
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot method for the synthesis of 1-aryl-2-aminoalkanol derivatives from the corresponding amides or nitriles†
Jan Otevrel‡
 *,
David Svestka‡ and
Pavel Bobal*
*,
David Svestka‡ and
Pavel Bobal*
Department of Chemical Drugs, Faculty of Pharmacy, Masaryk University, Palackeho 1946/1, 612 42 Brno, Czech Republic. E-mail: pbobal@icloud.com; otevrel@icloud.com
First published on 30th June 2020
Abstract
We have identified a novel one-pot method for the synthesis of β-amino alcohols, which is based on C–H bond hydroxylation at the benzylic α-carbon atom with a subsequent nitrile or amide functional group reduction. This cascade process uses molecular oxygen as an oxidant and sodium bis(2-methoxyethoxy)aluminum hydride as a reductant. The substrate scope was examined on 30 entries and, although the respective products were provided in moderate yields only, the above simple protocol may serve as a direct and powerful entry to the sterically congested 1,2-amino alcohols that are difficult to prepare by other routes. The plausible mechanistic rationale for the observed results is given and the reaction was applied to a synthesis of a potentially bioactive target.
Introduction
A 1,2-amino alcohol moiety is one of the most prominent structural components in a vast group of naturally occurring and synthetic molecules. The majority of the vic-amino alcohol-containing molecules are chiral and, although they are often used in their racemic forms, the desired properties or activities are usually associated with a single enantiomer.1 The above compounds in their enantiopure forms provide attractive synthetic intermediates for the bioactive agents and also important subunits for construction of the chiral auxiliaries, ligands or scaffolds for stereoselective syntheses.2 Hence the actively pursued research area of novel, more efficient, and selective synthetic transformations of these potent building blocks represents one of the ultimate goals in current organic chemistry.As the research in this field is vibrant, a number of possible routes for the β-amino alcohol synthesis including their enantioselective versions have been described and well-reviewed in the literature.2b,3 Accordingly, five general protocols have been established: (1) functional group interconversions of compounds with both heteroatoms already present in the molecule, such as a reduction of nitroaldols, cyanohydrins, azidohydrins, amino acids, amino ketones, hydroxy imines, etc.;4 (2) ring-opening of epoxides and aziridines;5 (3) addition of a single heteroatom to a molecule already containing oxygen or nitrogen functionality;2b (4) introduction of both heteroatoms in a single process, i.e. by aminohydroxylation or oxidative nitration;6 (5) C–C bond-forming reactions including 1,2-additions to α-hydroxy imines and α-amino aldehydes.7
Compared to other methods, the introduction of oxygen to a molecule already containing a nitrogen functionality is not a common approach for the synthesis of β-amino alcohols and hence remained considerably underexplored. Several reactions providing entry into this interesting pathway mainly comprise of the addition of O-nucleophiles to α,β- or β,γ-unsaturated amines3a,8 and nitroolefins,9 hydroboration–oxidation of enamines8 or the Schlenck ene reaction.10 Herein we disclose another, conceptually different method, which is based on a protocol including α-carbon atom hydroxylation and amide or nitrile functional group reduction.
In connection with our interest in the development of organocatalysts for the asymmetric aldol-type reactions, we have been continuously compelled to explore novel chiral auxiliaries for the catalyst design and screening. As part of the above synthetic efforts, we have prepared many chiral non-racemic amine scaffolds by reductions of the corresponding enantiopure amides.11
For such transformations, we normally utilized sodium bis(2-methoxyethoxy)aluminum hydride (SMEAH) as a reductant.12 In comparison to lithium aluminum hydride (LAH), SMEAH is much better soluble, easier to handle, insensitive to dry air, thermally stable (<205 °C), and non-violently reacting with water, which makes it an invaluable reducing agent for both laboratory and industrial use.13 Although it has been documented that SMEAH exhibited a unique reactivity and selectivity in some cases,13,14 we have been using it as an equivalent reagent to LAH for numerous amide reductions.
However, (S)-naproxamide subjected to the above reaction conditions gave a curious product, which was then, to our surprise, identified as 1,2-amino alcohol derivative (Fig. 1). From the initial insights, the formation of this unexpected substance seemed to be favored by a longer reaction time together with a large excess of SMEAH. As this represented, to the best of our knowledge, the only example of the organic transformation that enabled a direct conversion of the primary amide to the appropriate vic-amino alcohol derivative, it stimulated us into a research of this interesting phenomenon in more detail.
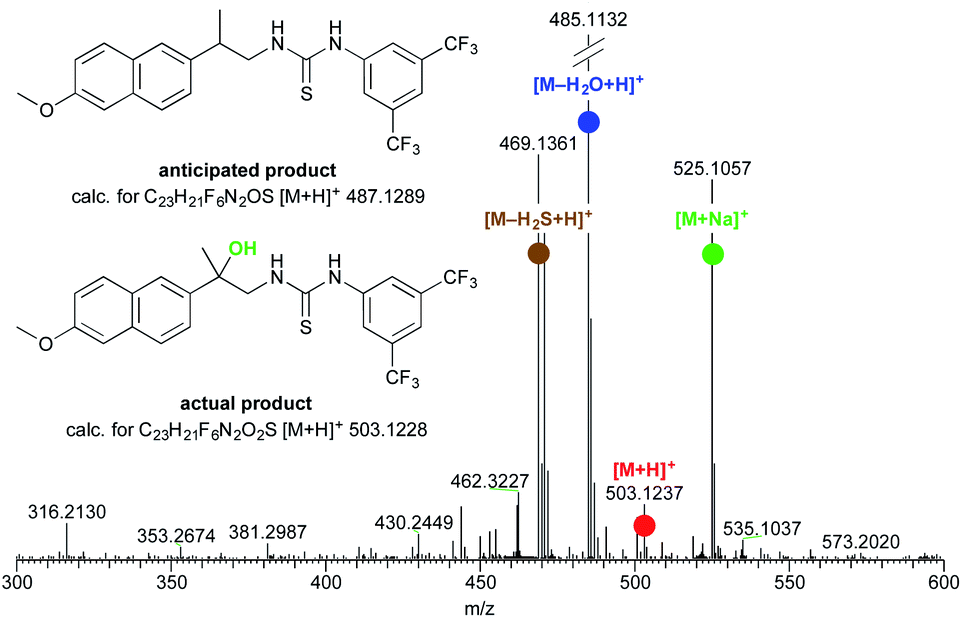 | ||
| Fig. 1 Partial HRMS spectrum showing the formation of the unexpected product. | ||
Results and discussion
The naproxamide was chosen as a model substrate for a further optimization study (Tables S1–S3†). At first, we investigated the loading of the hydride. Although the maximum yield of the β-amino alcohol was achieved with 20 equiv. of SMEAH, 15 equiv. were used in further optimization protocol as it represented the best balance between the hydride consumption and the product yield. Simultaneously the optimal reaction temperature was established to 25 °C. We found that either elevated or diminished reaction temperature led to a drop in the product yield. Moreover, even a slightly elevated reaction temperature (55 °C) caused a significant decrease in the 3a/4a product ratio. Next, several different complex metal hydrides were screened under the above reaction conditions. Accordingly, SMEAH provided the highest ratio of 3a/4a (84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16) and DIBAH gave almost pure 4a (>99%), whereas reductions with LAH and lithium dimethoxyaluminum hydride (LMAH) proceeded with the selectivity somewhere in between the two above-mentioned extremes (Fig. 2). Such results indicate the unique properties of SMEAH in this particular reduction, which justified its selection for further optimization steps.
:16) and DIBAH gave almost pure 4a (>99%), whereas reductions with LAH and lithium dimethoxyaluminum hydride (LMAH) proceeded with the selectivity somewhere in between the two above-mentioned extremes (Fig. 2). Such results indicate the unique properties of SMEAH in this particular reduction, which justified its selection for further optimization steps.
 | ||
| Fig. 2 Partial 1H NMR (400 MHz, DMSO-d6, 35 °C) spectra showing the 3a/4a ratio affected by the type of the complex metal hydride. | ||
Further, we examined other reaction solvents besides toluene, such as diethyl ether, glyme, dioxane, and THF. The highest selectivity towards the 1,2-amino alcohol formation was reached by performing the reduction in anhydrous THF (3a/4a = 92:8). Although a pretty good selectivity was obtained at this stage, we were still discouraged by a poor yield (21%) and a very long reaction time, which was necessary for completion of the reaction (>48 h). Finally, the optimization of the reaction atmosphere proved to be the major turning point. The switching of the previously used argon atmosphere to dry air and then oxygen dramatically enhanced both the reaction rate and the yield. Consequently, the desired product was furnished in 43% yield in less than 3 h even with the slightly improved selectivity (3a/4a = 94:6). Further attempts to increase the product yield were unsuccessful. Nevertheless, according to our experience, the analogous LAH reductions of 2-arylalkanoic acid amides or nitriles to amines also barely exceeded isolated yields of 50–60%.
In the following task, we investigated the scope of possible functional groups that undergo the β-amino alcohol-forming process. The obtained data is summarized in Scheme 1. Accordingly, the appropriate N-methylamide (1aa), hydrazide (1ab), hydroxamic acid (1ac), thioamide (1ad), and nitrile (2a) derivatives of the model compound were tested under the optimized reaction conditions. The above N-methylamide (1aa) provided a regular N-methylamine (4aa) instead of the desired N-methylamino alcohol, while the respective reaction of hydrazide (1ab) or hydroxamic acid (1ac) resulted only in a complete recovery of starting material. These experiments have confirmed that the corresponding N-substituted derivatives failed to provide β-amino alcohol products under the screening conditions. Amongst other tested functional groups, the best results in terms of the selectivity towards the formation of 3a were obtained with the amide (1a) and nitrile (2a) functions. Therefore these types of substrates were selected for the tests henceforth.
 | ||
| Scheme 1 The functional group scope of the SMEAH-mediated process. Notes: the reductions were performed at the 1.3 mmol scale; the yields refer to the isolated products; athe product ratio determined by RP-HPLC analysis; bthe product ratio determined by 1H NMR analysis. | ||
Following the optimization of the functional groups, the feasible substituents of the α-position of substrates were evaluated (Scheme 2). The α-oxidation of the compound 1b lacking the α-substituent turned out to be even more sensitive to the reaction atmosphere than 1a as the selectivity towards the 1,2-amino alcohol formation (3b/4b) was improved from 81:19 to 95:5 by replacing Ar by O2 (Fig. S10–11†). The control α-OH bearing substrate 1ba was reduced under the identical reaction conditions to the sole 3b in 36% yield. The SMEAH-mediated reduction of the α-methoxy and α-phenylsulfanyl functionalized derivatives 1bb and 1bc led at least partially to a displacement of the original α-substituent by OH group. On the contrary, the α-chlorine was preserved and 1bd yielded the regular 1,2-chloroamine 4bb in 57%. The α-alkyl and α-aryl derivatives 1c, 1cc, and 1d afforded the respective β-amino alcohols in excellent selectivities and modest yields. Surprisingly, reduction of the α-CF3 substituted 1ca provided the completely hydrodefluorinated derivative 3c identical to the product of the reaction of 1c. On the other hand, the corresponding α-OH analog thereof (1cb) did not exhibit any detectable defluorination under the above reaction conditions. According to these observations and the available literature,14d,15 we have postulated a plausible hypothesis that defluorination of the CF3 group of 1ca could be driven by a series of fluoride β-elimination – hydride addition events of the intermediary α-carbanion leading to 1c that is then converted to the β-amino alcohol 3c. This is also supported by the fact that the respective α,α-disubstituted derivative 1cb did not lose any of the fluorine atoms during the reduction.
 | ||
| Scheme 2 Variation of the α-substituent of substrates. Notes: the reductions were performed at the 1.3 mmol scale; the yields refer to the isolated products; athe product ratio determined by RP-HPLC analysis; bthe product ratio determined by 1H NMR analysis. | ||
Next, we tested also several nitrile derivatives 2b, 2ba, 2c, and 2d, which gave the results comparable with the amide partners regarding both the yield and the selectivity. The remaining amides 1e–h and also their nitrile counterparts 2e–h together with 1a–d and 2a–d are presented in Scheme 3.
 | ||
| Scheme 3 The substrate scope of the SMEAH-mediated process. Notes: the reductions were performed at the 1.3 mmol scale; the yields refer to the isolated products; athe product ratio determined by RP-HPLC analysis; bthe product ratio determined by 1H NMR analysis; cthe product yield isolated from the reaction involving substrate 1; dthe product yield and ratio obtained from the reaction with added NaCN (10 equiv.). | ||
In all the described experiments, the corresponding 1,2-amino alcohols were isolated by simple filtration of their oxalic acid salts. After isolation of the respective ammonium salts, the filtrate afforded the one-carbon-shorter alcohol as a predominant by-product in all the displayed entries. However, the corresponding dehomologated alcohols were isolated and purified only in the instance of the α-substituted derivatives (entries 1 and 3–6), where this method, in contrast to the α-unsubstituted 1b and 1g–h, can constitute at least an interesting route for the synthesis thereof.
As depicted in Schemes 1–3, the reaction tolerated well hydrogen, alkyl or aryl substituents at the benzylic position. Aromatic substitution may comprise alkyl and alkoxy groups. Aryl halides such as fluorine also remained practically untouched, however, in the case of aryl chloride, we have encountered dehalogenation on a significant level,16 which was also documented in the literature.17 In summary, it should be emphasized that although the reduction power of SMEAH is attenuated in comparison with the parent LAH due to the presence of two bulky and electron-withdrawing alkoxy groups, it is still a very powerful reducing agent and, as such, it is incompatible with almost any reducible functional group in a molecule. Thus, these properties considerably limit the chemoselectivity of the present process.13b,13c
To establish a plausible mechanistic proposal, a series of experiments with 1d, 2d, and other diphenyl substituted derivatives were performed. Initially, the respective products 3d and 4d were subjected to the standard reaction conditions (entries 1–2, Scheme 4). The full recovery of the unchanged material in both the cases supported that, according to our expectations, there is no detectable interconversion between the products and that the oxidation of the α-carbon precedes the functional group reduction step.
 | ||
| Scheme 4 Control experiments with 3d, 4d, 1d, 1da, 2d, and 2da. Notes: the yields were determined by RP-HPLC analysis using the external standard calibration. | ||
As mentioned above, the SMEAH-mediated reduction of 1d provided the oxalic acid salt of 3d as a sole product in the precipitate and 5c as a predominant side product in the filtrate.18
The subsequent analysis of the filtrate and particularly the fore fractions obtained during the purification of 5c by column chromatography (SiO2) n-heptane–EtOAc (9:1), revealed the minor and trace components 1da, 2d, 6, and 7, which were further confirmed by comparison with the authentic samples. The presence of 2d and 7 in the reaction mixture of 1d led us to assume that reduction of the primary amide likely involved intermediacy of the nitrile (Scheme 5).4 Moreover, if we consider that the respective nitrile 2d gave a similar product profile with the comparable selectivity in even better yield and shorter reaction time than the corresponding amide 1d (Scheme 4), it seems plausible to suppose that both 1d and 2d could share a common reduction pathway. The above hypothesis is also in connection with the aforementioned incapability of the secondary amides to undergo the present process.
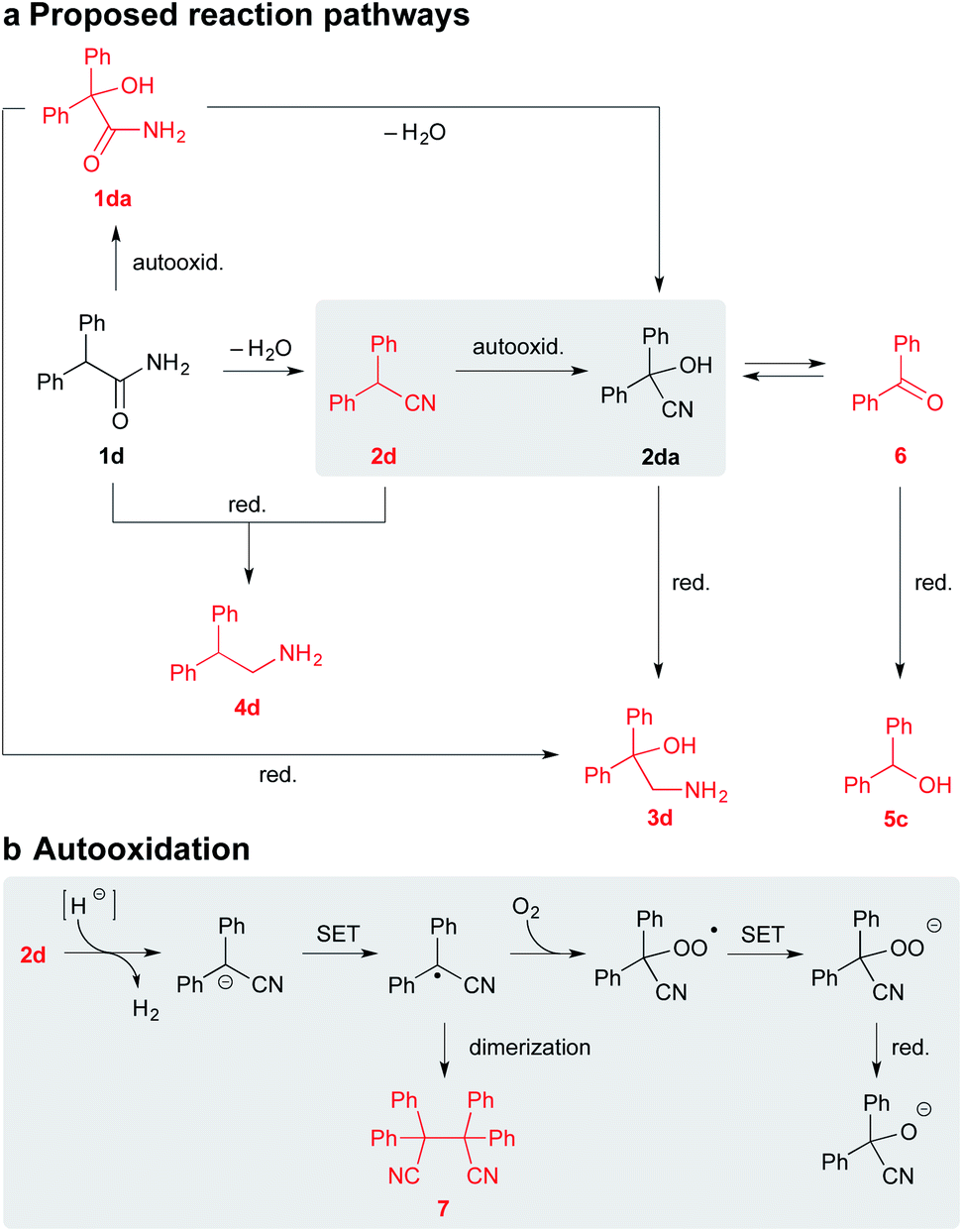 | ||
| Scheme 5 The working hypothesis of the possible reduction and oxidation pathways (a) and the plausible autooxidation process under basic conditions (b). Notes: the intermediates shown in red were successfully isolated from the reaction mixture of 1d; metal counterions were omitted for clarity. | ||
It has been reported earlier that a striking difference between the reduction properties of SMEAH and LAH towards nitriles was found with reductions of compounds carrying hydrogen atom at α-carbon, which proceeded unsatisfactorily with SMEAH. Hence the majority of the starting nitrile was always recovered on workup.19 These findings were also validated by us when the above reaction was conducted under the strictly oxygen-free conditions.
Therefore we have hypothesized that the nitrile intermediate 2d, which can be only α-deprotonated but not efficiently further reduced by SMEAH, is present in the reaction mixture as a colored resonance-stabilized nitrile anion. The small portion of the reduced substrate 2d is likely responsible for a formation of the minor by-product 4d, which can become more significant under elevated temperature.20 Under basic conditions, the electron-rich nitrile anions are susceptible to autooxidation via single-electron transfer (SET).21 Hence, the anion of 2d is probably trapped by O2 to the corresponding α-hydroperoxy nitrile, which is then rapidly reduced to the cyanohydrin 2da and the successive species (Scheme 5). This presumption was supported by the identification of the corresponding radical homocoupling product 7 from the above reaction mixture.
Since cyanohydrins are formed reversibly, they are considered to be unstable in basic media (entry 6, Scheme 4). Especially for cyanohydrins that are sterically hindered, the position of the equilibrium is unsatisfactory for the effective synthesis thereof. Therefore, cyanohydrins encumbered with bulky substituents are rather prone to the elimination of cyanide ion and the formation of the corresponding dehomologated carbonyl compounds.22
Our findings indicate that the moderate yields of β-amino alcohols in the present transformation are probably caused by the previously mentioned retro-cyanohydrination (Scheme 5). These speculations were also supported by isolation of traces of the ketone 6 from the SMEAH-driven reductions of 1d, 1da, 2d, and 2da.23
Under the above conditions, the presumptive α-hydroxy nitrile intermediate 2da can undergo either reduction to the desired β-amino alcohol 3d or expulsion of cyanide anion to 6. The resulting degraded carbonyl compound 6 is then readily reduced to the alcohol 5c by the hydride. To further prove this hypothesis, the separately prepared cyanohydrin 2da was subjected to identical reaction conditions. Accordingly, 4d and 5c were isolated as the major products. However, in comparison with the reaction starting from 1d or 2d, the desired β-amino alcohol 4d was obtained in a somewhat decreased yield (21%). Expectably, the yield of the corresponding alcohol 5c was slightly increased in this case (63%). Thus we suppose that the continuous reduction of cyanohydrin generated in situ during the present process is likely beneficial to the batch reduction of 2da, as it eliminates the excessive decomposition of this relatively labile compound by retro-cyanohydrination. Besides that, we have found that the formation of 5c was favored over 3d mainly under a diminished reaction temperature (<0 °C). This likely corresponds to the instability of 2da in a basic environment for the extended period necessary for the slower reduction thereof under a decreased temperature.
We further speculated whether the hypothetically employed decyanation of 2da might be suppressed by raising the concentration of free cyanide in the reaction medium according to Le Chatelier's principle. Indeed, the addition of powdered NaCN (10 equiv.) to the reaction mixture of 2d furnished a slightly increased yield of 3d (50% → 61%) after the prolonged reaction time (3 h) with the improved selectivity expressed by 3d/4d (92:8 → 99:1, Fig. S35–37†).
Although the residues of 1da were also detected in the SMEAH-mediated reduction involving 1d, the autooxidation of 1d to 1da presumably constitutes only a less important path in the overall process. Based on the control experiment with sodium hydride, the reaction time longer than 12 h was required for completion in this case, which is in contrast to the rapid autooxidation of 2d under the same conditions (entries 3 and 5, Scheme 4). Moreover, the analogous reaction of 1a with NaH under O2 atmosphere did not provide any product even after 24 h, which suggests that α-oxidation of nitriles is considerably faster in comparison with the parent primary amides. The subjection of α-hydroxy amide 1da to the control experiment with NaH resulted in the complete recovery of starting material (entry 4, Scheme 4). On the other hand, the SMEAH-driven reaction thereof afforded both 3d and 5c as the major products in accordance with 1d, 2d, and 2da. Hence we suppose that the possible reaction pathways of all the aforementioned derivatives with SMEAH can plausibly proceed as depicted in Scheme 5.
The analysis of the stereochemical course of the reaction involving (S)-naproxamide (S)-1a gave us evidence for complete racemization of the optically pure substrates lacking the α-hydroxy group during the reaction. It is in agreement with our assumption that the initial α-proton abstraction and mainly the subsequent radical isomerization led to the loss of the stereochemical integrity (Scheme 6).24 The respective alcohol 5a was isolated from the reaction mixture of (S)-1a as a slightly enriched (R)-isomer (6% ee).25 This phenomenon is not yet clearly understood by us but might be attributed to the asymmetric reduction of the corresponding ketone 6 by a non-racemic complex of the hydride with the substrate or some chiral intermediate. To the contrary, (S)-mandelamide (S)-1ba subjected to identical reaction conditions furnished (S)-3b with retention of its configuration, which suggests that the α-hydroxy-substituted stereocenter remained unaffected.
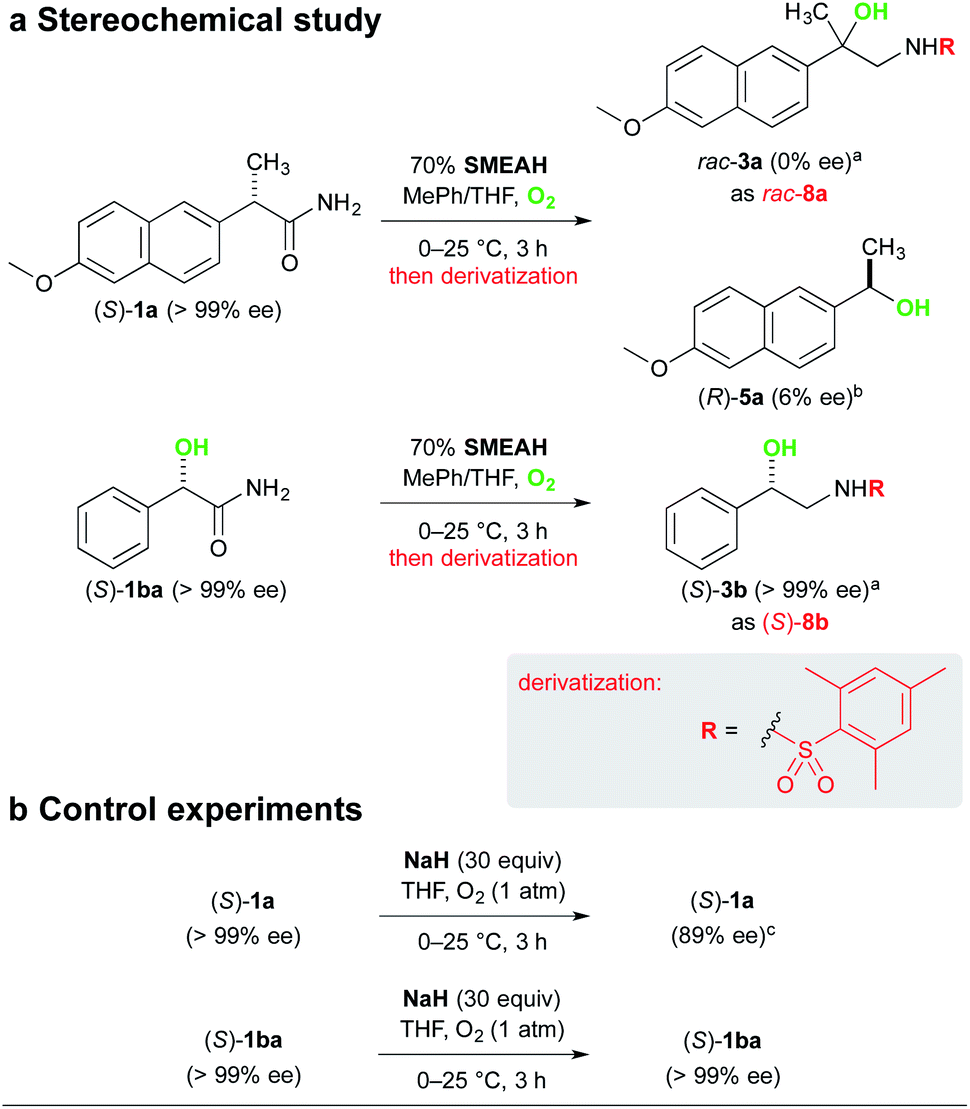 | ||
| Scheme 6 Analysis of the stereochemical outcome of the SMEAH-mediated process. Notes: adetermined by the CSP-HPLC of the 2-mesitylenesulfonyl derivative 8 (Chiralpak IA); bdetermined by the CSP-HPLC (Chiralpak IB); cspecific optical rotation decreased from +18 to +16 (c 0.5, MeOH). | ||
To showcase the synthetic utility of the developed process, we have prepared the racemic fluoro-analog of the biarylpropylsulfonamide potentiator of AMPA receptors, LY-503430, by our reaction (Scheme 7).26 Accordingly, the respective β-amino alcohol product 3e was subjected to the fluorodehydroxylation reaction with the Olah's reagent to afford the respective 1,2-fluoroamine 9 in 69% yield. This intermediate was further converted to the sulfonamide 10 using isopropylsulfonyl chloride (y. 37%).
 | ||
| Scheme 7 Synthesis of the LY-503430 analog. Conditions: (a) 65% HF-pyridine, CH2Cl2, rt, 24 h (y. 69%); (b) i-PrSO2Cl, Et3N, DMAP, CH2Cl2, 0 °C, 3 h (y. 37%). | ||
Conclusions
In summary, we have demonstrated that amides and nitriles bearing hydrogen atoms at benzylic α-carbon exhibit a unique reactivity towards SMEAH under O2 atmosphere. Performing the reaction under dry oxygen allows radical α-hydroxylation to precede amide or nitrile functional group reduction and thus both these steps can take place in a single cascade process. Although the respective products were isolated in the modest yields only with somewhat limited chemoselectivity, the above one-pot protocol could be used as a powerful entry to the 1,2-amino alcohols, especially the sterically encumbered ones, that are often difficult to prepare by other routes. The present method may also open up attractive prospective routes for future developments in 18O-labeling strategies or stereoselective synthesis thereof. The plausible mechanistic proposal for the observed results was given based on the isolation and identification of the stable intermediates and stereochemical evidence. Finally, the process was applied to the synthesis of a potentially bioactive target.Experimental
Materials and methods
If not stated otherwise, all experiments were performed standardly under open-vessel conditions. Moisture and air-sensitive reactions were done in oven-dried glassware (140 °C) under Ar atmosphere in anhydrous solvents. Solvents and reagents were purchased from commercial suppliers and used as received, if not stated otherwise. Flurbiprofen and naproxen were extracted from the commercially available formulations. Naproxamide (1a) and lithium dimethoxyaluminum hydride (LMAH) were prepared according to the literature.11c,27 Anhydrous solvents and reagents were absolutized as usual and distilled prior to use. The specific rotation was determined by an automatic polarimeter AA-10 (Optical Activity). Melting points were measured by a Böetius apparatus (Franz Küstner Nachf.) and are uncorrected. HPLC data were recorded on a Dionex UltiMate 3000 LC System and a Spectra-System (ThermoFisher Scientific). IR spectra were collected on a SmartMIRacle ATR (diamond) for a Nicolet Impact 410 FT-IR (ThermoFisher Scientific). NMR spectra were obtained from a JEOL ECZR-400 MHz (Jeol). NMR experiments were standardly performed at 25 °C (CDCl3) or 35 °C (DMSO-d6), chemical shifts are reported in δ parts per million (ppm) and J values in Hz, the signal of TMS or the residual solvent signals of CDCl3 or DMSO-d6 were used as a reference. GC/MS data were collected on a system consisting of a gas chromatograph Agilent 7890A and a mass spectrometer Agilent 5975C inert XL with TAD (Agilent Technologies). HRMS measurements were performed using a LTQ Orbitrap XL high-resolution mass spectrometer (ThermoFisher Scientific) and a Bruker Impact II Q-TOF high-resolution mass spectrometer (Bruker Daltonics).Synthetic procedures
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1: 3298m, 1645s, 1606m, 1552m, 1504m, 1390m, 1263m, 1210s, 1162m, 1030s, 924m, 892m, 855s, 812s, 690m; HRMS (ESI-Q-TOF) m/z: calcd for C15H18O2N [M + H]+ 244.1332, found 244.1339./cm−1: 3277m, 2958w, 1633s, 1606m, 1527w, 1480w, 1392m, 1263m, 1213s, 1162w, 1036m, 985w, 882m, 845s, 815m, 693s; HRMS (ESI-Orbitrap) m/z: calcd for C14H17N2O2 [M + H]+ 245.1285, found 245.1285./cm−1: 2900br, 1605s, 1503w, 1484m, 1392m, 1261m, 1213s, 1163m, 1068w, 1027s, 940w, 890w, 858s, 809s; HRMS (ESI-Orbitrap) m/z: calcd for C14H16NO3 [M + H]+ 246.1125, found 246.1131./cm−1: 3404w, 3140w, 1621s, 1600m, 1480m, 1425s, 1394m, 1264m, 1232m, 1207m, 1160m, 1025s, 962w, 852s, 817s, 763m, 696w; HRMS (ESI-Orbitrap) m/z: calcd for C14H14ONS [M − H]− 244.0802, found 244.0804./cm−1: 3348m, 3164m, 1633s, 1497w, 1413s, 1284w, 1184w, 1073w, 747m, 697s; HRMS (ESI-Orbitrap) m/z: calcd for C8H10NO [M + H]+ 136.0757, found 136.0756./cm−1: 3367m, 3170m, 1633s, 1496w, 1454w, 1411m, 1285w, 1182w, 1058w, 922w, 748m, 694s; HRMS (ESI-Orbitrap) m/z: calcd for C8H8NO2 [M − H]− 150.0561, found 150.0566./cm−1: 3461m, 3176w, 1695m, 1652s, 1466w, 1411m, 1327w, 1197m, 1086s, 983m, 950w, 813m, 769m, 704s; HRMS (ESI-Orbitrap) m/z: calcd for C9H12NO2 [M + H]+ 166.086, found 166.0865./cm−1: 3362m, 3174w, 1657s, 1481w, 1454w, 1439w, 1387m, 1237w, 1089w, 805w, 745m 733m; HRMS (ESI-Orbitrap) m/z: calcd for C14H12ONS [M − H]− 242.0645, found 242.0646./cm−1: 3396m, 3191m, 1650s, 1607m, 1497w, 1453w, 1405m, 1215m, 1178w, 1076w, 851m, 781w, 691m; HRMS (ESI-Orbitrap) m/z: calcd for C8H9NOCl [M + H]+ 170.0367, found 170.0374./cm−1: 3349m, 3176m, 1629s, 1496w, 1452w, 1403m, 1287w, 1030w, 850w, 694s; HRMS (ESI-Orbitrap) m/z: calcd for C9H12NO [M + H]+ 150.0913, found 150.0914./cm−1: 3448m, 3322w, 3193w, 1674s, 1616m, 1461w, 1403w, 1357m, 1325w, 1259s, 1149s, 1116s, 1097s, 1035w, 957w, 901m, 851m; HRMS (ESI-Orbitrap) m/z: calcd for C9H7NOF3 [M − H]− 202.048, found 202.0485./cm−1: 3503m, 3386w, 1697s, 1567m, 1258m, 1194m, 1167s, 1125s, 1080w, 983m, 943m, 769m, 745m; HRMS (ESI-Orbitrap) m/z: calcd for C9H7NO2F3 [M − H]− 218.0434, found 218.0436./cm−1: 3381m, 3174w, 2933m, 1651s, 1455w, 1413m, 1301w, 1126w, 816w, 755w, 719m, 697s; HRMS (ESI-Orbitrap) m/z: calcd for C12H17NO [M + H]+ 192.1383, found 192.1386./cm−1: 3386w, 3172w, 1651s, 1496m, 1405m, 1260m, 1103w, 1034w, 845w, 738m, 722m, 697s; HRMS (ESI-Orbitrap) m/z: calcd for C14H14NO [M + H]+ 212.1070, found 212.1075./cm−1: 3384w, 1682s, 1494w, 1445m, 1155w, 1049m, 903w, 755m, 696s; HRMS (ESI-Orbitrap) calcd for C14H12O2N [M − H]− 226.0874 m/z, found 226.0873 m/z./cm−1: 3360m, 3181m, 1656s, 1482m, 1422m, 1397s, 1282m, 1130w, 1090w, 1011w, 932m, 869w, 830w, 764m, 694s; HRMS (ESI-Orbitrap) m/z: calcd for C15H15NOF [M + H]+ 244.1132, found 244.1138./cm−1: 3342m, 3164m, 2958m, 1633s, 1515w, 1460m, 1406m, 1367m, 1265w, 1115w, 1070w, 999w, 841w; HRMS (ESI-Orbitrap) m/z: calcd for C13H20NO [M + H]+ 206.1539, found 206.1545./cm−1: 3361w, 3178w, 1659m, 1620m, 1512w, 1397m, 1291w, 1261w, 775s; HRMS (ESI-Orbitrap) m/z: calcd for C12H12NO [M + H]+ 186.0913, found 186.0916./cm−1: 3347m, 3157m, 1633s, 1405s, 1287m, 1258m, 1128w, 1040w, 828w, 760w, 694s; HRMS (ESI-Orbitrap) m/z: calcd for C6H8NOS [M + H]+ 142.0321, found 142.0325.:1) revealed a complete consumption of starting material (ca. 4 h), the reaction was quenched with water. The layers were separated, the organic phase was washed with a saturated aqueous solution of NaHCO3 and brine, dried over anhydrous Na2SO4, filtered, and evaporated in vacuo. Yield: 0.34 g (80%). Physical state: white powder. Mp 90–91 °C; 1H NMR (400 MHz, CDCl3) δ/ppm: 7.87–7.84 (m, 3H), 7.48 (dd, J = 8.5, 1.9 Hz, 1H), 7.34 (d, J = 2.5 Hz, 1H), 7.20 (dd, J = 9.0, 2.5 Hz, 1H), 4.40 (q, J = 7.2 Hz, 1H), 3.88 (s, 3H), 1.62 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ/ppm: 157.5, 133.6, 132.5, 129.2, 128.2, 127.6, 125.2, 125.1, 122.3, 119.1, 105.8, 55.2, 29.8, 20.5; IR (neat) /cm−1: 2962w, 1727w, 1602m, 1505w, 1482w, 1448w, 1393w, 1258m, 1212m, 1085w, 1022m, 927w, 890m, 856s, 816m; HRMS (ESI-Orbitrap) m/z: calcd for C14H12NO [M − H]− 210.0924, found 210.0926./cm−1: 3413br, 1494w, 1455m, 1261w, 1192w, 1025s, 932w, 819w, 762m, 698s; HRMS (ESI-Orbitrap) m/z: calcd for C8H8ON [M + H]+ 134.0600, found 134.0598./cm−1: 2241w, 1484w, 1452m, 1030w, 756m, 697s; HRMS (ESI-Orbitrap) m/z: calcd for C9H10N [M + H]+ 132.0808, found 132.0805./cm−1: 3364m, 2249w, 1492w, 1450m, 1410w, 1198w, 1177w, 1052m, 1032m, 769s, 746s; HRMS (ESI-Orbitrap) m/z: calcd for C14H10NO [M − H]− 208.0768, found 208.0774./cm−1: 2239w, 1563w, 1484m, 1450w, 1414w, 1375w, 1270w, 1215w, 1127w, 1085w, 1011w, 928w, 869w, 838m, 765m, 697s; HRMS (ESI-Orbitrap) m/z: calcd for C15H13NF [M + H]+ 226.1027, found 226.1026./cm−1: 2955s, 2868m, 2238w, 1513m, 1455m, 1240w, 1383m, 1168w, 1085m, 1022w, 846s, 797s; HRMS (ESI-Orbitrap) m/z: calcd for C13H18N [M + H]+ 188.1434, found 188.1431./cm−1: 2251w, 1598m, 1512m, 1396m, 1262w, 1018w, 792s, 776s; HRMS (ESI-Orbitrap) m/z: calcd for C12H10N [M + H]+ 168.0808, found 168.0806./cm−1: 2253w, 1412w, 1257w, 1041w, 851m, 831w, 703s; HRMS (ESI-Orbitrap) m/z: calcd for C6H6NS [M + H]+ 124.0215, found 124.0211.:1) and a solution of anhydrous oxalic acid (0.13 g, 1.1 equiv.) in 5 mL of Et2O–MeOH (5:1) was added dropwise. The resulting white precipitate was filtered off, washed several times with fresh Et2O–MeOH (5:1), then Et2O, and dried in vacuo. The above filtrate was neutralized by a saturated aqueous solution of NaHCO3 and repetitively extracted with Et2O. The combined organic extract was dried over anhydrous Na2SO4, filtered, and evaporated in vacuo to provide alcohol 5.48
/cm−1: 3298m, 1645s, 1606m, 1552m, 1504m, 1390m, 1263m, 1210s, 1162m, 1030s, 924m, 892m, 855s, 812s, 690m; HRMS (ESI-Q-TOF) m/z: calcd for C15H18O2N [M + H]+ 244.1332, found 244.1339./cm−1: 3277m, 2958w, 1633s, 1606m, 1527w, 1480w, 1392m, 1263m, 1213s, 1162w, 1036m, 985w, 882m, 845s, 815m, 693s; HRMS (ESI-Orbitrap) m/z: calcd for C14H17N2O2 [M + H]+ 245.1285, found 245.1285./cm−1: 2900br, 1605s, 1503w, 1484m, 1392m, 1261m, 1213s, 1163m, 1068w, 1027s, 940w, 890w, 858s, 809s; HRMS (ESI-Orbitrap) m/z: calcd for C14H16NO3 [M + H]+ 246.1125, found 246.1131./cm−1: 3404w, 3140w, 1621s, 1600m, 1480m, 1425s, 1394m, 1264m, 1232m, 1207m, 1160m, 1025s, 962w, 852s, 817s, 763m, 696w; HRMS (ESI-Orbitrap) m/z: calcd for C14H14ONS [M − H]− 244.0802, found 244.0804./cm−1: 3348m, 3164m, 1633s, 1497w, 1413s, 1284w, 1184w, 1073w, 747m, 697s; HRMS (ESI-Orbitrap) m/z: calcd for C8H10NO [M + H]+ 136.0757, found 136.0756./cm−1: 3367m, 3170m, 1633s, 1496w, 1454w, 1411m, 1285w, 1182w, 1058w, 922w, 748m, 694s; HRMS (ESI-Orbitrap) m/z: calcd for C8H8NO2 [M − H]− 150.0561, found 150.0566./cm−1: 3461m, 3176w, 1695m, 1652s, 1466w, 1411m, 1327w, 1197m, 1086s, 983m, 950w, 813m, 769m, 704s; HRMS (ESI-Orbitrap) m/z: calcd for C9H12NO2 [M + H]+ 166.086, found 166.0865./cm−1: 3362m, 3174w, 1657s, 1481w, 1454w, 1439w, 1387m, 1237w, 1089w, 805w, 745m 733m; HRMS (ESI-Orbitrap) m/z: calcd for C14H12ONS [M − H]− 242.0645, found 242.0646./cm−1: 3396m, 3191m, 1650s, 1607m, 1497w, 1453w, 1405m, 1215m, 1178w, 1076w, 851m, 781w, 691m; HRMS (ESI-Orbitrap) m/z: calcd for C8H9NOCl [M + H]+ 170.0367, found 170.0374./cm−1: 3349m, 3176m, 1629s, 1496w, 1452w, 1403m, 1287w, 1030w, 850w, 694s; HRMS (ESI-Orbitrap) m/z: calcd for C9H12NO [M + H]+ 150.0913, found 150.0914./cm−1: 3448m, 3322w, 3193w, 1674s, 1616m, 1461w, 1403w, 1357m, 1325w, 1259s, 1149s, 1116s, 1097s, 1035w, 957w, 901m, 851m; HRMS (ESI-Orbitrap) m/z: calcd for C9H7NOF3 [M − H]− 202.048, found 202.0485./cm−1: 3503m, 3386w, 1697s, 1567m, 1258m, 1194m, 1167s, 1125s, 1080w, 983m, 943m, 769m, 745m; HRMS (ESI-Orbitrap) m/z: calcd for C9H7NO2F3 [M − H]− 218.0434, found 218.0436./cm−1: 3381m, 3174w, 2933m, 1651s, 1455w, 1413m, 1301w, 1126w, 816w, 755w, 719m, 697s; HRMS (ESI-Orbitrap) m/z: calcd for C12H17NO [M + H]+ 192.1383, found 192.1386./cm−1: 3386w, 3172w, 1651s, 1496m, 1405m, 1260m, 1103w, 1034w, 845w, 738m, 722m, 697s; HRMS (ESI-Orbitrap) m/z: calcd for C14H14NO [M + H]+ 212.1070, found 212.1075./cm−1: 3384w, 1682s, 1494w, 1445m, 1155w, 1049m, 903w, 755m, 696s; HRMS (ESI-Orbitrap) calcd for C14H12O2N [M − H]− 226.0874 m/z, found 226.0873 m/z./cm−1: 3360m, 3181m, 1656s, 1482m, 1422m, 1397s, 1282m, 1130w, 1090w, 1011w, 932m, 869w, 830w, 764m, 694s; HRMS (ESI-Orbitrap) m/z: calcd for C15H15NOF [M + H]+ 244.1132, found 244.1138./cm−1: 3342m, 3164m, 2958m, 1633s, 1515w, 1460m, 1406m, 1367m, 1265w, 1115w, 1070w, 999w, 841w; HRMS (ESI-Orbitrap) m/z: calcd for C13H20NO [M + H]+ 206.1539, found 206.1545./cm−1: 3361w, 3178w, 1659m, 1620m, 1512w, 1397m, 1291w, 1261w, 775s; HRMS (ESI-Orbitrap) m/z: calcd for C12H12NO [M + H]+ 186.0913, found 186.0916./cm−1: 3347m, 3157m, 1633s, 1405s, 1287m, 1258m, 1128w, 1040w, 828w, 760w, 694s; HRMS (ESI-Orbitrap) m/z: calcd for C6H8NOS [M + H]+ 142.0321, found 142.0325.:1) revealed a complete consumption of starting material (ca. 4 h), the reaction was quenched with water. The layers were separated, the organic phase was washed with a saturated aqueous solution of NaHCO3 and brine, dried over anhydrous Na2SO4, filtered, and evaporated in vacuo. Yield: 0.34 g (80%). Physical state: white powder. Mp 90–91 °C; 1H NMR (400 MHz, CDCl3) δ/ppm: 7.87–7.84 (m, 3H), 7.48 (dd, J = 8.5, 1.9 Hz, 1H), 7.34 (d, J = 2.5 Hz, 1H), 7.20 (dd, J = 9.0, 2.5 Hz, 1H), 4.40 (q, J = 7.2 Hz, 1H), 3.88 (s, 3H), 1.62 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ/ppm: 157.5, 133.6, 132.5, 129.2, 128.2, 127.6, 125.2, 125.1, 122.3, 119.1, 105.8, 55.2, 29.8, 20.5; IR (neat) /cm−1: 2962w, 1727w, 1602m, 1505w, 1482w, 1448w, 1393w, 1258m, 1212m, 1085w, 1022m, 927w, 890m, 856s, 816m; HRMS (ESI-Orbitrap) m/z: calcd for C14H12NO [M − H]− 210.0924, found 210.0926./cm−1: 3413br, 1494w, 1455m, 1261w, 1192w, 1025s, 932w, 819w, 762m, 698s; HRMS (ESI-Orbitrap) m/z: calcd for C8H8ON [M + H]+ 134.0600, found 134.0598./cm−1: 2241w, 1484w, 1452m, 1030w, 756m, 697s; HRMS (ESI-Orbitrap) m/z: calcd for C9H10N [M + H]+ 132.0808, found 132.0805./cm−1: 3364m, 2249w, 1492w, 1450m, 1410w, 1198w, 1177w, 1052m, 1032m, 769s, 746s; HRMS (ESI-Orbitrap) m/z: calcd for C14H10NO [M − H]− 208.0768, found 208.0774./cm−1: 2239w, 1563w, 1484m, 1450w, 1414w, 1375w, 1270w, 1215w, 1127w, 1085w, 1011w, 928w, 869w, 838m, 765m, 697s; HRMS (ESI-Orbitrap) m/z: calcd for C15H13NF [M + H]+ 226.1027, found 226.1026./cm−1: 2955s, 2868m, 2238w, 1513m, 1455m, 1240w, 1383m, 1168w, 1085m, 1022w, 846s, 797s; HRMS (ESI-Orbitrap) m/z: calcd for C13H18N [M + H]+ 188.1434, found 188.1431./cm−1: 2251w, 1598m, 1512m, 1396m, 1262w, 1018w, 792s, 776s; HRMS (ESI-Orbitrap) m/z: calcd for C12H10N [M + H]+ 168.0808, found 168.0806./cm−1: 2253w, 1412w, 1257w, 1041w, 851m, 831w, 703s; HRMS (ESI-Orbitrap) m/z: calcd for C6H6NS [M + H]+ 124.0215, found 124.0211.:1) and a solution of anhydrous oxalic acid (0.13 g, 1.1 equiv.) in 5 mL of Et2O–MeOH (5:1) was added dropwise. The resulting white precipitate was filtered off, washed several times with fresh Et2O–MeOH (5:1), then Et2O, and dried in vacuo. The above filtrate was neutralized by a saturated aqueous solution of NaHCO3 and repetitively extracted with Et2O. The combined organic extract was dried over anhydrous Na2SO4, filtered, and evaporated in vacuo to provide alcohol 5.48
The reduction of 1a. The reduction of 1a (300 mg, 1.3 mmol) was performed according to GP. The precipitated product was obtained as an inseparable mixture of 3a·H2C2O4/4a·H2C2O4 in a 94
:6 ratio based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 40:60, 0.2 mL min−1, 40 °C, 230 nm, t1 = 3.43 min (3a), t2 = 5.67 min (4a). Yield: 180 mg (43%). Physical state: white hygroscopic powder. The crude alcohol 5a was isolated from the filtrate according to GP. Yield: 111 mg (41%). Physical state: beige solid.Analytical data for 3a·H2C2O4: Mp 226–228 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.94 (s, 1H), 7.82 (dd, J = 8.7, 2.8 Hz, 2H), 7.59 (d, J = 8.6 Hz, 1H), 7.31 (d, J = 1.9 Hz, 1H), 7.17 (dd, J = 8.9, 2.3 Hz, 1H), 3.87 (s, 3H), 3.13, 3.10 (q, AB, JAB = 13.0 Hz, 2H), 1.57 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.2, 157.3, 140.3, 133.3, 129.5, 128.0, 126.7, 124.0, 123.5, 118.6, 105.6, 71.0, 55.1, 49.1, 27.4; IR (neat) /cm−1: 3459w, 2961w, 1606m, 1515m, 1386w, 1265m, 1179s, 1025m, 898m, 854m, 812m, 702s; HRMS (ESI-Q-TOF) m/z: calcd for C14H18NO2 [M + H]+ 232.1332, found 232.1328.
Analytical data for 5a:49 Mp 93–95 °C; 1H NMR (400 MHz, CDCl3) δ/ppm: 7.75–7.71 (m, 3H), 7.48 (dd, J = 8.6, 1.6 Hz, 1H), 7.18–7.13 (m, 2H), 5.04 (q, J = 6.5 Hz, 1H), 3.93 (s, 3H), 2.26 (br s, 1H), 1.58 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ/ppm: 157.6, 140.9, 140.0, 129.4, 128.7, 127.1, 124.4, 123.7, 118.9, 105.6, 70.4, 55.3, 25.0; IR (neat) /cm−1: 3326br, 2961w, 1633w, 1605s, 1485m, 1462m, 1391w, 1260m, 1216m, 1162s, 1072m, 1028s, 961w, 930w, 890m, 853s, 815s, 750w, 673w; HRMS (ESI-Orbitrap) m/z: calcd for C13H15O2 [M + H]+ 203.1067, found 203.10664.
The reduction of (S)-1a. The reduction of (S)-1a (300 mg, 1.3 mmol) was performed according to GP and afforded precipitate of 3a·H2C2O4 and 4a·H2C2O4 in a 94
:6 ratio and the filtrate containing crude (R)-5a. The free bases of 3a and 4a were liberated from the corresponding oxalic acid salts by aqueous NaOH (2.5 M) and immediately back-extracted to toluene. The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated in vacuo. The obtained crude residue (67 mg) was dissolved in anhydrous CH2Cl2 (2 mL). DMAP (39 mg, 1 equiv.) and Et3N (88 μL, 2 equiv.) were added and the resulting solution was treated portionwise with 2-mesitylenesulfonyl chloride (67 mg, 1 equiv.) at 0 °C. The reaction mass was stirred under Ar at the same temperature for 3 h. Then the reaction mixture was diluted with water and repetitively extracted with CH2Cl2. The organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to provide the crude 8a, which was further purified by column chromatography (SiO2) n-hexane–EtOAc (4:1).Analytical data for rac-8a (0% ee): Yield: 100 mg (45%). Physical state: white powder. Mp 193–194 °C; [α]25D 0 (c 1.0, CHCl3). HPLC conditions: a Hypersil silica column (3 μm, 100 × 4.6 mm) used as a precolumn, which was connected via the standard blue PEEK capillary tubing (L 300 mm, ID 0.01′′, OD 1/16′′) to a Daicel Chiralpak IA column (5 μm, 250 × 4.6 mm), i-PrOH–n-heptane, 10:90, 0.5 mL min−1, 25 °C (230 nm), t1 = 36.36 min, t2 = 49.38 min. 1H NMR (400 MHz, CDCl3) δ/ppm: 7.71–7.69 (m, 2H), 7.60 (d, J = 8.6 Hz, 1H), 7.37 (dd, J = 8.6, 1.7 Hz, 1H), 7.22 (d, J = 2.4 Hz, 1H), 7.12 (dd, J = 9.0, 2.4 Hz, 1H), 6.97 (t, J = 6.2 Hz, 1H), 6.81 (s, 2H), 5.24 (s, 1H), 3.86 (s, 3H), 3.05 (dd, J = 6.2, 1.3 Hz, 2H), 2.40 (s, 6H), 2.18 (s, 3H), 1.44 (s, 3H); 13C NMR (100 MHz, CDCl3) δ/ppm: 157.0, 141.5, 141.1, 138.0, 134.2, 133.1, 131.4, 129.5, 127.9, 126.1, 124.2, 123.4, 118.3, 105.5, 72.7, 55.1, 53.5, 27.3, 22.5, 20.4; IR (neat) /cm−1: 3501w, 3296w, 2930w, 1605m, 1455w, 1378w, 1314s, 1261m, 1199m, 1171m, 1142s, 1084m, 1034m, 960w, 854s, 819s, 739w; HRMS (ESI-Orbitrap) m/z: calcd for C23H26NO4S [M − H]− 412.1588, found 412.1590.
Analytical data for (R)-5a were in accordance with a compound 5a reported above. [α]25D +5 (c 0.4, CH2Cl2), 6% ee; lit.49 [α]25D +26.7 (c 0.4, CH2Cl2), 70% ee. HPLC conditions: a Hypersil silica column (3 μm, 100 × 4.6 mm) used as a precolumn, which was connected via the standard blue PEEK capillary tubing (L 300 mm, ID 0.01′′, OD 1/16′′) to a Daicel Chiralpak IB column (5 μm, 250 × 4.6 mm), i-PrOH–n-heptane, 5:95, 0.5 mL min−1, 25 °C (230 nm), t1 = 28.50 min (S-isomer), t2 = 34.07 min (R-isomer).
The reduction of 1aa. The reduction of 1aa (316 mg, 1.3 mmol) was performed according to GP. 4aa·H2C2O4 was obtained in a pure form according to 1H NMR. Yield: 100 mg (24%). Physical state: white hygroscopic powder.
Analytical data for 4aa·H2C2O4: Mp 209–211 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.82–7.77 (m, 2H), 7.72 (s, 1H), 7.42 (d, J = 9.0 Hz, 1H), 7.30 (d, J = 2.3 Hz, 1H), 7.16 (dd, J = 8.9, 2.5 Hz, 1H), 3.87 (s, 3H), 3.29–3.17 (m, 3H), 2.54 (s, 3H), 1.34 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.3, 157.1, 137.6, 133.4, 129.0, 128.5, 127.2, 125.9, 125.4, 118.6, 105.8, 55.1, 54.4, 36.3, 33.1, 19.5; IR (neat) /cm−1: 2970br, 1715m, 1608s, 1485m, 1385w, 1263s, 1218s, 1162m, 1028s, 950w, 849s, 809m; HRMS (ESI-Q-TOF) m/z: calcd for C15H20NO [M + H]+ 230.1539, found 230.1545.
The reduction of 1ad. The reduction of 1ad (319 mg, 1.3 mmol) was performed according to GP with a reaction time of 30 min. The precipitated product was obtained as an inseparable mixture of 3a·H2C2O4/4a·H2C2O4 in a 62
:38 ratio based on 1H NMR and 63:37 according to RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 40:60, 0.2 mL min−1, 40 °C, 230 nm, t1 = 3.47 min (3a), t2 = 5.69 min (4a). Yield: 119 mg (29%). Physical state: white hygroscopic powder. The analytical data for 3a·H2C2O4 were in accordance with the product obtained by the reduction of 1a.Analytical data for 4a·H2C2O4 (the authentic sample was prepared by a DIBAH-mediated reduction of 1a): Mp 225–226 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.81–7.78 (m, 2H), 7.70 (s, 1H), 7.42 (dd, J = 8.5, 1.7 Hz, 1H), 7.30 (d, J = 2.5 Hz, 1H), 7.16 (dd, J = 8.9, 2.5 Hz, 1H), 3.87 (s, 3H), 3.19–3.03 (m, 3H), 1.33 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.2, 157.1, 137.7, 133.4, 129.0, 128.5, 127.1, 125.9, 125.4, 118.5, 105.8, 55.1, 44.9, 37.4, 19.2; IR (neat) /cm−1: 2959br, 1717w, 1606s, 1503m, 1458w, 1392w, 1263m, 1218s, 1197m, 1162m, 1030m, 928w, 849s, 815m, 705s; HRMS (ESI-Q-TOF) m/z: calcd for C14H18NO [M + H]+ 216.1383, found 216.1379.
The reduction of 1b. The reduction of 1b (176 mg, 1.3 mmol) was performed according to GP at 0 °C. The precipitated product was obtained as an inseparable mixture of 3b·H2C2O4/4b·H2C2O4 in a 95
:5 ratio based on RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 20:80, 0.2 mL min−1, 40 °C, 210 nm, t1 = 3.09 min (3b), t2 = 5.02 min (4b). Yield: 83 mg (28%). Physical state: white hygroscopic powder.Analytical data for 3b·H2C2O4 (in accordance with the authentic sample):50 Mp 232–235 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.41–7.35 (m, 4H), 7.33–7.28 (m, 1H), 4.81 (dd, J = 9.9, 2.8 Hz, 1H), 3.04 (dd, J = 12.6, 2.8 Hz, 1H), 2.84 (dd, J = 12.6, 9.9 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 163.7, 141.9, 128.3, 127.6, 125.8, 69.1, 45.7; IR (neat) /cm−1: 2884br, 1582s, 1446w, 1296m, 1061m, 1020m, 780m, 700m; HRMS (ESI-Orbitrap) m/z: calcd for C8H12NO [M + H]+ 138.0913, found 138.0915.
The reduction of 1ba. The reduction of 1ba (197 mg, 1.3 mmol) was performed according to GP at 0 °C. The precipitated product 3b·H2C2O4 was obtained in a pure form according to RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 20
:80, 0.2 mL min−1, 40 °C, 210 nm, t = 3.09 min (3b). Yield: 107 mg (36%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1b.
The reduction of (S)-1ba. The reduction of (S)-1ba (197 mg, 1.3 mmol) was performed according to GP at 0 °C and furnished the precipitate of (S)-3b·H2C2O4. The free base of (S)-3b was liberated and treated analogously to the product of (S)-1a to provide (S)-8b.
Analytical data for (S)-8b (>99% ee):51 Yield: 55 mg (48%). Physical state: colorless oil. [α]25D +40 (c 1.0, CHCl3). HPLC conditions: a Hypersil silica column (3 μm, 100 × 4.6 mm) used as a precolumn, which was connected via the standard blue PEEK capillary tubing (L 300 mm, ID 0.01′′, OD 1/16′′) to a Daicel Chiralpak IA column (5 μm, 250 × 4.6 mm), i-PrOH–n-heptane, 10:90, 0.5 mL min−1, 25 °C (230 nm), t = 57.51 min (S-isomer). 1H NMR (400 MHz, CDCl3) δ/ppm: 8.08 (s, 1H), 7.35–7.26 (m, 5H), 6.95 (s, 2H), 4.94 (dd, 7.1, 4.2 Hz, 1H), 4.79–4.76 (m, 1H), 3.20 (ddd, J = 13.2, 8.5, 4.5 Hz, 1H), 3.00 (ddd, J = 13.2, 8.5, 4.5 Hz, 1H), 2.62 (s, 6H), 2.29 (s, 3H); 13C NMR (100 MHz, CDCl3) δ/ppm: 142.4, 140.9, 139.1, 133.5, 132.1, 128.8, 128.4, 125.9, 72.8, 49.8, 23.0, 21.0; IR (neat) /cm−1: 3313w, 1603w, 1453w, 1318m, 1188w, 1149s, 1055m, 917w, 852w, 756w, 700m; HRMS (ESI-Orbitrap) m/z: calcd for C17H20O3NS [M − H]− 318.1169, found 318.1172.
The reduction of 1bb. The reduction of 1bb (215 mg, 1.3 mmol) was performed according to GP. The precipitated product was obtained as an inseparable mixture of 3b·H2C2O4/4ba·H2C2O4 in a 30
:70 ratio based on 1H NMR and 29:71 according to RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 20:80, 0.2 mL min−1, 40 °C, 210 nm, t1 = 3.10 min (3b), t2 = 8.17 min (4ba). Yield: 97 mg (31%). Physical state: white hygroscopic powder. The analytical data for 3b were in accordance with the product obtained by the reduction of 1b.Analytical data for 4ba·H2C2O4: 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.44–7.33 (m, 5H), 4.46 (dd, J = 8.7, 3.4 Hz, 1H), 3.19 (s, 3H), 3.03–2.94 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.0, 133.8, 128.6, 128.4, 126.7, 56.2, 44.3; HRMS (ESI-Orbitrap) m/z: calcd for C9H14NO [M + H]+ 152.1070, found 152.1069.
The reduction of 1bc. The reduction of 1bc (316 mg, 1.3 mmol) was performed according to GP at 0 °C. The precipitated product 3b·H2C2O4 was obtained in a pure form according to RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 20
:80, 0.2 mL min−1, 40 °C, 210 nm, t = 3.09 min (3b). Yield: 71 mg (24%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1b.
The reduction of 1bd. The reduction of 1bd (220 mg, 1.3 mmol) was performed according to GP at 0 °C. The precipitated product 4bb·H2C2O4 was obtained in a pure form according to 1H NMR. Yield: 182 mg (57%). Physical state: white hygroscopic powder.
Analytical data for 4bb·H2C2O4: Mp 173–175 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.48–7.37 (m, 5H), 4.28–4.25 (m, 1H), 3.75–3.66 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.2, 135.7, 128.5, 128.4, 127.4, 63.0, 56.0; IR (neat) /cm−1: 2968w, 1593m, 1529m, 1202s, 1057s, 765w; HRMS (ESI-Orbitrap) m/z: calcd for C8H11NCl [M + H]+ 156.0575, found 156.0570.
The reduction of 1c (194 mg, 1.3 mmol) was performed according to GP. The precipitated product was obtained as an inseparable mixture of 3c·H2C2O4/4c·H2C2O4 in a 95:5 ratio based on 1H NMR and 94:6 according to RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 30:70, 0.2 mL min−1, 40 °C, 210 nm, t1 = 2.77 min (3c), t2 = 4.76 min (4c). Yield: 101 mg (32%). Physical state: white hygroscopic powder. The crude alcohol 5b was isolated from the filtrate according to GP. Yield: 75 mg (47%). Physical state: yellowish oil.
Analytical data for 3c·H2C2O4: Mp 126–128 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.51–7.49 (m, 2H), 7.38–7.35 (m, 2H), 7.29–7.26 (m, 1H), 3.09, 2.98 (q, AB, JAB = 12.9 Hz, 2H), 1.49 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.4, 145.6, 128.1, 126.9, 124.9, 70.9, 49.1, 27.2; IR (neat) /cm−1: 3438br, 1713w, 1620w, 1514m, 1447w, 1394w, 1211m, 1062m, 760s; HRMS (ESI-Q-TOF) m/z: calcd for C9H14NO [M + H]+ 152.1070, found 152.1066.
Analytical data for 5b:52 1H NMR (400 MHz, CDCl3) δ/ppm: 7.37–7.32 (m, 4H), 7.29–7.25 (m, 1H), 4.88 (q, J = 6.5 Hz, 1H), 2.02 (s, 1H), 1.48 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ/ppm: 145.8, 128.5, 127.4, 125.3, 70.4, 25.1; IR (neat) /cm−1: 3329br, 2970w, 1493w, 1452w, 1368w, 1077m, 899m, 761m, 700s; HRMS (ESI) not detected.
The reduction of 1ca. The reduction of 1ca (264 mg, 1.3 mmol) was performed according to GP. The precipitated product 3c·H2C2O4 was obtained in a pure form based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 30
:70, 0.2 mL min−1, 40 °C, 210 nm, t = 2.79 min (3c). Yield: 98 mg (31%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1c.
The reduction of 1cb. The reduction of 1cb (285 mg, 1.3 mmol) was performed according to GP at 0 °C. The precipitated product 3ca·H2C242 was obtained in a pure form based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 30
:70, 0.2 mL min−1, 40 °C, 210 nm, t = 8.55 min (3ca). Yield: 130 mg (34%). Physical state: white hygroscopic powder.Analytical data for 3ca·H2C2O4: Mp 235–237 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.61–7.60 (m, 2H), 7.46–7.39 (m, 3H), 3.39, 3.37 (q, AB, JAB = 14.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.4, 135.1, 128.8, 128.3, 126.8, 125.1 (q, 1JCF = 287.1 Hz), 74.7 (q, 2JCF = 27.0 Hz), 43.1; 19F NMR (376 MHz, DMSO-d6) δ/ppm: −77.15 (s); IR (neat) /cm−1: 2900w, 1594m, 1506m, 1453m, 1299m, 1197m, 1152s, 1046m, 989m, 763s; HRMS (ESI-Q-TOF) m/z: calcd for C9H11F3NO [M + H]+ 206.0787, found 206.0795.
The reduction of 1cc. The reduction of 1cc (249 mg, 1.3 mmol) was performed according to GP. The precipitated product 3cb·H2C2O4 was obtained in a pure form based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 40
:60, 0.2 mL min−1, 40 °C, 210 nm, t = 5.43 min (3cb). Yield: 104 mg (28%). Physical state: white hygroscopic powder.Analytical data for 3cb·H2C2O4: Mp 138–139 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.46–7.44 (m, 2H), 7.38–7.34 (m, 2H), 7.28–7.25 (m, 1H), 3.12, 3.10 (q, AB, JAB = 13.0 Hz, 2H), 1.81–1.72 (m, 2H), 1.25–1.13 (m, 3H), 0.85–0.79 (m, 1H), 0.76 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.3, 143.1, 128.0, 126.8, 125.5, 73.5, 48.5, 24.7, 22.2, 13.8; IR (neat) /cm−1: 2930w, 1587m, 1504m, 1448m, 1218s, 1049w, 764w; HRMS (ESI-Q-TOF) m/z: calcd for C12H20NO [M + H]+ 194.1539, found 194.1546.
The reduction of 1d. The reduction of 1d (275 mg, 1.3 mmol) was performed according to GP. The precipitated product 3d·H2C2O4 was obtained in a pure form based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 40
:60, 0.2 mL min−1, 40 °C, 210 nm, t = 5.14 min (3d). Yield: 101 mg (26%). Physical state: white hygroscopic powder. The crude alcohol 5c was isolated from the filtrate according to GP. Yield: 146 mg (53%). Physical state: yellowish waxy solid.Analytical data for 3d·H2C2O4: Mp 182–184 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.50–7.48 (m, 4H), 7.36–7.33 (m, 4H), 7.27–7.24 (m, 2H), 3.68 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.1, 144.3, 128.2, 127.2, 125.8, 74.9, 47.3; IR (neat) /cm−1: 3443w, 1738m, 1614m, 1417m, 1224s, 715s; HRMS (ESI-Q-TOF) m/z: calcd for C14H15NO [M + H]+ 214.1226, found 214.1225.
Analytical data for 5c:52 Mp 60–61 °C; 1H NMR (400 MHz, CDCl3) δ/ppm: 7.39–7.31 (m, 8H), 7.28–7.24 (m, 2H), 5.84 (s, 1H), 2.23 (s, 1H); 13C NMR (100 MHz, CDCl3) δ/ppm: 143.8, 128.5, 127.5, 126.5, 76.2; IR (neat) /cm−1: 3378w, 1494m, 1446m, 1269w, 1181w, 1017m, 752m, 734s; HRMS (ESI-Orbitrap) m/z: calcd for C13H13O [M + H]+ 185.0961, found 185.0959.
The reduction of 1da. The reduction of 1da (295 mg, 1.3 mmol) was performed according to GP. The precipitated product 3d·H2C2O4 was obtained in a pure form based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 40
:60, 0.2 mL min−1, 40 °C, 210 nm, t = 5.11 min (3d). Yield: 150 mg (38%). The analytical data were in accordance with the product obtained by the reduction of 1d.
The reduction of 1e. The reduction of 1e (316 mg, 1.3 mmol) was performed according to GP. The precipitated product was obtained as an inseparable mixture of 3e·H2C2O4/4e·H2C2O4 in a 97
:3 ratio based on 1H NMR and >99:1 according to RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 50:50, 0.2 mL min−1, 40 °C, 210 nm, t = 3.89 min (3e). Yield: 172 mg (39%). Physical state: white hygroscopic powder. The crude alcohol 5d was isolated from the filtrate according to GP. Yield: 95 mg (41%). Physical state: white solid.Analytical data for 3e·H2C2O4: Mp 195–196 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.56–7.39 (m, 8H), 3.08 (q, AB, JAB = 13.5 Hz, 2H), 1.50 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 163.6, 158.9 (d, 1JCF = 245.6 Hz), 147.4 (d, 3JCF = 7.7 Hz), 134.8, 130.4, 128.60, 128.56, 127.8, 126.7 (d, 2JCF = 13.5 Hz), 121.6, 113.2 (d, 2JCF = 24.1 Hz), 70.7, 48.8, 27.4; 19F NMR (376 MHz, DMSO-d6) δ/ppm: −118.12 to −118.17 (m); IR (neat) /cm−1: 3465w, 2930w, 1615m, 1501m, 1410m, 1223m, 1107w, 1067m, 1009m, 882w, 831w, 766m; HRMS (ESI-Q-TOF) m/z: calcd for C15H17FNO [M + H]+ 246.1289, found 246.1290.
Analytical data for 5d:53 Mp 65–66 °C; 1H NMR (400 MHz, CDCl3) δ/ppm: 7.58–7.55 (m, 2H), 7.48–7.37 (m, 4H), 7.23–7.20 (m, 2H), 4.96 (q, J = 6.5 Hz, 1H), 2.08 (br s, 1H), 1.54 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ/ppm: 157.7 (d, 1JCF = 248.5 Hz), 147.4 (3JCF = 6.7 Hz), 135.6, 130.7 (3JCF = 3.9 Hz), 128.9 (3JCF = 2.9 Hz), 128.4, 127.9 (d, 2JCF = 13.5 Hz), 127.6, 121.2, 113.1 (d, 2JCF = 24.1 Hz), 69.6, 25.2; 19F NMR (376 MHz, CDCl3) δ/ppm: −117.56 to −117.61 (m); IR (neat) /cm−1: 3324br, 1581w, 1484m, 1416m, 1268m, 1153w, 1127w, 1070m, 1010m, 939w, 868m, 839m, 766s; HRMS (ESI-Orbitrap) m/z: calcd for C14H12F [M − H2O + H]+ 199.0918, found 199.0917.
The reduction of 1f. The reduction of 1f (267 mg, 1.3 mmol) was performed according to GP. The precipitated product was obtained as an inseparable mixture of 3f·H2C2O4/4f·H2C2O4 in a 93
:7 ratio based on 1H NMR and 94:6 according to RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 50:50, 0.2 mL min−1, 40 °C, 210 nm, t1 = 4.13 min (3f), t2 = 7.23 min (4f). Yield: 160 mg (42%). Physical state: white hygroscopic powder. The crude alcohol 5e was isolated from the filtrate according to GP. Yield: 100 mg (40%). Physical state: yellowish oil.Analytical data for 3f·H2C2O4: Mp 193–195 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.40–7.38 (m, 2H), 7.16–7.14 (m, 2H), 3.05, 2.97 (q, AB, JAB = 12.8 Hz, 2H), 2.44 (d, J = 7.1 Hz, 2H), 1.88–1.78 (m, 1H), 1.48 (s, 3H), 0.87 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 163.3, 142.7, 139.8, 128.6, 124.7, 70.7, 49.1, 44.1, 29.5, 27.2, 22.1; IR (neat) /cm−1: 3467m, 2954m, 1732w, 1615m, 1504s, 1365w, 1184s, 1063s, 1019s, 810w, 794m; HRMS (ESI-Q-TOF) m/z: calcd for C13H22NO [M + H]+ 208.1696, found 208.1689.
Analytical data for 4f·H2C2O4 (the authentic sample was prepared by a DIBAH-mediated reduction of 1f): Mp 190–191 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.19–7.11 (m, 4H), 3.00–2.95 (m, 3H), 2.43 (d, J = 6.9 Hz, 2H), 1.86–1.79 (m, 1H), 1.25–1.19 (m, 3H), 0.87 (d, J = 6.4 Hz, 6H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.0, 140.0, 139.6, 129.1, 126.7, 45.0, 44.1, 37.0, 29.4, 22.1, 19.1; IR (neat) /cm−1: 2866m, 1622m, 1574s, 1532s, 1446m, 1300s, 1186w, 1019w, 840w, 799w, 777s; HRMS (ESI-Q-TOF) m/z: calcd for C13H22N [M + H]+ 192.1747, found 192.1737.
Analytical data for 5e:54 1H NMR (400 MHz, CDCl3) δ/ppm: 7.28–7.25 (m, 2H), 7.13–7.11 (m, 2H), 4.86 (q, J = 6.5 Hz, 1H), 2.46 (d, J = 7.1 Hz, 2H), 2.15 (br s, 1H), 1.90–1.80 (m, 1H), 1.48 (d, J = 6.5 Hz, 3H), 0.90 (d, J = 6.5 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ/ppm: 143.0, 141.0, 129.2, 125.2, 70.2, 45.0, 30.2, 25.0, 22.3; IR (neat) /cm−1: 3344br, 2952s, 2923m, 2867m, 1650w, 1513w, 1465m, 1366m. 1077s, 1008m, 898m, 846m; HRMS (ESI-Orbitrap) m/z: calcd for C12H19O [M + H]+ 179.1430, found 179.1424.
The reduction of 1g. The reduction of 1g (241 mg, 1.3 mmol) was performed according to GP at 0 °C. The precipitated product was obtained as an inseparable mixture of 3g·H2C2O4/4g·H2C2O4 in a 95
:5 ratio based on RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 30:70, 0.2 mL min−1, 40 °C, 230 nm, t1 = 4.64 min (3g), t2 = 12.88 min (4g). Yield: 188 mg (52%). Physical state: white hygroscopic powder.Analytical data for 3g·H2C2O4: Mp 139–141 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 8.17 (d, J = 8.2 Hz, 1H), 7.99–7.96 (m, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.73 (d, J = 6.8 Hz, 1H), 7.61–7.53 (m, 3H), 5.60 (dd, J = 9.6, 2.3 Hz, 1H), 3.16 (dd, J = 12.9, 2.3 Hz, 1H), 2.92 (dd, J = 12.6, 9.6 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 163.6, 137.5, 133.2, 129.7, 128.7, 128.0, 126.3, 125.6, 125.4, 123.4, 122.7, 66.1, 45.4; IR (neat) /cm−1: 2899m, 1715w, 1652s, 1580m, 1524m, 1218m, 1150m, 1059m, 772s, 722s; HRMS (ESI-Q-TOF) m/z: calcd for C12H14NO [M + H]+ 188.1070 m/z, found 188.1072.
The reduction of 1h. The reduction of 1h (184 mg, 1.3 mmol) was performed according to GP at 0 °C. The precipitated product was obtained as an inseparable mixture of 3h·H2C2O4/4h·H2C2O4 in a 97
:3 ratio based on RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 20:80, 0.2 mL min−1, 40 °C, 230 nm, t1 = 2.83 min (3h), t2 = 5.32 min (4h). Yield: 87 mg (29%). Physical state: white hygroscopic powder.Analytical data for 3h·H2C2O4: Mp 168–170 °C; 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.47 (d, J = 4.8 Hz, 1H), 7.06–7.01 (m, 2H), 5.05 (d, J = 8.1 Hz, 1H), 3.13–3.10 (m, 1H), 2.95–2.90 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm: 164.5, 145.9, 126.9, 125.0, 123.9, 65.6, 45.8; IR (neat) /cm−1: 2892br, 1582s, 1446w, 1296m, 1232m, 1021w, 924w, 867w, 784w, 717m; HRMS (ESI-Orbitrap) m/z: calcd for C6H10NOS [M + H]+ 144.0478, found 144.0478.
The reduction of 2a. The reduction of 2a (275 mg, 1.3 mmol) was performed according to GP with a reaction time of 2 h. The precipitated product was obtained as an inseparable mixture of 3a·H2C2O4/4a·H2C2O4 in a 89
:11 ratio based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 40:60, 0.2 mL min−1, 40 °C, 230 nm, t1 = 3.42 min (3a), t2 = 5.65 min (4a). Yield: 167 mg (40%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1a.
The reduction of 2b. The reduction of 2b (152 mg, 1.3 mmol) was performed according to GP at 0 °C with a reaction time of 2 h. The precipitated product 3b·H2C2O4 was obtained in a pure form based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 20
:80, 0.2 mL min−1, 40 °C, 210 nm, t = 3.10 min (3b). Yield: 118 mg (40%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1b.
The reduction of 2ba. The reduction of 2ba (173 mg, 1.3 mmol) was performed according to GP at 0 °C with a reaction time of 1.5 h. The precipitated product 3b·H2C2O4 was obtained in a pure form based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 20
:80, 0.2 mL min−1, 40 °C, 210 nm, tR = 3.10 min (3b). Yield: 99 mg (34%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1b.
The reduction of 2c. The reduction of 2c (171 mg, 1.3 mmol) was performed according to GP with a reaction time of 2 h. The precipitated product was obtained as an inseparable mixture of 3c·H2C2O4/4c·H2C2O4 in a 94
:6 ratio based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 30:70, 0.2 mL min−1, 40 °C, 210 nm, t1 = 2.78 min (3c), t2 = 4.79 min (4c). Yield: 103 mg (33%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1c.
The reduction of 2d. The reduction of 2d (251 mg, 1.3 mmol) was performed according to GP with a reaction time of 2 h. The precipitated product was obtained as an inseparable mixture of 3d·H2C2O4/4d·H2C2O4 in a 92
:8 ratio based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 40:60, 0.2 mL min−1, 40 °C, 210 nm, t1 = 5.14 min (3d), t2 = 7.90 min (4d). Yield: 195 mg (50%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1d.
The reduction of 2da. The reduction of 2da (272 mg, 1.3 mmol) was performed according to GP with a reaction time of 1.5 h. The precipitated product 3d·H2C2O4 was obtained in a pure form based on both 1H NMR and RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 40
:60, 0.2 mL min−1, 40 °C, 210 nm, t = 5.16 min (3d). Yield: 83 mg (21%). The analytical data were in accordance with the product obtained by the reduction of 1d.
The reduction of 2e. The reduction of 2e (293 mg, 1.3 mmol) was performed according to GP with a reaction time of 2 h. The precipitated product was obtained as an inseparable mixture of 3e·H2C2O4/4e·H2C2O4 in a 91
:9 ratio based on 1H NMR, 89:11 based on 19F NMR and 93:7 according to RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 50:50, 0.2 mL min−1, 40 °C, 210 nm, t1 = 3.88 min (3e), t2 = 6.50 min (4e). Yield: 141 mg (33%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1e.
The reduction of 2f. The reduction of 2f (243 mg, 1.3 mmol) was performed according to GP with a reaction time of 2 h. The precipitated product was obtained as an inseparable mixture of 3f·H2C2O4/4f·H2C2O4 in a 96
:4 ratio based on 1H NMR and 97:3 according to RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 50:50, 0.2 mL min−1, 40 °C, 210 nm, t1 = 4.23 min (3f), t2 = 7.76 min (4f). Yield: 182 mg (47%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1f.
The reduction of 2g. The reduction of 2g (217 mg, 1.3 mmol) was performed according to GP at 0 °C with a reaction time of 2 h. The precipitated product was obtained in a pure form based on RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 30
:70, 0.2 mL min−1, 40 °C, 230 nm, t = 4.61 min (3g). Yield: 183 mg (51%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1g.
The reduction of 2h. The reduction of 2h (160 mg, 1.3 mmol) was performed according to GP at 0 °C with a reaction time of 2 h. The precipitated product was obtained as an inseparable mixture of 3h·H2C2O4/4h·H2C2O4 in a 96
:4 ratio based on RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH 9.30), 20:80, 0.2 mL min−1, 40 °C, 230 nm, t1 = 2.84 min (3h), t2 = 5.54 min (4h). Yield: 74 mg (24%). Physical state: white hygroscopic powder. The analytical data were in accordance with the product obtained by the reduction of 1h.
:7). Yield: 90 mg (69%). Physical state: brown oil. 1H NMR (400 MHz, CDCl3) δ/ppm: 7.58–7.56 (m, 2H), 7.49–7.45 (m, 3H), 7.41–7.37 (m, 1H), 7.20–7.15 (m, 2H), 3.11–3.05 (AB-part of ABX system, JAB = 14.5 Hz, JAX = 14.8 Hz, JBX = 27.8 Hz, 2H), 1.69 (d, 3JHF = 22.3 Hz, 3H), 1.29 (br s, 2H); 13C NMR (100 MHz, CDCl3) δ/ppm: 159.6 (d, 1JCF = 246.6 Hz), 144.2 (dd, JCF = 22.6, 7.2 Hz), 135.2, 130.8, 128.9, 128.4, 128.0 (d, 2JCF = 13.5 Hz), 127.7, 120.2 (dd, JCF = 9.6, 2.9 Hz), 112.6 (dd, JCF = 25.1, 10.6 Hz), 97.6 (d, 1JCF = 173.4 Hz), 52.0 (d, 2JCF = 25.1 Hz), 24.9 (d, 2JCF = 24.1 Hz); 19F NMR (376 MHz, CDCl3) δ/ppm: −117.09 to −117.14 (m, 1F), −156.63 to −156.92 (X-part of ABX system, 1F); IR (neat) /cm−1: 1581w, 1484m, 1408m, 1281w, 1184w, 1075w, 1010w, 830m, 767s; HRMS (ESI-Orbitrap) m/z: calcd for C15H16N2F [M + H]+ 248.1245, found 248.1254.:1). Yield: 47 mg (37%). Physical state: white solid. Mp 86–88 °C; 1H NMR (400 MHz, CDCl3) δ/ppm: 7.56–7.54 (m, 2H), 7.50–7.56 (m, 3H), 7.42–7.38 (m, 1H), 7.22–7.17 (m, 2H), 4.47 (t, J = 6.3 Hz, 1H), 3.67–3.51 (m, 2H), 3.09 (sept, J = 6.8 Hz, 1H), 1.77 (d, 3JHF = 22.5 Hz, 3H), 1.34 (d, J = 6.8 Hz, 3H); 1.31 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ/ppm: 159.7 (d, 1JCF = 248.5 Hz), 142.9 (dd, JCF = 22.2, 7.7 Hz), 135.0, 131.0, 128.9, 128.7 (d, 2JCF = 13.5 Hz), 128.5, 127.9, 120.1 (dd, JCF = 9.6, 3.4 Hz), 112.5 (dd, JCF = 25.1, 10.6 Hz), 96.4 (d, 1JCF = 176.3 Hz), 54.0, 52.4 (d, 2JCF = 23.1 Hz), 24.5 (d, 2JCF = 24.1 Hz), 16.5 (d, 3JCF = 7.7 Hz); 19F NMR (376 MHz, CDCl3) δ/ppm: −116.42 to −116.52 (m, 1F), −153.04 to −153.33 (m, 1F); IR (neat) /cm−1: 3137w, 2933w, 1484w, 1450w, 1409m, 1308s, 1187w, 1116s, 1072m, 915m, 872w, 839m, 770s, 725s; HRMS (ESI-Orbitrap) m/z: calcd for C18H20O2NF2S [M − H]− 352.1188, found 352.1191.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
CIISB research infrastructure project LM2015043 funded by MEYS CR is gratefully acknowledged for the financial support of the measurements at the Proteomics Core Facility. We wish to thank Peter Zubac for the generous gift of several 2-arylalkanoic acids.Notes and references
- (a) L. A. Nguyen, H. He and C. Pham-Huy, Int. J. Biomed. Sci., 2006, 2, 85 CAS; (b) S. W. Smith, Toxicol. Sci., 2009, 110, 4 CrossRef CAS PubMed; (c) W. H. Brooks, W. C. Guida and K. G. Daniel, Curr. Top. Med. Chem., 2011, 11, 760 CrossRef CAS PubMed; (d) M. Hyneck, J. Dent, J. B. Hook, E. J. Ariens and D. S. Davies, in Chirality in Drug Design and Synthesis, ed. C. Brown, Academic Press, San Diego, 1st edn, 2013, pp. 1–51 Search PubMed.
- (a) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835 CrossRef CAS PubMed; (b) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561 CrossRef CAS; (c) H.-S. Lee and S. H. Kang, Synlett, 2004, 1673 CrossRef CAS; (d) S. Giorgio Della, R. Alessio and L. Alessandra, Curr. Org. Chem., 2011, 15, 2147 CrossRef; (e) H. Nakano, I. A. Owolabi, M. Chennapuram, Y. Okuyama, E. Kwon, S. Seki, M. Tokiwa and M. Takeshita, Heterocycles, 2018, 97, 647 CrossRef CAS.
- (a) O. K. Karjalainen and A. M. P. Koskinen, Org. Biomol. Chem., 2012, 10, 4311 RSC; (b) T. Sehl, Z. Maugeri and D. Rother, J. Mol. Catal. B: Enzym., 2015, 114, 65 CrossRef CAS; (c) P. Gupta and N. Mahajan, New J. Chem., 2018, 42, 12296 RSC.
- Vol. 8: Reduction, in Comprehensive Organic Synthesis, ed. J. Clayden, Elsevier Science, Amsterdam, 2nd edn, 2014 Search PubMed.
- (a) J. Cossy, D. Gomez Pardo, C. Dumas, O. Mirguet, I. Déchamps, T.-X. Métro, B. Burger, R. Roudeau, J. Appenzeller and A. Cochi, Chirality, 2009, 21, 856 CrossRef PubMed; (b) T.-X. Métro, B. Duthion, D. Gomez Pardo and J. Cossy, Chem. Soc. Rev., 2010, 39, 89 RSC; (c) C. Weng, H. Zhang, X. Xiong, X. Lu and Y. Zhou, Asian J. Chem., 2014, 26, 3761 CrossRef CAS; (d) D. S. Bhagavathula, G. Boddeti and R. Venu, J. Chem., 2017, 6, 27 CAS.
- (a) T. J. Donohoe, C. K. A. Callens, A. Flores, A. R. Lacy and A. H. Rathi, Chem.–Eur. J., 2011, 17, 58 CrossRef CAS PubMed; (b) T. Taniguchi, A. Yajima and H. Ishibashi, Adv. Synth. Catal., 2011, 353, 2643 CrossRef CAS; (c) D. Hofmann, C. de Salas and M. R. Heinrich, ChemSusChem, 2015, 8, 3167 CrossRef CAS PubMed; (d) M. M. Heravi, T. B. Lashaki, B. Fattahi and V. Zadsirjan, RSC Adv., 2018, 8, 6634 RSC.
- O. N. Burchak and S. Py, Tetrahedron, 2009, 65, 7333 CrossRef CAS.
- G. B. Fisher, C. T. Goralski, L. W. Nicholson, D. L. Hasha, D. Zakett and B. Singaram, J. Org. Chem., 1995, 60, 2026 CrossRef CAS.
- (a) A. Kamimura and N. Ono, Tetrahedron Lett., 1989, 30, 731 CrossRef CAS; (b) S.-C. Tsay, H. V. Patel and J. R. Hwu, Synlett, 1998, 939 CrossRef CAS; (c) P. Diner, M. Nielsen, S. Bertelsen, B. Niess and K. A. Jørgensen, Chem. Commun., 2007, 3646 RSC; (d) A. Russo and A. Lattanzi, Adv. Synth. Catal., 2008, 350, 1991 CrossRef CAS.
- (a) H.-G. Brunker and W. Adam, J. Am. Chem. Soc., 1995, 117, 3976 CrossRef; (b) W. Adam and H.-G. Brunker, Synthesis, 1995, 1066 CrossRef CAS.
- (a) J. Otevrel and P. Bobal, Synthesis, 2017, 49, 593 CAS; (b) J. Otevrel and P. Bobal, J. Org. Chem., 2017, 82, 8342 CrossRef CAS PubMed; (c) J. Otevrel, D. Svestka and P. Bobal, Org. Biomol. Chem., 2019, 17, 5244 RSC.
- J. Vit, B. Casensky and J. Machacek, Patent FR 1515582, Czechoslovak Academy of Sciences, 1968; Chem. Abstr., 1969, 70, 115009.
- For selected reviews, see: (a) O. Strouf, B. Casensky and V. Kubanek, Sodium Dihydrido-bis(2-methoxyethoxo)-aluminate (SDMA): A Versatile Organometallic Hydride, Elsevier, Amsterdam, 1985 Search PubMed; (b) J. Malek, Org. React., 1985, 34, 1 CAS; (c) J. Malek, Org. React., 1988, 36, 249 CAS; (d) M. Gugelchuk, L. F. Silva III, R. S. Vasconcelos and S. A. P. Quintiliano, Bis(2-methoxyethoxy)aluminum Hydride, in Encyclopedia of Reagents for Organic Synthesis, ed. P. Fuchs, J. Bode, A. Charette and T. Rovis, John Wiley, New York, 2007 Search PubMed; (e) A. Rathi, Synlett, 2010, 1140 CrossRef CAS; (f) M. B. Smith, Organic Synthesis, Elsevier Science, Cambridge, 2016, pp. 339–344 Search PubMed.
- For recent seminal works, see: (a) N. Bajwa and M. P. Jennings, J. Org. Chem., 2008, 73, 3638 CrossRef CAS PubMed; (b) F. von Kieseritzky and J. Lindstrom, Tetrahedron Lett., 2011, 52, 4558 CrossRef CAS; (c) K. Miyamoto, M. M. Hoque and S. Ogasa, J. Org. Chem., 2012, 77, 8317 CrossRef CAS PubMed; (d) J. Wu, J. Xiao, W. Dai and S. Cao, RSC Adv., 2015, 5, 34498 RSC; (e) R. Sun, J. Liu, S. Yang, M. Chen, N. Sun, H. Chen, X. Xie, X. You, S. Li and Y. Liu, Chem. Commun., 2015, 51, 6426 RSC.
- (a) H. H. Jeon, J. B. Son, J. H. Choi and I. H. Jeong, Tetrahedron Lett., 2007, 48, 627 CrossRef CAS; (b) T. Konno, M. Kishi, T. Ishihara and S. Yamada, J. Fluorine Chem., 2013, 156, 144 CrossRef CAS; (c) G. Jin, J. Zhang, W. Wu and S. Cao, J. Fluorine Chem., 2014, 168, 240 CrossRef CAS.
- e.g. 2-(4-chlorophenyl)-2-hydroxyacetonitrile subjected to the standard reaction conditions provided a mixture of 2-amino-1-phenylethan-1-ol and 2-amino-1-(4-chlorophenyl)ethan-1-ol in a 26:74 ratio according to RP-HPLC. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–50 mM HCOONH4 (pH = 9.30), 20:80, 0.2 mL min−1, 40 °C, 210 nm.
- (a) M. Kraus, Collect. Czech. Chem. Commun., 1972, 37, 3052 CrossRef CAS; (b) M. Czakoova, J. Hetflejs and J. Vcelak, React. Kinet. Catal. Lett., 2001, 72, 277 CrossRef CAS.
- HPLC purity was >93%. HPLC conditions: an YMC-Triart C18 column (3 μm, 150 × 2.0 mm), MeCN–H2O, 50:50 → 90:10, 0.2 mL min−1, 40 °C, 210 nm.
- (a) V. Bazant, M. Capka, M. Cerny, V. Chvalovsky, K. Kochloefl, M. Kraus and J. Malek, Tetrahedron Lett., 1968, 9, 3303 CrossRef; (b) M. Cerny, J. Malek, M. Capka and V. Chvalovsky, Collect. Czech. Chem. Commun., 1969, 34, 1033 CrossRef CAS.
- This was revealed during optimization of the reaction conditions (entry 5, Table S1†).
- (a) M. S. Kharasch and G. Sosnovsky, Tetrahedron, 1958, 3, 97 CrossRef CAS; (b) M. S. Kharasch and G. Sosnovsky, Tetrahedron, 1958, 3, 105 CrossRef CAS; (c) J. S. Selikson and D. S. Watt, J. Org. Chem., 1975, 40, 267 CrossRef; (d) C. Walling, Autoxidation in Active Oxygen in Chemistry, ed. C. S. Foote, J. S. Valentine, A. Greenberg and J. F. Liebman, Springer, Dordrecht, 1995, pp. 24–65 Search PubMed.
- U. Bergstrasser, S. J. Collier, Y. Ito, S. Kanemasa and S. I. Murahashi, Three Carbon–Heteroatom Bonds: Nitriles, Isocyanides, and Derivatives in Science of Synthesis: Houben-Weyl Methods of Molecular Transformations, ed. S. I. Murahashi, Georg Thieme, Stuttgart, 2004, vol. 19 Search PubMed.
- Traces of analogous ketones were isolated also from the reaction mixtures of 1a, 1c, 1d, 1e, and the respective nitriles.
- M. Moller, M. Husemann and G. Boche, J. Organomet. Chem., 2001, 624, 47 CrossRef CAS.
- It is noteworthy that rac-1a provided 5a in the racemic form.
- (a) D. M. Bender, B. E. Cantrell, A. H. Fray, W. D. Jones, W. D. Miller, D. Mitchell, R. L. Simon, H. Zarrinmayeh and D. M. Zimmerman, Patent WO 2000066546, Eli Lilly, 2000Chem. Abstr., 2000, 133, 350055; (b) J. A. Clemens and D. Lodge, Patent WO 2002032389, Eli Lilly, 2002Chem. Abstr., 2002, 136, 340487.
- L. H. Sommer, C. L. Frye, G. A. Parker and K. W. Michael, J. Am. Chem. Soc., 1964, 86, 3271 CrossRef CAS.
- M. Amir, H. Kumar and S. A. Javed, Arch. Pharm., 2007, 340, 577 CrossRef CAS PubMed.
- N. Ohtsuka, M. Okuno, Y. Hoshino and K. A. Honda, Org. Biomol. Chem., 2016, 14, 9046 RSC.
- F. A. J. Kerdesky, C. D. W. Brooks, K. I. Hulkower, J. B. Bouska and R. L. Bell, Bioorg. Med. Chem., 1997, 5, 393 CrossRef CAS PubMed.
- T. Tu, Z. Wang, Z. Liu, X. Feng and Q. Wang, Green Chem., 2012, 14, 921 RSC.
- T. Kanda, A. Naraoka and H. Naka, J. Am. Chem. Soc., 2019, 141, 825 CrossRef CAS PubMed.
- T. Mecca, S. Superchi, E. Giorgio and C. Rosini, Tetrahedron: Asymmetry, 2001, 12, 1225 CrossRef CAS.
- M. Kimura, A. Kuboki and T. Sugai, Tetrahedron: Asymmetry, 2002, 13, 1059 CrossRef CAS.
- W. A. Bonner, J. Am. Chem. Soc., 1954, 76, 6350 CrossRef CAS.
- N. Iqbal and E. J. Cho, Adv. Synth. Catal., 2015, 357, 2187 CrossRef CAS.
- E. V. Bellale, D. S. Bhalerao and K. G. Akamanchi, J. Org. Chem., 2008, 73, 9473 CrossRef CAS PubMed.
- G. Nemeth, E. Rakoczy and G. Simig, J. Fluorine Chem., 1996, 76, 91 CrossRef CAS.
- J. A. Dale, D. L. Dull and S. H. Mosher, J. Org. Chem., 1969, 34, 2543 CrossRef CAS.
- F. Li, X. Zou and N. Wang, Adv. Synth. Catal., 2015, 357, 1405 CrossRef CAS.
- B. Guo, J. G. de Vries and E. Otten, Chem. Sci., 2019, 10, 10647 RSC.
- H.-M. Xia, F.-L. Zhang, T. Ye and Y.-F. Wang, Angew. Chem., Int. Ed., 2018, 57, 11770 CrossRef CAS PubMed.
- N. H. Khan, S. Agrawal, R. I. Kureshy, S. H. R. Abdi, S. Singh and R. V. Jasra, J. Organomet. Chem., 2007, 692, 4361 CrossRef CAS.
- P. G. Gassman and J. J. Talley, Org. Synth., 1981, 60, 14 CrossRef CAS.
- T. Hiyama, M. Inoue and K. Saito, Synthesis, 1986, 645 CrossRef CAS.
- R. Shang, D.-S. Ji, L. Chu, Y. Fu and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 4470 CrossRef CAS PubMed.
- The same results were obtained with 60% SMEAH in toluene (6.4 mL, 15 equiv.).
- The free base of the respective aminoalcohol 3 was liberated from the corresponding oxalic acid salt on demand by addition of an aqueous solution of NaOH (2.5 M) and back-extraction to toluene.
- J. Xiao, Z. Z. Wong, Y. P. Lu and T. P. Loh, Adv. Synth. Catal., 2010, 352, 1107 CrossRef CAS.
- A. Dornow and W. Sassenberg, Justus Liebigs Ann. Chem., 1955, 594, 185 CrossRef CAS.
- V. M. Balaramnavar, R. Srivastava, N. Rahuja, S. Gupta, A. K. Rawat, S. Varshney, H. Chandasana, Y. S. Chhonker, P. K. Doharey, S. Kumar, S. Gautam, S. P. Srivastava, R. S. Bhatta, J. K. Saxena, A. N. Gaikwad, A. K. Srivastava and A. K. Saxena, Eur. J. Med. Chem., 2014, 87, 578 CrossRef CAS PubMed.
- C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816 CrossRef CAS PubMed.
- J. V. Castell, M. J. Gomez-Lechon, M. A. Miranda and I. M. Morera, J. Photochem. Photobiol., B, 1992, 13, 71 CrossRef CAS.
- D. P. Riley, D. P. Getman, G. R. Beck and R. M. Heintz, J. Org. Chem., 1987, 52, 287 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI contains optimization, mechanistic experiments, HPLC, NMR, GC/MS, and HRMS data for compounds 1–10. See DOI: 10.1039/d0ra04359a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |