
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic methodology towards allylic trans-cyclooctene-ethers enables modification of carbohydrates: bioorthogonal manipulation of the lac repressor†
Mark A. R.
de Geus
 ,
G. J. Mirjam
Groenewold
,
Elmer
Maurits
,
Can
Araman
* and
Sander I.
van Kasteren
*
,
G. J. Mirjam
Groenewold
,
Elmer
Maurits
,
Can
Araman
* and
Sander I.
van Kasteren
*
Leiden Institute of Chemistry, The Institute for Chemical Immunology, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: m.c.araman@lic.leidenuniv.nl; s.i.van.kasteren@lic.leidenuniv.nl
First published on 8th September 2020
Abstract
The inverse electron-demand Diels–Alder (IEDDA) pyridazine elimination is one of the key bioorthogonal bond-breaking reactions. In this reaction trans-cyclooctene (TCO) serves as a tetrazine responsive caging moiety for amines, carboxylic acids and alcohols. One issue to date has been the lack of synthetic methods towards TCO ethers from functionalized (aliphatic) alcohols, thereby restricting bioorthogonal utilization. Two novel reagents were developed to enable controlled formation of cis-cyclooctene (CCO) ethers, followed by optimized photochemical isomerization to obtain TCO ethers. The method was exemplified by the controlled bioorthogonal activation of the lac operon system in E. coli using a TCO-ether-modified carbohydrate inducer.
Introduction
Bioorthogonal bond cleavage reactions have garnered significant interest in recent years.1–4 Amongst these new “click-to-release” reactions, the inverse electron-demand Diels–Alder (IEDDA) pyridazine elimination has shown particular promise for bioorthogonal utilization.5 The method employs a trans-cyclooctene (TCO) carrying an allylic substituent that upon [4 + 2] cycloaddition with a 1,2,4,5-tetrazine results in the formation of a 4,5-dihydropyridazine.6 This 4,5-tautomer can rearrange to form two new tautomers of which the 1,4-tautomer can release the allylic payload, followed by rearomatization to the pyridazine (Fig. 1A).7,8 The excellent biocompatibility and high bimolecular reaction rate of the IEDDA pyridazine elimination9 has given rise to a multitude of in vitro and in vivo applications such as the regulation of protein activity10–12 and the activation of pro-drugs, in which spatiotemporal control is achieved by antibody–drug conjugates (ADCs),13–15 nanoparticles,16 enzymatic supramolecular self-assembly17 or hydrogel injection.18,19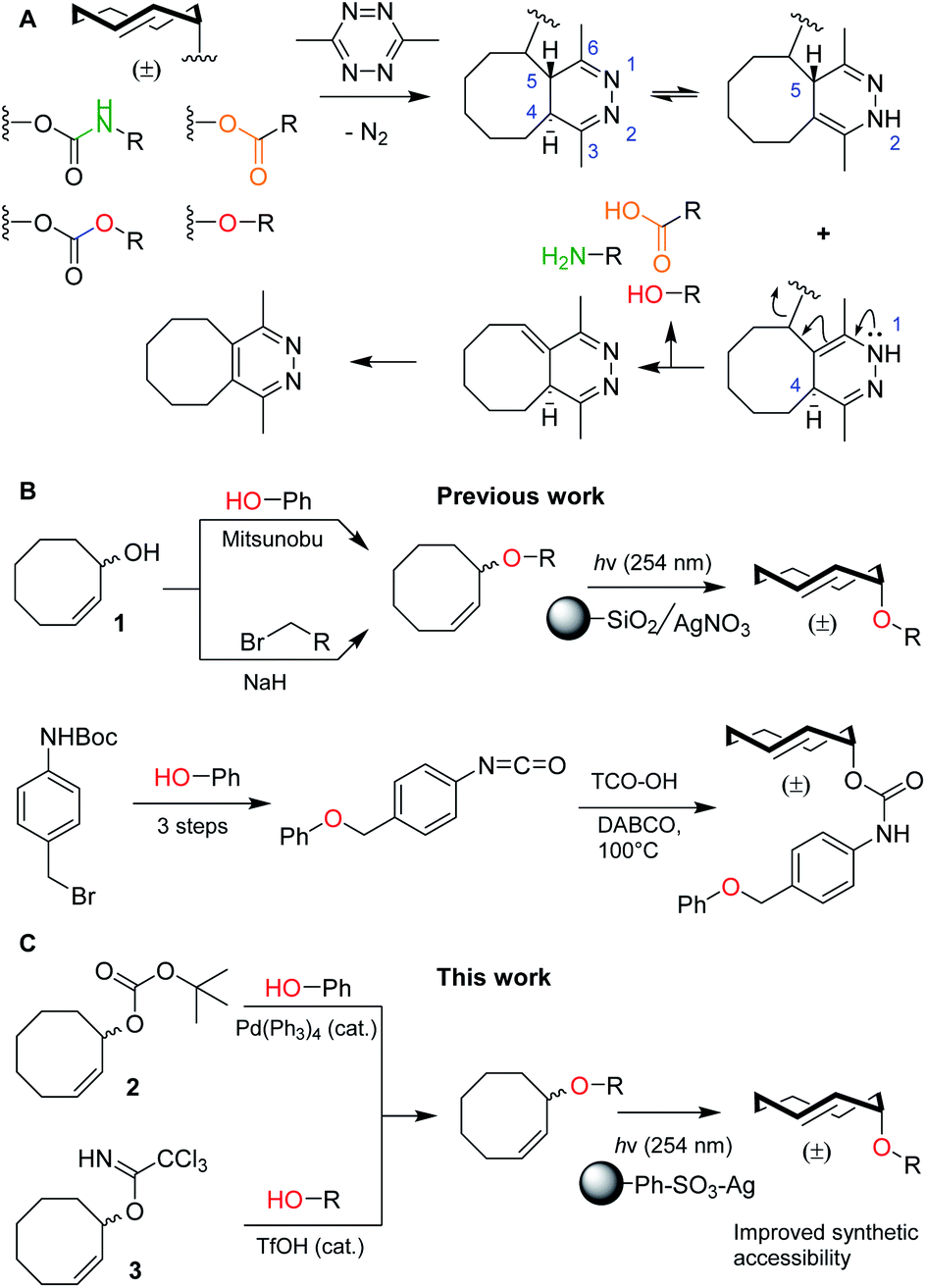 | ||
| Fig. 1 (A) Overview of the inverse electron-demand Diels–Alder (IEDDA) pyridazine elimination reaction, including the current scope of this method to decage carbamates, carbonates, ethers and esters to obtain amines (green), alcohols (red) and carboxylic acids (orange), respectively. (B) Overview of the current synthetic methods to obtain allylic TCO ethers, including methods from Robillard and co-workers (top)8 and Bernardes and co-workers (bottom).21 (C) Novel synthetic methods described in this work, including reagents 2 and 3. | ||
To date, nearly all applications for this reaction have relied on the protection of (primary) amines as TCO-carbamates (Fig. 1A, green). Recently, Robillard8 and Bernardes20,21 showed that release of other functional groups, such as carboxylic acids (from TCO-esters) and alcohols (from TCO-carbonates or TCO-ethers), is indeed possible (Fig. 1A, orange and red). Whilst TCO-esters and carbonates suffer from reduced hydrolytic stability in biological systems,8,20,21 TCO-ethers are particularly appealing due to their high stability and surprisingly fast decaging kinetics when compared to vinyl ether analogues.8,22–24 Unfortunately, widespread use of TCO-ethers is constricted by their challenging synthesis. Formation of the crucial cis-cyclooctene (CCO) ether bond, employing either Mitsunobu chemistry or nucleophilic substitution of a primary alkyl bromide with 1, is followed by photochemical isomerization25 to the TCO-ether and isolation of the desired axial isomer (Fig. 1B, top).8 One exception is the direct alkylation of axial TCO-OH to form a benzylic TCO-ether.8 Additionally, Bernardes and co-workers developed a self-immolative linker in which a TCO-carbamate is connected to a benzyl ether (Fig. 1B, bottom).21 Although 1 was used as a Mitsunobu substrate with moderate success,8 this method essentially limits the scope to phenolic nucleophiles without including the formation of ethers from aliphatic alcohols.26 Furthermore, direct nucleophilic substitution with 1 is limited to primary positions,8 in which the desired aliphatic alcohol requires an additional transformation into a leaving group. The self-immolative linker21 requires at least four synthetic steps from the substrate, which to date has also been limited to phenols. Taken together, synthesis and utilization of TCO-ethers derived from functionalized aliphatic alcohols encountered in biological systems is currently unfeasible. This led us to develop novel synthetic methods for the (regioselective) installation of TCO-ethers in biomolecules as caging moieties, based on reagents 2 and 3, which we present here (Fig. 1C). Furthermore, we applied our method to enable the synthesis and bioorthogonal decaging of a TCO-ether modified carbohydrate, as no TCO-protected variants of these biomolecules currently exist.
Results and discussion
We envisioned a two-step procedure in which the use of electrophilic cyclooctene reagents secured formation of a CCO-ether bond under mild conditions, followed by photochemical isomerization25 to the desired TCO-ether. Palladium catalysis enables mild, decarboxylative conversion of allyl carbonates to allyl ethers via a reactive π-allyl cation species.27,28 We found a ring strained variant of this reaction was feasible for phenols by transforming para-nitrophenyl carbonate 4 into cyclooctene ether 5 under Pd(PPh3)4 catalysis (50 °C) in 92% yield (Scheme 1). Based on this initial observation, we reasoned the palladium-catalyzed method for direct allylation by Grover and co-workers29 would enable cyclooctene ether formation in a single step. We designed cyclooctene tert-butyl carbonate reagent 2, which decarboxylates upon coordination with a palladium catalyst (Scheme 2A). The spectator tert-butoxide formed in this step29 ensures rapid deprotonation of the phenol nucleophile, which can subsequently attack the π-allyl electrophile to form the ether bond. We examined the cyclooctene ether formation with N-trifluoroacetyl-protected L-tyrosine methyl ester 6 using reagent 2 (1.2 equivalents) under Pd(PPh3)4 catalysis (80 °C) instead of the previously reported Mitsunobu procedure (12% yield),8 obtaining cyclooctene ether 7 in 80% yield (Scheme 2A).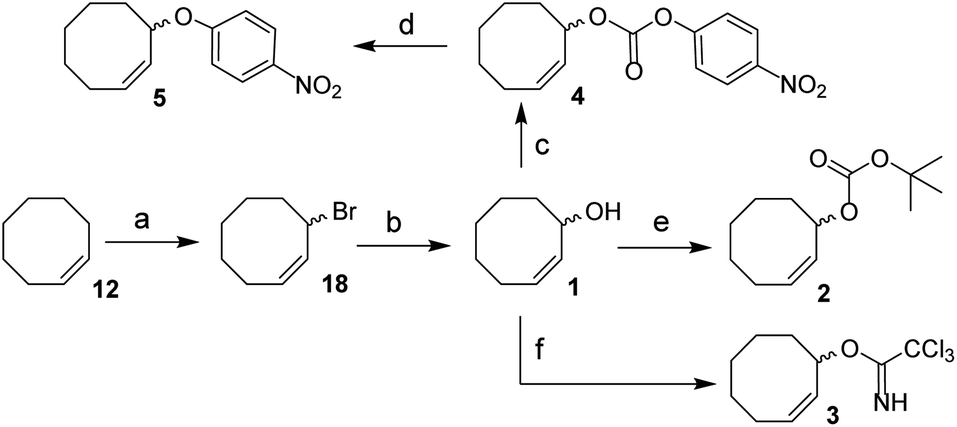 | ||
| Scheme 1 Synthesis of 1, 2, 3, 4 and 5 from cis-cyclooctene (12). Reagents/conditions: (a) NBS, AIBN, cyclohexane, reflux, 71%; (b) acetone, H2O, NaHCO3, reflux, 83%; (c) 4-nitrophenyl chloroformate, pyridine, DCM, 0 °C to rt, 83%; (d) Pd(PPh3)4, toluene, 50 °C, 92%; (e) NaHMDS, Boc-ON, THF, 0 °C to rt, 81%; (f) trichloroacetonitrile, DBU, NaHCO3, DCM, 0 °C to rt, ∼80% (Table S1†). | ||
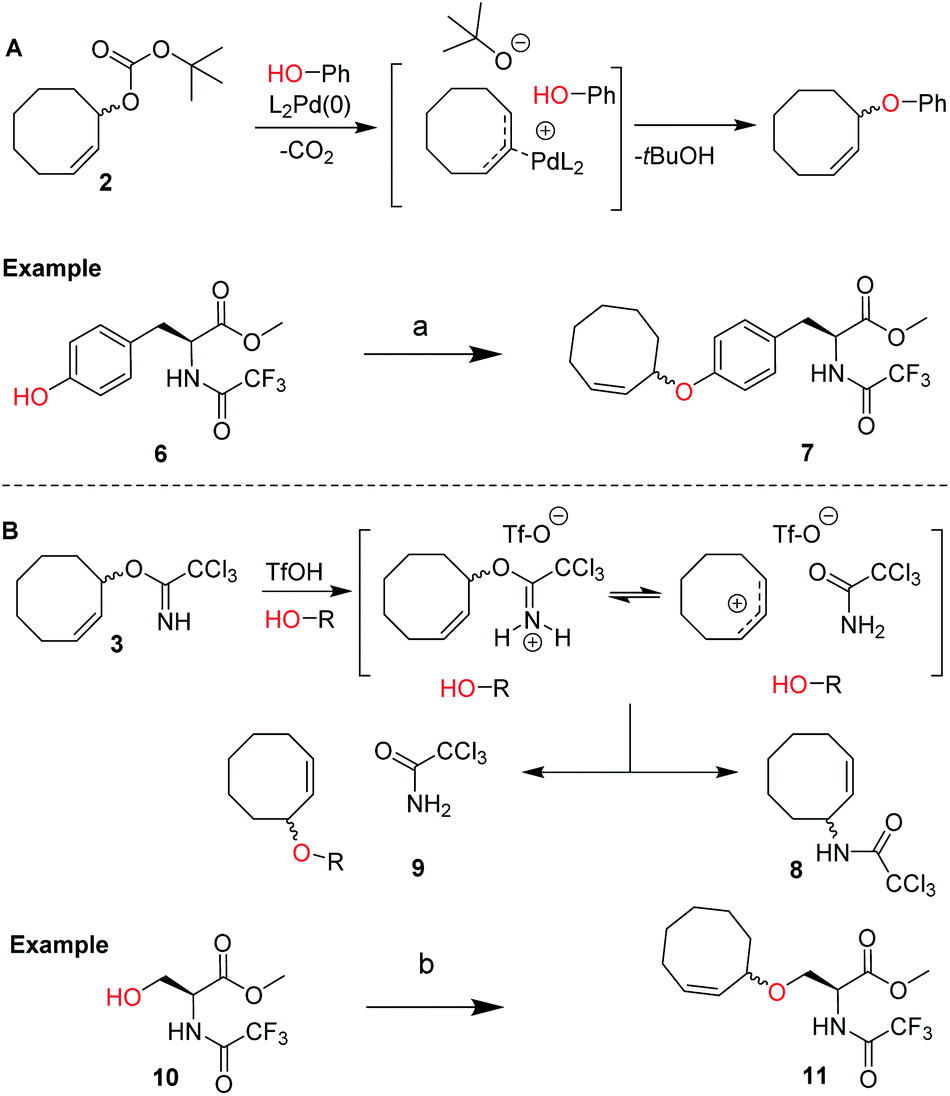 | ||
| Scheme 2 (A) Design of cyclooctene tert-butyl carbonate reagent 2 for the palladium catalyzed installation of cyclooctene ethers on phenols, including the proposed mechanism based on the work of Grover and co-workers.19 Reagents/conditions: (a) 2, Pd(PPh3)4, dioxane, 80 °C, 80%; (B) design of cyclooctene trichloroimidate reagent 3 for the Lewis acid triggered formation of cyclooctene ethers from aliphatic alcohols. Proposed mechanism and products observed upon treating 3 with catalytic triflic acid are shown. Reagents/conditions: (b) 3, TfOH, DCM, −35 °C to 0 °C, 46% (Table S2†). | ||
In parallel, we designed a second reagent (3) for the synthesis of cyclooctene ethers from aliphatic alcohols (Scheme 2B). Lewis acid triggered activation of cyclooctene trichloroimidate 3 can result in elimination and rearrangement pathways, including the formation of ionic intermediates.30,31 Cyclooctene trichloroamide 8 (see ESI† for characterization), trichloroacetamide (9) and the desired cyclooctene ether can be formed from these reactive intermediates. N-trifluoroacetyl-protected L-serine methyl ester 10 was alkylated using 2 equivalents of 3 under triflic acid catalysis (−35 °C to 0 °C) to obtain 11 in 46% yield (Scheme 2B, Table S2†). Furthermore, reagents 2 and 3 were both synthesized from cis-cyclooctene (12) in 3 steps (∼48% yield) with a common intermediate (1) and a single chromatographic purification (Scheme 1, Table S1†).
Carbohydrates orchestrate a diverse array of biological processes and as such, spatiotemporal control over these biological activities would be a powerful addition to the “click-to-release” toolkit. A classic example of a biological process directed by glycans is the switching of the lac operon.32,33 It is a regulatory element that is used to control nutrient-dependent transcription in E. coli. When lactose concentration is low, the activity of the operon is inhibited by a repressor element.34 When the concentration of lactose increases, it will bind to the repressor, leading to a decreased affinity of the lactose–repressor complex for the operon.35 This results in enhanced gene transcription for proteins under control of this promotor. This system has been extensively used for the expression of recombinant proteins in E. coli and the synthetic lac operon inducer isopropyl β-D-1-thiogalactopyranoside (IPTG, 13) is a key reagent here. Control over its activity has previously been attempted using both photochemical36 and hypoxia-triggered37 induction strategies. We therefore deemed it an excellent model carbohydrate to test IEDDA pyridazine elimination on carbohydrates. Initial experiments with peracetylated IPTG (14) under conditions typically used for photochemical isomerization of cyclooctenes confirmed the high affinity of the thioacetal functionality towards the silver nitrate used for enrichment of the TCO-isomers upon photoisomerization (Scheme 3). We therefore designed a novel inducer for the lac operon, substituting the sulfur with oxygen to obtain an O-glycoside, isopropyl β-D-1-galactopyranoside (IPG, 15, Scheme 4).
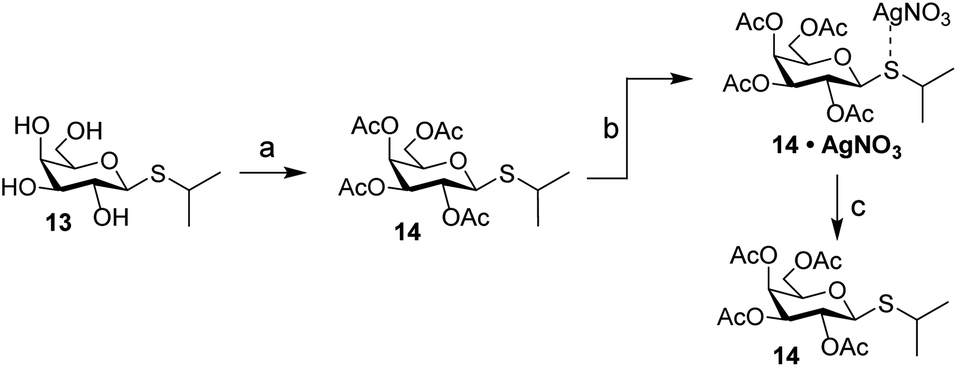 | ||
| Scheme 3 Evaluation of the fate of peracetylated IPTG (14) during photoisomerization conditions. Reagents/conditions: (a) Ac2O, pyridine, rt, 100%; (b) methyl benzoate, hv (254 nm), Et2O, heptane, rt; (c) NH4OAc (aq), DCM, rt, 61%. | ||
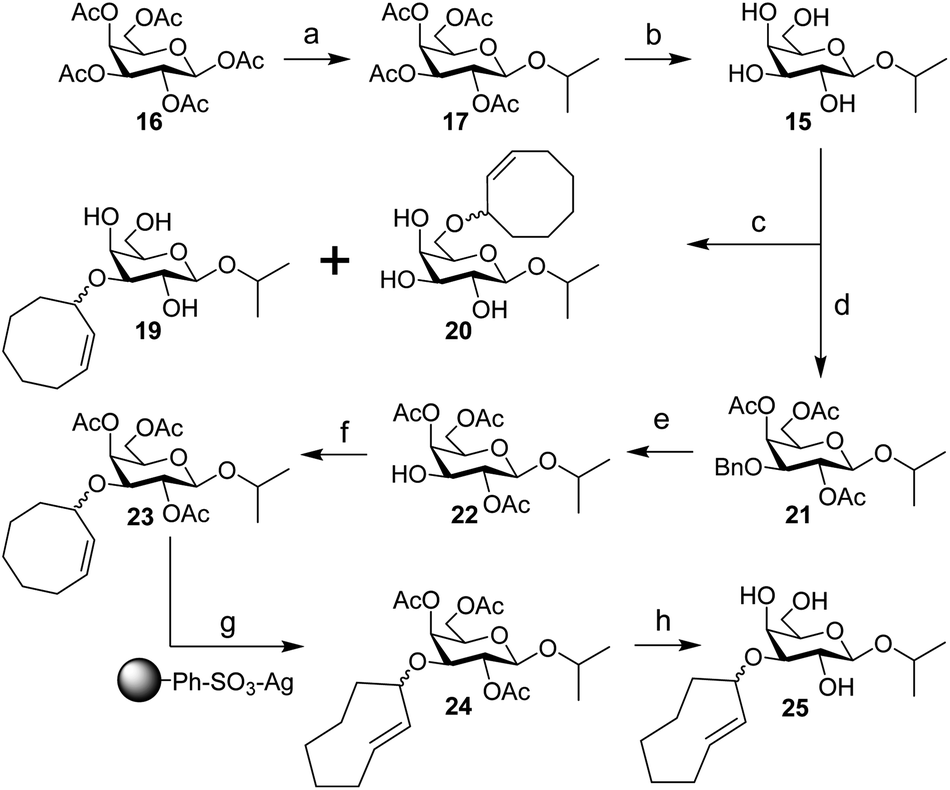 | ||
| Scheme 4 Synthesis of IPG (15), 3-CCO-IPG (19), 6-CCO-IPG (20), and 3-TCO-IPG (25) from peracetylated β-D-galactopyranoside (16). Reagents/conditions: (a) (i) HBr, AcOH, DCM, 0 °C to rt; (ii) 2-propanol, I2, DCM, 0 °C to 4 °C, 70% over 2 steps; (b) NaOMe, MeOH, DCM, rt, 90%; (c) (i) Bu2SnO, toluene, 105 °C; (ii) 18, CsF, toluene, 105 °C, ∼20% (19), ∼20% (20) (Table S3†); (d) (i) Bu2SnO, toluene, 105 °C; (ii) benzyl bromide, TBABr, toluene, 70 °C; (iii) Ac2O, pyridine, rt, 91% over 2 steps; (e) Pd(OH)2/C, H2, EtOAc, rt, 86%; (f) 3, TfOH, DCM, −40 °C to rt, ∼30% (Table S5†); (g) methyl benzoate, hv (254 nm), Et2O/isopropanol, rt; (h) NaOMe, MeOH, rt, ∼30% over 2 steps (Table S6†). | ||
Starting from peracetylated β-D-galactopyranoside 16, installation of the beta isopropyl group on the anomeric center was achieved in a one-pot, two-step procedure in 70% yield. The ‘disarmed’ pentaacetate was first transformed into an anomeric bromide, which was subsequently activated with stoichiometric I2 in the presence of isopropanol to obtain 17, according to a method reported by Field and colleagues.38 Deacetylation gave IPG (15) in 90% yield. Stannylene acetal-mediated alkylation of IPG (15) with (Z)-3-bromocyclooct-1-ene (18) in the presence of CsF or TBAI as an additive at 105 °C was initially investigated (Table S3†). A mixture of 3-CCO-IPG (19, ∼20% yield) and 6-CCO-IPG (20, ∼20% yield) was obtained and purified via (laborious) chromatographic separation. Decomposition of 18 at high temperature into 1,3-cyclooctadiene and hydrogen bromide39 limited its use. Attempts to alkylate 6-TBS and 4,6-DTBS functionalized derivatives of 15 in a regioselective manner with 18 were unsuccessful (Table S4†).
Instead, IPG (15) was regioselectively benzylated using organotin chemistry followed by acetylation to obtain 2,4,6-OAc-3-OBn-IPG (21) in 91% yield over two steps. Hydrogenation of 21 in the presence of Pearlman's catalyst afforded 2,4,6-OAc-IPG (22) in 86% yield. Alkylation of 22 could be achieved in the timespan of hours at low temperature (≤0 °C), typically employing 4 equivalents of trichloroimidate 3 and 10 mol% of triflic acid (Table S5†). The crude reaction mixture was washed with aqueous NaOH (to remove 9)40 before purification by silica gel chromatography to obtain 23 in ≥30% yield. Singlet sensitized photoisomerization of 23 to 24 (Table S6†) was executed with the general flow setup described by Fox and co-workers25 using silver(I) exchanged tosic acid silica gel (TAg silica) as the stationary phase.41 Irradiation (λ = 254 nm) of mixture of 23 and methyl benzoate in Et2O for ±24 h, whilst continuously circulating the reaction mixture over the stationary phase, was followed by treatment of the stationary phase with NH3 in MeOH to obtain (partially deacetylated) 24. Zemplén deacetylation of 24 afforded 25 after extractive desalting in ≥30% yield over 2 steps. This final product, 3-TCO-IPG (25), was exclusively obtained as the desired axial TCO isomer and its purity was optimized by carefully increasing the polarity of the photoisomerization reaction mixture with isopropanol (2–5%; Fig. S1†).
We next evaluated whether the TCO-modified IPG 25 could be used to control the function of the parent molecule (15) in a biological setting (Fig. 2A). For this we transformed E. coli to express the model protein ovalbumin (OVA) under control of the lac operon. Overnight cultures were inoculated in LB medium supplemented with 1% glucose to inhibit leaky expression. Next, cultures were grown in fresh LB medium (no glucose) before adding (caged) inducers (1 mM) and tetrazine (2.5 mM) in DMSO. We first tested whether IPG (15) could induce expression, which it did in a comparable level to IPTG (13; Fig. 2B, S2 and Table S7†). Furthermore, up to 10% DMSO was tolerated for IPG (15) induced OVA expression (Fig. S3 and Table S8†).
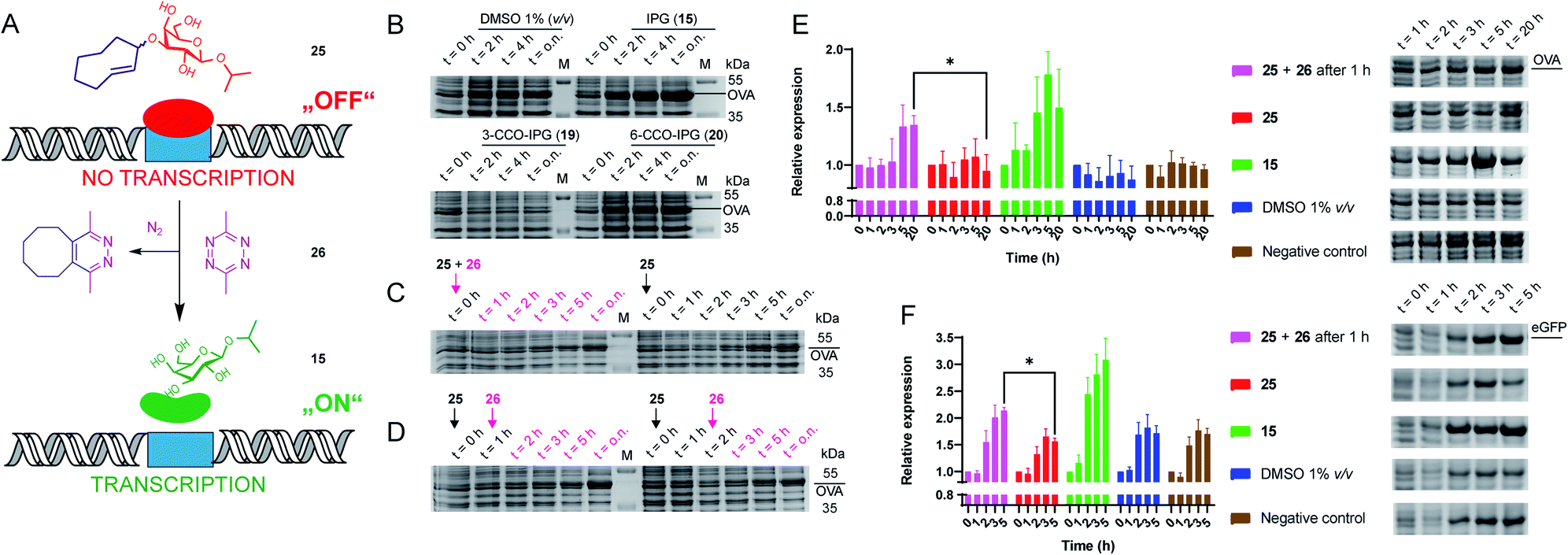 | ||
| Fig. 2 Recombinant expression of proteins with caged IPG variants. (A) Schematic representation of the chemical control over lac operon activity, mediated by 3-TCO-IPG (25), which does not induce expression. IEDDA pyridazine elimination with 3,6-dimethyl-tetrazine (26) liberates IPG (15), which induces overexpression of the protein of interest. (B) Recombinant OVA expression, comparing the effects of DMSO (negative control, 1% v/v), IPG (15, positive control, 1 mM), 3-CCO-IPG (19, 1 mM) and 6-CCO-IPG (20, 1 mM). The respective compounds were added at t = 0 h after which protein expression was followed over time. (C and D) Temporal control of OVA expression via addition of tetrazine 26 (2.5 mM) in the presence of 3-TCO-IPG (25, 1 mM). (C) Direct addition of 25 and 26 at t = 0 h (left) or addition of 25 at t = 0 h (right). (D) Addition of 26 1 h (left) or 2 h (right) after adding 25 at t = 0 h. (E and F) Replicate expression experiments for OVA (2E; N = 3) and eGFP_A206K (2F; N = 4) comparing decaging conditions (1 mM 25 at t = 0 and 2.5 mM 26 at 1 = 1 h) with addition of 25 (1 mM), 15 (1 mM), DMSO (1% v/v) and a negative control. Mean quantification values (Coomassie) were plotted with SD as error bars. Representative gel sections are shown to serve as an example. Full gels for all replicate experiments are shown in the ESI (Fig. S6–S12†). Relative band intensity was measured via densitometry for all experiments. For eGFP (2F), quantification was based on the Coomassie signal of the monomeric protein. *Significant difference based on an unpaired, two-tailed t-test (P < 0.05). | ||
To assess the functionality of the TCO-protecting group, we confirmed 3-TCO-IPG (25) did not affect expression levels in absence of a deprotection agent (Fig. S4 and Table S9†). Subsequently, we analyzed whether deprotection with the prokaryote-compatible 3,6-dimethyl-tetrazine (26) could be used to switch on recombinant protein expression.12,42 We found that 26 caused a minor delay on IPG (15) induced OVA expression without reducing overall expression levels (Fig. S5 and Table S10†).
Therefore, deprotection of 3-TCO-IPG (25) with 26 was studied in a time course experiment (Fig. 2C and D). Simultaneous addition of both 25 and 26 at the same time did not result in a controlled overexpression. However, a preincubation of 1 or 2 hours with 25 before addition of 26 did result in tightly controlled overexpression of OVA. This was particularly striking at late time points (Fig. 2D and Table S11†). Replicate experiments (N = 3) confirmed these findings (Fig. 2E, S6–S8 and Tables S12–S15†). Control compounds 3-CCO-IPG (19) and 6-CCO-IPG (20) failed to induce overexpression (Fig. 2B and Table S16†). A reduction in expression compared to the DMSO background was even observed for 19 (Fig. S4 and Table S9†), but this was not a property of the CCO-moeity in general.
The method was next applied to the expression of two other proteins, eGFP (Fig. 2F, S9–S12 and Tables S17–S21;†N = 4) and DsRed2 (Fig. S13†). Again, overexpression after addition of 26 was observed for both proteins based on Coomassie quantification (Fig. 2F and Table S22†). For eGFP, a significant overexpression was found after 5 hours (Fig. 2F). Taken together, it appears this chemical method can be utilized as a general tool for temporally controlled gene expression in E. coli.
Conclusions
In conclusion, we report the synthesis of TCO-ethers using two novel reagents (2–3), providing facile access to TCO-protected phenols and aliphatic alcohols, respectively. This methodology enables access to the modification of complex biomolecules as TCO-ethers. A carbohydrate probe, 3-TCO-IPG (25), made in this manner was successfully used to chemically modulate lac operon activity, thereby providing a first example of control over glycan activity by IEDDA pyridazine elimination. The spatiotemporal control available to this method also bodes well for other applications in which carbohydrate/receptor interactions direct biological processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Jessica E. Pigga and Joseph M. Fox (University of Delaware) for useful discussions concerning the TAg silica gel. S. I. V. K. was the recipient of an ERC Starting Grant of the European Research Council (ERC-2014-StG-639005). C.A. acknowledges the Netherlands Organization for Scientific Research (NWO)-CW-ECHO Grant program 2016 for financial support.Notes and references
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS.
- J. Tu, M. Xu and R. M. Franzini, ChemBioChem, 2019, 20, 1615–1627 CrossRef CAS.
- K. Neumann, A. Gambardella and M. Bradley, ChemBioChem, 2019, 20, 872–876 CrossRef CAS.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef CAS.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS.
- J. C. T. Carlson, H. Mikula and R. Weissleder, J. Am. Chem. Soc., 2018, 140, 3603–3612 CrossRef CAS.
- R. M. Versteegen, W. ten Hoeve, R. Rossin, M. A. R. de Geus, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2018, 57, 10494–10499 CrossRef CAS.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- J. Li, S. Jia and P. R. Chen, Nat. Chem. Biol., 2014, 10, 1003–1005 CrossRef CAS.
- G. Zhang, J. Li, R. Xie, X. Fan, Y. Liu, S. Zheng, Y. Ge and P. R. Chen, ACS Cent. Sci., 2016, 2, 325–331 CrossRef CAS.
- J. Zhao, Y. Liu, F. Lin, W. Wang, S. Yang, Y. Ge and P. R. Chen, ACS Cent. Sci., 2019, 5, 145–152 CrossRef CAS.
- R. Rossin, R. M. Versteegen, J. Wu, A. Khasanov, H. J. Wessels, E. J. Steenbergen, W. ten Hoeve, H. M. Janssen, A. H. A. M. van Onzen, P. J. Hudson and M. S. Robillard, Nat. Commun., 2018, 9, 1484 CrossRef.
- R. Rossin, S. M. J. van Duijnhoven, W. ten Hoeve, H. M. Janssen, F. J. M. Hoeben, R. M. Versteegen and M. S. Robillard, Bioconjugate Chem., 2016, 27, 1697–1706 CrossRef CAS.
- S. Du, D. Wang, J.-S. Lee, B. Peng, J. Ge and S. Q. Yao, Chem. Commun., 2017, 53, 8443–8446 RSC.
- I. Khan, L. M. Seebald, N. M. Robertson, M. V. Yigit and M. Royzen, Chem. Sci., 2017, 8, 5705–5712 RSC.
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 5032 CrossRef.
- J. M. Mejia Oneto, I. Khan, L. Seebald and M. Royzen, ACS Cent. Sci., 2016, 2, 476–482 CrossRef CAS.
- M. Czuban, S. Srinivasan, N. A. Yee, E. Agustin, A. Koliszak, E. Miller, I. Khan, I. Quinones, H. Noory, C. Motola, R. Volkmer, M. Di Luca, A. Trampuz, M. Royzen and J. M. Mejia Oneto, ACS Cent. Sci., 2018, 4, 1624–1632 CrossRef CAS.
- S. Davies, L. Qiao, B. L. Oliveira, C. D. Navo, G. Jiménez-Osés and G. J. L. Bernardes, ChemBioChem, 2019, 20, 1541–1546 CAS.
- S. Davies, B. L. Oliveira and G. J. L. Bernardes, Org. Biomol. Chem., 2019, 17, 5725–5730 RSC.
- E. Jiménez-Moreno, Z. Guo, B. L. Oliveira, I. S. Albuquerque, A. Kitowski, A. Guerreiro, O. Boutureira, T. Rodrigues, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2017, 56, 243–247 CrossRef.
- K. Neumann, S. Jain, A. Gambardella, S. E. Walker, E. Valero, A. Lilienkampf and M. Bradley, ChemBioChem, 2017, 18, 91–95 CrossRef CAS.
- H. Wu, S. C. Alexander, S. Jin and N. K. Devaraj, J. Am. Chem. Soc., 2016, 138, 11429–11432 CrossRef CAS.
- M. Royzen, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 3760–3761 CrossRef CAS.
- S. Fletcher, Org. Chem. Front., 2015, 2, 739–752 RSC.
- F. Guibe and Y. S. M’leux, Tetrahedron Lett., 1981, 22, 3591–3594 CrossRef CAS.
- J. Muzart, Tetrahedron, 2005, 61, 5955–6008 CrossRef CAS.
- A. R. Haight, E. J. Stoner, M. J. Peterson and V. K. Grover, J. Org. Chem., 2003, 68, 8092–8096 CrossRef CAS.
- F. Cramer and N. Hennrich, Chem. Ber., 1961, 94, 976–989 CrossRef CAS.
- H.-P. Wessel, T. Iversen and D. R. Bundle, J. Chem. Soc., Perkin Trans. 1, 1985, 2247 RSC.
- F. Jacob and J. Monod, J. Mol. Biol., 1961, 3, 318–356 CrossRef CAS.
- M. Lewis, C. R. Biol., 2005, 328, 521–548 CrossRef CAS.
- C. E. Bell and M. Lewis, Nat. Struct. Biol., 2000, 7, 209–214 CrossRef CAS.
- M. Lewis, J. Mol. Biol., 2013, 425, 2309–2316 CrossRef CAS.
- D. D. Young and A. Deiters, Angew. Chem., Int. Ed., 2007, 46, 4290–4292 CrossRef CAS.
- S. L. Collins, J. Saha, L. C. Bouchez, E. M. Hammond and S. J. Conway, ACS Chem. Biol., 2018, 13, 3354–3360 CrossRef CAS.
- K. P. R. Kartha, M. Aloui and R. A. Field, Tetrahedron Lett., 1996, 37, 8807–8810 CrossRef.
- S. Kobayashi, K. Fukuda, M. Kataoka and M. Tanaka, Macromolecules, 2016, 49, 2493–2501 CrossRef CAS.
- M. Heuckendorff and H. H. Jensen, Carbohydr. Res., 2017, 439, 50–56 CrossRef CAS.
- A. Darko, S. J. Boyd and J. M. Fox, Synthesis, 2018, 50, 4875–4882 CrossRef CAS.
- L. Liu, Y. Liu, G. Zhang, Y. Ge, X. Fan, F. Lin, J. Wang, H. Zheng, X. Xie, X. Zeng and P. R. Chen, Biochemistry, 2018, 57, 446–450 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03216f |
| This journal is © The Royal Society of Chemistry 2020 |