-FADT before (a) and after (b) light exposure, and TES-FADT before (c) and after (d) light exposure. Measured in CDCl3 at 298 K. | ||
The most likely decomposition product is the endoperoxide, as has been discussed for acenes in general.19 This reaction is thought to occur through a number of reaction pathways. In short, the acene excited state can react with dissolved O2 to form reactive oxygen species, which then react with ground-state acene molecules. Thus, the energy level of the acene LUMO will dictate the formation of the reactive oxygen species and the energy of the acene HOMO will dictate the reaction of the acene with reactive oxygen species. The photostability follows the order of both the HOMO and LUMO energies, with TEC-FADT showing the fastest degradation. Given the fairly modest changes in orbital energies, the difference in photostability is quite stark, with TEC-FADT exhibiting a half-life of around 3 hours while TES-FADT did not reach 50% of its initial absorbance during the experimental timeframe. TEG-FADT exhibited photoreaction rates intermediate between TEC- and TES-FADT, as might be expected from the trends in optoelectronic properties in Table 1. Calculated reaction enthalpies (see Table S6, ESI†) show that the endoperoxide formation is the most favourable for TEC-FADT by 6.7 kJ mol−1 over TES-FADT, and that endoperoxide formation is more favourable than an alternative photodimerization pathway in each case. The photoreaction was seen to be reversible since gentle heating returned the FADT chromophore for all three derivatives.
NMR spectroscopy provides further information on the photodecomposition reactions, since exposing the samples to light produced additional peaks in the aromatic region (Fig. 2). Notably, only a single symmetrically substituted product is formed, presumably the endoperoxide, with upfield shifts of the benzenoid proton signals. After 5 days under identical illumination for all samples, TEC-FADT mostly converted to the endoperoxide, while the TES-FADT sample remained a mixture of FADT and endoperoxide. The photoreaction is likely slowed down due to the increased concentration for NMR versus absorption spectroscopy.18 Very dilute NMR samples showed almost complete photoconversion in much shorter time (see Fig. S16, ESI†). While these reactions are more rapid for TEC-FADT than either the silicon or germanium derivatives in solution, the molecular stabilities of all three materials are expected to greatly increase in the polycrystalline environment of transistor devices, which is the main application of this class of materials.
Additional molecular properties that are influenced by the group 14 substitution are the molecular vibrations observed in the IR spectrum (Fig. 3). A key vibration is the alkynyl C–C stretch, occurring around 2200 cm−1. This peak is much more intense in TES-FADT than in TEC-FADT, and comes with a small change to lower vibrational frequency due to the increased mass of the silicon atom. Other changes are observed at lower frequencies, where many absorptions are stronger for TES-FADT than TEC-FADT. Peaks around 700 and 1000 cm−1 are almost completely absent for TEC-FADT. DFT computed IR spectra largely agree with the experimental spectra, although shifted to higher wavenumber. Visualisation of the vibrations corresponding to the peaks at 700 and 1000 cm−1 reveal a complex mixture of core aromatic stretches as well as some involvement from the alkyne and group 14 element (Fig. S20, ESI†). It is likely that all of the vibrations are influenced by the change in electron density of the FADT core by electron withdrawing silicon, as well as an increased mass for vibrations involving silicon directly. The polarisation of Si–C bonds also leads to a larger change in bond dipole moment upon atomic displacement, thus the absorptions are generally more intense in TES-FADT than in TEC-FADT. The spectrum for TEG-FADT is very similar to that of TES-FADT (Fig. S21, ESI†). Differences in molecular vibrations could be particularly influential in the impact of dynamic disorder in solid state organic semiconductors, where low-frequency vibrations cause large fluctuations in the charge transport ability of the material.20
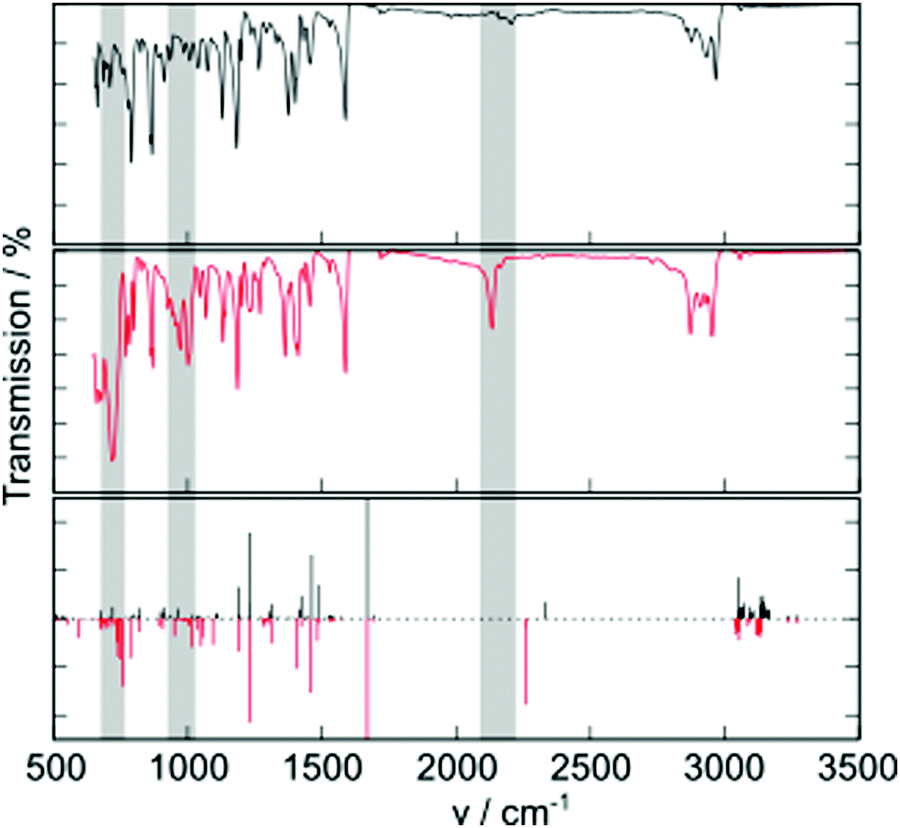 | ||
| Fig. 3 Experimental FTIR spectra of TEC-FADT (top) and TES-FADT (middle), and computed vibrational frequencies and intensities (bottom, TEC-FADT in black and TES-FADT in red) using tuned ωB97XD/6-31G*. Computed intensities for TEC-and TES-FADT are depicted with opposite sign for clarity. | ||
Solid state properties
Of prime interest to the development of the side chains built only of carbon and hydrogen is the packing motifs found in single-crystal structures of the functionalised acenes. Careful choice of solubilising chains allows materials to be crystal engineered such that orbital overlap between adjacent π systems is maximised for intermolecular charge transfer. TES-FADT adopts a 2D brickwork pattern10 which makes for an ideal charge transport pathway in thin-film transistor devices. TEG-FADT adopts an almost identical packing configuration to TES-FADT except for the rotation of one of the ethyl chains.21 In contrast, TEC-FADT adopts a 1D slipped-stack pattern (Fig. 4), where side chains from one stack interdigitate into the side chains of a neighbouring stack, preventing further π–π interaction. The 2D packing of TES-FADT by contrast has distinct boundaries between side chains of adjacent stacks. While the 1D slipped stack still offers a network of molecules for charge transport, it is also more likely to be affected by static and dynamic disorder effects and presence of impurities. TEC-FADT crystals contained disorder in the thiophene rings (a ratio of 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35% occupancies), due to the flipping of a molecule as well as the presence of both syn and anti isomers.
:35% occupancies), due to the flipping of a molecule as well as the presence of both syn and anti isomers.
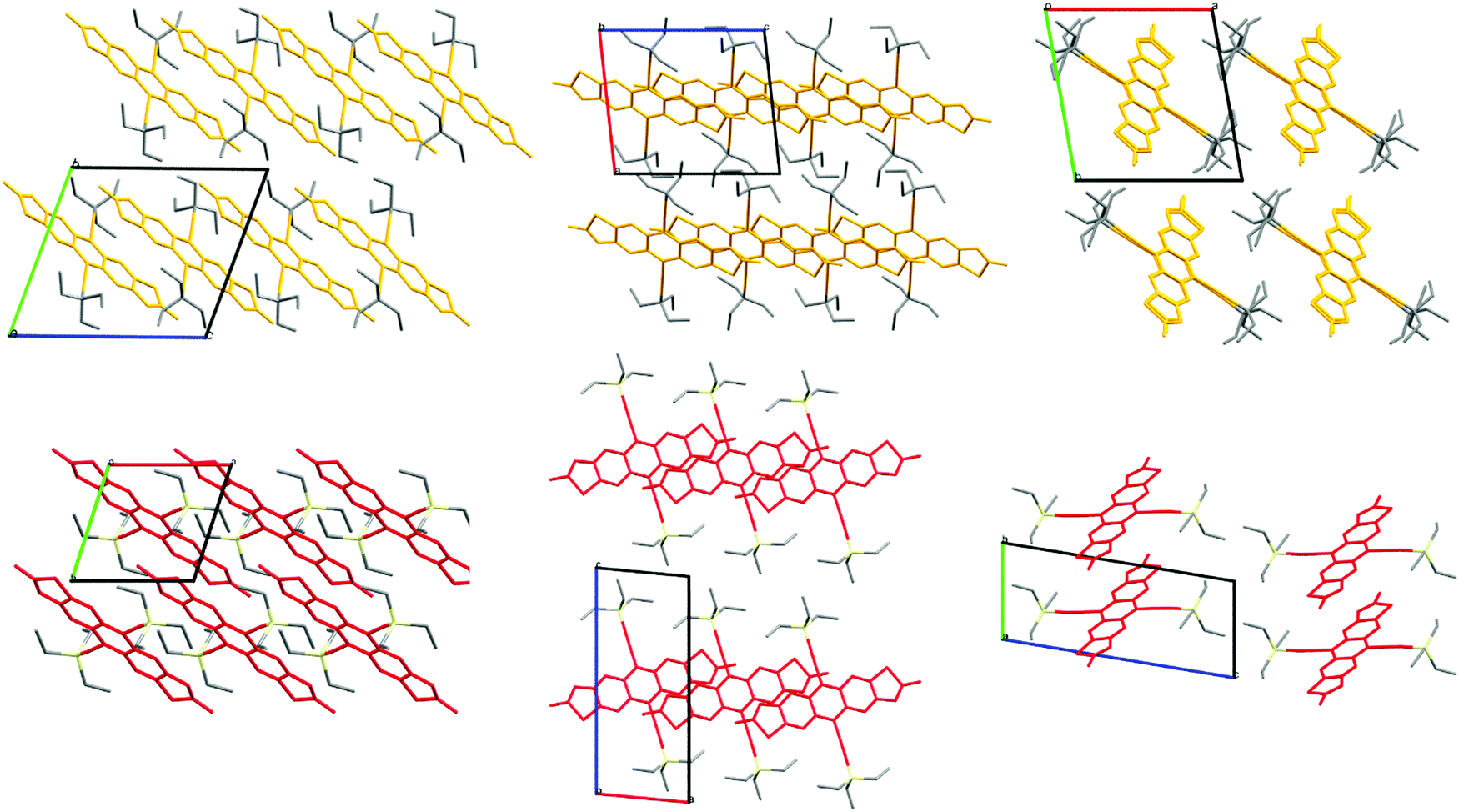 | ||
| Fig. 4 Crystal packing of TEC-FADT (orange, 1D, interdigitated side chains) and TES-FADT (red, 2D, interstack boundaries). | ||
Computational models of solid-state interactions
To rationalise the difference in packing between TEC and TES FADT molecules, a series of density functional theory (DFT) calculations were performed. First, the unit cells of TES-FADT and TEC-FADT were optimised using plane-wave DFT, and then the alkynyl substituent was swapped to create hypothetical TES-FADT in 1D stacking and TEC-FADT in 2D stacking structures which were further optimised. Binding energies (see Table S7, ESI†) of the respective structures show that indeed TEC-FADT is more stable in its 1D packing (−343.2 vs. −323.0 kJ mol−1), while TES-FADT is more stable in a 2D packing (−337.2 vs. −332.0 kJ mol−1). However these energies are close enough that similar thin film polymorphs may be accessible, as in the case of germyl substituted pentacene derivatives.Symmetry adapted perturbation theory (SAPT0) calculations were performed on close pairs of molecules in the relaxed crystal structures. In the 1D packing, TEC-FADT was more stable than TES-FADT in the alkyl–π interaction, as well as the in-plane H–F interaction (See Table S8, ESI†). Of more interest are the differences in pairwise interaction in the desired 2D packing configuration (Fig. 5). The largest differences are all within the brickwork stacks, whereas alkyl–alkyl interactions between stacks are similar for both TES and TEC. Within the π-stacking framework, three different molecular interactions possess a larger stabilisation for the TES groups of close to 1 kcal mol−1 each over the TEC equivalent interactions. This is mainly due to an increased dispersion stabilisation energy, larger than the difference in exchange repulsion energy, therefore leading to an overall stronger interaction. In one of the π stacking directions (Fig. 5, black), the exchange energy is actually smaller for the TES group due to the increased Si–C bond length moving the alkyl atoms further away from the neighbouring molecule. The more remote interaction (Fig. 5, red) provides 1 kcal mol−1 more stabilisation for TES than TEC even though the interaction involves only longer distance alkyl–alkyl interactions. There is also some extra stabilisation from electrostatics with the TES side chain for each pairwise interaction, which might originate from the polarisation of the Siδ+–Cδ− bond and interactions of these partial charges with neighbouring molecules. The findings from SAPT0 calculations can also be rationalised by estimation of the side chain size. The longest atomic distances within the TEC side chain are found to be 0.3 Å shorter than TES in both DFT and crystallographic measurements. Intermolecular PM7 calculations (See ESI† for details) predict that the overall size of the TEC group is around 5.2 Å compared to around 5.8 Å for TES. While the maximum stabilisation at optimum intermolecular distance appears similar, longer range stabilisation is slightly higher for TES due to increased size and electron density. Therefore most of the differences in packing can be summarised as follows: The TEC side chain is slightly smaller than TES and has lower electron density. This results in lower dispersion stabilisation at the distances needed for 2D packing. Bond polarisation also offers some electrostatic stabilisation with TES side groups that is not available in TEC-FADT.
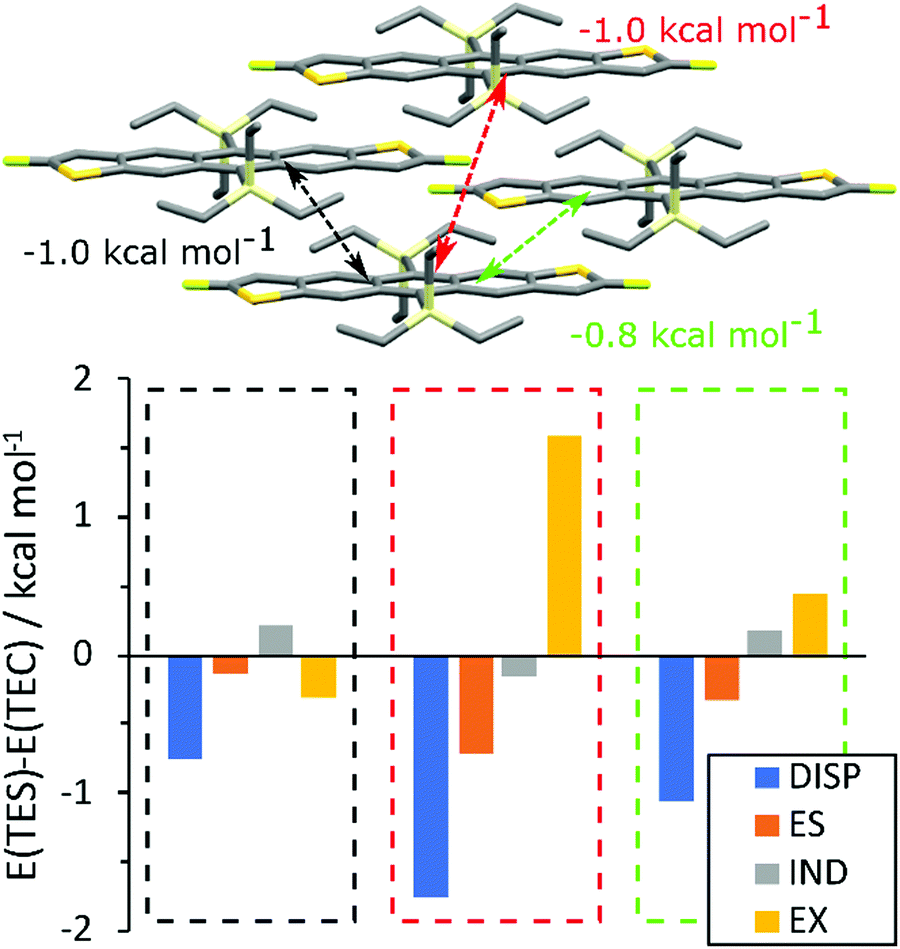 | ||
| Fig. 5 Difference in SAPT0 interaction energies (E(TES) min E(TEC)) for relaxed 2D packing structures. | ||
The crystal packing of TEG-FADT is very similar to that of TES-FADT, perhaps not surprising given that the germyl and silyl side groups are similar in size – (maximum side chain lengths are only 0.04 Å different from X-ray analysis) and side chain polarity (similar Hirshfeld atomic charges in Table 2). One main difference between TES and TEG side chains is the electron density due to the larger electron count of the heavier element, which would be expected to increase the magnitudes of both dispersion stabilisation and exchange repulsion, and result in slight molecular re-alignment to maximise the overall non-covalent binding. SAPT0 analysis of relaxed 2-dimensional packing (See Fig. S23, ESI†) reveal that one π-stack exhibits a stronger non-covalent interaction in TEG-FADT by 0.4 kcal mol−1, with the other remaining equally attractive for both TES and TEG side chains. The third more distant sidechain–sidechain interaction is slightly more stabilised in TES-FADT (0.15 kcal mol−1) due to reduced exchange repulsion. Overall, these differences in pairwise interactions are much smaller than the difference between silicon and carbon derived side chains.
Conclusions
To investigate the influence of the group 14 atom in alkynyl substituted organic semiconductors, we have synthesised a carbon equivalent to the triethylsilyl (TES) group. Attaching to a difluoroanthradithiophene core results in a π system which bears subtle changes in its electronic structure from the silyl derivative. Silicon and germanium act as electron withdrawing groups by resonance and hyperconjugation, while carbon side chains are slightly electron donating. This electron donation and adjustment of molecular orbital energies results in decreased photostability for TEC-FADT, or conversely, the inclusion of silyl groups in organic semiconductor design over the last decade has aided the photostability of these materials. The changes in electronic structure and bond polarity also impact vibrational spectra, hinting that the dynamic disorder in thin films for transistor application may be affected. Despite the same ethyl substituents, differences in overall solubilizing group size and changes in bond polarisation affect the solid-state packing. All of our findings are supported by theoretical calculations. Using a general synthetic route outlined here, we are currently synthesising a range of carbon alkynes to crystal engineer acene molecules into packing that is desirable for charge transport, and to further investigate how this atomistic substitution may play an important role on bulk material properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The D8 Venture diffractometer was funded by the NSF (MRI CHE1625732). M.B acknowledges KY-WV LSAMP - grant number NSF HRD 1826763f for funding. Synthetic efforts were supported by the National Science Foundation under Cooperative Agreement No. 1849213. Computational analysis was supported by NSF DMREF (DMREF-1627428).References
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, Functionalized Pentacene: Improved Electronic Properties from Control of Solid-State Order, J. Am. Chem. Soc., 2001, 123(38), 9482–9483, DOI:10.1021/ja0162459.
- L. Zhang, A. Fonari, Y. Liu, A.-L. M. Hoyt, H. Lee, D. Granger, S. Parkin, T. P. Russell, J. E. Anthony, J.-L. Brédas, V. Coropceanu and A. L. Briseno, Bistetracene: An Air-Stable, High-Mobility Organic Semiconductor with Extended Conjugation, J. Am. Chem. Soc., 2014, 136(26), 9248–9251, DOI:10.1021/ja503643s.
- L. Zhang, A. Fonari, Y. Zhang, G. Zhao, V. Coropceanu, W. Hu, S. Parkin, J.-L. Brédas and A. L. Briseno, Tri-Isopropylsilylethynyl-Functionalized Graphene-Like Fragment Semiconductors: Synthesis, Crystal Packing, and Density Functional Theory Calculations, Chem. – Eur. J., 2013, 19(52), 17907–17916, DOI:10.1002/chem.201303308.
- D. T. Chase, A. G. Fix, B. D. Rose, C. D. Weber, S. Nobusue, C. E. Stockwell, L. N. Zakharov, M. C. Lonergan and M. M. Haley, Electron-Accepting 6,12-Diethynylindeno[1,2-b]Fluorenes: Synthesis, Crystal Structures, and Photophysical Properties, Angew. Chem., Int. Ed., 2011, 50(47), 11103–11106, DOI:10.1002/anie.201104797.
- L. R. Weiss, S. L. Bayliss, F. Kraffert, K. J. Thorley, J. E. Anthony, R. Bittl, R. H. Friend, A. Rao, N. C. Greenham and J. Behrends, Strongly Exchange-Coupled Triplet Pairs in an Organic Semiconductor, Nat. Phys., 2016, 13(2), 176–181, DOI:10.1038/nphys3908.
- K. J. Thorley, T. W. Finn, K. Jarolimek, J. E. Anthony and C. Risko, Theory-Driven Insight into the Crystal Packing of Trialkylsilylethynyl Pentacenes, Chem. Mater., 2017, 29(6), 2502–2512, DOI:10.1021/acs.chemmater.6b04211.
- J. C. Sorli, Q. Ai, D. B. Granger, K. Gu, S. Parkin, K. Jarolimek, N. Telesz, J. E. Anthony, C. Risko and Y. L. Loo, Impact of Atomistic Substitution on Thin-Film Structure and Charge Transport in a Germanyl-Ethynyl Functionalized Pentacene, Chem. Mater., 2019, 31(17), 6615–6623, DOI:10.1021/acs.chemmater.9b00546.
- W. Ried, W. Donner and W. Schlegelmilch, Äthinierungsreaktionen, XVIII. Mono- Und Dianlagerungen von Monosubstituierten Acetylen-Derivaten an Unsubstituiertes Und Substituiertes Anthrachinon, Chem. Ber., 1961, 94(4), 1051–1058, DOI:10.1002/cber.19610940423.
- W. A. Chalifoux, M. J. Ferguson, R. McDonald, F. Melin, L. Echegoyen and R. R. Tykwinski, Adamantyl-Endcapped Polyynes, J. Phys. Org. Chem., 2012, 25(1), 69–76, DOI:10.1002/poc.1874.
- S. Subramanian, K. P. Sung, S. R. Parkin, V. Podzorov, T. N. Jackson and J. E. Anthony, Chromophore Fluorination Enhances Crystallization and Stability of Soluble Anthradithiophene Semiconductors, J. Am. Chem. Soc., 2008, 130(9), 2706–2707, DOI:10.1021/ja073235k.
- F. C. Whitmore and C. E. Lewis, Abnormal Grignard Reactions. XIV. Sterically Hindered Aliphatic Carbonyl Compounds. IV. Methyl Triethylcarbinyl Ketone and Its Bromomagnesium Enolate, J. Am. Chem. Soc., 1942, 64(7), 1618–1619, DOI:10.1021/ja01259a039.
- J. E. Dubois and P. Bauer, Metathetical Transposition of Bis-Tert-Alkyl Ketones. 1. A Model for a Study of Group Migration, J. Am. Chem. Soc., 1976, 98(22), 6993–6999, DOI:10.1021/ja00438a042.
- B. Stulgies, P. Prinz, J. Magull, K. Rauch, K. Meindl, S. Rühl and A. de Meijere, Six- and Eightfold Palladium-Catalyzed Cross-Coupling Reactions of Hexa- and Octabromoarenes, Chem. – Eur. J., 2005, 11(1), 308–320, DOI:10.1002/chem.200400723.
- W. Priester and R. West, Polylithium Compounds. 9. Sequential Derivatizations of C3Li4, MeC3Li3, and Me2C3Li2 with Diethyl Sulfate and Trimethylchlorosilane, J. Am. Chem. Soc., 1976, 98(26), 8421–8425, DOI:10.1021/ja00442a018.
- A. N. Egorockkin, S. E. Skobeleva and T. G. Mushtina, The First Ionization Potentials and Conjugation in Benzene Derivatives Containing Organosilicon, Organogermanium, Organotin, and Organolead Substituents, Russ. Chem. Bull., 1997, 46(1), 65–70, DOI:10.1007/bf02495349.
- A. J. Fleisher, C. D. Schaeffer, B. A. Buckwalter and C. H. Yoder, 1H,13C, And73Ge NMR Spectral Analysis of Substituted Aryltrimethylgermanes, Magn. Reson. Chem., 2006, 44(2), 191–194, DOI:10.1002/mrc.1746.
- W. Fudickar and T. Linker, Why Triple Bonds Protect Acenes from Oxidation and Decomposition, J. Am. Chem. Soc., 2012, 134(36), 15071–15082, DOI:10.1021/ja306056x.
- L. Abu-Sen, J. J. Morrison, A. B. Horn and S. G. Yeates, Concentration- and Solvent-Dependent Photochemical Instability of 6,13-Bis(Triisopropysilylethynyl)Pentacene. Adv, Opt. Mater., 2014, 2(7), 636–640, DOI:10.1002/adom.201400003.
- J. Zhang, Z. C. Smith and S. W. Thomas, Electronic Effects of Ring Fusion and Alkyne Substitution on Acene Properties and Reactivity, J. Org. Chem., 2014, 79(21), 10081–10093, DOI:10.1021/jo501696d.
- G. Schweicher, G. D’Avino, M. T. Ruggiero, D. J. Harkin, K. Broch, D. Venkateshvaran, G. Liu, A. Richard, C. Ruzié, J. Armstrong, A. R. Kennedy, K. Shankland, K. Takimiya, Y. H. Geerts, J. A. Zeitler, S. Fratini and H. Sirringhaus, Chasing the “Killer” Phonon Mode for the Rational Design of Low-Disorder, High-Mobility Molecular Semiconductors, Adv. Mater., 2019, 31(43), 1902407, DOI:10.1002/adma.201902407.
- Y. Mei, M. A. Loth, M. Payne, W. Zhang, J. Smith, C. S. Day, S. R. Parkin, M. Heeney, I. McCulloch, T. D. Anthopoulos, J. E. Anthony and O. D. Jurchescu, High Mobility Field-Effect Transistors with Versatile Processing from a Small-Molecule Organic Semiconductor, Adv. Mater., 2013, 25(31), 4352–4357, DOI:10.1002/adma.201205371.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2083385. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00447f |
| This journal is © The Royal Society of Chemistry 2021 |