
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanically tunable elastomer and cellulose nanocrystal composites as scaffolds for in vitro cell studies†
Senay
Ustunel‡
 ab,
Marianne E.
Prévôt‡
a,
Grace A. R.
Rohaley
a,
Caitlyn R.
Webb
ab,
Benjamin
Yavitt
c,
Guillaume
Freychet
c,
Mikhail
Zhernenkov
c,
Ron
Pindak
c,
Eric
Schaible
d,
Chenhui
Zhu
d,
Torsten
Hegmann
aefg,
Robert J.
Clements
*bef and
Elda
Hegmann
*abef
ab,
Marianne E.
Prévôt‡
a,
Grace A. R.
Rohaley
a,
Caitlyn R.
Webb
ab,
Benjamin
Yavitt
c,
Guillaume
Freychet
c,
Mikhail
Zhernenkov
c,
Ron
Pindak
c,
Eric
Schaible
d,
Chenhui
Zhu
d,
Torsten
Hegmann
aefg,
Robert J.
Clements
*bef and
Elda
Hegmann
*abef
aMaterials Science Graduate Program, Advanced Materials and Liquid Crystal Institute, Kent State University, Kent (OH) 44242-0001, USA. E-mail: ehegmann@kent.edu
bDepartment of Biological Sciences, Kent State University, Kent (OH) 44242-0001, USA. E-mail: rclement@kent.edu
cBrookhaven National Laboratory, National Synchrotron Light Source-II, Upton, NY 11973, USA
dAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
eBrain Health Research Institute, Kent State University, Kent (OH) 44242-0001, USA
fBiomedical Sciences Program, Kent State University, Kent (OH) 44242-0001, USA
gDepartment of Chemistry and Biochemistry, Kent State University, Kent (OH) 44242-0001, USA
First published on 15th December 2020
Abstract
Considering the range of properties that various materials offer for tissue engineering it has come clear that no one size fits all, as no one material can be fully effective for all types of cell and ensuing tissues. Scaffolds need to address the delicate balance between cell-scaffold interactions and the particular requirements of different cell types. To address the specific needs for the controlled growth of tissues it is imperative to match scaffold stiffness and elasticity to cells and tissues of interest to promote regeneration success. We here report an efficient method for creating scaffolds of tunable elasticity by generating a range of composites based on ε-caprolactone-D,L-lactide-based elastomer with cellulose nanocrystals (CNC). Two specific composites with different Young's modulus (E) values (∼5 MPa and ∼15 MPa) were selected and fully evaluated by tensile tests, Fourier Transform-Infrared (FT-IR), Scanning Electron Microscopy (SEM), contact angle measurements, and X-ray scattering. As a proof of concept this work studies how matching the scaffold's mechanical properties to neuroblastomas and fibroblasts cells affects cell behavior. Specifically, the composite with lower E, by design with less CNC content, is more suitable for neuroblastomas, whereas the one with higher E via higher CNC content is more suited for human dermal fibroblasts. The approach of matching cells with appropriate mechanical environments can provide important insights into fundamental cell behaviors.
1. Introduction
Prior research in tissue engineering (TE) has demonstrated that the interactions between cells and scaffolds are complex and delicate, and different tissues have particular needs, indicating that not all scaffolds can be fully functional for all cell types.1 Every cell in our body differs by its location, function, shape, size and mechanical properties. Also, every material implanted inside the body will face a dynamic, cell-specific environment and needs to be able to sustain mechanical stress and dynamic pressures while protecting and interacting with the surrounding tissues.2,3 At the same time substrate stiffness and porous architecture also affects cell behavior.4–6 Among several materials studied for TE, natural and synthetic polymeric scaffolds offer versatility in geometries and pore sizes and interconnections for mimicking and/or promoting the natural extracellular matrix (ECM).7,8 In tissue repair, different proteins of the ECM affect the integrity, adhesion, migration, and trigger signaling within the injury, as well as the timeline of these events.9,10 Physical and mechanical properties of ECM are distinct in different tissues.11 Mechanical properties of cells are established by cytoskeletal elements which consist of actin filaments, intermediate filaments, and microtubules12 and each of them responds differently to deformations.13 Cell–ECM interactions are also important with integrins and other adhesion molecules anchoring cells to ECM constituents.14 Midwood et al. reported the timeline for cells to reach maturation and tissue start to regenerate and remodel to be about 15 days for three different cell lines (fibroblasts, myofibroblasts, endothelial cells).15The stiffness range of living tissue varies from the Pa to the GPa range, have been reported depending on the measurement method.16 Youngs Moduli (YM, E) values range from grey matter (indentation ∼ 3 kPa, tensile ∼ 2 MPa), to liver and kidney (indentation ∼ 190 kPa, tensile ∼ 10 MPa) to skin (indentation ∼ 85 kPa, tensile ∼ 30 MPa) and muscle (indentation ∼ 7 kPa, tensile ∼ 480 MPa).16 Several reports17,18 indicate that elastomers and Liquid Crystal Elastomers (LCEs) can have E within the range of several soft tissues in the human body.
Considering their similar contractile and expansion properties, LCEs are promising as cell culture scaffolds, as they can mimic key features of endogenous tissue, promoting the development of the ECM and functioning as longitudinal multi-responsive cell scaffolds.19–21 In prior studies, our team succeeded in creating a series of porous film-cell culture side-chain smectic-A liquid crystal elastomer (SmA-LCE) based scaffolds where the mechanical properties were adjusted to fit the mechanical needs of skeletal muscle cells (C2C12s),22 and human dermal fibroblasts (hDFs).23 We also reported nematic globular LCEs to be adequate for cell lines such as C2C12s, hDFs and neuroblastomas (SH-SY5Y).24,25 We then created foam-like architectures showing four-times higher cell-proliferation capability compared to conventional porous templated films,26 and we later demonstrated that this design supports long-term neuronal cultures for over 60 days.27,28 We have shown that cells sense the anisotropy of LCE scaffolds thereby promoting cell orientation and alignment without the use of external stimuli.23,27
To further the search for mechanically compatible materials we began to study the most abundant natural biopolymer on earth, cellulose. There are many studies that have shown tuning of tensile strength and E values using cellulose nanocrystals (CNC) additives in different polymer matrices such as polylactic acid (PLA), poly(vinyl alcohol) (PVA) and poly(ethylene glycol) (PEG).29–31 It has been widely used in different applications due to its biocompatibility, biodegradability, and renewable nature; in addition, tons are produced annually.32–36 Thus, the last decade has seen a significant interest in the use of cellulose fibers for biomedical applications such as drug delivery,37 imaging agents,38 and biosensors.39–41 These treatments involve acid hydrolysis to produce rod-like crystalline shapes, with shape varying between 50 to 160 nm in length and 3 to 50 nm in diameter depending on source and treatment.42–44 Acid hydrolysis is performed by some of the most common acids–hydrochloric, sulfuric and phosphoric acid, to produce CNCs. The acid used reacts differently with the functional groups on the nanocrystals, affecting their colloidal stability. For example, sulfuric acid-derived CNCs have high colloidal dispersibility because this process creates a negatively charged surface due to conversion of hydroxy groups into sulfate ester groups; in aqueous dispersion these are stabilized by electrostatic repulsion.45 It was reported that CNC dispersions produced by sulfuric acid treatment form a lyotropic liquid crystalline (LLC) phase, which was later identified as a chiral nematic phase in aqueous dispersions.44,46,47
Cellulose and its derivatives’, specifically CNCs, mechanical properties improve stiffness and strength of polymer composites promoting the creation of novel polymer nanocomposites and lubricants48,49 or improving shape memory effects of polymer matrices.50 In addition to synthetic polymers, chemically modified cellulose derivatives such as cellulose acetate, hydroxypropyl cellulose, and carboxymethyl cellulose have been used as additives for functional biomaterials to create films, membranes, electrospun into nanofibers51 as well as formed into aerogel films,52 hydrogels,53 and three-dimensional porous foams.54–57
Fernandes et al. presented several studies that showed cellulose and its derivatives are biocompatible and have tunable biodegradable properties.58 Li et al. created CNC-reinforced collagen composite films and showed that the mechanical properties of these films improved wound healing on skin. In vitro test results of 3T3 fibroblast cell cultures showed no indication of cytotoxicity, further supporting the biocompatibility of CNCs.59 Additionally, as previously stated, use in drug delivery applications show their biocompatibility and biodegradability. Since CNCs have been recognized for exhibiting low cytotoxicity for a variety of animal and human cell-types,60–65 CNC/polymer composite platforms have shown high potential to support cell attachment and proliferation and can serve as an additive in a cellulose acetate propionate 3D matrix to mimic vascular tissue.66–69 CNCs were used for the production and orientation of myotubes; by first creating CNC monolayers that oriented muscle tissue cells, myoblasts (C2C12), which later differentiated to form radial motives of myotubes following the pattern of CNCs.70,71 Cellulose and chitin nanofibril-based hydrogel scaffolds, designed to be applied for bone tissue, exhibited attachment of human mesenchymal stem cells and induced differentiation into osteogenic-like cells. Moreover, CNC-reinforced polyacrylamide/sodium alginate/silica glass hybrid hydrogels showed potential for MC3T3-E1 osteoblast precursor cell adhesion and proliferation72 as well as an increase of the elastic modulus by up to 114% with 9% by weight of CNC in PBAT (poly(butylene adipate-co-terephthalate)) composites for L929 mouse fibroblasts.73
Moving beyond simple in vitro studies of plant-derived cellulose scaffolds, we aim to demonstrate that cell scaffolds with a suitable mechanical environment can potentially induce a controlled, growth and proliferation. In doing so, we should be able to shorten tissue growth and wound-healing times, which is usually challenging when using traditional 2D cell cultures. Such an approach would also contribute to providing cell-friendly 3D scaffolds in the form of polycaprolactone (PCL)-based elastomers–CNC composites designed for the particular needs of different types of cells and tuned according to the cell response observed.
All tissues in our body are exposed to mechanical forces deriving from interstitial flow and tissue movement. Cells are extremely sensitive to their environment and are affected by even the slightest changes in their environment and are capable of sensing the (mechanical) properties of their supportive scaffold, including both stiffness and strength.74,75 Cells are also known to apply stress onto their 3D scaffold matrix during tissue development.76 The scaffold stiffness then determines the contraction of ECM and more generally regulates cell behaviors and the migration that allows reorganization.
Here we present a biocompatible and biodegradable caprolactone-based elastomer–CNC composite that combines the swelling capability and degree of flexibility of elastomers with cell-customized tensile E properties, enhanced by CNC additives.77 The approach described here uses two different concentrations of CNCs, 5 wt% and 40 wt%, as reinforcers for ε-caprolactone-based elastomers to tune the mechanical properties of the resultant CNC/PCL composite to match two distinct cell lines, SH-SY5Y (Human Bone Marrow Neuroblastoma) and hDF (Human Dermal Fibroblast) cells, respectively. We compare the growth and proliferation of both cultures in the resultant composites to evaluate the importance of bulk tensile mechanical properties in the design of appropriate 3D scaffolds in regenerative medicine. In addition, we demonstrate that cells sense a mechanically matched scaffold environment ultimately influencing cellular functions and behaviors, including developmental processes, complex multicellular interactions, division rate, and cellular crosstalk.
2. Results and discussion
2.1. Scaffold mechanical properties
Materials deform when exposed to force, and the ratio between force and consequent deformation is known as Young's modulus, which describes the stiffness of the material. In the human body, for example, brain tissue is less stiff than muscle tissue and muscle tissue is less stiff than bone. When designing the mechanical properties of tissue building blocks, extracellular matrix (ECM) and cell type should be considered. They both have intrinsic mechanical properties, and the ECM moreover contains molecules that have roles in cellular signaling affecting mechanical properties and cell–cell interactions.Mechanical properties should be considered in combination with porosity and permeability where high porosity and adequate stiffness should be balanced.78 Biological tissues usually show a large spectrum of mechanical properties,5 so finding a mechanically optimized material to cover all tissues can be challenging. Our proposed binary composite platform consists of a CNC constituent added at several ratios (from 5 wt% to 50 wt%) to a polymeric scaffold material, enabling the tunability of the mechanical properties to match tissues of interest. In addition, we demonstrate that when used in combination with the salt-leaching method, this approach rapidly and efficiently results in a homogeneous, highly porous, interconnected architecture with 250 μm pore size that permits cell infiltration and proliferation.79–81
Native CNCs are mechanically strong, with axial elastic moduli varying from 110 to 220 GPa and tensile strengths ranging from 7.5 to 7.7 GPa,82–86 and this strength derives from their structure, namely microfibrils held together by hydrogen bonding. Considering this, we added CNCs to our 6A-PCL as reinforcement nanofiller to produce scaffold architectures with stiffer mechanical properties.
The synthesis and characterization of the polycaprolactone (PCL)-based elastomer (Scheme 1) and the resulting 3D scaffold are detailed in the Methods section. In a random ring opening polymerization, ε-caprolactone (ε-CL), (D,L)-lactide (D,L-LA), and dipentaerythritol (6-arm initiator) were polymerized using tin(II) 2-ethylhexanoate as a catalyst to obtain a 6-arm star block-copolymer (6A-PCL). The hydrophilic nature of cellulose, together with the presence of features of various length scales in the composite, can cause aggregation that may reduce the pronounced expected enhancement of the mechanical properties.58 To overcome agglomeration of the CNC constituents, both surfactants87 and surface modifications88 have been previously explored. We proceeded to disperse CNCs using ε-caprolactone as a solvent prior to crosslinking. This technique allowed us to create composites containing up to 50 wt% of CNCs before observing cluster formation. Observations by SEM validated the homogenization throughout the resulting elastomer composite (see Fig. S1 in ESI†). CNCs are incorporated into the thermally polymerizable pre-polymer solution, which can then be designed into different shapes (see ESI,† Fig. S2).
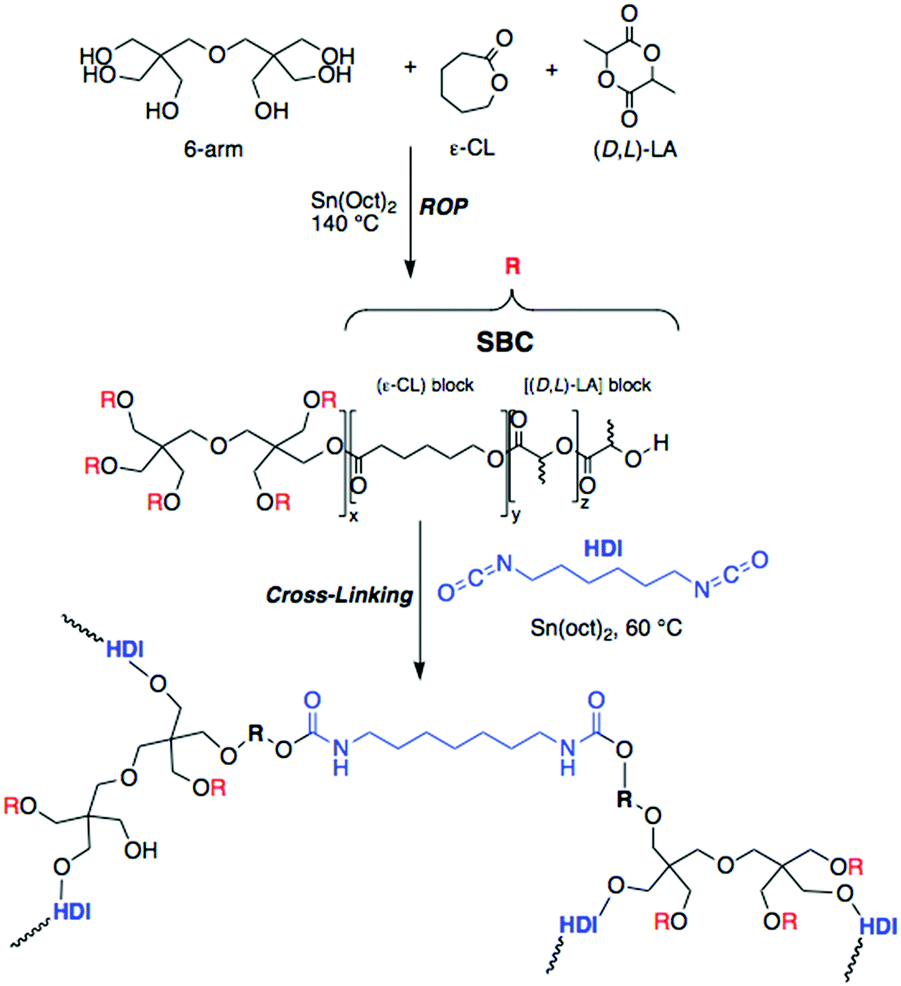 | ||
| Scheme 1 Synthetic path for 6-arm ε-caprolactone-based star block-copolymer elastomer (6A-PCL) by ring opening polymerization (ROP). | ||
Fig. 1 summarizes the uniaxial tensile stretching E obtained from the 6A-PCL/CNC nanocomposites, with different ratios of CNCs ranging from 0 wt% CNC content (pure 6A-PCL) to a composite containing 50 wt% of CNCs. The addition of CNCs effectively increases the bulk stiffness of the composite relative to the pure 6A-PCL-based elastomer and this dependency is exponential. We observe an 870% increase of the uniaxial tensile stretching E when the 6A-PCL/CNC contains 50 wt% of CNCs. The elastomer nanocomposite platform allows tuning of the bulk mechanical properties of the elastomer films to match tensile Es reported for soft biological tissues (tensile moduli values of soft tissues range from about 560 MPa for tendons to about 2 MPa for spinal cord containing gray matter).1,9 This type of tissue is able to resist deformation from a given tensile stress in the direction of fiber orientation. Values for skin tissues have been reported from 21 MPa to 39 MPa.89,90 This tensile E order of magnitude can be mimicked with a 6A-PCL/CNCs composite containing 40 wt% to 50 wt% of CNCs (respectively 14.6 ± 0.3 MPa and 22.9 ± 0.6 MPa). Tensile deformation for spinal cord is significantly lower, between 0.4 MPa to 3.6 MPa,91–94 which corresponds to the value obtained for the 6A-PCL/CNCs composite with 5 wt% CNCs (3.9 ± 0.1 MPa). Hence, by changing the percent by weight of CNC, we can tune the E of the scaffold to create an environment that is more favorable for the needs of each cell type.
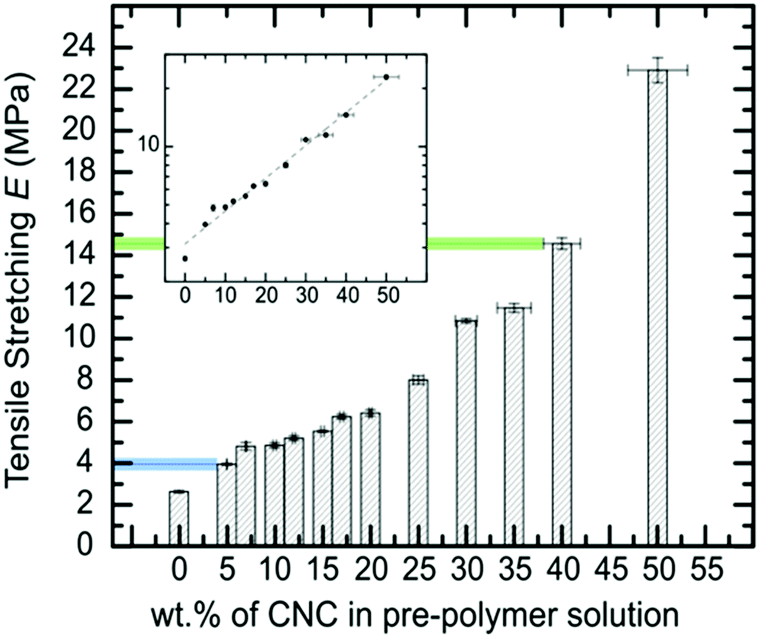 | ||
| Fig. 1 Evolution of the tensile Young's modulus of 6A-PCL/CNC composite films with the addition of CNCs (data measured by tensile stretching). The values in blue and green match reported tensile deformation values of brain and skin tissues, respectively. The inset shows the E in log10 scale, to emphasize the exponential dependency found with the addition of CNCs. | ||
2.2. Biodegradation behavior and ensuing changes in mechanical properties
Besides providing a mechanical and morphological environment for the tissue, the scaffold must be biocompatible and bioresorbable, its degradation rate should match that of tissue formation, typically until the injured or diseased tissue portion is completely replaced by newly formed healthy tissue.95 Sun reported the use of a hydrogel scaffold containing dextran-isocyanatoethyl methacrylate-ethylamine (DexIEME) to treat third degree burn scars. Scar tissue was replaced with a hydrogel scaffold and observed full skin regeneration in vivo after 5 weeks, with an increase in hair follicles compared to hairless untreated scarred skin.96 Bini et al. created a peripheral nerve conduit made of micro-braided PLGA biodegradable polymer fibers, and nerve generation was observed three weeks after the implementation.97 Nervous tissue is a complex environment and the process of restoration involves a variety of events that occur over time. Thus, the integrity of the tissue after implantation should be maintained for an extended period. Smith et al. studied tissue engineered nerve grafts and noticed a significant density of myelinated host axons at week 16.98,99 We have previously reported that the degradation of the ε-caprolactone-D,L-lactide polymer backbone, constituted of ester linkages, involves both an autocatalytic hydrolysis and an enzymatic action.6,100 Biodegradability studies of 6A-PCL in physiological buffers indicated that these scaffolds are intact for 11 to 15 weeks, after which degradation sets in at an exponential rate.6,12 Furthermore, caprolactone–lactide co-polymers are known to degrade primarily via bulk acid-catalyzed hydrolysis, which affect the whole sample via a bulk erosion mechanism.100,101 The degradation rate also depends on the degree of crosslinking.28 Therefore, this parameter as well as the amount of pre-polymer were kept constant in both sets of experiments, resulting in similar rates of degradation as shown in Fig. 2 and 3. Cellulose, being a linear polymer of β-1,4 linked glucose units, is degraded by cellulases, which are enzymes that hydrolyze β-1,4-linkages.102 This enzyme appears to be absent in humans. Additionally, the structure of cellulose itself practically excludes the process of enzymatic hydrolysis. As a result, cellulose composites become structurally stiffer, making them more resistant to degradation. The particularly slow degradation rate of cellulose has been reported by Märtson et al., which found that more than 60 weeks were necessary for viscose cellulose sponges implanted subcutaneously into rats to degrade.103 Here we focused on hydrolysis degradation, without enzymatic considerations, and the films tested contained no cells. In vitro degradation was measured by the amount of weight loss over time in DMEM media (Fig. 3). The approximately 15 days to form,15 will be ensured before the degradation of the support occurs (Fig. S3, see ESI†).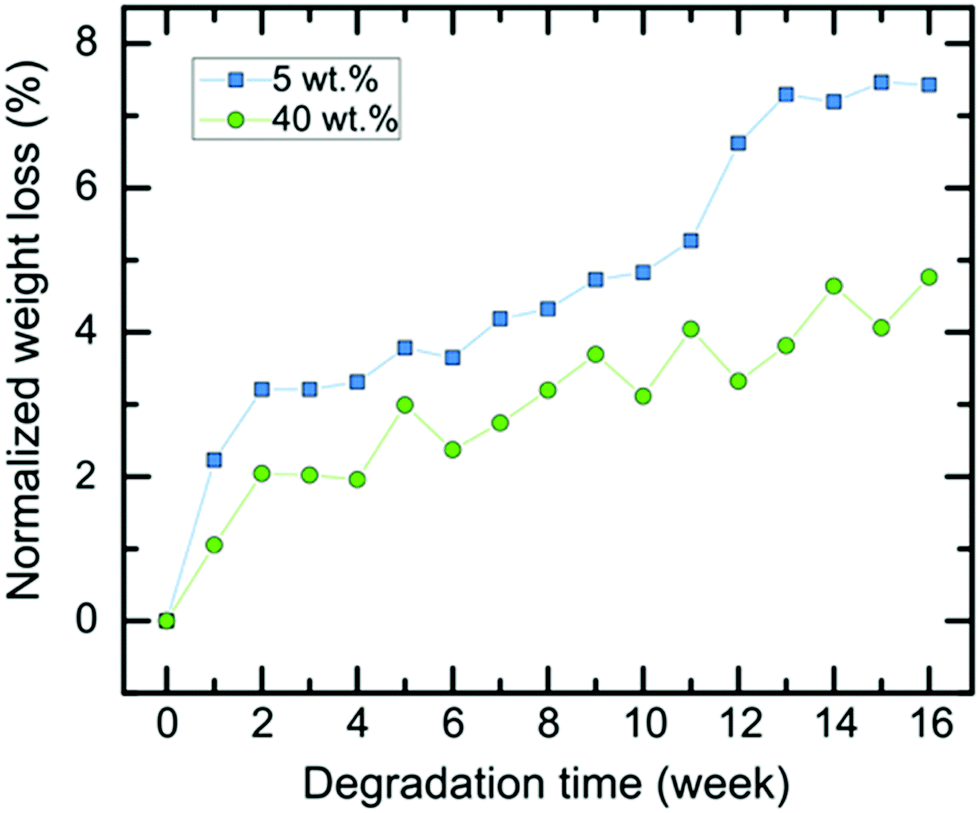 | ||
| Fig. 2 Normalized degradation curves of 6A-PLC/CNC nanocomposite scaffolds in DMEM media for 5 wt% (blue curve) and 40 wt% CNCs (green curve). | ||
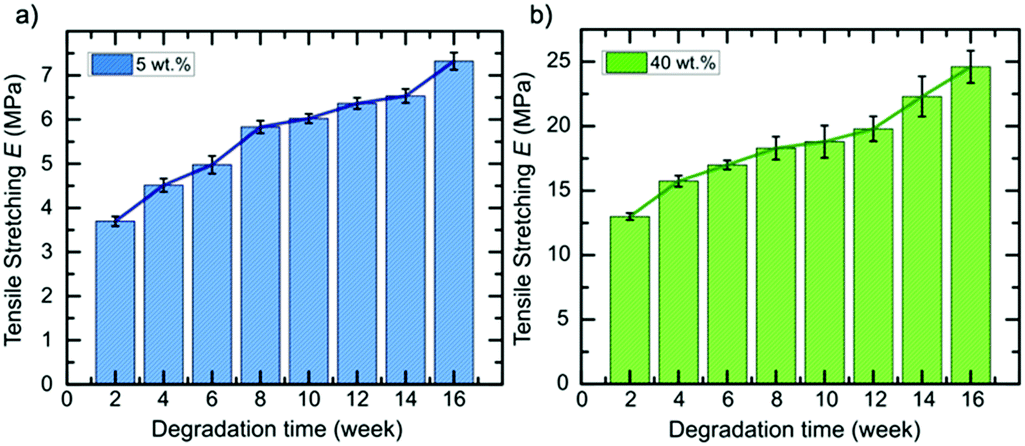 | ||
| Fig. 3 Evolution of the tensile stretching E of 6A-PCL/CNC nanocomposite scaffolds containing different wt% of CNCs in media solution as the degradation occurs. The composites contain (a) 5 wt% and (b) 40 wt% of CNCs, respectively. | ||
Next, we extended our degradation study to the mechanical strength resistance of both 5 wt% and 40 wt% composite scaffolds intended for cell culturing (in vitro hydrolytic degradation studies, see Experimental section for details). Samples were kept in DMEM media prior testing. We examined the evolution of the tensile E values as the degradation occurs (Fig. 3). A report on similar PCL-based elastomers without any cellulose content showed that the tensile E of such elastomers decreases with time according to a logarithmic regression.28 Here, we observed that the elastomer–CNC composites become stiffer over time causing their E to double after 16 weeks. We observed an increase in water content on day 3 of 2 (±1)% for the 5 wt% CNC and 13 (±2)% for the 40 wt% CNC composites, respectively. Comparing both datasets in Fig. 2 and 3 and focusing on weeks 10 to 14 (with a discernable change in slope of the weight of the studied composites increased periodically due to the formation of degradation products within the matrix that permitted the penetration of water into the polymer matrix via osmosis. These resulting non-toxic degradation products were then leached out of the elastomer block. In Fig. 3, we observe that the normalized weight loss at 40 wt% CNCs (green dataset) exhibits a low but steady rate of degradation, whereas at 5 wt% CNCs (blue curve) weight loss slopes upward after week 10, likely due to an increase in water absorption, indicative of a bulk hydrolysis mechanism in the PCL-based elastomer.100 However, at the end of week 16, the total change in weight of both composites did not vary more than 10%, indicating that cell growth and the development of ECM, requiring loss with time), we observed that for the 5 wt% CNC composite the E only increased from about 6 MPa to 6.5 MPa (8.3% change) whereas for the 40 wt% CNC composite the values increased from 18.8 to 22.3 MPa (18.6% change). Over time and with continued hydrolysis of 6A-PCL, we propose that 6A-PCL progressively contributes less to the value of E, especially at 40 wt% of CNC content, and that the CNC counterpart on the contrary becomes dominant in the bulk, likely due to hydrogen bonding between the CNCs and the acid as well as hydroxy groups of the remaining 6A-PCL fragments. As the degradation proceeds using DMEM cell media, the bulk mechanical integrity is maintained, and the order of magnitude of the E at week 16 continues to match the targeted tissues for 5 wt% and 40 wt% CNCs, respectively: 7.3 ± 0.2 MPa for the composite against [0.4–3.6] MPa for brain and 25 ± 1 MPa against [21–39] MPa for skin. We further studied the impact of CNC on our composites using FT-IR (see Fig. 4). There was a clear difference between both composites, especially at 40 wt% CNC content. At this higher CNC content, the hydrogen-bonding band at around 3500 cm−1 with the peak becoming broader and more intense as 6A-PCL degraded. Free O–H bonding is generally characterized by a single peak at 3600 cm−1 whereas a hydrogen bonding band is represented by a wide intense band from 3000 to 3600 cm−1.104 As 6A-PCL degrades, there are more opportunities for the CNCs to hydrogen-bond with themselves and with the degradation fragments of 6A-PCL, and these hydrogen bonds increase the stiffness of the material. This effect would indeed be more pronounced the higher the concentration of the CNCs.
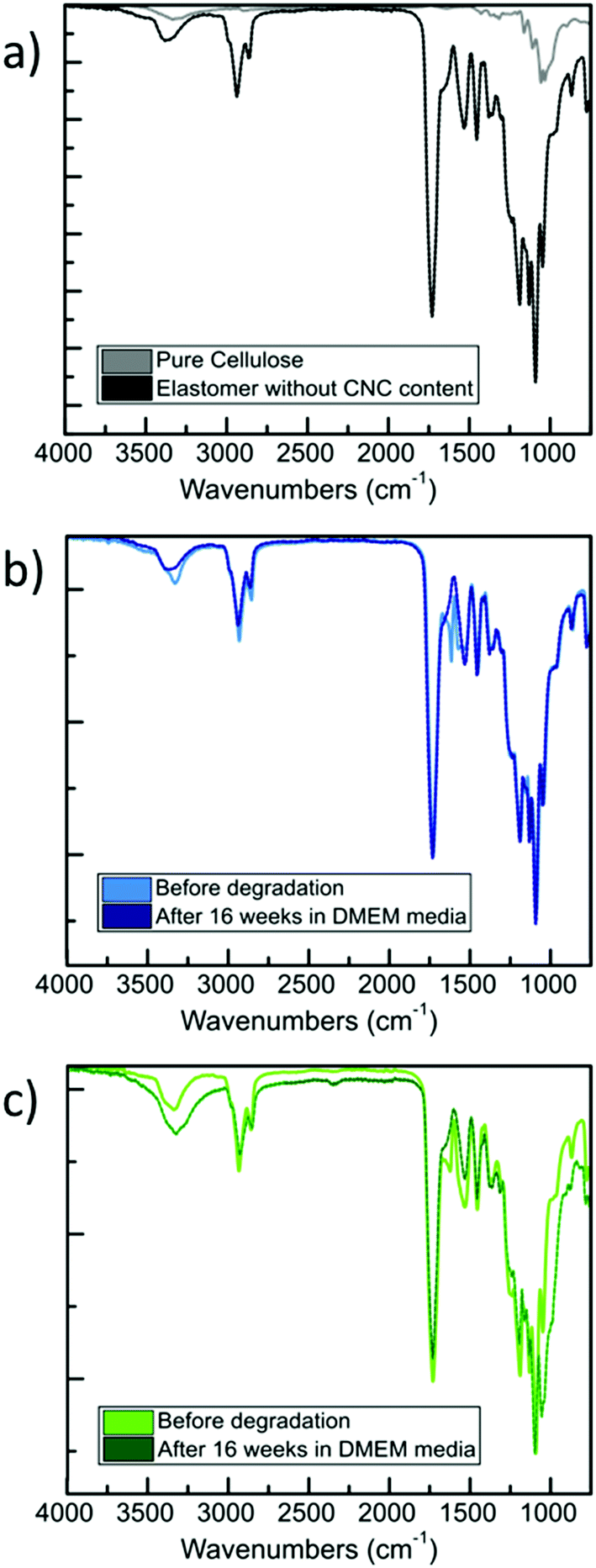 | ||
| Fig. 4 FT-IR spectra for: (a) pure elastomer and neat CNCs; (b) 6A-PCL-LCE matrix containing 5 wt% CNC before and after 16 weeks in vitro degradation; (c) 6A-PCL-LCE matrix containing 40 wt% CNC before and after 16 weeks in vitro degradation. | ||
Our mechanical results suggest that our composite provides a biomechanically stable platform that should allow cells to fully regenerate. With the addition of cells, while the tendency to degrade should remain the same, we expect a faster degradation of the 6A-PCL elastomer due to a dual process of hydrolysis and enzymatic (from cells) degradation. Furthermore, reported methods could be used to allow cellulose decomposition in vivo, for example by introducing modifications such as aldehyde groups on the cellulose chain105,106 or targeted enzyme delivery.107
2.3. X-Ray scattering measurements
CNCs have been reported to self-organize into a chiral nematic LC phase.108 In order to assess whether this was occurring in our material, we used X-ray scattering to analyze the structure 6A-PCL/CNC composites. A q-range from 0.024 Å−1 to 2.19 Å−1 (corresponding to 26.2–0.29 nm) was scanned, corresponding to distances, d, that are several times the width of a CNC particle (2.3 to 4.5 nm according to CelluForce product specification). Fig. 5 depicts the X-ray scattering data from 6A-PCL matrices containing 0 wt% and 50 wt% CNC, that covers the full range of our composite materials. The amorphous peak at 1.3 Å−1 corresponds to the 4.8 Å spacing of the polymer backbone. The CNC spacing of 300 Å reported by Lagerwall et al.108 does not disrupt the polymer spacing.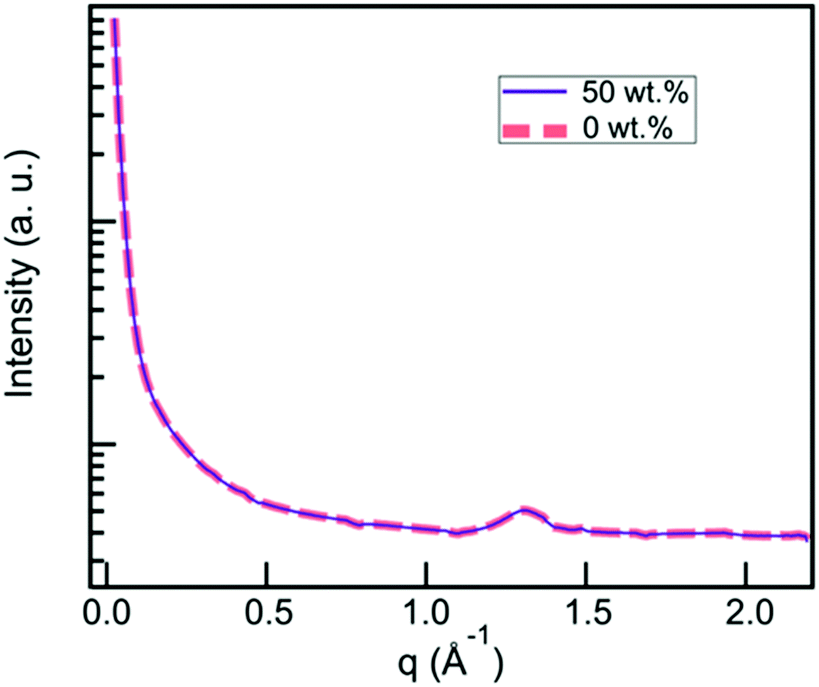 | ||
| Fig. 5 X-Ray scattering data from 6A-PCL matrices containing 0 wt% CNC (grey dashed curve) and 50 wt% CNC (black dashed curve), showing a broad halo indicative of the materials being amorphous. | ||
2.4. Cellular response and behavior on mechanically matched elastomers
As cells are sensitive to their environment, material stiffness is a critical factor in ensuring proper cell attachment and cellular responses.109 Cellular proliferation, differentiation, and migration can all be dramatically changed by varying the elastic moduli and porosity of the cellular environment.110 Epithelial cells and fibroblasts were the first cell lines to be used to show varying responses to stiff and soft collagen-coated polyacrylamide substrates.111 Also, there are many studies that report the effects of cell growth and response by changing the stiffness of hydrogels.112,113 Guvendiren et al. studied spreading, proliferation, motility, and differentiation of hMSC (Human Mesenchymal Stem Cells) on soft (∼3 kPa) and stiff (∼30 kPa) hydrogels.114We here used human dermal fibroblast (hDF) and neuroblastoma (SH-SY5Y) cell lines to compare cell growth and proliferation within two different 3D foam scaffolds. hDFs generate ECM that forms the connective tissue of the skin and are model cells for studying skin biology.15,115 Undifferentiated and differentiated SH-SY5Y cell lines are used as a model for neuron-like cells for in vitro studies.116 Based on the previous study, we created two 6A-PCL-CNC composite foams, one with 5 wt% of CNC content that had a stiffness well matched to the mechanical properties of neuroblastomas, and another with 40 wt% of CNC content that had a stiffness well matched to the mechanical properties of dermal fibroblasts. Cells were seeded on the scaffolds, and after two weeks, cell viability and number of cells were measured using on the scaffold to see distribution and proliferation using fluorescence confocal microscopy after two weeks of cell proliferation.
Confocal imaging was performed to see how cells attached and distributed around and in 6A-PCL/CNC composites containing 5 wt% as well as 40 wt% CNC. Nuclei of SH-SY5Y and hDF cells were stained with propidium iodine stain and are shown in red. Optical image stacks were acquired and projected as a 3D image. Scanning electron microscopy (SEM) was used to show the porous structure of foam composites (see Fig. S2, see ESI†). MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed at different weeks. Cells were also fixed and stained, and Fig. 6 shows the attachment and cell proliferation of SH-SY5Y cells through and inside the pores of the 5 wt% of CNC content composite, which has E value matching brain/gray matter tissue. However, at 40 wt% of CNC content, where E values matched hDF cells, SH-SY5Y cells were not found proliferating throughout the scaffold. Instead, they clustered at certain places, probably attaching to ECM produced by clusters of cells, and they showed a significantly slower rate of proliferation.
 | ||
| Fig. 6 Confocal images of SH-SY5Y cells seeded into (a) 5 wt% and (b) 40 wt% of CNC in 6A – PCL/CNC composite foams after two weeks of proliferation. Cell nuclei are shown in bright red (propidium iodide staining, circles). The number of nuclei counted are 740 and 233 for 5 wt%, and 40 wt% 6A – PCL/CNC, respectively. Circles show cluster of cells of cell nuclei. | ||
Fig. 7 shows the opposite effect for hDF cells within the same time frame. hDF cells were found to proliferate in higher number within the 40 wt% of CNC in 6A-PCL/CNC composite foam than on the lower 5 wt% CNC, demonstrating that hDF cells prefer to grow and proliferate faster on a mechanically matched scaffold (see Fig. S4 and S5 for additional confocal images; see ESI†). hDF cells on 40 wt% of CNC in 6A-PCL/CNC and SHSY-5Y on 5 wt% of CNC in 6A-PCL/CNC composite foam were also imaged after 8 weeks of seeding (Fig. S6, see ESI†), to further demonstrate cell growth and proliferation for long periods of time, with cells proliferating throughout and upon the scaffold as can be further observed in Fig. S6 (see ESI†). We fluorescently stained cell and did not see significant morphological changes compared to typical cultures (see Fig. S7 in ESI† for more confocal images).
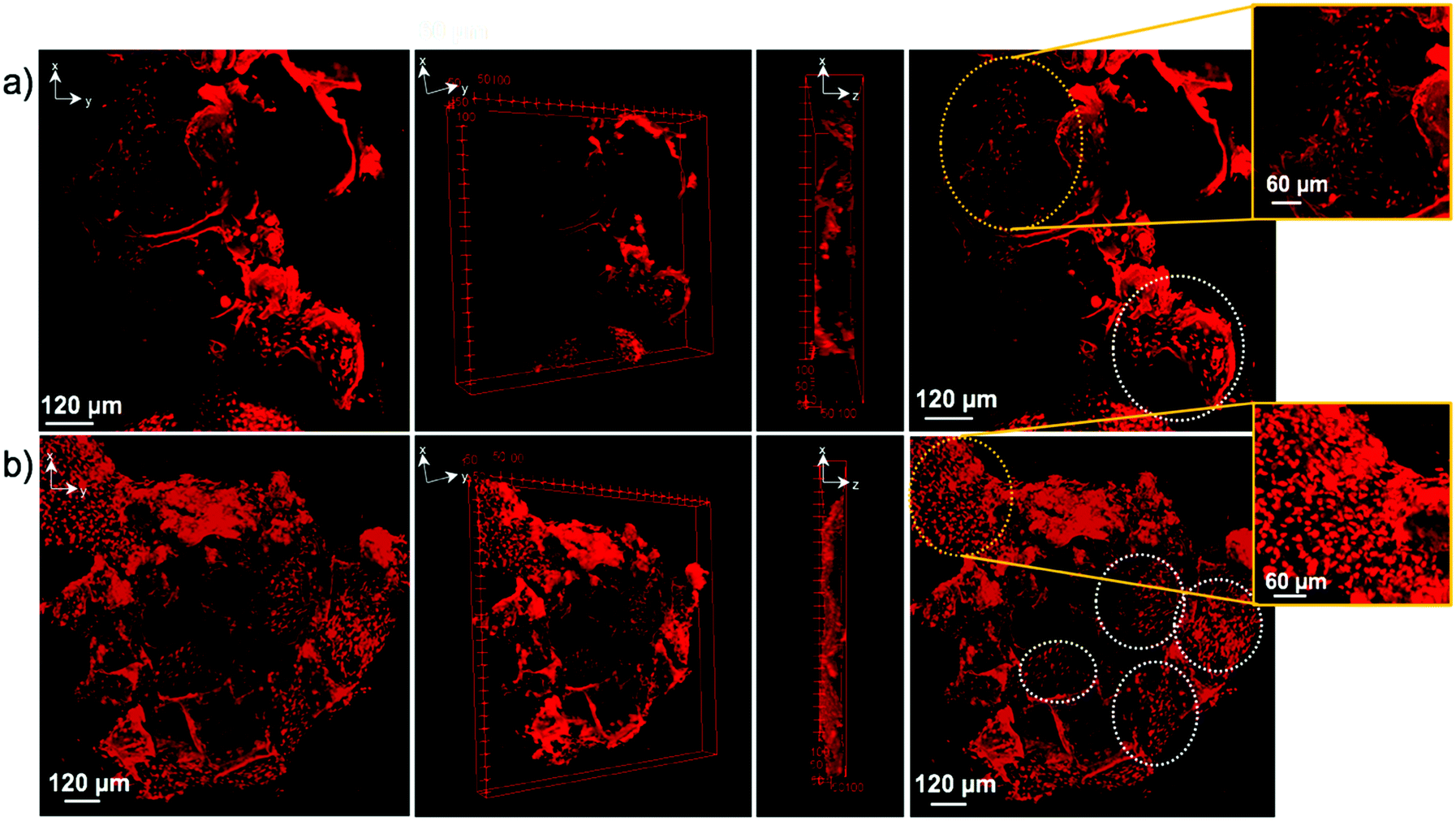 | ||
| Fig. 7 Confocal images of hDF cells seeded into (a) 5 wt% and (b) 40 wt% 6A-PCL/CNC composite foams after two weeks of proliferation. Cell nuclei are shown in bright red (propidium iodide staining arrows), with larger cell clusters being shown inside of circles. Number of nuclei are 291 and 1227 for 5 wt%, and 40 wt% 6A – PCL/CNC, respectively. Circles show clusters of cell nuclei. | ||
We then used MTT assays as a method to observe cell viability, proliferation, and cytotoxicity for the two different composites. The mostly water soluble MTT turns into insoluble formazan, which is spectrophotometrically detected, and serves as a quantitative way to monitor the number of cells that proliferate and continue their metabolic activities on the scaffolds.
hDF and SH-SY5Y cells were seeded on both composites, as shown in the graphs in Fig. 8 for both cell lines (weeks 2, 4 and 6 of cell growth). The numbers of cells are slightly different according to their environment's stiffness. After four weeks, more drastic changes can be observed. The proliferation rate of SH-SY5Y cells is higher in the 5 wt% composite that matches the stiffness of brain tissue and cells proliferated slower in the 40 wt% composite. Again, the opposite effect is observed for hDF cells, with values ranging from 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for the 5 wt% to 19000 for the 40 wt% composite, respectively. Similarly, the number of SH-SY5Y cells after two weeks for both composites was in reverse order. For hDF cells, rate of cell division and proliferation is not as fast as SH-SY5Y cells on 5 wt% composite. However, the cell number is larger when using the 40 wt% foam-composite compared to the one based on 5 wt% composite.
000 for the 5 wt% to 19000 for the 40 wt% composite, respectively. Similarly, the number of SH-SY5Y cells after two weeks for both composites was in reverse order. For hDF cells, rate of cell division and proliferation is not as fast as SH-SY5Y cells on 5 wt% composite. However, the cell number is larger when using the 40 wt% foam-composite compared to the one based on 5 wt% composite.
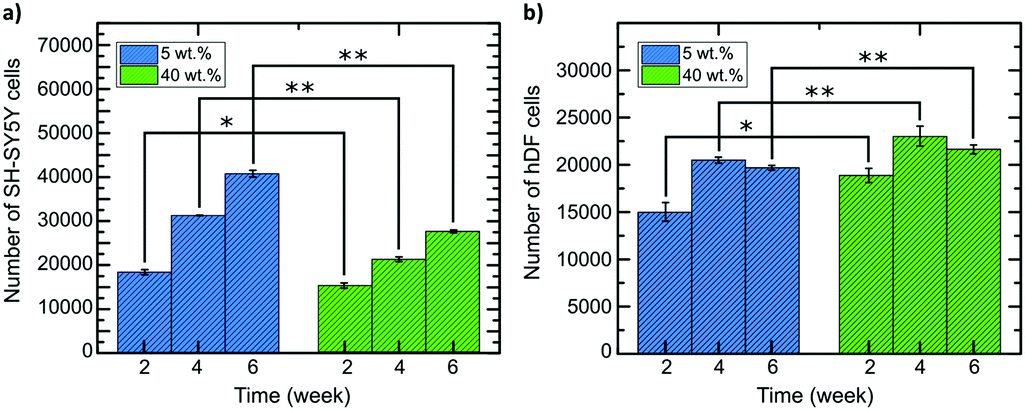 | ||
| Fig. 8 MTT measurements of (a) SH-SY5Y and (b) hDF cell proliferation within the different composites, 5 wt% and 40 wt%. The error bars represent SEM of the triplicate measurements and *p ≤ 0.05 and ** p ≤ 0.01. | ||
As demonstrated by the microscopy and the MTT assays data, cell attachment as well as cell growth and proliferation rate can be improved by choosing scaffolds with mechanical properties matched to the target tissues of interest. More efficient cell growth and proliferation will yield increasingly stable cultures that efficiently expand with high viability, reducing experimental time and material costs, resulting in more reliable and faster studies. Enhancing viability within the grown cell populations will also permit longitudinal in vitro studies over longer time periods, permitting dynamic experiments that model different phases of disease or tissue function. Beyond in vitro applications, providing a method to better match the requirements of endogenous environments for bio-implantation and matching this with tissue-tuned degradation will promote the eventual incorporation with endogenous tissues.
2.5. Contact angle measurements
To further study and determine the viability of cells on and within the composites we tested their surface hydrophobic/hydrophilic nature. It has been reported that some types of cells prefer to adhere to more hydrophilic surfaces, while other types of cells favor a more hydrophobic surface.117 We have previously reported that human myoblasts (C2C12s) showed higher cell density on a more hydrophobic environment (θc = 133.5°) versus a more hydrophilic environment (θc = 51.7°).22 Contact angle measurements showed that the composites become more hydrophilic as the CNC wt% content increases. At 0 wt% CNCs the contact value was determined at θc = 77.5°, the value for 5 wt% CNCs was only slightly lower at θc = 73.3°, whereas for 40 wt% CNCs the value was θc = 64.5° (see Fig. 9, see Table S1 in ESI† for all composite values). The value of θc = 64.5° is in accordance with what is reported from Chang et al. where fibroblasts in general have increased adhesion within values of θc = 60–80°.118 | ||
| Fig. 9 Contact angle images for composites containing (a) 0 wt% CNCs (not used in cell study), (b) 5 wt% CNCs, (c) 25 wt% CNCs (not used in cell study), and (d) 40 wt% CNCs. Graph summarizes some of the composites prepared and their respective contact angle measurements. | ||
The value for 5 wt% CNCs at θc = 73.3°is slightly on the hydrophobic value, however, also falls within the range of 40–80° as found by Bartolo et al.119 It has also been reported that protein absorption increases at values above θc = 60°,120 which could be a key for neural like cell maturation such as myelin formation. Thus, the 5 wt% CNCs composite while is slightly on the hydrophobic values, it is a good environment for neural like cells to grow, ideal to promote protein absorption, while maintaining a soft E nature for cell proliferation.
3. Conclusions
We here reported a pathway to create materials that are cell-mechanically and morphologically tuned and can support a large range of physiological tissues. We have shown that scaffold stiffness has a significant impact on cell proliferation and growth. Matching the E of tissues is an ideal method to ensure the most efficient cell proliferation and demonstrates that not all scaffolds can be fully efficient for all cell types. Previous TE studies (in 2D and 3D) have shown that cells grow within mechanically unmatched scaffolds, however, there is typically a need to add components (collagen-based, D-lysine, or Matrigel among others) to ensure cell attachment and “bridging” before they can produce their own ECM. This usually requires longer cell attachment and proliferation times.Here we have shown that matching scaffold stiffness to cells of interest can potentially reduce proliferation/expansion times, improve regeneration, and enhance viability. Importantly, the incorporation of CNCs does not adversely affect the amorphous nature of the PCL-D,L-lactide-based elastomer. We also studied the effect of CNCs on the 6A-PCL-LCE anisotropy at different CNC weight ratios and found no disruption of the SmA phase (see Fig. S8, see ESI†) even at 50 wt% CNCs. This may make this study directly transferable to the 6A-PCL-LCE-based scaffolds reported earlier27,28 to compare how stiffness and LC anisotropy can potentially further enhance cell growth and organization. Further studies will then focus on designing a physical template for cells that is responsive to stimuli, further promoting, directing and signaling cell growth.
4. Experimental section
All air sensitive manipulations were carried out under nitrogen gas. ε-Caprolactone (ε-CL, purchased from Alfa Aesar) was dried over calcium hydride (from Sigma-Aldrich) and distilled under reduced pressure. D,L-Lactide was used as received (from Alfa Aesar).Dipentaerythritol, triethylamine, stannous 2-ethylhexanoate and hexamethylene diisocyanate (HDI) were used as received (Sigma-Aldrich). All solvents used for the synthesis and purification were EMD Millipore grade purified by a Pure-Solv solvent purification system (Innovative Technology Inc. CNCs (chemical name cellulose hydrogen sulfate sodium salt, powder form) and were purchased from CelluForce. Salt crystals (NaCl) were purchased from BDH).
For SH-SHY5Y cells (from ATCC®), 10% Fetal Bovine Serum (from VWR), 1% penicillin–streptomycin (from Gibco) in DMEM:F12 media (from Gibco) were prepared as media. Trypsin–EDTA (0.25%) (from composite scaffold). Cell culture media for hDF was 10% Fetal Bovine Serum (from VWR), 1% penicillin–streptomycin (from Gibco) in DMEM (from Gibco). MTT assay kit was purchased from Abcam. Propidium iodide (from Invitrogen) was used to stain nuclei of cells. Triton X-100 (from Sigma Aldrich) was used to permeabilize cells prior to staining. 4′,6-Diamidino-2-phenylindole (DAPI) (purchased from Invitrogen) was used to satin nuclei and primary anti-Tubulin β 3 (TUBB3) antibody (from BioLegend) was used to stain cell body.
1H and 13C NMR of copolymers and elastomers were recorded in CDCl3 at room temperature on a Bruker DMX 400 MHz instrument and referenced internally to residual peaks at 7.26 (1H).
Scanning electron microscopy (SEM) was performed on freeze fractured elastomer foams, gold coated (700 Å) using a sputter coater (Hummer VI-A, Anatech Ltd, VA) at 10 mA DC for 3 min. Images were acquired using a Quanta 450 FEG SEM.
Uniaxial tensile testing (Linkam TST350, 20 N load cell) was performed under ambient conditions at a strain rate of 40% min−1. Fifteen tensile samples per elastomer were examined for mechanical response. The elastic modulus (E) was determined from the slope of the stress–strain curve between 0.10% and 0.35% strain.
For in vitro hydrolytic degradation studies, the samples were placed into a 15 mL tissue culture tube, which was then filled with 12 mL of media solution and kept in the fridge. They were washed gently with distilled water, wiped before use, and weighed to calculate the mass loss. This standard procedure was conducted for 16 weeks. For the studies of the integrity of mechanical properties, five tensile samples per elastomer were examined by uniaxial tensile testing every two weeks for 16 weeks in the same conditions as described for the mechanical properties.
4.1. Preparation of 6-arm-poly(ε-CL-co-D,L-LA) pre-polymer
Solvent free polymerization was carried out in a sealed silanized 20 mL ampoule. Into a dry silanized ampoule, dipentaerythritol (1.27 g, 0.005 mol) and ε-CL (5.54 mL, 0.05 mol) were transferred and mixed until homogeneous. D,L-LA (7.21 g, 0.05 mol) was transferred to the ampoule, the ampoule filled with nitrogen, then placed in an oven at 140 °C for 2 h, allowing the D,L-LA to melt. The mix was then stirred using a vortex mixer and 100 μL of stannous-2-ethylhexanoate was added. The ampoule was flushed with dry nitrogen, flame sealed under vacuum, and left in an oven at 140 °C for 24 h. The co-polymer was then purified by precipitation from dichloromethane solution into cold methanol. 1H NMR (CDCl3, 400 MHz), δ [ppm]: 5.31–5.04 (m, COCHCH3), 4.41–4.25 (m, OCH(CH2)2 initiator), 4.24–4.12 (m, CH2O), 4.11–4.00 (t, J = 4.6 Hz, CH2O), 3.65–3.49 (m, CHCH2), 3.18–2.67 (broad, s, OH), 2.43–2.31 (m, CH2CH2CH2OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1.77–1.63 (m, –CH2CH2CH2–), 1.63–1.34 (m, CH2, CH3).
O), 1.77–1.63 (m, –CH2CH2CH2–), 1.63–1.34 (m, CH2, CH3).
4.2. Preparation of composite 6A-PCL/cellulose films
For the elastomer preparation, pre-polymer (6A-PCL):solvent (ε-CL):crosslinking (HDI) ratio of 4:2:1 was maintain throughout the study according to the following method. First, 6A-PCL SBC and ε-CL were vortexed until homogenous. CNCs was mixed with ε-CL and integrated into the pre-polymer by weight percentage of the SBC, then the mixture was mechanically stirred until homogeneous. HDI was added and the polymer was vortexed. The composite was then poured onto a silanized glass slide and placed in the oven at 65 °C for 2 h. Once crosslinked, the elastomeric composite was peeled from the glass slide and cut into 5x12mm pieces using a dog-bone template.
4.3. Preparation of porous 3D scaffold by a salt leaching method
According to the method given for the preparation of films, 6A-PCL SBC and ε-CL were added first, then CNCs were added and later followed by HDI. Before crosslinking, salt was added. For the scaffold to reach 85% porosity, the amount of salt added corresponded to the mass of (6A-PCL SBC + ε-CL + HDI) multiplied by 5.6. The salt and the previous mixture were mechanically stirred until the salt was covered by the liquid mixture and subsequently poured in a mold and pressed. The foam was placed in the oven at 65 °C for 2 h. Once crosslinked, the elastomeric composite foam was immersed in distilled water for 3 days until the salt was completely removed and finally dried. Foams can be subjected to a gentle tactile compression test to ensure that the foam is free of salt.4.4. Cell culturing
Elastomers were cut into 2 mm × 2 mm × 2 mm dimensions. To sterilize them they were washed by 70% ethanol and PBS (Phosphate Buffer Solution) and they were left under UV light for 15 minutes. 2 × 104 cell per mL were added to each elastomer. hDF (Human Dermal Fibroblast) cells (ATCC® PCS-201-010) and SH-SY5Y (ATCC® CRL-2266) were seeded into 5% and 40% by weight 6A-PCL/CNC composites. Media for cells were changed every 48 hours.4.5. Confocal imaging
4% paraformaldehyde (PFA) was used to fix cells on the scaffold and 0.01% Triton X was used to permeabilize cell membrane. Propidium iodide (1:1000 in PBS) was used to stain cell nuclei. 4′,6-Diamidino-2-phenylindole (DAPI) was used to stain cell nuclei (1:1000 in PBS) on hDF cells grown on 40 wt% CNCs whereas cell body was stained with primary anti-Tubulin β 3 (TUBB3) Antibody. Fluorescence confocal microscopy was carried out using an Olympus FV1000 and FV3000 and image stacks combined and analyzed using ImageJ.121
4.6. Viability assay
After one week of proliferation on the composites, a sample was taken in another well and trypsin was added. After 5 minutes incubation with trypsin, serum included media was added to stop trypsin activation and the media-trypsin mixture was collected and centrifuged to collect cell pellets. Cell pellets were dispersed in media and plated on 96 well plate and allowed to attach for 24 hours. After attachment MTT assay procedures were applied; 50 μL serum free media and 50 μL MTT reagent were added to each well and they were incubated for 3 hours. After that media-reagent mixture was discarded and 150 μL MTT solution was added to each well, the plate was shaken for 15 minutes on orbital share before reading the absorbance at 590 nm (SpectraMax M4).Small angle X-ray scattering (SAXS) data were collected at beamline 7.3.3122 of the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory and also at the SMI beamline of the National Synchrotron Light Source II (NSLS-II), at the Brookhaven National Laboratory (BNL). At the ALS, 10 keV X-rays were passed through the sample and the scattered X-rays were collected with a Pilatus3.2M detector at a distance of 0.574 m. A silver behenate standard was used to calibrate the detector distance and beam center, and the calibrated 2D images were converted to radially integrated 1D curves using the NIKA123 SAXS data reduction software. SAXS patterns of 6A-PCL-LCE matrix containing CNC were obtained at the Soft Matter Interfaces (SMI, Beamline 12-ID) at the National Synchrotron Light Source II, with a beam energy of 16 keV and a spot size of 2.5 × 20 μm. Scattering patterns were collected on an in-vacuum Pilatus 1 M detector, consisting of 0.172 mm square pixels in a 941 × 1043 array, at a detector-to-sample distance of 3.2 meters. Exposure time was set to one second.
Contact angle measurements of the sample the surfaces were characterized by the sessile drop method using a contact angle goniometer (Rame-Hart-250 Goniometer, Rame-Hart, Inc., USA). An automated dispensing system was used to dispense a 200 μl droplet of deionized water on the surfaces. The water contact angle was measured 15 s after placing the droplet to allow time for equilibration.
Authors contributions
All authors have given approval to the final version of the manuscript. SU and MEP contributed equally to this manuscript. EH and TH conceived the idea of doping the elastomers with CNCs. MEP, SU, GARR, and CW synthesized and characterized the elastomers; SU performed all cell and confocal studies under supervision of RJC. SU and RJC analyzed all confocal data. SU, MEP, CZ and ES collected and analyzed all ALS data. CZ, ES, and EH directed all ALS experiments. SU, MEP, CW, BY, GF, MZ, RP, and EH collected and analyzed all BNL data. MZ, RP, and EH directed all BNL experiments. SU, MEP, and EH prepared an early manuscript draft. EH wrote all beamline proposals. TH, RJC, and EH directed the research and wrote the manuscript with contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank KSU for financial support of the KSU Collaborative Research Grant and the National Science Foundation (CHE-1263087). SAXS/WAXS measurements were funded from beamline time applications at Advanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, Beamline 7.3.3 of the Advanced Light Source is supported by the Director of the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, and SMI beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The authors also want to thank the Liquid Crystal Characterization Facility at the Advanced Materials and Liquid Crystal Institute (AMLCI) where the SEM data was acquired. The authors are grateful of the Advanced Light Source (ALS) beamline 7.3.3 and National Synchrotron Light Source II (NSLS-II) SMI beamline for their support and help in collecting and analyzing SAXS data. The authors thank Prof. Min-Ho Kim (KSU, Department of Biological Sciences) for generously donating hDF cells for our study and PhD student Ahlam Nemati (TH research group) for helping collect degradation weights.Notes and references
- M. E. Furth and A. Atala, Tissue Engineering: Future Perspective, in Principles of Tissue Engineering, ed. R. Lanza, R. Langer and J. Vacanti, 4th edn, 2014, ch. 6, pp. 83–123 Search PubMed.
- Y. Wang, G. A. Ameer, B. J. Sheppard and R. Langer, Nat. Biotechnol., 2002, 20, 602–606 CrossRef CAS.
- A. Agrawal, O. Adetiba, H. Kim, H. Chen, J. Jacot and R. Verduzco, J. Mater. Res., 2015, 30, 453–462 CrossRef CAS.
- D. E. Discher, Science, 2005, 310, 1139–1143 CrossRef CAS.
- F. Rehfeldt, A. J. Engler, A. Eckhardt, F. Ahmed and D. E. Discher, Adv. Drug Delivery Rev., 2007, 59, 1329–1339 CrossRef CAS.
- K. A. Beningo and Y. L. Wang, Trends Cell Biol., 2002, 12, 79–84 CrossRef CAS.
- P. Bartolo, M. Domingos, A. Gloria and J. Ciurana, CIRP Ann., 2011, 60, 271–274 CrossRef.
- M. Prévôt and E. Hegmann, From Biomaterial, Biomimetic, and Polymer to Biodegradable and Biocompatible Liquid Crystal Elastomer Cell Scaffolds, in ACS Symposium Series, ed. Y. Ito, X. Chen and I.-K. Kang, ACS, Washington, DC, 2017, vol. 1253, ch. 1, pp. 3–45 Search PubMed.
- R. O. Hynes, Science, 2009, 326, 1216–1219 CrossRef CAS.
- E. Carletti, M. Stoppato, C. Migliaresi and A. Mottaa, The Functional Role of Extracellular Matrix, in Scaffolds for Tissue Engineering: Biological Design, Materials, and Fabrication, ed. C. Migliaresi and A. Motta, Pan Stanford, Singapore, 2014, pp. 21–56 Search PubMed.
- B. Yue, J. Glaucoma, 2014, 23, S20–S23 CrossRef.
- D. Fletcher and R. Mullins, Nature, 2010, 463, 485–492 CrossRef CAS.
- S. Kumar, I. Z. Maxwell, A. Heisterkamp, T. R. Polte, T. P. Lele, M. Salanga, E. Mazur and D. E. Ingber, Biophys. J., 2006, 90, 3762–3773 CrossRef CAS.
- H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, Cell-Matrix Adhesion, in Molecular Cell Biology, ed. W. H. Freeman, 4th edn, 2000, Section 22.2, Cell-Matrix Adhesion Search PubMed.
- K. S. Midwood, L. V. Williams and J. E. Schwarzbauer, Tissue repair and the dynamics of the extracellular matrix, Int. J. Biochem. Cell Biol., 2004, 36, 1031–1037 CrossRef CAS.
- C. T. McKee, J. A. Last, P. Russell and C. J. Murphy, Tissue Eng., Part B, 2011, 17, 155–164 CrossRef.
- M.-H. Li and P. Keller, Philos. Trans. R. Soc., A, 2006, 364, 2763–2777 CrossRef CAS.
- D. L. Thomsen III, P. Keller, J. Naciri, R. Pink, H. Jeon, D. Shenoy and B. R. Ratna, Macromolecules, 2001, 34, 5868–5875 CrossRef.
- C. Ohm, M. Brehmer and R. Zentel, Adv. Polym. Sci., 2012, 250, 49–94 CrossRef CAS.
- H. R. Brand, H. Finkelmann, D. Demus, J. W. Goodby, G. W. Gray, H. W. Spiess and V. Vill, Physical properties of liquid crystalline elastomers, Handbook of Liquid Crystals, Wiley, New York, 2008, vol. 3, pp. 277–302 Search PubMed.
- W. H. E. de Jeu, Liquid crystal elastomers: Materials and applications, Springer, New York, 2012 Search PubMed.
- A. Sharma, A. Neshat, C. J. Mahnen, A. D. Nielsen, J. Snyder, T. L. Stankovich, B. G. Daum, E. M. LaSpina, G. Beltrano, Y. Gao, S. Li, B.-W. Park, R. J. Clements, E. J. Freeman, C. Malcuit, J. A. McDonough, L. T. J. Korley, T. Hegmann and E. Hegmann, Macromol. Biosci., 2015, 15, 200–214 CrossRef CAS.
- A. Sharma, T. Mori, C. J. Mahnen, H. R. Everson, M. T. Leslie, A.d. Nielsen, L. Lussier, C. Zhu, C. Malcuit, T. Hegmann, J. A. McDonough, E. J. Freeman, L. T. J. Korley, R. J. Clements and E. Hegmann, Macromol. Biosci., 2017, 17, 1600278 CrossRef.
- T. Bera, E. J. Freeman, J. A. McDonough, R. J. Clements, A. Aladlaan, D. W. Miller, C. Malcuit, T. Hegmann and E. Hegmann, ACS Appl. Mater. Interfaces, 2015, 7, 14528–14535 CrossRef CAS.
- T. Bera, C. Malcuit, R. J. Clements and E. Hegmann, Front. Mater., 2016, 3, 31 Search PubMed.
- Y. Gao, T. Mori, S. Manning, Y. Zhao, A. D. Nielsen, A. Sharma Neshat, C. J. Mahnen, H. R. Everson, S. Crotty, R. J. Clements, C. Malcuit and E. Hegmann, ACS Macro Lett., 2016, 5, 4–9 CrossRef CAS.
- T. Mori, R. Cukelj, M. E. Prévôt, S. Ustunel, A. Story, Y. Gao, K. Diabre, J. A. McDonough, E. J. Freeman, E. Hegmann and R. J. Clements, Macromol. Rapid Commun., 2020, 41, 1900585 CrossRef CAS.
- M. E. Prévôt, H. Andro, S. L. M. Alexander, S. Ustunel, C. Zhu, Z. Nikolov, S. T. Rafferty, M. T. Brannum, B. Kinsel, L. T. J. Korley, E. J. Freeman, J. A. McDonough, R. J. Clements and E. Hegmann, Soft Matter, 2018, 14, 354–360 RSC.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS.
- S. Mueller, J. Sapkota, A. Nicharat, T. Zimmermann, P. Tingaut, C. Weder and E. J. Foster, J. Appl. Polym. Sci., 2015, 132, 41740 CrossRef.
- M. Jorfi, M. N. Roberts, E. J. Foster and C. Weder, ACS Appl. Mater. Interfaces, 2013, 5, 1517–1526 CrossRef CAS.
- S. Bottan, F. Robotti, P. Jayathissa, A. Hegglin, N. Bahamonde, J. A. Heredia-Guerrero, I. S. Bayer, A. Scarpellini, H. Merker, N. Lindenblatt, D. Poulikakos and A. Ferrari, ACS Nano, 2015, 9, 206–219 CrossRef CAS.
- J. M. Dugan, R. F. Collins, J. E. Gough and S. J. Eichhorn, Acta Biomater., 2013, 9, 4707–4715 CrossRef CAS.
- E. Entcheva, H. Bien, L. Yin, C.-Y. Chung, M. Farrell and Y. Kostov, Biomaterials, 2004, 25, 5753–5762 CrossRef CAS.
- M. Märtson, J. Viljanto, T. Hurme, P. Laippala and P. Saukko, Biomaterials, 1999, 20, 1989–1995 CrossRef.
- A. D. French, N. R. Bertoniere, R. M. Brown, H. Chanzy, D. G. Gray, K. Hattori and W. Glasser, in Kirk-Othmer Encyclopedia of Chemical Technology, ed. A. Seidel, John Wiley & Sons Inc., New York, 5th edn, 2004, vol. 5, pp. 360–394 Search PubMed.
- J. K. Jackson, K. Letchford, B. Z. Wasserman, L. Ye, W. Y. Hamad and H. M. Burt, Int. J. Nanomed., 2011, 6, 321–330 CAS.
- X.-M. Fan, H.-Y. Yu, D.-C. Wang, J. Yao, H. Lin, C.-X. Tang and K. C. Tam, ACS Appl. Mater. Interfaces, 2019, 11, 48192–48201 CrossRef CAS.
- J. V. Edwards, N. Prevost, K. Sethumadhavan, A. Ullah and B. Condon, Cellulose, 2013, 20, 1223–1235 CrossRef CAS.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- B. G. Rånby, Acta Chem. Scand., 1949, 3, 649–650 CrossRef.
- B. L. Peng, N. Dhar, H. L. Liu and K. C. Tam, Can. J. Chem. Eng., 2011, 89(5), 1191–1206 CrossRef CAS.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS.
- M. Giese, L. K. Blusch, M. K. Khan and M. J. MacLachlan, Angew. Chem., Int. Ed., 2015, 54, 2888–2910 CrossRef CAS.
- N. Grishkewich, N. Mohammed, J. Tang and K. C. Tam, Curr. Opin. Colloid Interface Sci., 2017, 29, 32–45 CrossRef CAS.
- R. Marchessault, F. Morehead and N. Walter, Nature, 1959, 184, 632–633 CrossRef CAS.
- J.-F. Revol, H. Bradford, J. Giasson, R. H. Marchessault and D. G. Gray, Int. J. Biol. Macromol., 1992, 14, 170–172 CrossRef CAS.
- N. W. Awang, D. Ramasamy, K. Kadirgama, G. Najafi and N. A. C. Sidik, Int. J. Heat Mass Transfer, 2019, 131, 1196–1204 CrossRef CAS.
- J. J. Valle-Delgado, L. S. Johansson and M. Österberg, Colloids Surf., B, 2016, 138, 86–93 CrossRef CAS.
- A. Nicharat, A. Shirole, E. J. Foster and C. Weder, J. Appl. Polym. Sci., 2017, 134, 45033 CrossRef.
- J. E. Sanders, Y. Han, T. S. Rushing and D. J. Gardner, Nanomaterials, 2019, 9, 805 CrossRef.
- T. Or, S. Same, A. Esteve, D. A. Osorio, K. J. De France, J. Vapaavuori, T. Hoare, A. Cerf, E. D. Cranston and J. M. Moran-Mirabal, ACS Appl. Nano Mater., 2019, 2, 4169–4179 CrossRef CAS.
- M. Bhattacharya, M. M. Malinen, P. Lauren, Y.-R. Lou, S. W. Kuisma, L. Kanninen, M. Lille, A. Corlu, C. GuGuen-Guillouzo, O. Ikkala, A. Laukkanen, A. Urtti and M. Yliperttula, J. Controlled Release, 2012, 164, 291–298 CrossRef CAS.
- R. M. A. Domingues, M. E. Gomes and R. L. Reis, Biomacromolecules, 2014, 15, 2327–2346 CrossRef CAS.
- S. Xie, X. Zhang, M. P. Walcott and H. Lin, Eng. Sci., 2018, 2, 4–16 Search PubMed.
- J. G. Torres-Rendon, T. Femmer, L. De Laporte, T. Tigges, K. Rahimi, F. Gremse, S. Zafarnia, W. Lederle, S. Ifuku, M. Wessling, J. G. Hardy and A. Walther, Adv. Mater., 2015, 27, 2989–2995 CrossRef CAS.
- I. Th, A. S. Shaheen, S. Montaser and S. Li, Int. J. Biol. Macromol., 2019, 121, 814–821 CrossRef.
- E. M. Fernandes, R. A. Pires, J. F. Mano and R. L. Reis, Prog. Polym. Sci., 2013, 38, 1415–1441 CrossRef CAS.
- W. Li, R. Guo, Y. Lan, Y. Zhang, W. Xue and Y. Zhang, J. Biomed. Mater. Res., Part A, 2014, 102, 1131–1139 CrossRef.
- T. Kovacs, V. Naish, B. O'Connor, C. Blaise, F. Gagné, L. Hall, V. Trudeau and P. Martel, Nanotoxicology, 2010, 4, 255–270 CrossRef CAS.
- M. Roman, S. Dong, A. Hirani and Y. W. Lee, Cellulose Nanocrystals for Drug Delivery, in Polysaccharide Materials: Performance by Design, ed. K. J. Edgar, T. Heinze and C. M. Buchanan, ACS, Washington, DC, 2009, pp. 81–91 Search PubMed.
- S. Dong, A. A. Hirani, K. R. Colacino, Y. W. Lee and M. Roman, Cytotoxicity and Cellular Uptake of Cellulose Nanocrystals, Nano Life, 2012, 2, 1241006 CrossRef.
- K. A. Mahmoud, J. A. Mena, K. B. Male, S. Hrapovic, A. Kamen and J. H. T. Luong, ACS Appl. Mater. Interfaces, 2010, 2, 2924–2932 CrossRef CAS.
- K. B. Male, A. C. W. Leung, J. Montes, A. Kamen and J. H. T. Luong, Nanoscale, 2012, 4, 1373 RSC.
- J. K. Jackson, K. Letchford, B. Z. Wasserman, L. Ye, W. Y. Hamad and H. M. Burt, Int. J. Nanomedicine, 2011, 6, 321–330 CAS.
- P. Pooyan, R. Tannenbaum and H. Garmestani, J. Mech. Behav. Biomed. Mater., 2012, 7, 50–59 CrossRef CAS.
- R. Dash, M. Foston and A. J. Ragauskas, Carbohydr. Polym., 2013, 91, 638–645 CrossRef CAS.
- N. Lin, C. Bruzzese and A. Dufresne, ACS Appl. Mater. Interfaces, 2012, 4, 4948–4959 CrossRef CAS.
- Y. Wang, C. Chang and L. Zhang, Macromol. Mater. Eng., 2010, 295, 137–145 CrossRef CAS.
- J. M. Dugan, J. E. Gough and S. J. Eichhorn, Biomacromolecules, 2010, 11, 2498–2504 CrossRef CAS.
- J. M. Dugan, R. F. Collins, J. E. Gough and S. J. Eichhorn, Acta Biomater., 2013, 9, 4707–4715 CrossRef CAS.
- A. Kumar, K. M. Rao and S. S. Han, Chem. Eng. J., 2017, 317, 119–131 CrossRef CAS.
- K. S. Rahimi, R. Aeinehvand, K. Kim and J. U. Otaigbe, Biomacromolecules, 2017, 18, 2179–2194 CrossRef.
- N. Wang, K. Naruse, D. Stamenovic, J. J. Fredberg, S. M. Mijailovich, I. M. Tolic-Norrelykke, T. Polte, R. Mannix and D. E. Ingber, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7765–7770 CrossRef CAS.
- L. G. Griffith and M. A. Swartz, Nat. Rev. Mol. Cell Biol., 2006, 7, 211–224 CrossRef CAS.
- S. van Helvert, C. Storm and P. Friedl, Nat. Cell Biol., 2018, 20, 8–20 CrossRef CAS.
- L. Thompson, J. Azadmanjiri, M. Nikzad, I. Sbarski, J. Wang and A. Yu, Rev. Adv. Mater. Sci., 2019, 58, 1–16 Search PubMed.
- Y. Chen, S. Zhou and Q. Li, Biomaterials, 2011, 32, 5003–5014 CrossRef CAS.
- R. K. Shaha, D. R. Merkel, M. P. Anderson, E. J. Devereaux, R. R. Patel, A. H. Torbati, N. Willett, C. M. Yakacki and C. P. Frick, J. Mech. Behav. Biomed. Mater., 2020, 107, 103757 CrossRef CAS.
- A. A. Alzahrani, M. Saed, C. M. Yakacki, H. B. Song, N. Sowan, J. J. Walston, P. K. Shah, M. K. McBride, J. W. Stansbury and C. N. Bowman, Polym. Chem., 2018, 9, 121–130 RSC.
- A. L. DiRienzo, C. M. Yakacki, M. Frensemeier, A. S. Schneider, D. L. Safranski, A. J. Hoyt and C. P. Frick, J. Mech. Behav. Biomed. Mater., 2014, 30, 347–357 CrossRef CAS.
- F. L. Dri, L. G. Hector Jr., R. J. Moon and P. D. Zavattieri, Cellulose, 2013, 20, 2703–2718 CrossRef CAS.
- T. Nishino, K. Takano and K. Nakamae, J. Polym. Sci., Part B: Polym. Phys., 1995, 33(11), 1647–1651 CrossRef CAS.
- A. Sturcová, G. R. Davies and S. J. Eichhorn, Biomacromolecules, 2005, 6, 1055–1061 CrossRef.
- N. Lin, J. Huang and A. Dufresne, Nanoscale, 2012, 4, 3274–3294 RSC.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- E. Fortunati, M. Peltzer, I. Armentano, L. Torre, A. Jiménez and J. M. Kenny, Carbohydr. Polym., 2012, 90, 948–956 CrossRef CAS.
- B. Braun and J. R. Dorgan, Biomacromolecules, 2009, 10, 334–341 CrossRef CAS.
- H. G. Vogel, Connect. Tissue Res., 1978, 6, 161–166 CrossRef CAS.
- I. Ozyazgan, N. Liman, N. Dursun and N. Güneş, Maturitas, 2002, 43, 65–74 CrossRef.
- J. T. Maikos, R. A. I. Elias and D. I. Shreiber, J. Neurotrauma, 2008, 25, 38–51 CrossRef.
- F. Velardi, F. Fraternali and M. Angelillo, Biomech. Model. Mechanobiol., 2006, 5, 53–61 CrossRef CAS.
- L. E. Bilston and L. E. Thibault, Ann. Biomed. Eng., 1996, 24, 67–74 CrossRef CAS.
- K. Miller and K. Chinzei, J. Biomech., 2002, 35, 483–490 CrossRef.
- B. M. Holzapfel, J. C. Reichert, J.-T. Schantz, U. Gbureck, L. Rackwitz, U. Nöth, F. Jakob, M. Rudert, J. Groll and D. W. Hutmacher, Adv. Drug Delivery Rev., 2013, 65, 581–603 CrossRef CAS.
- G. Sun, Adv. Healthcare Mater., 2017, 6, 1700659 CrossRef.
- T. B. Bini, S. Gao, X. Xu, S. Wang, S. Ramakrishna and K. W. Leong, J. Biomed. Mater. Res., Part A, 2004, 68, 286–295 CrossRef CAS.
- B. J. Pfister, J. Neurosci., 2004, 24, 7978–7983 CrossRef CAS.
- B. J. Pfister, A. Iwata, A. G. Taylor, J. A. Wolf, D. F. Meaney and D. H. Smith, J. Neurosci. Methods, 2006, 153, 95–103 CrossRef.
- H. M. Younes, E. Bravo-Grimaldo and B. G. Amsden, Biomaterials, 2004, 25, 5261–5269 CrossRef CAS.
- I. Castilla-Cortázar, J. Más-Estellés, J. M. Meseguer-Dueñas, J. L. Escobar Ivirico, B. Marí and A. Vidaurre, Polym. Degrad. Stab., 2012, 97, 1241–1248 CrossRef.
- S. Lakhundi, R. Siddiqui and N. Khan, Parasites Vectors, 2015, 8, 23 CrossRef.
- M. Märtson, J. Viljanto, T. Hurme, P. Laippala and P. Saukko, Biomaterials, 1999, 20, 1989–1995 CrossRef.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, 3rd edn, 2004 Search PubMed.
- J. M. Dugan, J. E. Gough and S. J. Eichhorn, Nanomedicine, 2013, 8, 287–298 CrossRef CAS.
- E. Lam, K. B. Male, J. H. Chong, A. C. Leung and J. H. Luong, Trends Biotechnol., 2012, 30, 283–290 CrossRef CAS.
- P. Béguin and J. P. Aubert, FEMS Microbiol. Rev., 1994, 13, 25–58 CrossRef.
- J. P. F. Lagerwall, C. Schütz, M. Salajkova, J. H. Noh, J. H. Park, G. Scalia and L. Bergström, NPG Asia Mater., 2014, 6, e80–e91 CrossRef CAS.
- R. G. M. Breuls, T. U. Jiya and T. H. Smit, Toorthj, 2008, 2, 103–109 CrossRef.
- K. Ye, X. Wang, L. Cao, S. Li, Z. Li, L. Yu and J. Ding, Nano Lett., 2015, 15, 4720–4729 CrossRef CAS.
- R. J. Pelham and Y.-I. Wang, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 13661–13665 CrossRef CAS.
- A. X. Sun, H. Lin, M. R. Fritch, H. Shen, P. G. Alexander, M. DeHart and R. S. Tuan, Acta Biomater., 2017, 58, 302–311 CrossRef CAS.
- W. J. Hadden, J. L. Young, A. W. Holle, M. L. McFetridge, D. Y. Kim, P. Wijesinghe, H. Taylor-Weiner, J. H. Wen, A. R. Lee, K. Bieback, B. N. Vo, D. D. Sampson, B. F. Kennedy, J. P. Spatz, A. J. Engler and Y. S. Choi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5647–5652 CrossRef CAS.
- M. Guvendiren and J. A. Burdick, Nat. Commun., 2012, 3, 792 CrossRef.
- K. K. Sethi, I. V. Yannas, V. Mudera, M. Eastwood, C. McFarland and R. A. Brown, Wound Repair Regen., 2002, 10, 397–408 CrossRef.
- J. Kovalevich and D. Langford, Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology, in Neuronal Cell Culture, ed. S. Amini and M. K. White, Methods in Molecular Biology, Humana Press, Totowa, NJ, 2013, vol. 1078, pp. 9–21 Search PubMed.
- W. Zang, D. S. Choi, Y. H. Nguyen, J. Chang and L. Qin, Sci. Rep., 2013, 3, 2332 CrossRef.
- H.-I. Chang and Y. Wang, Cell Responses to Surface and Architecture of Tissue Engineering Scaffolds, Regenerative Medicine and Tissue Engineering – Cells and Biomaterials, ed. D. Eberli, 2011, ISBN: 978-953-307-663-8 Search PubMed.
- L. D. Bartolo, M. Rende, S. Morelli, G. Biuseppina, S. Salerno, A. Piscioneri, A. Gordano, A. Di Vito, M. Canonaco and E. Drioli, J. Membr. Sci., 2008, 325, 139–149 CrossRef.
- L.-C. Xu and C. A. Siedlecki, Biomaterials, 2007, 28, 3273–3283 CrossRef CAS.
- W. S. Rasband, ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/index.html.
- A. Hexemer, W. Bras, J. Glossinger, E. Schaible, E. Gann, R. Kirian, A. MacDowell, M. Church, B. Rude and H. A. Padmore, J. Phys.: Conf. Ser., 2010, 247, 012007 CrossRef.
- J. Ilavsky, Nika: software for two-dimensional data reduction, J. Appl. Crystallogr., 2012, 45, 324–328 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional confocal, SEM, MTT, and X-ray data. See DOI: 10.1039/d0ma00676a |
| ‡ Equal contribution to the manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |