
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel quercetin and apigenin-acetamide derivatives: design, synthesis, characterization, biological evaluation and molecular docking studies†
Daniel Isikaa,
Mustafa Çeşmeb,
Francis J. Osongaa and
Omowunmi A. Sadik*a
aDepartment of Chemistry and Environmental Science, Sensors Mechanisms Research & Technology (The SMART Center), New Jersey Institute of Technology, 161 Warren Street, University Heights, Newark, NJ 07102, USA. E-mail: sadik@njit.edu
bDepartment of Chemistry, Faculty of Art and Sciences, Kahramanmaras Sutcu Imam University, Kahramanmaras, 46040, Turkey
First published on 30th June 2020
Abstract
Flavonoids exhibit essential but limited biological properties which can be enhanced through chemical modifications. In this study, we designed, synthesized, and characterized two novel flavonoid derivatives, quercetin penta-acetamide (1S3) and apigenin tri-acetamide (2S3). These compounds were confirmed using (1H, 13C) NMR, UV-Vis, and FT-IR characterizations. Their interaction with fish sperm DNA (FS-DNA) at physiological pH was investigated by UV-Vis and fluorescence spectrophotometry. The binding constant (Kb) for the UV-Vis experiment was found to be 1.43 ± 0.3 × 104 M−1 for 1S3 and 2.08 ± 0.2 × 104 M−1 for 2S3. The binding constants (KSV) for the fluorescence quenching experiment were 1.83 × 104 M−1 and 1.96 × 104 M−1 for 1S3 and 2S3, respectively. Based on molecular modeling and docking studies, the binding affinities were found to be −7.9 and −9.1 kcal mol−1, for 1S3 and 2S3, respectively. The compound–DNA docked model correlated with our experimental results, and they are groove binders. Furthermore, mutagenicity potential was examined. 1S3 and its metabolites showed no mutagenic activity for both TA98 and TA100 strains. 2S3 did not show any mutagenic activity for the strain TA 98, while its metabolites were only active at high doses. Both 2S3 and its metabolites showed mutagenic activity in the TA100 strain.
1. Introduction
Flavonoids are a class of polyphenolic plant metabolites structurally consisting of 15-carbons where two benzene rings are linked by a three-carbon chain to afford a C6–C3–C6 carbon skeleton. They possess anticancer,1 antibacterial,2 antifungal3 and anti-inflammatory4 properties. They exist in various classes notably flavones (such as apigenin) and flavanols (such as quercetin); structures are shown in Fig. 1. Apigenin occurs in vegetables and fruits.5 It has anticancer,5–7 anti-inflammatory8 and antibacterial9 properties. Quercetin occurs in medicinal plants, fruits and vegetables.5 It exhibits anti-cancer,10 antibacterial,11 gastroprotective,12 antiviral13 and anti-inflammatory14 properties.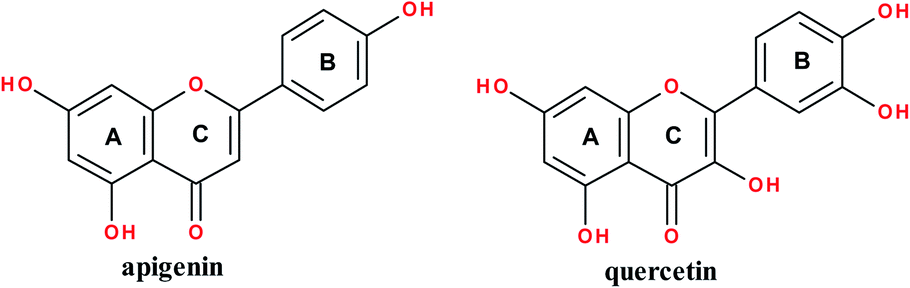 | ||
| Fig. 1 Structure of apigenin and quercetin. | ||
Biological applications of flavonoids are limited by low bioavailability,15 fast body clearance,16 low aqueous solubility10,17 enzyme degradation18 and fast body metabolism.10 To address these limitations, quercetin has been modified by the functionalization of A- and B-rings and chemical manipulation of the phenolic hydroxyl groups.10
Quercetin penta-phosphate and quercetin sulfonic acid are water soluble and effective in the reduction of Cr4+ ions.19,20 Phosphorylated flavonoids have improved aqueous solubility and metal chelation.20,21 They are reducing, stabilizing and capping agents in the synthesis of gold, palladium and silver nanoparticles with enhanced antimicrobial properties.21–23
In this study, we chemically modified quercetin and apigenin to yield quercetin- and apigenin-acetamide derivatives. The rationale is that the acetamide group comprises of a sp3 hybridized carbon which, when incorporated into the flavonoid molecule, will induce flexibility and enhance the overall lipophilicity of the resultant derivative. The amide group would potentially increase hydrogen bonding capability and interaction sites, of the derivatives. The viability of these derivatives to applications in biological systems was evaluated by biological and molecular docking studies. Acetamide derivatives possess antimicrobial24 and anti-inflammatory25 activities. Acetamides can inhibit hepatitis C virus26 and are possible analgesics.27 Modafinil, levetiracetam, acetaminophen and acetazolamide are some acetamide-based drugs.
Nucleic acids are powerful tools in the recognition and monitoring of vital compounds.28–31 The monitoring of DNA–drug interactions using spectroscopic methods is based on the fact that the fluorescence of free ligands interacts with DNA in the electronic absorption spectra.32,33 The Ames test34 is used to determine the mutations occurring by chemicals at the cell level.
To the best our knowledge, this is the first time the synthesis of 1S3 and 2S3 is being reported. DNA-binding of these compounds was investigated using fluorescence quenching and UV-Vis spectroscopy. The atomic details of these ligands–DNA interactions were investigated through the molecular docking method. The results based on the in silico DNA–ligand interaction studies show that 1S3 and 2S3 ligands are groove binders and that H-bond interactions play a principal role in the stability of these ligand–DNA complexes. This research exhibits the details of mutagenicity, the mode of binding interaction, binding affinity, main interaction forces of ligands with FS-DNA and structure of ligand–FS-DNA complex. The interaction of FS-DNA with the two synthesized compounds is significant in pharmaceutical drug discovery. Based on the in silico molecular docking studies, the binding score of the DNA interaction of the compounds is quite satisfactory. When the results obtained from the DNA interaction and the mutagenicity tests are evaluated, it can be concluded that these compounds can be used in biological applications and in vivo studies. This is after further considering that these compounds are not toxic.
2. Materials and methods
2.1 Reagents and solutions
High purity quercetin and apigenin were purchased from Indofine Chemical Company, Hillsborough, NJ, USA. Ethyl chloroacetate, dimethylformamide, thionyl chloride, lithium hydroxide monohydrate, anhydrous potassium carbonate, sodium chloride, ethyl acetate, hexane, acetone, ammonia solution, silica gel, methyl sulfoxide-d6, sodium bicarbonate, and anhydrous sodium sulfate were purchased from Millipore Sigma (Burlington, MA, USA). All chemicals were of analytical grade and were used without further purification. Fish sperm DNA (74782) and Tris–HCl (RES3098T-B7) were supplied by Sigma-Aldrich Chemie GmbH (Germany). Ethidium bromide (EtBr) and sodium chloride (≥99.5%) were purchased from Sigma Aldrich, USA. Other chemicals and reagents purchased from Sigma or Merck and used in this work were all of the analytical grades.2.2 Apparatus
Absorption measurements were carried out using a double-beam spectrophotometer (PerkinElmer, Lambda 25, USA) and PG spectrophotometer (T80+, UK) equipped with 1.0 cm quartz cells. Fluorescence spectra were collected on a PerkinElmer LS55 luminescence spectrophotometer. All samples were prepared in spectrophotometric grade solvents and analyzed in a 1.0 cm optical path quartz cuvette. Infrared spectra were recorded on a PerkinElmer spectrometer (Spectrum 400) equipped with ATR accessory. A pH meter (Thermo Scientific Orion Star A215 pH/conductivity benchtop) was used for the adjustment of pH values. The 1H and 13C NMR data were recorded on a Bruker Avance III HD 400 MHz Spectrometer.2.3 Synthesis of the compounds
Quercetin, 1S0, (604 mg, 2 mmol, 1 eq.) was dissolved in dry DMF (10 mL) in an oven-dried round-bottomed flask forming a brown solution. Anhydrous K2CO3 (2070 mg, 15 mmol, 7.5 eq.) was added and the mixture was stirred for 1 hour at room temperature. Ethyl chloroacetate (1716 mg, 14 mmol, 7 eq.) was slowly added into the reaction mixture, which was vigorously stirred at room temperature, under nitrogen gas. The progress of the reaction was monitored by TLC and the reaction was stopped after 37 hours. The final reaction mixture was filtered, and the excess solvent was removed in a rotavapor. The resultant crude solid product was dissolved in ethyl acetate and washed with 5% sodium bicarbonate, brine and finally water. The final product was purified by flash column chromatography using silica gel in (hexane/ethyl acetate/acetone, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1, v/v/v) to afford 1S1, a light yellow solid, 73% yield, Rf = 0.22. The structure of this compound was verified by 1H and 13C NMR characterizations.
:2:1, v/v/v) to afford 1S1, a light yellow solid, 73% yield, Rf = 0.22. The structure of this compound was verified by 1H and 13C NMR characterizations.
1S1: δ1H (400 MHz, DMSO-d6): 7.82–7.67 (2H, m), 7.14–7.02 (1H, m), 6.92–6.84 (1H, m), 6.47 (1H, d, J = 2.3 Hz), 5.03–4.71 (10H, m), 4.24–4.08 (10H, m), 1.27–1.13 (15H, m). δ13C (400 MHz, DMSO-d6): 171.71, 168.64, 168.57, 168.40, 168.17, 167.93, 161.68, 158.58, 157.69, 151.03, 149.30, 146.60, 138.57, 123.23, 122.47, 114.65, 113.60, 108.94, 97.83, 94.83, 67.60, 65.69, 65.65, 65.20, 65.05, 60.85, 60.73, 60.63, 60.45, 14.01, 13.99, 13.91.
:1, v/v) to yield a brown solid, 992.9 mg, 94%. Rf = 0.16 (hexane/ethyl acetate, 1:1, v/v). The structure of the intermediate product was confirmed using 1H and 13C NMR characterizations.2S1: δ1H NMR (400 MHz, DMSO-d6) δ 7.99 (d, J = 9.0 Hz, 2H), 7.10 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 2.3 Hz, 1H), 6.71 (s, 1H), 6.46 (d, J = 2.3 Hz, 1H), 4.98–4.87 (m, 6H), 4.25–4.13 (m, 6H), 1.27–1.18 (m, 9H). δ13C NMR (400 MHz, DMSO-d6) δ 175.38, 168.36, 168.27, 168.00, 161.53, 160.13, 159.66, 158.83, 158.52, 127.72, 123.63, 115.04, 109.04, 106.94, 98.11, 95.24, 65.66, 64.99, 64.73, 60.84, 60.74, 60.69, 14.01.
1S1 (732 mg, 1 mmol, 1 eq.) was dissolved in THF/H2O (1:2). Into the resultant reaction mixture, LiOH·H2O (419.6 mg, 10 mmol, 10 eq.) was added. The reaction mixture was stirred at room temperature and the progress of the reaction was monitored by TLC for 24 hours. The excess lithium hydroxide was neutralized with 1 N HCl to afford a light-yellow precipitate which was filtered off to obtain a yellow residue. The final product was concentrated in vacuo and left in a line vacuum overnight to achieve 1S2 as a fine light-yellow solid, 515 mg, 87% yield. The structure was confirmed using 1H NMR characterization.
1S2: δH (400 MHz, DMSO-d6): 13.07 (5H, s), 7.81 (1H, d, J = 2.1 Hz), 7.74 (1H, dd, J = 8.6, 2.1 Hz), 7.06 (1H, d, J = 8.8 Hz), 6.86 (1H, d, J = 2.2 Hz), 6.45 (1H, d, J = 2.3 Hz), 4.87–4.80 (6H, m), 4.79 (2H, s), 4.70 (2H, s).
:1. LiOH·H2O (252 mg, 6 mmol, 6 eq.) was slowly added into the reaction mixture at room temperature, the reaction was monitored by TLC for 14 hours. The reaction mixture was neutralized with 1 M HCl to form a light yellow precipitate. The precipitate was filtered off and purified by flash column chromatography on silica gel (methanol/water, 9:1, v/v). The final product was concentrated en vacuo and left in a line vacuum overnight to achieve 2S2, as a fine light-yellow solid, 395.4 mg, 89% yield. The structure of the product was confirmed using 1H characterization.2S2: δH (400 MHz, DMSO-d6): 13.15 (3H, s), 8.06–7.97 (2H, m), 7.13–7.05 (2H, m), 6.93 (1H, d, J = 2.3 Hz), 6.75 (1H, s), 6.49 (1H, d, J = 2.3 Hz), 4.87–4.79 (6H, m).
1S2 (414.7 mg, 0.7 mmol, 1 eq.) was mixed with excess thionyl chloride in an oven-dried round-bottomed flask. The reaction mixture was vigorously stirred under reflux conditions; the reaction was monitored by TLC and was stopped after 13 hours. The reaction mixture was concentrated under reduced pressure to form a yellow powder, the acyl chloride intermediate product. This product was slowly transferred into ice-cold aqueous ammonium hydroxide. The reaction mixture was stirred at this temperature for 2 hours and at room temperature for 4 hours. The excess solvents were removed in a rotavapor followed by the purification in a flash column chromatography. The final product was a light brown solid, 329 mg, 80% yield. The products were confirmed by 1H NMR, 13C NMR and FT-IR characterizations.
1S3: δH (400 MHz, DMSO-d6): 8.07 (1H, s), 7.88–7.65 (4H, m), 7.61 (1H, s), 7.56–7.33 (6H, m), 7.12 (1H, d, J = 8.6 Hz), 6.88 (1H, s), 6.67 (1H, s), 4.59 (8H, dd, J = 16.0, 10.2 Hz), 4.33 (2H, s).
δ13C (400 MHz, DMSO-d6): 172.93, 170.04, 169.94, 169.66, 169.57, 169.52, 168.92, 162.45, 157.67, 152.63, 149.91, 147.22, 139.17, 122.97, 122.75, 114.59, 113.71, 108.43, 98.29, 94.94, 70.24, 68.12, 67.77, 67.45, 67.03.
FT-IR: (ATR, cm−1): 3411 ν(N–H), 3310 ν(N–H), 1682 ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)ketone, 1601 ν(CO)amide UV-Vis, λmaxnm, (ε m−1 cm−1) (DI water as solvent): 255 (1.80 × 104), 300 (9.2 × 103), 345 (1.71 × 104). Conductivity: 11.3 μS.
O)ketone, 1601 ν(CO)amide UV-Vis, λmaxnm, (ε m−1 cm−1) (DI water as solvent): 255 (1.80 × 104), 300 (9.2 × 103), 345 (1.71 × 104). Conductivity: 11.3 μS.
2S3: 1H NMR (400 MHz, DMSO-d6) δ 8.25 (1H, s), 8.02 (2H, d, J = 8.4 Hz), 7.68 (1H, s), 7.60 (2H, s), 7.45 (2H, d, J = 17.4 Hz), 7.12 (2H, d, J = 8.6 Hz), 6.92 (1H, s), 6.76 (1H, s), 6.66 (1H, d, J = 2.1 Hz), 4.64–4.51 (6H, m). 13C NMR (400 MHz, DMSO-d6) δ 176.93, 170.30, 169.88, 169.46, 162.79, 161.06, 160.97, 159.12, 158.13, 128.33, 123.84, 115.75, 108.91, 107.30, 99.17, 95.74, 68.39, 67.48, 67.20.
FT-IR: (ATR, cm−1): 3391 ν(N–H), 3311 ν(N–H), 1674 ν(CO)ketone, 1587 ν(CO)amide UV-Vis λmaxnm, (ε m−1 cm−1) (DMSO as solvent): 260 (1.8 × 104), 290 (1.59 × 104), 357 (2.36 × 104). Conductivity: 7.9 μS.
2.4 DNA interaction study
The stock solution of FS-DNA was prepared in a Tris–HCl buffer with a pH of 7.4 and a concentration of 50 mM then stored in the refrigerator at 4 °C for a maximum of 72 hours. In all experiments, the FS-DNA concentration was determined using the absorption band at 260 nm, and the molar absorption coefficient reported as 6600 L mol−1 cm−1 the relevant calculations were made based on this concentration.38 To determine that FS-DNA did not contain protein impurity; the purity of the prepared stock FS-DNA solution was confirmed by the UV absorption rate (A260/A280 > 1.8) as a result of measurements obtained at 260 and 280 nm.39 Similarly, stock solutions of the ligands were prepared in deionized water and then diluted to desired concentrations by Tris–HCl buffer. The EtBr used in photoluminescence studies was prepared at a concentration of 3.0 × 10−3 mol L−1 in ethanol and used for the experiment immediately.| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/[Kb(εb − εf)] | (1) |
:EtBr (10:1) for 15 minutes at 25 °C. The binding of EtBr molecules to the double helix structure of DNA by intercalation mode causes a significant increase in the fluorescence intensity of EB. Subsequently, various amounts of ligand solutions (0–70 μM) were added to this mixture, allowing the final mixture to incubate for 15 minutes. Fluorescence spectra of the resulting final solutions were displayed in the range of 500–700 nm with an excitation wavelength of 520 nm. Fluorescence quenching efficiency was evaluated according to the following equation (Stern–Volmer equation, eqn (2)).41| I0/I = KSV[Q] + 1 | (2) |
2.5 Mutagenicity assay
2.6 Docking and molecular modelling studies
The synthesized ligands, 1S3 and 2S3, were sketched in drawing BIOVIA Draw 2019 software and their energies minimized by Chem3D 19. Molecular modeling studies of these ligands were realized using HyperChem 8.03.43 The molecular docking analysis was operated on the ligands against 1BNA by utilizing the PyRx docking software (academic licensed version 0.9.8).44 To evaluate the binding and interaction of the two compounds to DNA, we employed the Dickerson–Drew dodecamer (DDD) with the sequence d(CGCGAATTCGCG)2 as a model system.45 The high-resolution structure of the free DNA was downloaded from the PDB database (PDB code: 1BNA) (http://www.rcsb.org/pdb). The heteroatoms were removed from 1BNA.pdb, to remove any free ligand from the complex receptor. DNA was prepared by the addition of Kollman united atom charges, polar hydrogens, and solvation parameters. The default exhaustiveness value for both positions was set to 8 to achieve the highest level of accuracy for binding conformational analysis. Ligands were arranged by combining non-polar hydrogens and assigning the Gasteiger charge. In order to dock the synthesized ligands to DNA, a blind docking (placement) was conducted with 128 lattice points throughout the X, Y, and Z axes to find the binding site of complexes on DNA with a grid point range of 0.375 Å. In the next step, the center of the grid box was located at the binding site, and the second docking was performed using a cubic box with 60 × 60 × 60 dimensions. The search for best conformers was done using a Lamarckian genetic algorithm (LGA).46 The docked complexes (1S3–DNA and 2S3–DNA) were assessed on the lowest binding energy (kcal mol−1) values. The graphical depictions and related calculations of all the docking complexes were carried out and monitored using Discovery Studio 2020.3. Results and discussion
3.1 Synthesis and NMR study of the compounds
Apigenin (4′,5,7-trihydroxyflavone) structure consists of 2-phenylchromen-4-one with three hydroxyl groups at positions 4′, 5 and 7. It has three rings: two benzene rings (ring A and B) and a pyran ring, ring C. Quercetin (2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one) structure consist of 2-phenylchromen-4-one with five hydroxyl groups at positions 3, 3′, 4′, 5 and 7. It has three rings: two benzene rings (ring A and B) and a pyran ring, ring C; Fig. 1.Quercetin and apigenin-acetamide derivatives were synthesized and characterized, starting from the flavonoid molecules. The structures of the intermediates and the products are shown in Fig. 2 and Table 1. The NMR characterization data conformed with the structures of both the key intermediates and the final products. The modification process was designed based on the chemistry of the phenol hydroxyl groups. These hydroxyl groups were sequentially derivatized into the desired acetamide group through an ester, carboxylic acid and acyl chloride intermediates.
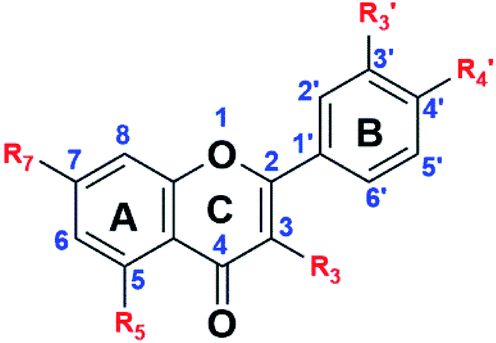 | ||
Fig. 2 General structure of the flavonoids, the intermediates and the final products. R3,  R5 and R7 variables are shown in Table 1. R5 and R7 variables are shown in Table 1. | ||
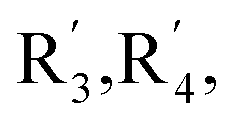 R5 and R7 variables in Fig. 2
R5 and R7 variables in Fig. 2
| R3 |
|
|
R5 | R7 | |
|---|---|---|---|---|---|
| 1S0 | OH | OH | OH | OH | OH |
| 1S1 | OCH2COOCH2CH3 | OCH2COOCH2CH3 | OCH2COOCH2CH3 | OCH2COOCH2CH3 | OCH2COOCH2CH3 |
| 1S2 | OCH2COOH | OCH2COOH | OCH2COOH | OCH2COOH | OCH2COOH |
| 1S3 | OCH2CONH2 | OCH2CONH2 | OCH2CONH2 | OCH2CONH2 | OCH2CONH2 |
| 2S0 | H | H | OH | OH | OH |
| 2S1 | H | H | OCH2COOCH2CH3 | OCH2COOCH2CH3 | OCH2COOCH2CH3 |
| 2S2 | H | H | OCH2COOH | OCH2COOH | OCH2COOH |
| 2S3 | H | H | OCH2CONH2 | OCH2CONH2 | OCH2CONH2 |

The hydroxyl groups in the quercetin and apigenin molecules, S0, were converted into ester intermediates, S1, through an ether-bond formation. This step was achieved through the Williamson ether synthesis. In step 2, the ester intermediate (S1) was converted into a carboxylic acid intermediate (S2) in an ester-hydrolysis reaction. In the final step, step 3, the carboxylic acid derivative was converted into an acyl chloride, which was subsequently amidated to achieve the acetamide derivative, S3. The general steps followed are shown in Scheme 1.
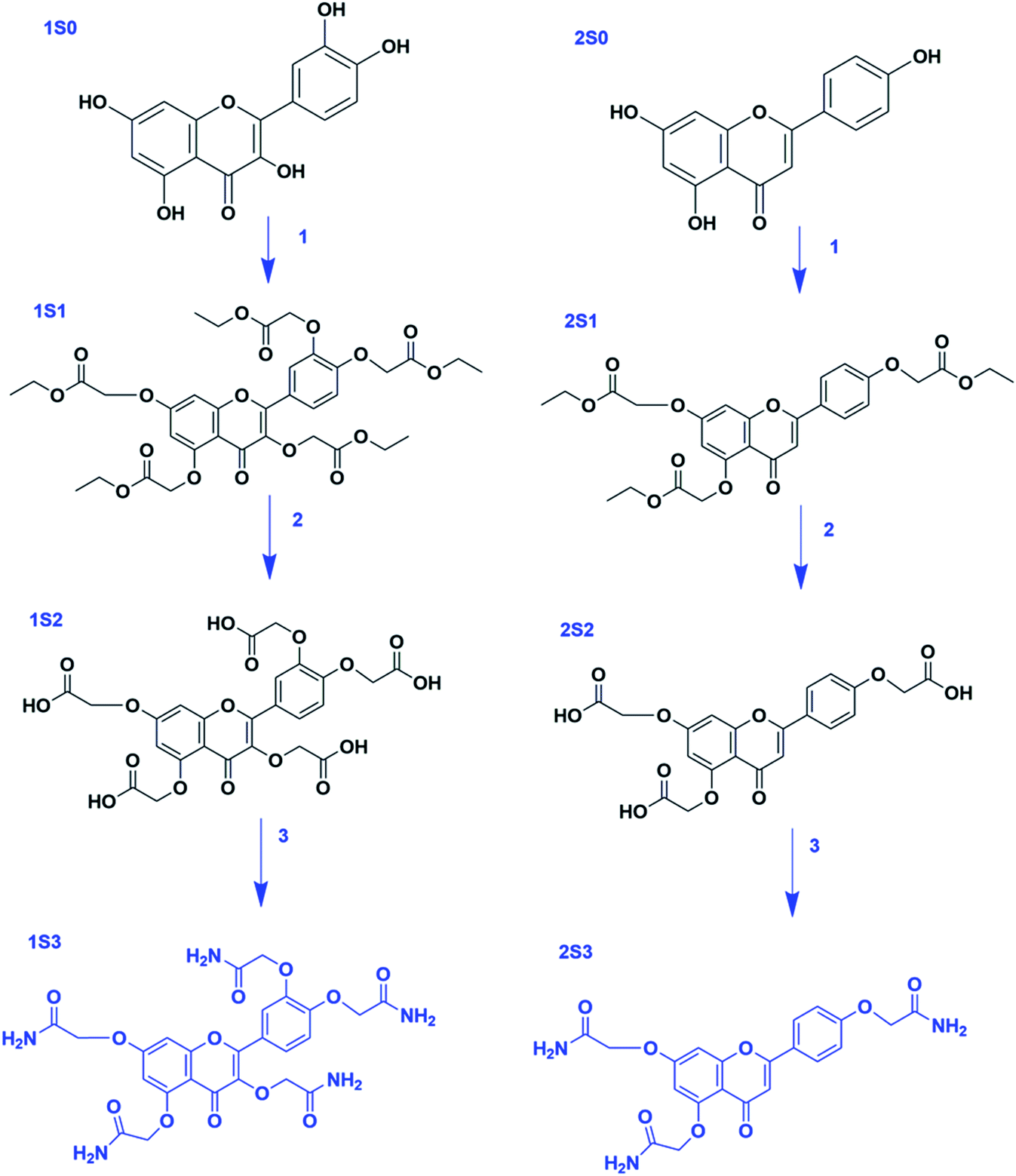 | ||
| Scheme 1 Steps in the synthesis of 1S3 & 2S3 derivatives. Reagents and reaction conditions: step 1 (S0 → S1) ethyl chloroacetate, K2CO3 (excess), DMF, rt. Step 2 (S1 → S2) LiOH·H2O, THF/H2O, rt. Step 3 (S2 → S3) SOCl2 (excess), reflux, 75 °C; NH4OH (excess), 0 °C – rt. | ||
Step 1 was achieved through the Williamson ether synthesis reaction. Hypothetically, this reaction proceeds through an SN2 reaction pathway. Anhydrous potassium carbonate was used to deprotonate the flavonoid hydroxyl groups yielding poly phenoxide ions. These anions served as the nucleophile, whereas ethyl chloroacetate served as the substrate for the reaction. Excess potassium carbonate was used to ensure optimum deprotonation of the flavonoid hydroxyl groups into the corresponding nucleophiles.
The S1 derivatives were confirmed using 1H and 13C NMR characterizations. In 1S1, 1H NMR confirmed 5 aromatic, 10 methylene (ether), 10 methylene (ester), and 15 methyl (ester) protons, by integration (Fig. S1†). 2S1 1H NMR integration confirmed 7 aromatic, 6 methylene (ether), 6 methylene (ester) and 9 methyl (ester) protons (Fig. S2†).
In step 2, the flavonoid-ester intermediate (S1) was converted into a flavonoid-carboxylic acid derivative (S2) through a saponification reaction. The ethoxy (–OCH2CH3) group of the flavonoid ester derivative was cleaved in an ester hydrolysis reaction. The formation of the S2 derivatives was confirmed using 1H NMR characterization. The 1H NMR integration confirmed 5-COOH protons, 5 aromatic protons and 10 methylene protons in 1S2, Fig. S3.† In 2S2, 3-COOH protons, 7 aromatic and 6 methylene protons were confirmed by the integration of the 1H NMR, Fig. S4.†
Step 3 involved the conversion of the S2 intermediate into the desired flavonoid acetamide derivative (S3). This was done by first converting the –COOH group into an acyl chloride (–COCl) by refluxing the carboxylic acid derivative with thionyl chloride. The acyl chloride intermediate, with a better leaving group, was finally converted into S3 product using an aqueous ammonium solution. The S3 product was characterized using 1H & 13C NMR. The 1H NMR confirmed the amide, aromatic and the methylene protons.
In 1S3, 10 amide (–CONH2) protons, 5 aromatic protons and 10 methylene protons were positively confirmed by the integration of the 1H NMR, Fig. S5.† The additional carbons the CH2 and the amide carbons were confirmed from the 13C NMR spectrum, as shown in Fig. S6.† The 2S3 1H NMR integration confirmed 6-CONH2, 7 aromatic and 6 methylene protons, Fig. S7.†
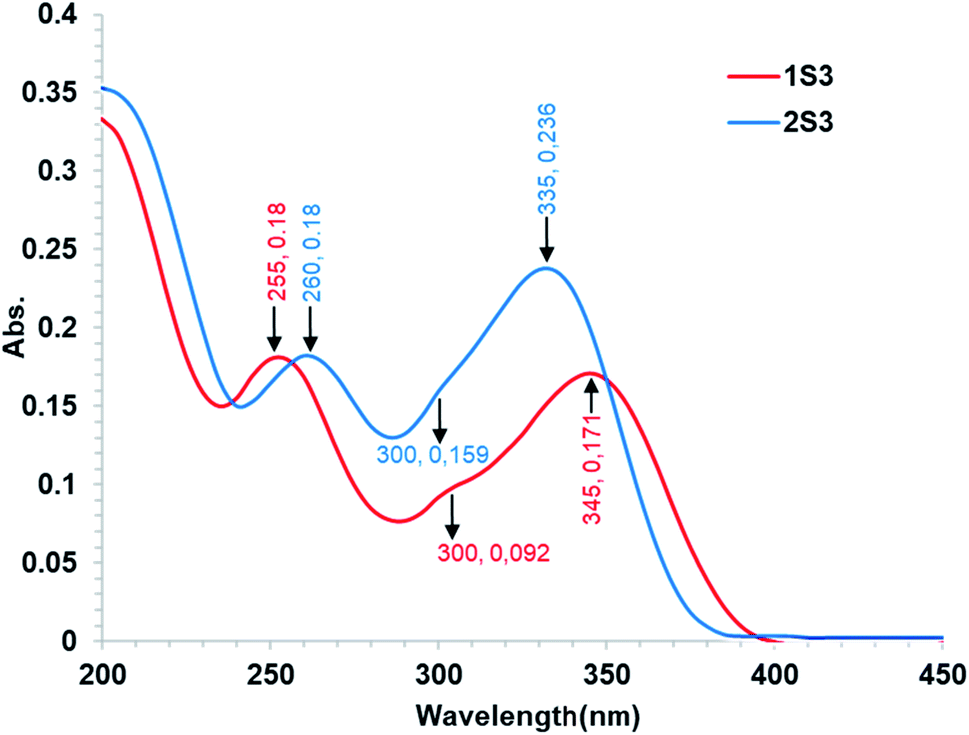 | ||
| Fig. 3 Absorption spectra of 10 μmol L−1 1S3 and 2S3 in Tris–HCl buffer (0.1 mol L−1, pH 7.4). | ||
O stretches were relatively identified in the FR-IR spectra. The ketone CO stretching absorptions were recorded at 1682 and 1674 cm−1 for 1S3 and 2S3, respectively. The amide CO stretching absorptions were recorded at 1601 and 1587 cm−1 for 1S3 and 2S3, respectively. The key FT-IR spectra peaks are shown in Table S1.† The FT-IR spectra of the two compounds are shown in Fig. S8.†3.2 Compound–DNA interaction study
To examine the interaction between the ligands with FS-DNA, 1S3 and 2S3 UV-Vis absorption spectra were recorded in the absence and presence of different DNA concentrations. The UV-Vis spectra of 1S3 and 2S3 in the absence and presence of FS-DNA are given in Fig. 4. In the absence of DNA, 1S3 showed two different absorbance peaks at 255 and 335 nm, respectively. As the absorption in the 255 nm band increased with the addition of DNA, the absorption at 335 nm decreased and a partial red shift was observed. Similarly, the absorption of the band at 260 nm in 2S3 increased with the addition of DNA, while the absorption of the band at 345 nm decreased and a slight redshift was observed. This can be explained by a decrease in the number of chromophores in the solution medium and conformational changes in the DNA as a result of DNA–ligand interaction. Furthermore, as a result of interaction with DNA in both compounds, approximately 300 nm isosbestic point occurred. This situation is a strong evidence48 of the formation of a new DNA–ligand complex on the interaction of compounds and DNA affinity. Based on variations in the absorbance bands, the binding constant (Kb) for the ligand–DNA was calculated using the Benesi–Hildebrand equation (eqn (1)) and the relevant values are summarized in Table 2. The binding constant was found to be 1.43 ± 0.3 × 104 M−1 for 1S3 and 2.08 ± 0.2 × 104 M−1 for 2S3. When these values are compared with the classical intercalation agent ethidium bromide, which is known to interact with DNA (although the EtBr–DNA (Kb = 1.4 × 106) complex is significantly lower than binding) it can be said that it falls within the range of groove binding51 constant from binding modes when DNA interacts with a small molecule.
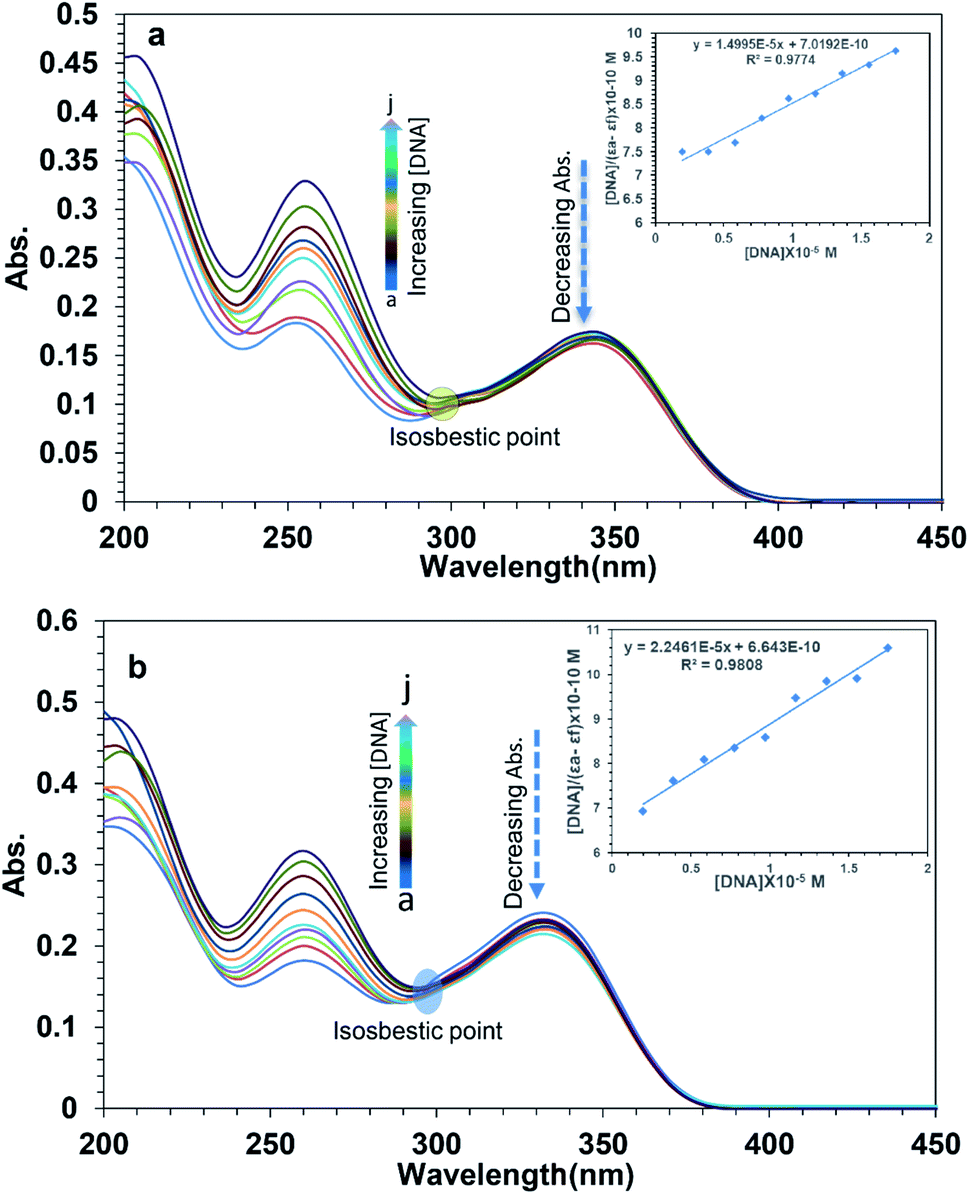 | ||
| Fig. 4 Absorption spectra of 10 μmol L−1 1S3 (a) and 10 μmol L−1 2S3 (b) in the presence of FS-DNA at different concentrations (a: 0.0, b: 1.94, c: 3.89 d: 5.84, e: 7.78, f: 9.73, g: 11.68, h: 13.63, i: 15.58, and j: 17.52 μmol L−1 in Tris–HCl buffer (0.1 mol L−1, pH 7.4)). The inset showing the plot of [DNA/(εa − εf)] versus [DNA]. | ||
| Property | 1S3 | 2S3 |
|---|---|---|
| Absorbance (nm) | 335 | 345 |
| Kb (M−1) | 1.43 ± 03 × 104 | 2.08 ± 02 × 104 |
| Hypochromism (%) | 22.54 | 20.91 |
| Isosbestic point (nm) | 300 | 301 |
| Shift (nm) | 3 | 3 |
The fluorescence spectra of 1S3 and 2S3 in the absence and presence of FS-DNA were shown in Fig. 5. As 1S3 and 2S3 are gradually titrated into the EtBr–DNA complex, the maximum emission of the solution is reduced. This data has been validated by the interaction of the ligands with FS-DNA, which causes the formation of non-fluorescent complexes in both compounds and quenches the endogenous fluorescence. According to the eqn (2), the KSV values of the compounds have been calculated to be 1.83 × 104 M−1 and 1.96 × 104 M−1 for 1S3 and 2S3, respectively. KSV values calculated from the Stern–Volmer equation have lower than the values of ethidium bromide, acridine orange, and methylene blue, which are well known to have intercalation interaction with DNA.53 Therefore, for both 1S3 and 2S3 compounds, it cannot be said that it interacts with DNA by intercalation and displaces EtBr.54 The decrease in the fluorescence intensity of EtBr–DNA is an indication that these compounds may be partially intercalated, but this interaction is also unstable in line with the KSV values obtained and the fluorescence intensity of magnitude.55
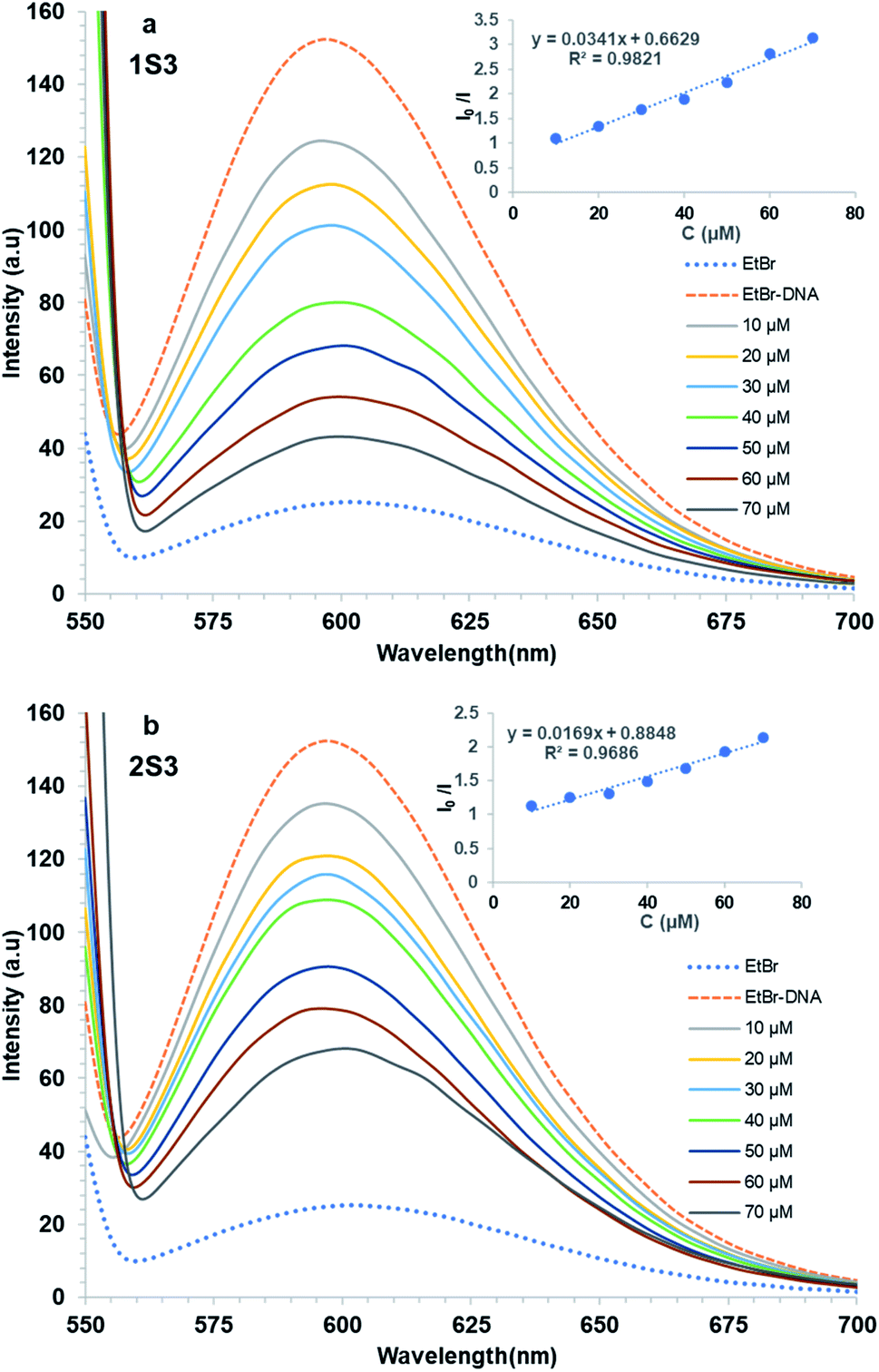 | ||
| Fig. 5 Quenched Fluorescence spectra of FS-DNA bound to the EtBr system with the addition of compounds 1S3 (a) and 2S3 (b). Inset: I0/I versus [DNA] (μM). | ||
3.3 Mutagenicity properties of the compounds
Salmonella, a microsome test system, is used to detect point mutations in DNA. In this test, TA98 and TA100 strains were obtained from the LT2 ancestral strain of Salmonella typhimurium by a mutation in vitro.56 This test was developed by Ames in 1971.34,42,56–58 Among these strains, TA100 had base change mutation, and TA98 had frameshift mutation. Due to these mutations, these strains are unable to synthesize histidine (his−). The principle of the experiment is based on the correction of these mutations by treating these mutated strains (TA98 and TA100) with various chemicals, i.e., calculating the histidine expression frequencies of the strains (his+). Point mutation and frameshift mutations are the basis of many genetic diseases observed in humans. There is strong evidence that it induces tumor formation in humans and animals due to base change mutations in tumor suppressor genes in both oncogenes and somatic cells.34 Frameshift mutations occur as a result of adding/removing the base as much as 1, 2, and its multiples occurring in DNA. The mRNA synthesized from this mutated DNA molecule and the protein chain synthesized then make major changes in the amino acid sequence (in the entire chain). Therefore, if a chemical frame-shift mutation is induced, major changes occur in the protein as a result. However, since point mutations occur as a result of the structural change of one of the bases in the DNA molecule, much fewer changes occur in the mRNA synthesized from this DNA molecule and subsequently the synthesized protein molecule. Compound 1S3 did not cause any detectable increase in the number of revertant colonies in both TA98 and TA100 strains, both in the presence (+S9 mixture) and the absence of the metabolic activation system (−S9 mixture). This shows that both the 1S3 and its metabolites do not have any mutagenic activity.Our study reveals that compound 2S3 did not cause an increase in the number of revertant colonies in the absence of a metabolic activation system for the TA98 strain. However, this compound caused a statistically significant increase in the number of revertant colonies in high doses (0.40, 0.20 and 0.10 mg per plate) in the presence of metabolic activation system.34 In addition, 2S3 significantly increased the number of revertant colonies for TA100 strain both in the presence and the absence of the activation system. This shows that both the 2S3 and its metabolites are potentially mutagenic for the TA100 strain at high doses. It is evident that strain TA98 shows frameshift mutation and strain TA100 shows base change mutation.58 In summary, 1S3 does not have any mutagenic potential in both TA98 and TA100 strains. Compound 2S3 does not show any mutagenic activity in the TA98 strain; however, its metabolites have a mutagenic potential at high doses. In addition, both the 2S3 itself and its metabolites have mutagenic activity in the TA100 strain. The values of both compounds are given in detail in Fig. 6 and Table 3. According to the data that we obtained from this study, we can conclude that compound 1S3 by itself and its metabolites are not mutagenic because they do not cause frameshifts and point mutations. On the other hand, compound 2S3 by itself does not induce the frame-shift mutation, but it induces metabolites at high doses. It further induces the point mutation of both 2S3 itself and its metabolites. Therefore, we can conclude that it has mutagenic potential.
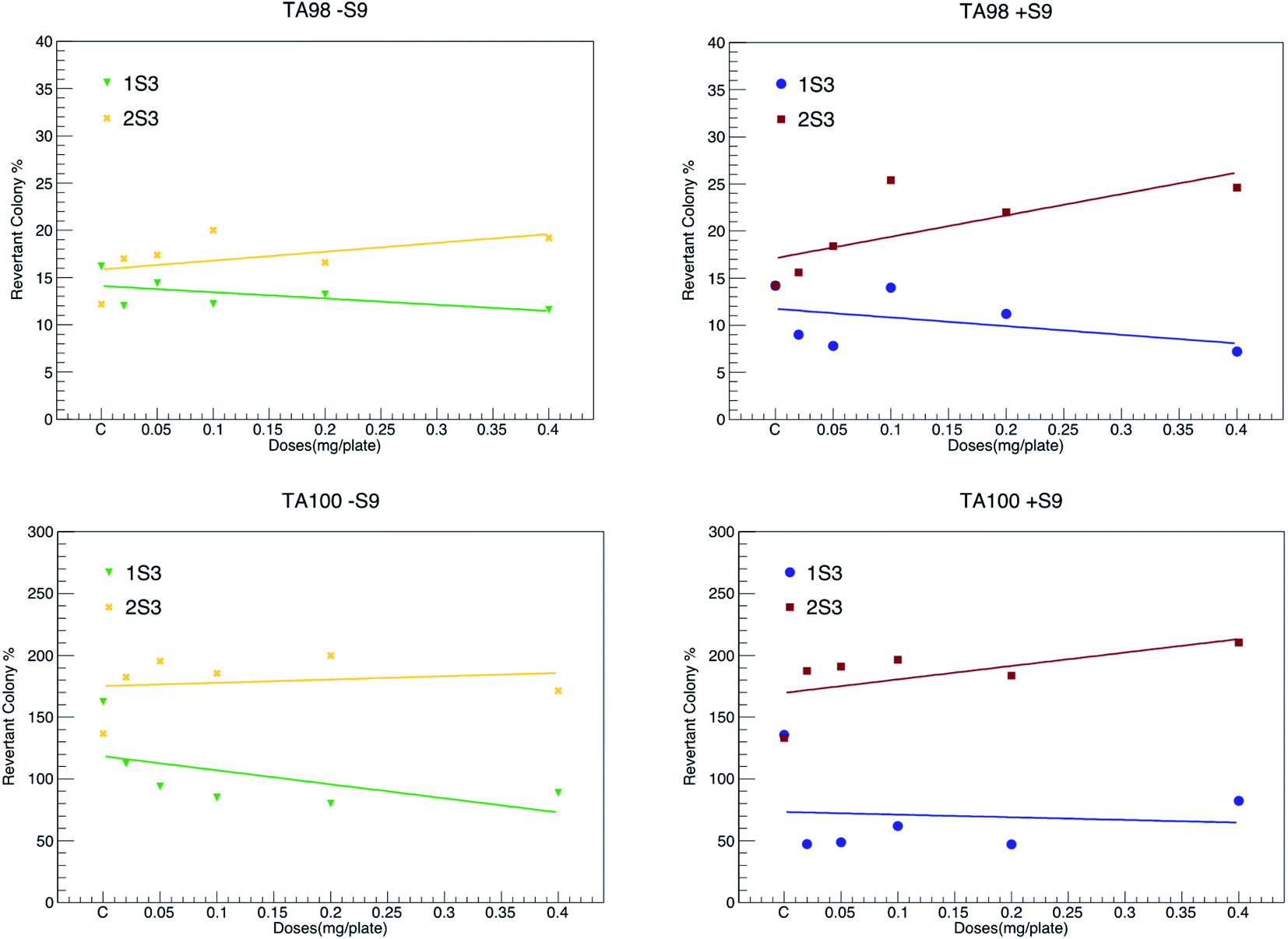 | ||
| Fig. 6 Comparison graphs showing the changes in revertant colony percentages in the presence (+S9) and absence (−S9) of the metabolic activation system for TA98 and TA100 strains of compounds 1S3 and 2S3. | ||
| Test substances | Concentration, mg per plate | TA98 | TA100 | ||
|---|---|---|---|---|---|
| −S9 | +S9 | −S9 | +S9 | ||
| a (a) Significant from control. (b) Significant from positive control, (1) P ≤ 0.05; (2) P ≤ 0.01; (3) P ≤ 0.001, NPD: 4-nitro-o-phenylenediamine, 2-AF: 2-aminoflourene, SA: sodium azide. | |||||
| Spontaneous control | — | 16.20 ± 1.77 | 14.20 ± 2.26 | 162.60 ± 12.23 | 135.80 ± 7.35 |
| NPD | 200 μg mL−1 | 4260 ± 291.75 | — | — | — |
| 2-AF | 20 μg mL−1 | — | 2504.60 ± 397.74 | — | 3207.40 ± 385.93 |
| SA | 1 μg mL−1 | — | — | 1497.2 ± 230.28 | — |
| 1S3 | 0.40 | 11.60 ± 1.63 a1b3 | 7.20 ± 1.68 a1b3 | 88.80 ± 14.07 a2b3 | 82.20 ± 5.37 a3b3 |
| 0.20 | 13.20 ± 1.01 a1b3 | 11.20 ± 2.97 b3 | 80.20 ± 10.65 a2b3 | 47.00 ± 6.73 a3b3 | |
| 0.10 | 12.20 ± 0.66 a2b3 | 14.00 ± 2.60 b3 | 85.00 ± 7.17 a3b3 | 62.00 ± 4.95 a3b3 | |
| 0.05 | 14.40 ± 2.20 b3 | 7.80 ± 1.11 a2b3 | 94.00 ± 8.43 a3b3 | 48.80 ± 12.85 a2b3 | |
| 0.02 | 12.00 ± 1.41 a1b3 | 9.00 ± 0.44 a3b3 | 112.40 ± 3.28 a3b3 | 47.00 ± 7.16 a3b3 | |
| Spontaneous control | 16.20 ± 1.77 | 14.20 ± 2.81 | 136.80 ± 5.95 | 133.20 ± 4.56 | |
| NPD | 3808.40 ± 221.78 | — | — | — | |
| 2-AF | — | 4135.80 ± 181.49 | — | 3518.40 ± 385.76 | |
| SA | — | — | 1506.40 ± 114.82 | — | |
| 2S3 | 0.40 | 19.20 ± 2.08 b3 | 24.60 ± 2.11 a2b3 | 171.40 ± 8.55 a1b3 | 210.40 ± 2.31 a3b3 |
| 0.20 | 16.60 ± 1.80 b3 | 22.00 ± 1.37 a2b3 | 200.00 ± 8.44 a2b3 | 183.60 ± 9.35 a2b3 | |
| 0.10 | 20.00 ± 2.25 b3 | 25.40 ± 1.83 a2b3 | 185.60 ± 7.20 a2b3 | 196.60 ± 15.75 a2b3 | |
| 0.05 | 17.40 ± 1.12 b3 | 18.40 ± 1.74 b3 | 195.40 ± 6.96 a3b3 | 190.80 ± 11.16 a2b3 | |
| 0.02 | 17.00 ± 1.00 b3 | 15.60 ± 2.52 b3 | 182.40 ± 9.15 a2b3 | 187.40 ± 18.93 a1b3 | |
3.4 Molecular docking studies
Molecular docking studies have gained an increasing interest in the research of the binding interaction mechanisms of biological macromolecules with small molecules. Molecular docking is a valuable technique in rational drug design to predict the most stable structure, binding tendency, and binding mode of the receptor–ligand complex so that interaction details can be better recognized and examined during new drug discovery and development process. This method is often used as virtual search tools in crucial steps of drug design and development. Molecular docking studies have been conducted to find the preferred location of ligands on DNA. In general, the more negative the binding energy resulting from the interaction, the more stable the small molecule and the complex formed by the DNA, the stronger the interaction between the small molecule and the DNA.30 The docking energy values and inhibition constants of the ligands during their interaction with DNA are given in Table 4.| Ligand | Binding affinity (kcal mol−1) | Kb (M−1) | Inhibition constant at 298.15 K (μM) |
|---|---|---|---|
| 1S3 | −7.9 | 1.74 × 104 | 1.92 |
| 2S3 | −9.1 | 2.23 × 104 | 2.34 |
According to the docking models, 1S3 binds to the major groove of DNA, while 2S3 binds to the minor groove. The binding affinities were found to be −7.9 and −9.1 kcal mol−1 according to the best pose conditions-for the 1S3 and 2S3, respectively. These scores relate to the minimum binding energy and the maximum docking cluster. These results are compatible with the spectroscopic results obtained. In addition, docking studies have shown that H-bond interactions play the main role in the stability of ligand–DNA complexes. The energetically most convenient conformation of the docked pose of DNA–ligand complexes is depicted in Fig. 7 as a 3D. When the 2D plot of the interaction between DNA with 1S3 and 2S3 is examined (Fig. 8), for 1S3, DNA appears to have 6 different H-bond interactions with both A and B chain. Considering the nucleotide-binding and the molecular structure of 1S3, it is seen that the hydrogen bonds formed in the 1S3–DNA complex occur via the nucleotides of the guanine and adenine and the oxygen in the amide groups of the compound. For 2S3, a significant change has occurred in the conformation of the molecule to exhibit the ability to bind along the minor groove, and this conformational change is closely related to the structure of the DNA minor groove. Besides, this situation can be described as an important indication that the DNA–2S3 complex is formed. The interaction between the ligand and DNA for the DNA–2S3 complex occurred via amide groups attached to the aromatic ring, as in 1S3. However, apart from the conventional hydrogen bond, carbon–H bond and Pi-donor-H bond interactions were observed. For the 1S3–DNA and 2S3–DNA complexes, the data showing the bonds, bond types, and bond lengths formed between the nucleotides and ligands are summarized in Table 5. For the 1S3–DNA and 2S3–DNA complexes, the docked model illustrations are given in detail in the ESI, Fig. S10–S19.†
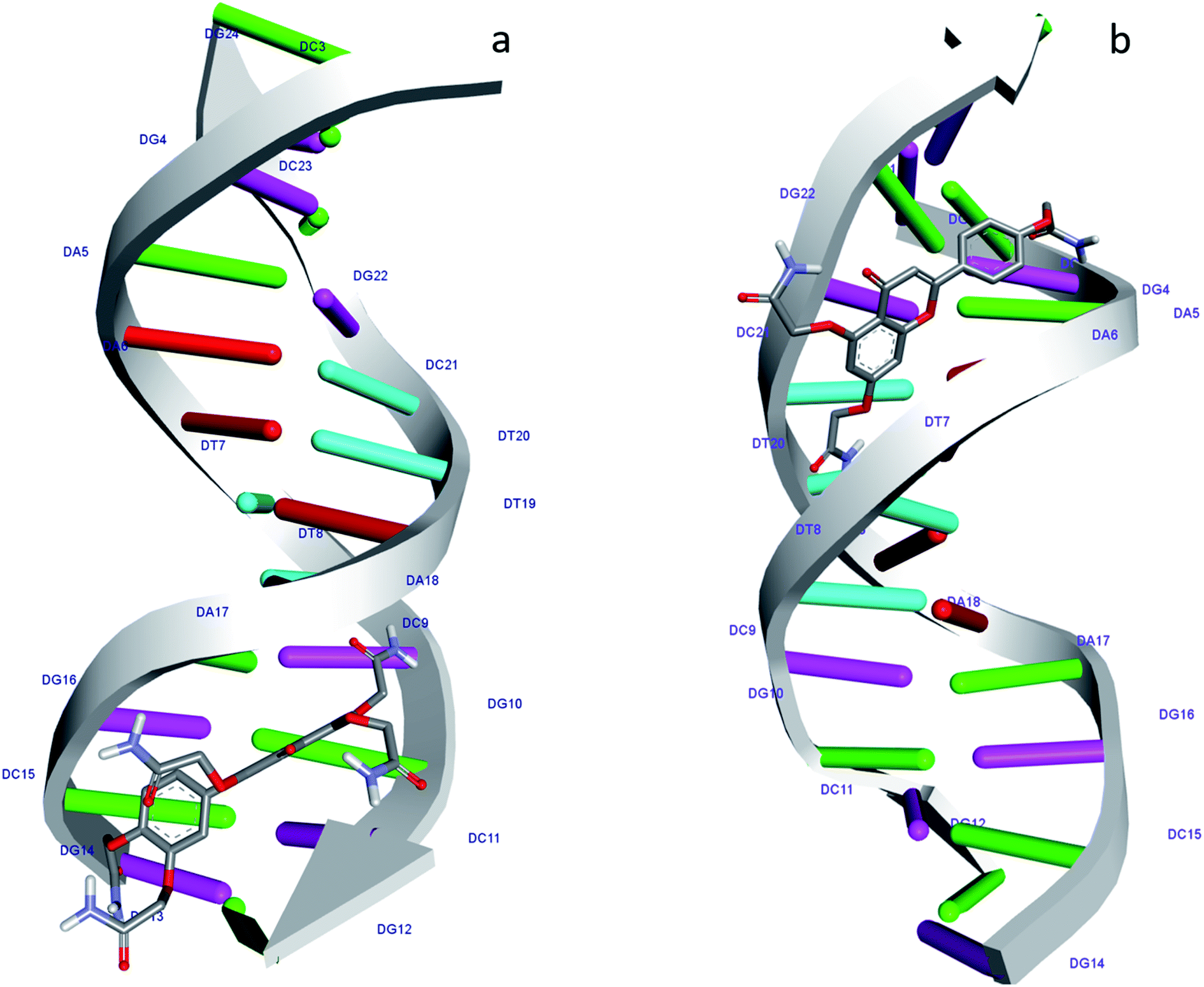 | ||
| Fig. 7 Molecular docked structure of 1S3 (a) and 2S3 (b) complexed with DNA. Surface representation showing the binding of 1S3 and 2S3 with the dodecamer duplex sequence, d(CGCGAATTCGCG)2 (PDB ID: 1BNA). | ||
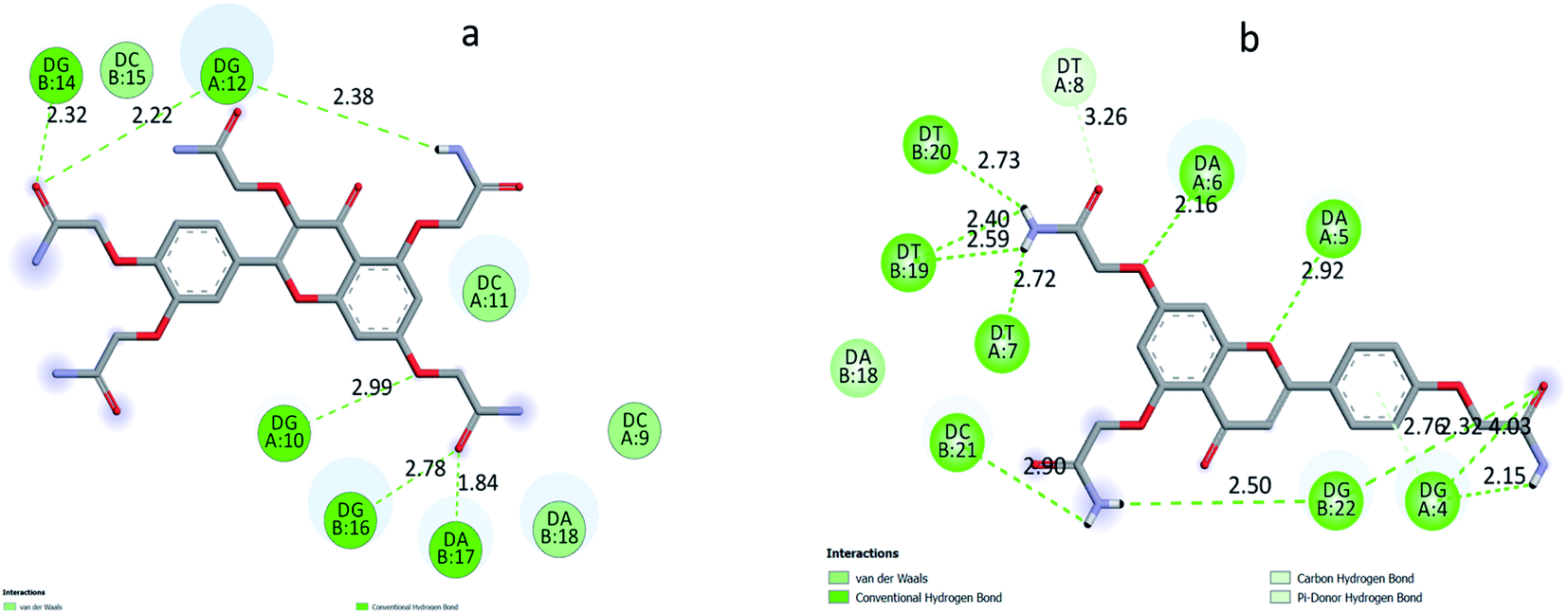 | ||
| Fig. 8 2D plot of interaction between the 1S3 (a) and 2S3 (b) with the dodecamer duplex sequence, d(CGCGAATTCGCG)2 (PDB ID: 1BNA). | ||
| Interactions | Distance | Bonding types | From | To | |
|---|---|---|---|---|---|
| 1S3–DNA complex | A:DG10:H3 – :1S3:O35 | 2.98657 | Conventional H bond | A:DG10:H3 | :1S3:O35 |
| A:DG12:H22 – :1S3:O30 | 2.21828 | Conventional H bond | A:DG12:H22 | :1S3:O30 | |
| B:DG14:H3 – :1S3:O30 | 2.32444 | Conventional H bond | B:DG14:H3 | :1S3:O30 | |
| B:DG16:H22 – :1S3:O39 | 2.77904 | Conventional H bond | B:DG16:H22 | :1S3:O39 | |
| B:DA17:H3 – :1S3:O39 | 1.84249 | Conventional H bond | B:DA17:H3 | :1S3:O39 | |
| :1S3:H57 – :1S3:O25 | 2.51626 | Conventional H bond | :1S3:H57 | :1S3:O25 | |
| :1S3:H60 – :1S3:O26 | 2.439 | Conventional H bond | :1S3:H60 | :1S3:O26 | |
| :1S3:H47 – A:DG12:OP1 | 2.37572 | Conventional H bond | :1S3:H47 | A:DG12:OP1 | |
| :1S3:H48 – :1S3:O11 | 2.24041 | Conventional H bond | :1S3:H48 | :1S3:O11 |
| Interactions | Distance | Types | From | To | |
|---|---|---|---|---|---|
| 2S3–DNA complex | :2S3:H42 – A:DG4:O4′ | 2.15093 | Conventional H bond | :2S3:H42 | A:DG4:O4′ |
| :2S3:H50 – A:DT7:O2 | 2.71946 | Conventional H bond | :2S3:H50 | A:DT7:O2 | |
| :2S3:H50 – B:DT19:O2 | 2.59064 | Conventional H bond | :2S3:H50 | B:DT19:O2 | |
| :2S3:H51 – B:DT19:O2 | 2.40101 | Conventional H bond | :2S3:H51 | B:DT19:O2 | |
| :2S3:H51 – B:DT20:O2 | 2.73073 | Conventional H bond | :2S3:H51 | B:DT20:O2 | |
| :2S3:H46 – B:DG22:OP1 | 2.49963 | Conventional H bond | :2S3:H46 | B:DG22:OP1 | |
| :2S3:H47 – :2S3:O11 | 1.92188 | Conventional H bond | :2S3:H47 | :2S3:O11 | |
| :2S3:H47 – B:DC21:O3′ | 2.90098 | Conventional H bond | :2S3:H47 | B:DC21:O3′ | |
| A:DG4:H22 – :2S3:O22 | 2.92642 | Conventional H bond | A:DG4:H22 | :2S3:O22 | |
| A:DG4:H3 –:2S3:O22 | 2.4405 | Conventional H bond | A:DG4:H3 | :2S3:O22 | |
| A:DA5:H3 – :2S3:O7 | 2.92123 | Conventional H bond | A:DA5:H3 | :2S3:O7 | |
| A:DA6:H3 – :2S3:O28 | 2.16167 | Conventional H bond | A:DA6:H3 | :2S3:O28 | |
| B:DG22:H22 – :2S3:O22 | 2.32328 | Conventional H bond | B:DG22:H22 | :2S3:O22 | |
| A:DT8:C5′ – :2S3:O32 | 3.25681 | Carbon–H bond | A:DT8:C5′ | :2S3:O32 | |
| A:DG4:H22 – :2S3 | 2.75747 | Pi-donor-H bond | A:DG4:H22 | :2S3 |
4. Conclusion
In the present study, we have successfully synthesized two new flavonoid acetamide derivatives namely: 2,2′,2′′-((2-(3,4-bis(2-amino-2-oxoethoxy)phenyl)-4-oxo-4H-chromene-3,5,7-triyl)tris (oxy))triacetamide (1S3) and 2,2′-((2-(4-(2-amino-2-oxoethoxy)phenyl)-4-oxo-4H-chromene-5,7-diyl)bis(oxy))diacetamide (2S3). To the best our knowledge, this is the first time the synthesis of these derivatives is being reported. DNA-binding of these compounds/ligands was investigated using fluorescence quenching and UV-Vis spectroscopy. Furthermore, the atomic details of these ligands–DNA interactions were investigated using the molecular docking method. The results based on the in silico DNA–ligand interaction studies show that 1S3 and 2S3 ligands are groove binders and that H-bond interactions have played a principal role in the stability of these ligand–DNA complexes. The present research exhibits the details of the mode of binding interaction, binding affinity, main interaction forces of ligands with FS-DNA and structure of ligand–FS-DNA complex. The flavonoid acetamide derivatives were tested for mutagenicity effect. Therefore, the results obtained from this study, focusing on the interaction mechanisms of flavonoid acetamide-derived ligands on FS-DNA, both experimental and molecular docking, will present useful information to entirely comprehend the mechanism of action and pharmacokinetics of these flavonoid derivatives.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge the National Science Foundation Grant # IOS-1543944 and the Bill & Melinda Gates Foundation for funding.References
- W. Ren, Z. Qiao, H. Wang, L. Zhu and L. Zhang, Med. Res. Rev., 2003, 23, 519–534 CrossRef CAS PubMed.
- T. P. T. Cushnie and A. J. Lamb, Int. J. Antimicrob. Agents, 2011, 38, 99–107 CrossRef CAS PubMed.
- R. M. De Conti Lourenço, P. Da Silva Melo and A. B. A. De Almeida, in Antifungal Metabolites from Plants, Springer-Verlag, Berlin Heidelberg, 2013, pp. 283–300 Search PubMed.
- A. García-Lafuente, E. Guillamón, A. Villares, M. A. Rostagno and J. A. Martínez, Inflammation Res., 2009, 58, 537–552 CrossRef PubMed.
- A. N. Panche, A. D. Diwan and S. R. Chandra, J. Nutr. Sci., 2016, 5(1) DOI:10.1017/jns.2016.41.
- T. Der Way, M. C. Kao and J. K. Lin, J. Biol. Chem., 2004, 279, 4479–4489 CrossRef PubMed.
- A. Kowalczyk, A. Bodalska, M. Miranowicz and K. Karłowicz-Bodalska, Adv. Clin. Exp. Med., 2017, 26, 1143–1146 CrossRef PubMed.
- X. Zhang, G. Wang, E. C. Gurley and H. Zhou, PLoS One, 2014, 9(9), e107072 CrossRef PubMed.
- R. Liu, H. Zhang, M. Yuan, J. Zhou, Q. Tu, J. J. Liu and J. Wang, Molecules, 2013, 18, 11496–11511 CrossRef CAS PubMed.
- A. Massi, O. Bortolini, D. Ragno, T. Bernardi, G. Sacchetti, M. Tacchini and C. De Risi, Molecules, 2017, 22, 1270 CrossRef PubMed.
- F. A. Ramos, Y. Takaishi, M. Shirotori, Y. Kawaguchi, K. Tsuchiya, H. Shibata, T. Higuti, T. Tadokoro and M. Takeuchi, J. Agric. Food Chem., 2006, 54, 3551–3557 CrossRef CAS PubMed.
- A. V. Anand David, R. Arulmoli and S. Parasuraman, Pharmacogn. Rev., 2016, 10, 84–89 CrossRef PubMed.
- J. Johari, A. Kianmehr, M. R. Mustafa, S. Abubakar and K. Zandi, Int. J. Mol. Sci., 2012, 13, 16020–16045 CrossRef PubMed.
- N. Rani, L. P. T. Velan, S. Vijaykumar and A. Arunachalam, Chin. J. Integr. Med., 2015, 9, 1–16 Search PubMed.
- M. Russo, C. Spagnuolo, I. Tedesco, S. Bilotto and G. L. Russo, Biochem. Pharmacol., 2012, 83, 6–15 CrossRef CAS PubMed.
- W. Wang, C. Sun, L. Mao, P. Ma, F. Liu, J. Yang and Y. Gao, Trends Food Sci. Technol., 2016, 56, 21–38 CrossRef CAS.
- D. Patel, S. Shukla and S. Gupta, Int. J. Oncol., 2007, 30, 233–245 CAS.
- M. Saibu, S. Sagar, I. Green, F. Ameer and M. Meyer, Anticancer Res., 2014, 34, 4077–4086 CAS.
- V. A. Okello, S. Mwilu, N. Noah, A. Zhou, J. Chong, M. T. Knipfing, D. Doetschman and O. A. Sadik, Environ. Sci. Technol., 2012, 46, 10743–10751 CrossRef CAS PubMed.
- F. J. Osonga, J. O. Onyango, S. K. Mwilu, N. M. Noah, J. J. J. J. Schulte, M. An and O. A. Sadik, Tetrahedron Lett., 2017, 58, 1474–1479 CrossRef CAS.
- F. J. Osonga, I. Yazgan, V. Kariuki, D. Luther, A. Jimenez, P. Le and O. A. Sadik, RSC Adv., 2016, 6, 2302–2313 RSC.
- F. J. Osonga, S. Kalra, R. M. Miller, D. Isika and O. A. Sadik, RSC Adv., 2020, 10, 5894–5904 RSC.
- F. J. Osonga, A. A. A. Akgul, I. Yazgan, A. A. A. Akgul, R. Ontman, V. M. M. Kariuki, G. B. Eshun and O. A. Sadik, RSC Adv., 2018, 8, 4649–4661 RSC.
- R. V. Patel, P. Kumari, D. P. Rajani and K. H. Chikhalia, Med. Chem. Res., 2013, 22, 195–210 CrossRef CAS.
- O. Kouatly, A. Geronikaki, C. Kamoutsis, D. Hadjipavlou-Litina and P. Eleftheriou, Eur. J. Med. Chem., 2009, 44, 1198–1204 CrossRef CAS PubMed.
- S. Harper, B. Pacini, S. Avolio, M. Di Filippo, G. Migliaccio, R. Laufer, R. De Francesco, M. Rowley and F. Narjes, J. Med. Chem., 2005, 48, 1314–1317 CrossRef CAS PubMed.
- P. S. Nayak, B. Narayana, B. K. Sarojini, K. Hegde and K. S. Shashidhara, Med. Chem. Res., 2014, 23, 4280–4294 CrossRef CAS.
- K. Al-Jorani, A. Rüther, M. Martin, R. Haputhanthri, G. B. Deacon, H. L. Li and B. R. Wood, Sensors, 2018, 18(12), 4297 CrossRef PubMed.
- R. Hajian, P. Hossaini, Z. Mehrayin, P. M. Woi and N. Shams, J. Pharm. Anal., 2017, 7, 176–180 CrossRef PubMed.
- J. H. Shi, K. L. Zhou, Y. Y. Lou and D. Q. Pan, Spectrochim. Acta, Part A, 2018, 193, 14–22 CrossRef CAS PubMed.
- N. Shahabadi, F. Shiri and S. Hadidi, Spectrochim. Acta, Part A, 2019, 219, 195–201 CrossRef CAS PubMed.
- R. K. Gilpin and C. S. Gilpin, Anal. Chem., 2011, 83, 4489–4507 CrossRef CAS PubMed.
- R. B. Silverman and M. W. Holladay, The Organic Chemistry of Drug Design and Drug Action, Elsevier, 3rd edn, 2014 Search PubMed.
- B. N. Ames, J. McCann and E. Yamasaki, Mutat. Res., Environ. Mutagen. Relat. Subj., 1975, 31, 347–363 CAS.
- K. Lee, J. H. Lee, S. K. Boovanahalli, Y. Jin, M. Lee, X. Jin, J. H. Kim, Y.-S. Hong and J. J. Lee, J. Med. Chem., 2007, 50, 1675–1684 CrossRef CAS PubMed.
- Y. Xia, M.-S. Won, J.-E. Kang, S.-K. Park, K.-H. Lee, H.-M. Kim and K. Lee, Bull. Korean Chem. Soc., 2010, 31, 3826–3829 CrossRef CAS.
- T. Li, G. Liu, H. Li, X. Yang, Y. Jing and G. Zhao, Bioorg. Med. Chem., 2012, 20, 2316–2322 CrossRef CAS PubMed.
- J. Lakshmipraba, S. Arunachalam, R. V. Solomon, P. Venuvanalingam, A. Riyasdeen, R. Dhivya and M. A. Akbarsha, J. Biomol. Struct. Dyn., 2015, 33, 877–891 CrossRef CAS PubMed.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- V. L. Elena and L. E. Vîjan, Optoelectron. Adv. Mater., Rapid Commun., 2009, 3, 60–64 Search PubMed.
- L. Liu, C. Zhang, Y. Yu and F. Chen, Microchim. Acta, 2018, 185(11), 514 CrossRef PubMed.
- D. M. Maron and B. N. Ames, Mutat. Res., Environ. Mutagen. Relat. Subj., 1983, 113, 173–215 CAS.
- M. M. 8. HyperChem™ Professional Chemistry Software Search PubMed.
- S. Dallakyan and A. J. Olson, in Global Food Security Governance, ed. J. E. Hempel, C. H. Williams and C. C. Hong, Springer New York, New York, NY, 2015, vol. 1263, pp. 243–250 Search PubMed.
- H. R. Drew, R. M. Wing, T. Takano, C. Broka, S. Tanaka, K. Itakura and R. E. Dickerson, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2179–2183 CrossRef CAS PubMed.
- S. K. Tripathi, R. Muttineni and S. K. Singh, J. Theor. Biol., 2013, 334, 87–100 CrossRef CAS PubMed.
- B. Li, D. H. Robinson and D. F. Birt, J. Pharm. Sci., 1997, 86, 721–725 CrossRef CAS PubMed.
- M. Sirajuddin, S. Ali and A. Badshah, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS PubMed.
- X. Xu, D. Wang, X. Sun, S. Zeng, L. Li and D. Sun, Thermochim. Acta, 2009, 493, 30–36 CrossRef CAS.
- A. Shah, E. Nosheen, S. Munir, A. Badshah, R. Qureshi, Z. U. Rehman, N. Muhammad and H. Hussain, J. Photochem. Photobiol., B, 2013, 120, 90–97 CrossRef CAS PubMed.
- J. H. Shi, J. Chen, J. Wang and Y. Y. Zhu, Spectrochim. Acta, Part A, 2015, 136, 443–450 CrossRef CAS PubMed.
- A. Jamshidvand, M. Sahihi, V. Mirkhani, M. Moghadam, I. Mohammadpoor-Baltork, S. Tangestaninejad, H. Amiri Rudbari, H. Kargar, R. Keshavarzi and S. Gharaghani, J. Mol. Liq., 2018, 253, 61–71 CrossRef CAS.
- S. Nafisi, A. A. Saboury, N. Keramat, J.-F. F. Neault and H.-A. A. Tajmir-Riahi, J. Mol. Struct., 2007, 827, 35–43 CrossRef CAS.
- Y. Sha, X. Chen, B. Niu and Q. Chen, Chem. Biodiversity, 2017, 14(10), e1700133 CrossRef PubMed.
- C. Gan, X. Huang, J. Zhan, X. Liu, Y. Huang and J. Cui, Spectrochim. Acta, Part A, 2020, 227, 117525 CrossRef CAS PubMed.
- L. Snyder, J. Peters, W. Champness and T. Henkin, Molecular Genetics of Bacteria, 2012 Search PubMed.
- K. Mortelmans and E. Zeiger, Mutat. Res., 2000, 455, 29–60 CrossRef CAS PubMed.
- A. Kayraldiz, F. F. Kaya, S. Canimoĝlu and E. Rencüzoĝullari, Ann. Microbiol., 2006, 56, 129–133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04559d |
| This journal is © The Royal Society of Chemistry 2020 |