
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective production of bio-based aromatics by aerobic oxidation of native soft wood lignin in tetrabutylammonium hydroxide†
Takashi Hosoya a,
Kohei Yamamotoa,
Hisashi Miyafuji*a and
Tatsuhiko Yamadab
a,
Kohei Yamamotoa,
Hisashi Miyafuji*a and
Tatsuhiko Yamadab
aGraduate School of Life and Environmental Sciences, Kyoto Prefectural University, 1-5 Shimogamo-hangi-cho, Sakyo-ku, Kyoto 606-8522, Japan. E-mail: miyafuji@kpu.ac.jp
bForestry and Forest Products Research Institute, 1 Matsunosato, Tsukuba, Ibaragi 305-8687, Japan
First published on 20th May 2020
Abstract
Aerobic oxidation of native soft wood lignin in an aqueous solution of Bu4NOH facilitates efficient production of vanillin (4-hydroxy-3-methoxybenzaldehyde), which is one of the platform chemicals in industry. Oxidation of Japanese cedar (Cryptomeria japonica) wood flour at 120 °C for 4 h under O2 in Bu4NOH-based aqueous solutions produced vanillin in 23.2 wt% yield based on the Klason lignin content of the starting material. This yield was comparable to that in alkaline nitrobenzene oxidation of the same material (27.2%), which indicated that our aerobic oxidation exploited the full potential of the wood flour for vanillin production. Further mechanical investigation with lignin model compounds suggested that the vanillin formation occurred mainly through following successive reactions: alkaline-catalyzed degradation of β-ether linkages in middle units of lignin polymer to form a glycerol end group, oxidation of the glycerol end group by O2 to a HCα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety, and release of vanillin from the HCαO end. One of the reasons for the high performance of Bu4NOH for the vanillin production was explained by the general understanding in organic chemistry that Bu4OH is a stronger base than simple alkali, e.g. NaOH. The other more fundamental mechanical aspect was that Bu4N+ suppressed disproportionation of the vanillin precursor (the CαHO end group) probably due to strong interaction between the cation and the HCαO end group.
O moiety, and release of vanillin from the HCαO end. One of the reasons for the high performance of Bu4NOH for the vanillin production was explained by the general understanding in organic chemistry that Bu4OH is a stronger base than simple alkali, e.g. NaOH. The other more fundamental mechanical aspect was that Bu4N+ suppressed disproportionation of the vanillin precursor (the CαHO end group) probably due to strong interaction between the cation and the HCαO end group.
Introduction
Vanillin (3-methoxy-4-hydroxybenzaldehyde) is a major target compound to be produced from soft wood lignin. Quite a few lignin depolymerization methods such as degradation with various oxidants,1–14 catalytic degradation,6,12–15 and pyrolytic processes16 make it clear that vanillin is one of the major minima in the potential energy map in those lignin degradation reactions. Also, vanillin is a platform compound in the chemical industry, and is a source of several medicines, polymer materials, etc.17,18Until the 1990s industrial vanillin production was based mainly on aerobic oxidation of lignosulfonate, a major component of waste water from sulfite pulping.3,6,19–21 This vanillin production was carried out in alkaline media under compressed air with the yield of vanillin being ∼5% based on the original lignosulfonate.3,6,7 However, management of waste water containing sulfur compounds is a major problem of this biomass-based vanillin production process. Due to this drawback, recent vanillin production is almost completely substituted with phenol-based ones, where phenol is manufactured from fossil resources. Considering recent environmental requirements, however, the biomass-based vanillin production is worth being re-investigated and re-developed.
Our previous studies have reported that selective vanillin production can be made by aerobic oxidation of several soft wood lignin samples, e.g. soft wood flour, milled wood lignin, and several types of technical lignins including lignosulfonate, at 120 °C in the presence of tetrabutylammonium ion Bu4N+ and OH−.5,8 When Japanese cedar (Cryptomeria japonica) wood flour is degraded in 1.25 mol L−1 Bu4NOH aq. at 120 °C under air, vanillin and vanillic acid are produced in 15.4 and 3.9%, respectively, based on the Klason lignin content of the wood.8 These yields are considerably higher than those obtained in NaOH aq. with the same OH− concertation,5,8 indicating that the Bu4NOH solution exhibits much better performance for vanillin production than the corresponding NaOH aq.
The Bu4NOH-based oxidation method has several advantages over the process with lignosulfonate: this method employs reaction temperature (120 °C) significantly lower than that of the lignosulfonate process (∼170 °C);3 the process can utilize non-sulfur containing low materials such as wood flour and soda-lignin as well as lignosulfonate. Also, the use of molecular oxygen as an ideal oxidation reagent in this process should be highly emphasized.4b On the other hand, there are several issues to be further managed: considerably long reaction times (up to 72 h) and unclear mechanical aspects of the reaction involved in the aerobic oxidation, especially the roles of Bu4N+ in overall reactions. It is also noted that the highest yield of the target compound obtained with the Bu4NOH-based method is still lower than those obtained in the alkaline nitrobenzene (AN) oxidation,8 which is used as a benchmark to evaluate the performance of lignin oxidation processes.
The first half of this article handles the above issues of reaction time and the yields of vanillin and vanillic acid. We will report the reaction time required to reach the maximum vanillin and vanillic acid yields is significantly shortened along with improved yields of the compounds, when the reaction was carried out with the introduction of pure O2 to the reaction system. In the second haft of this article, we will report results obtained with lignin model compounds (see Scheme 1 for their structures) and discuss the reasons for the high performance of Bu4NOH, including the roles of Bu4+ in the oxidation process.
 | ||
| Scheme 1 β-O-4 type dimeric lignin model compounds employed in this study. | ||
Experimental
Materials
1.5 mol L−1 Bu4NOH aq. was purchased from the Sigma-Aldrich Co. The solution was diluted to 1.25 mol L−1 with distilled water and used as reaction solution. Vanillin, vanillic acid, veratraldehyde, veratric acid, veratryl alcohol, and sodium hydroxide were purchased from FUJIFILM Wako Pure Chemical Co. The particle size of Japanese cedar wood flour was 90–180 μm. Lignin model compounds, veratryl glycerol-β-guaiacyl ether (LM1) and guaiacyl glycerol-β-guaiacyl ether (LM2), were purchased from Tokyo-Kasei Co. and used without purification (see Scheme 1 for the structures of LM1 and LM2).Degradation of samples and analysis of reaction mixture
Staring material (the wood flour or the lignin model compound, 3.0–20 mg) was placed in a 10 mL glass tube. After adding the reaction solution (1.25 mol L−1 Bu4NOH aq. or 1.25 mol L−1 NaOH aq., 1.0–2.0 mL), the tube was tightly sealed. In several experiments, the concentration of OH− of the Bu4NOH aq. was increased to 3.75 mol L−1 by the addition of solid NaOH (200 mg in the case of 2.0 mL of the reaction solution). In the experiments that were performed under O2 or N2, the tube was flushed with the corresponding gas and immediately sealed. The tube was then heated at 120 °C in an oil bath with stirring. At a certain reaction time, the tube was cooled with cold water and then 1,5-dihydroxy-1,2,3,4-tetrahydronaphthalene/ethanol solution (2.0 g L−1, 800 μL) was added as an internal standard. A sample (50 μL) of the resulting reaction mixture was taken and added to acetonitrile (450 μL) containing 1.5% acetic acid. The solution was filtered and used directly for high-performance liquid chromatography (HPLC) analysis. The conditions for the HPLC analysis were described in our previous studies.5,8Gas chromatography/mass spectrometric (GC/MS) analysis was also carried out for the reaction mixture. Prior to the GC/MS analysis, the pH of the reaction solution (1.0 mL) after the degradation experiment was adjusted to ∼5 by the addition of saturated citric acid aq. The solution was then extracted with ethyl acetate (2.0 mL) and the organic layer was dried with anhydrous Na2SO4 and evaporated under reduced pressure. The residue was acetylated in 1.0 mL of acetic anhydride/pyridine (1/1, v/v) and directly used for the GC/MS analysis. The GC/MS analysis was carried out with Shimadzu GCMS-QP2010 ultra apparatus under the following conditions: column, Agilent J&W DB-5ms; injector temperature, 250 °C; profile of column oven temperature, 60 °C (1 min), 60 → 300 °C (1 → 48 min), 300 °C (10 min); carrier gas, helium; flow rate, 1.5 mL min−1; emission current, 20 μA; ionization time, 2.0 ms.
Identification of enol ethers, 2-methoxy-4-[2-(2-methoxyphenoxy)vinyl]phenol
Model compound LM2 (20 mg) was degraded in 1.25 mol L−1 Bu4NOH aq. (2.0 mL) at 120 °C for 1 h according to similar manner described above. After cooling the tube with cold water, saturated citric acid aq. was added to the reaction solution to adjust the pH to ∼5. The reaction mixture was then extracted with ethyl acetate (6 mL) and the organic layer was dried with anhydrous Na2SO4. Evaporation of ethyl acetate under reduced pressure gave crude target compounds. The products were purified with silica gel column with the eluent being ethyl acetate/n-hexane (1/1, v/v) to obtain cis/trans-2-methoxy-4-[2-(2-methoxyphenoxy)vinyl]phenol (EEs, 8.0 mg). trans-2-Methoxy-4-[2-(2-methoxyphenoxy)vinyl]phenol: 1H NMR (400 MHz, 1.25 mol L−1 NaOD/D2O): δ (ppm) = 3.50 (s, 3H), 3.62 (s, 3H), 6.00 (d, J = 11.8 Hz, 1H), 6.33 (d, J = 9.5 Hz, 1H), 6.54 (dd, J1 = 8.0 Hz, J2 = 2.1 Hz, 1H), 6.65 (d, J = 1.5 Hz, 1H), 6.78 (dt, J1 = 7.1 Hz, J2 = 2.1 Hz, 1H), 6.83 (d, J = 12.0, 1H), 6.90 (m, 3H). cis-2-Methoxy-4-[2-(2-methoxyphenoxy)vinyl]phenol: 1H NMR (400 MHz, 1.25 mol L−1 NaOD/D2O): δ (ppm) = 3.48 (s, 3H), 3.64 (s, 3H), 5.47 (d, J = 6.8 Hz, 1H), 6.22 (d, J = 6.8 Hz, 1H), 6.34 (d, J = 8.3 Hz, 1H), 7.16 (d, J = 1.5 Hz, 1H), the other protons were not discernable due to their overlapping with the signals from the trans-isomer.The HPLC analysis was performed for isolated EEs under the analytical conditions identical to those presented above to the same internal standard being employed. The obtained calibration curves were used for quantification of EEs formed by the degradation of LM2.
Results and discussion
Vanillin production from wood flour with introduction of O2
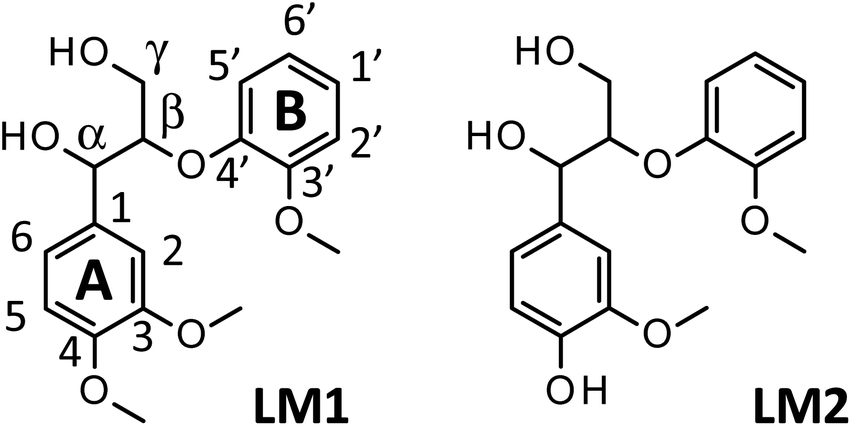
Our previous study reported that aerobic oxidation of the wood flour (14 mg) in 1.25 mol L−1 Bu4NOH aq. (2.0 mL) under air for 43 h produced vanillin and vanillic acid in 15.4 and 3.9 wt% yields, respectively, based on the Klason lignin amount of the wood flour.8 The 1.25 mol L−1 aqueous solution of Bu4NOH is the molten form of the Bu4NOH·30H2O crystal. To further enhance the degradation, we carried out similar degradation experiments after flushing the reaction system (sealed 10 mL test tube) with pure O2. We will hereafter call this “degradation under O2” for convenience, although the atmosphere of the reaction system is not fully substituted with O2. Note that 1.25 mol L−1 is the best concentration of Bu4N+ in terms of vanillin production according to our previous study:5 increase in [Bu4N+] to more than 1.25 mol L−1 scarcely boosts the vanillin yield and also results in degradation of Bu4N+ probably into BuOH and Bu3N.Fig. 1A shows changes in the yield of vanillin during the degradation in the Bu4NOH aq. under O2 along with our previous results obtained under air in the same reaction medium (Fig. 1C). It becomes clear that the vanillin formation is significantly faster under O2: the time required for reaching the maximum vanillin yield was 8 h under O2 whereas the reaction under air needed 43 h. The maximum yield of vanillin was also increased to 19.8 wt% (8 h) from 15.4 wt% (43 h) by the O2 substitution. After reaching the maximum yield in 8 h, the yield of vanillin was decreased to 17.0 wt% at 20 h and this degradation of vanillin was more significant under O2 than under air. This indicates that vanillin is somewhat reactive toward O2 and reaction time should be carefully controlled when the vanillin production is carried out under O2 instead of air.
 | ||
| Fig. 1 Changes with time in yields of vanillin (●), vanillic acid (■), and their total yields (△) during degradation of Japanese cedar wood flour (14 mg) in 1.25 mol L−1 Bu4NOH aq. (2.0 mL) under O2 (A), under O2 with the addition of NaOH(s) (200 mg) (B), and under air8 (C) at 120 °C. The total amount of Klason lignin and acid-soluble lignin of the wood flour was 34.3 wt%. | ||
Fig. 1 also shows the yield of vanillic acid (3-methoxy-4-hydroxybenzoic acid) along with that of vanillin, as vanillic acid is the second major product, as reported previously.5,8 The reaction under O2 tends to form more vanillin than vanillic acid, when it is compared to the one under air (Fig. 1A and C). It is thus stated that O2 introduction made the reaction more vanillin selective. One of the possible reasons for this selectivity change is that vanillic acid is more reactive toward O2 than vanillin and readily release CO2. As shown in Fig. 1A, the maximum yield of vanillic acid was achieved at 4 h in the Bu4NOH aq. under O2 and then the yield gradually decreased. Note that increase in the yield of vanillic acid was not observed during the degradation of vanillin from 8 to 20 h, which suggested oxidation of vanillin to vanillic acid was not important under the employed conditions.
We then carried out the degradation of the wood flour (14.0 mg) in 1.25 mol L−1 Bu4NOH aq. (2.0 mL) under O2 with the addition of NaOH(s) (200 mg) to increase the OH− concentration of the reaction solution to 3.75 mol L−1, as vanillin formation is significantly enhanced in stronger alkaline media.5 In this alkaline-enhanced solution, as shown in Fig. 1B, the reaction time required for the maximum yield of vanillin was further shortened to 4 h. The yield of vanillin was also improved to 23.2 wt% by the addition of NaOH. We thus tried further addition of NaOH to the solution, but when the concentration of OH− was set at 5.0 mol L−1 by the addition of 300 mg of NaOH, significant decrease in the vanillin yield was observed.
Table 1 summarizes the maximum yield of vanillin obtained from the wood flour in the above Bu4NOH-based reaction solutions along with that obtained in alkaline nitrobenzene (AN) oxidation of the same wood flour. AN oxidation is one of the most selective methods to convert various lignin samples to the corresponding monomeric benzaldehydes (vanillin in the case of soft wood lignins) and used as a benchmark to measure the performance of lignin conversion methods. The maximum vanillin yield (23.0 wt%) and the shortest reaction time (4 h) were both achieved when the reaction was carried out under O2 in the Bu4NOH aq. with the addition of NaOH. Considering that our previous study about the reaction under air required 43 h reaction time to achieve much lower maximum vanillin yield (15.4 wt%), the O2 introduction investigated in this study had drastic effects for improving the performance of our vanillin production method. Also, the 23.0 wt% vanillin yield obtained in the NaOH-added Bu4NOH aq. under O2 was comparable to those in AN oxidation (Table 1). This also indicates that our Bu4NOH-based reaction system well exploits vanillin forming potential of the wood flour.
| Medium | Atmosphere | Reaction timea (h) | Yield (wt%) | ||
|---|---|---|---|---|---|
| Vanillin | Vanillic acid | Total | |||
| a The reaction times that result in the maximum vanillin yield are selected for each column. For AN oxidation, the reaction time was 2.5 h according to the standard procedure.b Reaction solution was prepared by the addition of NaOH(s) (200 mg) to 1.25 mol L−1 Bu4NOH aq. (2.0 mL).c These are the results presented in our previous study,8 where 1.25 mol L−1 Bu4NOH aq. (2.0 mL) was used without the NaOH addition. | |||||
| Bu4NOH aq. + NaOH(s)b | O2 | 4.0 | 23.2 | 1.2 | 24.4 |
| Bu4NOH aq.8c | Air | 43 | 15.4 | 3.9 | 19.3 |
| AN oxidation8 | Air | 2.5 | 27.2 | 1.2 | 28.4 |
Mechanical investigation with lignin model compounds
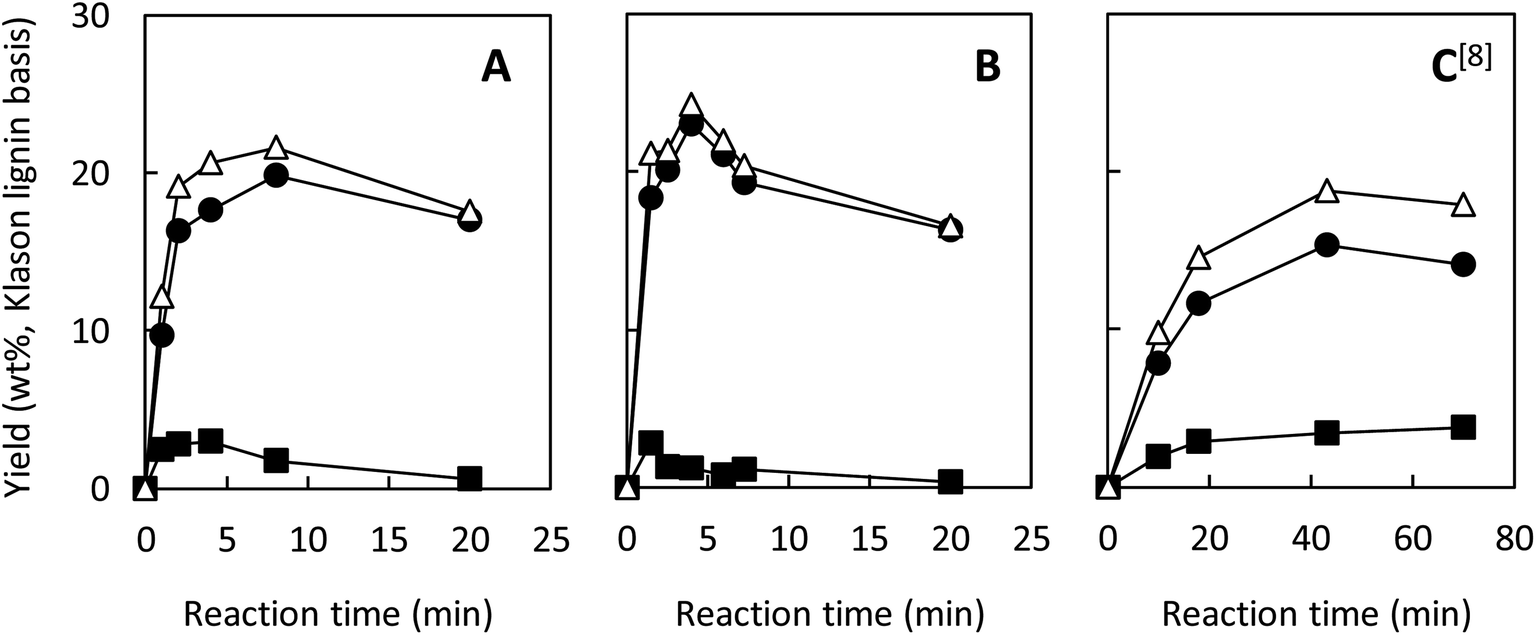
We have so far shown that vanillin is effectively produced by aerobic oxidation of the wood flour in the Bu4NOH-based solutions. Since such effective vanillin production cannot be achieved in a simple alkali solution (e.g. NaOH aq.) with the same OH− concentration as that of the Bu4N+-based one,5,8 it is clear that chemical nature specific for Bu4NOH plays pivotal roles in the vanillin formation. To investigate the mechanisms underlying the vanillin production in the Bu4NOH solution, degradation of several types of lignin model compounds (mainly LM1 and LM2, see Scheme 1) was carried out in 1.25 mol L−1 Bu4NOH and NaOH solutions and the reaction behaviors were carefully compared. In these model experiments, we carried out the degradation under air (not under O2) to minimize degradation of the products.Fig. 2 presents a HPLC chromatogram of the reaction mixture obtained from the degradation of LM1 (20 mg) in the Bu4NOH aq. (2.0 mL) under air, where β-O-4 is the most abundant linkage type in native lignin. LM1 formed vanillin along with veratraldehyde (3,4-dimethoxybenzaldehyde), veratryl alcohol (3,4-dimethoxybenzyl alcohol), veratric acid (3,4-dimethoxybenzoic acid), and guaiacol. The compounds with the veratryl moiety and vanillin are most likely to be formed from the A ring of LM1 and guaiacol is derived from the B ring (see Scheme 1 for the definition of the A- and B-rings).
 | ||
| Fig. 2 HPLC chromatogram of the reaction mixture obtained after the degradation of LM1 (20 mg) in 1.25 mol L−1 Bu4NOH aq. (2.0 mL) under air for 4 h. | ||
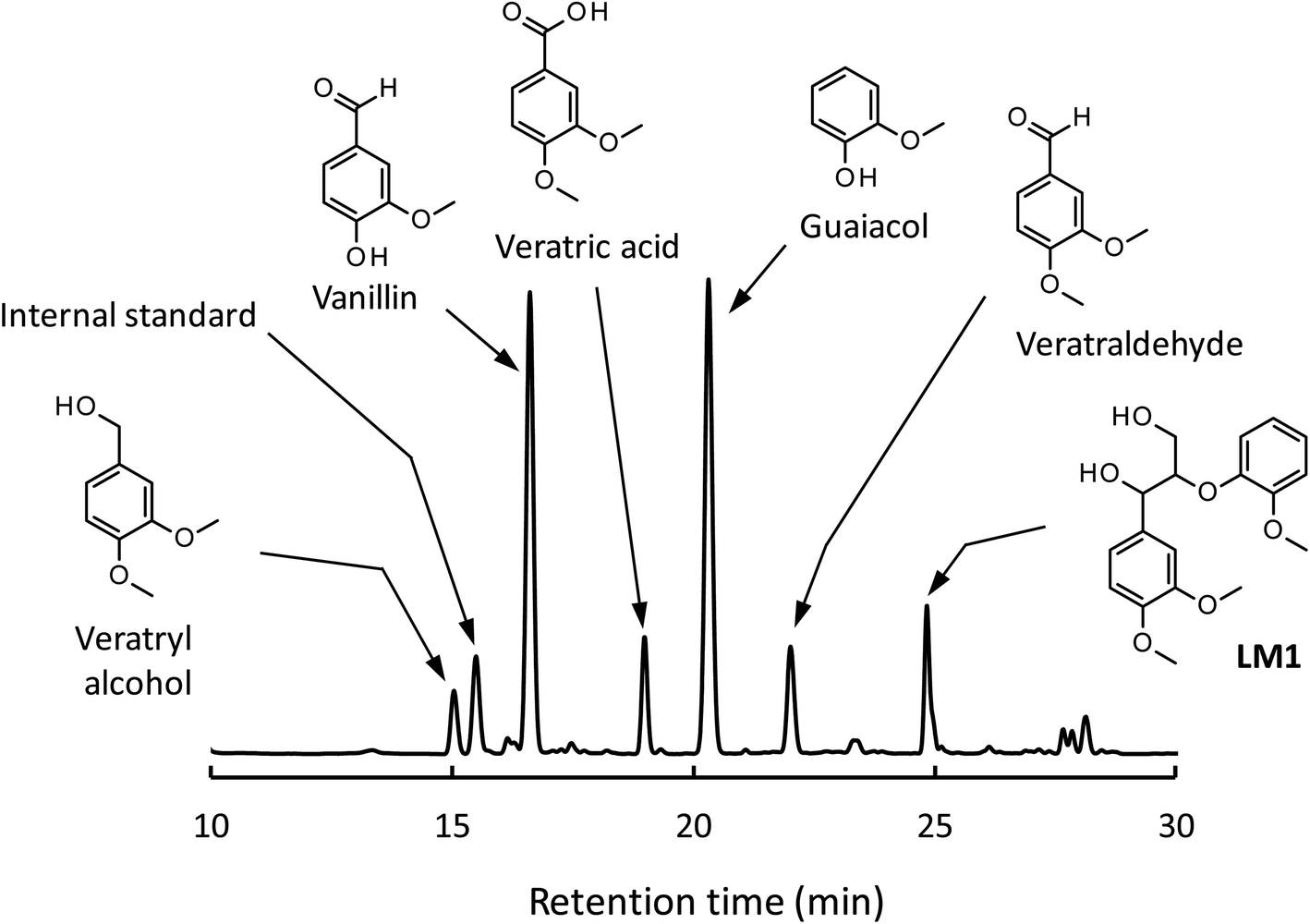
As shown in Fig. 3A, degradation of LM1 for short reaction times gave guaiacol and small amount of the A-ring-derived compounds. At the 1 h reaction time (the second plot from the left), for instance, guaiacol was formed in 12.3 mol% yield and the A-ring-derived compounds, mainly veratraldehyde, were produced in 7.2 mol% yield, with 86.4% of LM2 remaining intact. This suggests that the initial step of the degradation is alkaline-catalyzed β-ether cleavage shown as pathway A in Scheme 2, as reported in many studies.22–25 This β-ether cleavage results in the formation of veratryl glycerol from the A-ring along with the releasement of guaiacol from the B-ring. Veratryl glycerol was not detected in the HPLC analysis probably due to its very high polarity, but the presence of significant amount of the compound in the reaction mixture was strongly suggested from the GC/MS analysis of the acetylated reaction mixture (see Fig. S1 in ESI†). The yields of the A-ring-derived compounds were gradually increased at prolonged reaction times (Fig. 3A). Veratraldehyde was predominant in the A-ring derived products until 6 h and vanillin then became the major product with almost full consumption of LM1. At 43 h vanillin and veratraldehyde were finally formed in 15.7 mol% and 2.2 mol% yields, respectively, with the LM1 recovery being 3.7%. Note that the required 43 h reaction time is in good agreement with the experimental fact that vanillin yield became almost constant after 43 h in the degradation of the wood flour in the Bu4NOH aq. under air (see Fig. 1C).8
 | ||
Fig. 3 Changes with time in recovery of LM1 ( ), yields of guaiacol ( ), yields of guaiacol ( ), veratraldehyde ( ), veratraldehyde ( ), vanillin ( ), vanillin ( ), veratric acid ( ), veratric acid ( ), veratryl alcohol ( ), veratryl alcohol ( ), and the total yield of the A-ring-derived products ( ), and the total yield of the A-ring-derived products ( ) during degradation of LM1 (20 mg) at 120 °C under air in 1.25 mol L−1 Bu4NOH aq. (2.0 mL) (A) and in 1.25 mol L−1 NaOH aq. (2.0 mL) (B). ) during degradation of LM1 (20 mg) at 120 °C under air in 1.25 mol L−1 Bu4NOH aq. (2.0 mL) (A) and in 1.25 mol L−1 NaOH aq. (2.0 mL) (B). | ||
 | ||
| Scheme 2 Reaction pathways from LM1 to major products detected in this study. | ||
The above results suggest that, as presented in Scheme 2, veratryl glycerol formed by the β-ether cleavage is gradually oxidized into veratraldehyde by O2 via pathway B. However, our attempts to detect any intermediates in this aerobic oxidation step were not successful. Veratraldehyde then undergoes a demethylation reaction on the 4-OMe group to finally form vanillin via pathway C. The detailed mechanism of this demethylation is not clear at the moment. One of the possibilities is a simple SN2 reaction on the methyl group by OH−, but a mechanism involving a nucleophilic addition of OH− on the 4-position of the benzene ring followed by the elimination of MeO− cannot be ruled out due to the presence of a strong electron-withdrawing CHO group at the 1-position. It is also noted that the demethylation of the 3-OMe group is disfavored because of the lack of the influence of the CHO group at the 3-position.
Fig. 2 and 3 indicate the reaction mixture also contains considerable amount of veratric acid and veratryl alcohol. It is likely that these two compounds are produced through disproportionation of veratraldehyde via Cannizzaro reaction shown in pathway D in Scheme 2. This consideration is supported by the fact that the amount of these three compounds increased with the gradual decrease in the amount of veratraldehyde (Fig. 3A). There also might be another pathway where LM1 directly gave veratraldehyde, pathway E in Scheme 2, as suggested from the result that the sum of the guaiacol yield and the LM1 recovery was less than 100% at 43 h reaction time. However, this pathway must be minor compared to the veratraldehyde formation via the pathways A and B, since the guaiacol yield is quantitative at least at the initial stage of the reaction, as discussed above. It is also noted that guaiacyl moieties including guaiacol degrade in the presence of O2 (ref. 26–30) and this is probably one of the reasons for the small yield of the compound at the prolonged reaction times.
We then performed the degradation of LM1 in 1.25 mol L−1 NaOH aq., a simple alkaline solution with the same OH− concentration as that of the Bu4NOH aq. As shown in Fig. 3B, the degradation velocity of LM1 was lower in the NaOH aq. than in the Bu4NOH aq. The maximum yield of vanillin in the NaOH aq. (0.8 mol%, 6 h) was also much lower in the NaOH solution. One of the plausible reasons for this is that the activity of OH− is greater in the Bu4NOH aq. than in the NaOH aq. even when their OH− concentrations are the same. In other words, OH− in the Bu4NOH aq. is less influenced by the counter cation Bu4N+, which renders the Bu4NOH solution more basic over the NaOH aq. The fast degradation of LM1 will result in smooth formation of the veratryl glycerol, which is the major origin of vanillin (Scheme 2).
There is another mechanical aspect that accounts for the better performance of Bu4NOH for vanillin production. Table 2 summarizes the yields of the major products quantified after the degradation of LM1 for 43 h. In the Bu4NOH aq. LM1 formed the aldehyde products (veratraldehyde and vanillin) in 17.9 mol% (18.6 mol%) yield, where the yield in the parenthesis is the one based on degraded LM1. The other non-aldehyde A-ring derived products (veratric acid and veratryl alcohol) were formed in 15.8 mol% yield (16.5 mol%) in total, which was similar to that of the aldehyde products. These results suggest that the reactions via the pathways C and D almost evenly occur in the Bu4NOH aq. It is also noted that total yield of the four compounds was 33.7 mol% (35.1 mol% based on degraded LM1) and the rest (64.9 mol%) undetected products probably involve considerable amount of veratryl glycerol (see above discussion and Fig. S1 in ESI†).
| Aldehydes | Non-aldehydes | Total | |||||
|---|---|---|---|---|---|---|---|
| Vanillin | Veratraldehyde | Total | Veratryl alcohol | Veratric acid | Total | ||
| a The value in the parenthesis shows the yield based on the amount of degraded LM1: 96.3% and 42.9% in the Bu4NOH aq. and the NaOH aq. based on the initial amount of LM1, respectively. | |||||||
| Bu4NOH aq. | 15.7 (16.3)a | 2.2 (2.3) | 17.9 (18.6) | 9.1 (9.5) | 6.7 (7.0) | 15.8 (16.5) | 33.7 (35.1) |
| NaOH aq. | 0.4 (0.9) | 5.2 (12.1) | 5.6 (13.0) | 3.9 (9.1) | 5.4 (12.6) | 9.3 (21.7) | 14.9 (34.7) |
The degradation of LM1 in the NaOH aq., on the other hand, formed relatively small amounts of the four A-ring-derived products (the total yield: 14.9 mol% after 43 h degradation), as summarized in Table 2. This is because complete degradation of LM1 was not achieved in the NaOH aq. even after 43 h degradation (see Fig. 3B). The total yield in the NaOH aq. eventually became comparable to that in Bu4NOH when it was calculated based on the degraded LM1: 35.1 mol% in the Bu4NOH aq. and 34.7 mol% in the NaOH aq. It is therefore likely that reactivity of veratryl glycerol toward O2 to form veratraldehyde – in other words the velocity of the reactions involved in the pathway B – is almost constant no matter whether the reaction was carried out in the Bu4NOH aq. or the NaOH aq. However, as shown in Table 2, the total yield of the aldehyde products in NaOH aq. based on degraded LM1 (13.0 mol%) was smaller than that in Bu4NOH (18.6 mol%) with the total yields of the non-aldehyde products in NaOH aq. (21.7 mol%) being greater than that in Bu4NOH (16.5 mol%). These different yields of the aldehyde and the non-aldehyde products suggest that the pathway C is more favored over the pathway D in the Bu4NOH aq., which results in the increased yield of vanillin.
To check this idea, we degraded veratraldehyde (5.0 mg) at 120 °C for 4 h under N2 either in the Bu4NOH aq. or the NaOH aq. (1.0 mL) and compared the product distribution between the degradations in the two reaction media. As summarized in Table 3, the degradation of veratraldehyde in Bu4NOH resulted in the formation of vanillin, veratryl alcohol, and veratric acid in very similar yields. On the other hand, the degradation in NaOH aq. gave much smaller amount of vanillin and more amount of veratryl alcohol and veratric acid. This is in complete agreement with the above consideration that the demethylation pathway C (Scheme 2) is more favored over the disproportionation pathway D in the Bu4NOH aq. We also carried out similar degradation in Bu4NCl-added NaOH aq., where the 1.25 mol L−1 Bu4NOH aq. was mimicked by the addition of the corresponding amount of Bu4NCl to 1.25 mol L−1 NaOH aq. The addition of Bu4NCl also increased the selectivity toward vanillin as indicated from the higher vanillin yield (20.2 mol%) based on the degraded veratraldehyde and the lower yield of veratryl alcohol and veratric acid (29.5 and 32.3 mol%, respectively) than those obtained in the simple NaOH aq. Interestingly, in this Bu4NCl-added system, the degradation of veratraldehyde was considerably suppressed: much more starting material (57.0%) was recovered in the Bu4NCl-added NaOH aq. These results suggest that Bu4N+ slows down both pathways C and D, and along with that, makes the degradation more vanillin-oriented. Similar recoveries of the starting material obtained between the Bu4NOH and the NaOH aq. – they are 11.5 and 8.6%, respectively – would be explained by the idea that the slowing-down effect by Bu4N+ was compensated by the strong basicity of Bu4NOH (see above).
| Yield | Recovery | |||
|---|---|---|---|---|
| Vanillin | Veratryl alcohol | Veratric acid | ||
| a The value in the parenthesis shows the yield based on the amount of degraded veratraldehyde.b Solid Bu4NCl was added to the NaOH solution so that the concentration of Bu4N+ in the reaction solution became 1.25 mol L−1. | ||||
| Bu4NOH aq. | 30.6 (33.5)a | 28.1 (30.7) | 31.2 (34.1) | 8.6 |
| NaOH aq. | 5.9 (6.7) | 38.3 (43.3) | 41.6 (47.0) | 11.5 |
| NaOH aq. + Bu4NCl(s)b | 8.7 (20.2) | 12.7 (29.5) | 13.9 (32.3) | 57.0 |
To obtain deeper insights into the effect of Bu4N+, we carried out 1H NMR analyses of veratraldehyde in 1.25 mol L−1 NaOD/D2O-based solutions. As summarized in Table 4, the signal of all protons of veratraldehyde considerably shifted to lower magnetic fields by 20 to 112 Hz, when Bu4NCl was added to the NaOD/D2O. This shift scarcely occurred when the same molar amount of NaCl was added to the solution, which strongly suggested that the shift was caused by Bu4N+ and not by Cl−. It is therefore likely that veratraldehyde is in a close contact with Bu4N+ in the solutions containing Bu4N+. The largest shift observed for the aldehyde proton (112 Hz) suggests strong interaction between Bu4N+ and the aldehyde group. The disproportionation reaction is eventually suppressed when both reactants are in the “cage” of Bu4N+. On the other hand, in the case of the demethylation (pathway C in Scheme 2), one of the reactants is OH−, which is a small molecule and hence can more easily squeeze itself into the “cage”. Although further investigation is necessary to elucidate more detailed mechanism, this idea can reasonably explain the above experimental facts. According to these mechanisms, it can be stated that Bu4N+ does not have a specific catalytic site, but the interaction between the aldehyde group and Bu4N+ is a major origin of the good performance of Bu4NOH for the vanillin production. Also, as shown in Fig. 3, the yield of guaiacol in the degradation in the Bu4NOH aq. is extremely higher than that in the NaOH aq. and this result may be also explained with the idea that guaiacol was protected from the O2 attack by the cage effect caused by the presence of Bu4N+.
|
||||||
|---|---|---|---|---|---|---|
| C2–H (d) | C5–H (d) | C6–H (dd) | Cα–H (s) | C3–OCH3 (s) | C4–OCH3 (s) | |
| a Bu4NCl and NaCl were added to the NaOD solution with the concentration of the additives being set at 1.25 mol L−1.b Changes in the chemical shift (Hz) caused by the additive were shown in the parenthesis. | ||||||
| 1.25 mol L−1 NaOD/D2O | 7.16 | 6.89 | 7.32 | 9.33 | 3.64 | 3.69 |
| 1.25 mol L−1 NaOD/D2O + Bu4NCl | 7.29 (+52)b | 7.09 (+80) | 7.50 (+72) | 9.61 (+112) | 3.69 (+20) | 3.75 (+24) |
| 1.25 mol L−1 NaOD/D2O + NaCl | 7.18 (+8) | 6.93 (+40) | 7.36 (+16) | 9.38 (+20) | 3.67 (+12) | 3.73 (+16) |

Fig. 4 shows a typical HPLC chromatogram of the reaction mixture obtained after the degradation of the phenolic lignin model LM2 (5.0 mg) in 1.25 mol L−1 Bu4NOH aq. (1.0 mL) at 120 °C under air. In the case of LM2, vanillin and vanillic acid were detected as major A-ring-derived products along with guaiacol as B-ring-derived one. We also found two large peaks at 37.6 and 39.0 min, which were assigned as cis- and trans-isomers of the enol ether EE with 1H NMR analysis after their isolation from the reaction mixture with column chromatography (see experimental section for details). The formation of the trans-isomer was favored in the degradation. Note that EE are reported as major products in degradation of phenolic lignin models under alkaline conditions in many previous studies.22,31–33
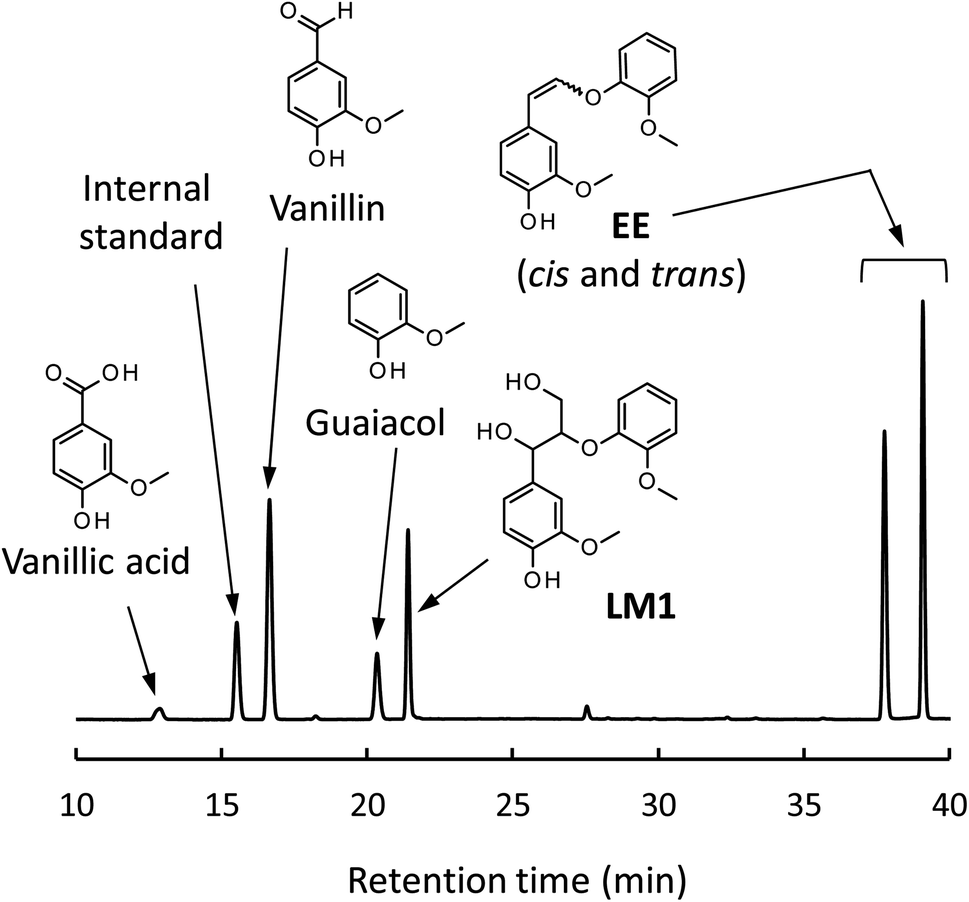 | ||
| Fig. 4 HPLC chromatogram of the reaction mixture obtained after the degradation of LM2 (5 mg) in 1.25 mol L−1 Bu4NOH aq. (1.0 mL) under air for 1 h. | ||
In the degradation of LM2, as shown in Fig. 5A, vanillin, guaiacol, and EEs were produced from the initial stage of the degradation in the Bu4NOH aq. After 2 h reaction time, the yield of EEs started to decrease with increase in the yield of vanillin and guaiacol. At 4 h reaction time, vanillin and guaiacol were produced in 59.5 and 61.9 mol% yields, respectively (see also Table 5). Vanillic acid started to form after 15 min and the yield reached 12.7 mol% at 4 h, although the compound was not detected in the reaction mixture before 15 min. These results suggest that LM2 is first degraded to EE as presented in pathway F in Scheme 3 and EE is then degraded into vanillin and vanillic acid along with the releasement of guaiacol (pathway G). The pathway F is a simple alkaline degradation process via quinone methide formation followed by succeeding elimination of Cγ as formaldehyde, as extensively investigated previously.22,31–33 The pathway G requires O2 according to the previous studies reporting that EE is substantially stable under N2 even at elevated temperatures (∼170 °C) but readily to be oxidized into vanillin and guaiacol in the presence of O2.26,28 Note that, according to comparison between the results in Fig. 3 and 5, the degradation of LM2 was much faster than that of LM1 and this will be discussed in the next section.
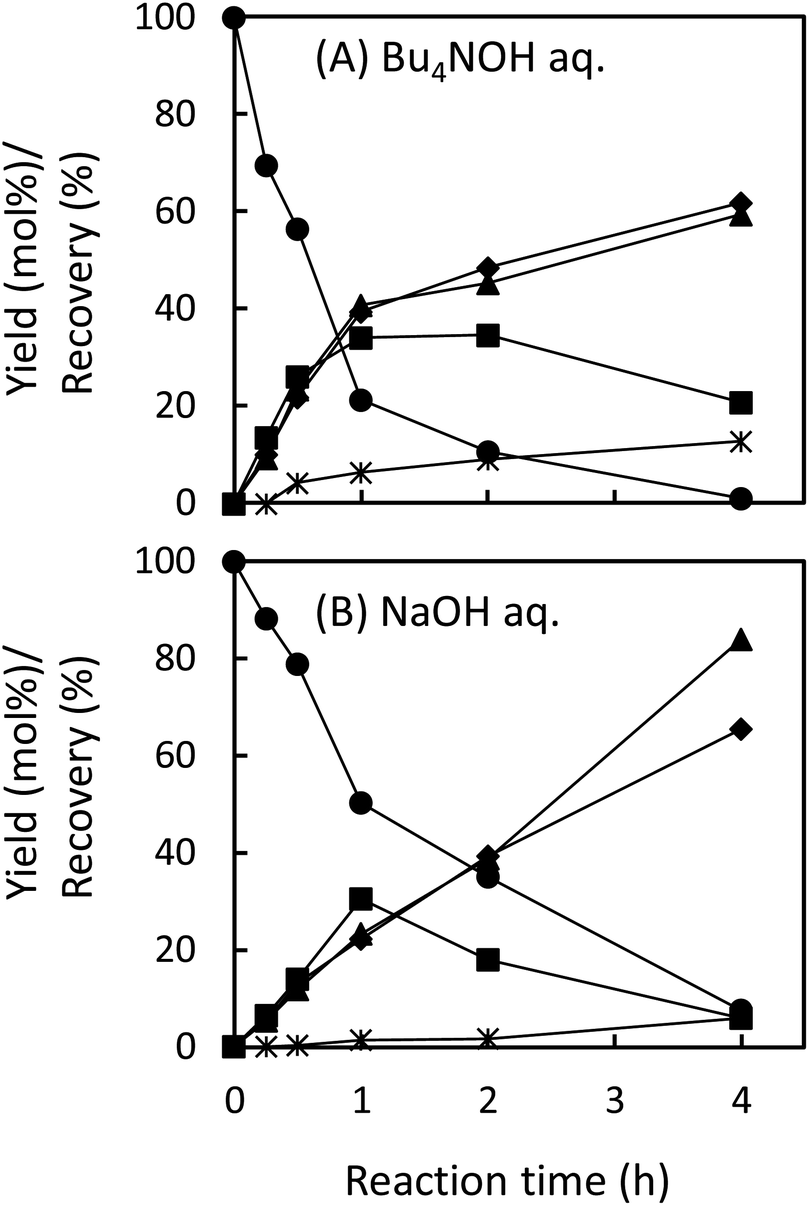 | ||
Fig. 5 Changes with time in recovery of LM2 ( ), yields of guaiacol ( ), yields of guaiacol ( ), vanillin ( ), vanillin ( ), vanillic acid ( ), vanillic acid ( ), and enol ethers EEsa ( ), and enol ethers EEsa ( ) during degradation of LM2 (5.0 mg) at 120 °C under air in 1.25 mol L−1 Bu4NOH aq. (1.0 mL) (A) and in 1.25 mol L−1 NaOH aq. (1.0 mL) (B). a The yields of EEs are shown as the sum of the cis- and trans-isomers. ) during degradation of LM2 (5.0 mg) at 120 °C under air in 1.25 mol L−1 Bu4NOH aq. (1.0 mL) (A) and in 1.25 mol L−1 NaOH aq. (1.0 mL) (B). a The yields of EEs are shown as the sum of the cis- and trans-isomers. | ||
| Staring material | Reaction medium | Yield (mol%) | Recovery (%) | |||
|---|---|---|---|---|---|---|
| Vanillin | Vanillic acid | Guaiacol | EE | |||
| LM2 | Bu4NOH aq. | 59.5 | 12.7 | 61.9 | 7.0 | 1.0 |
| LM2 | NaOH aq. | 83.8 | 5.9 | 65.5 | 6.0 | 7.4 |
| EE | Bu4NOH aq. | 43.6 | 22.2 | 41.4 | — | 29.2 |
| EE | NaOH aq. | 51.8 | 6.5 | 49.7 | — | 17.7 |
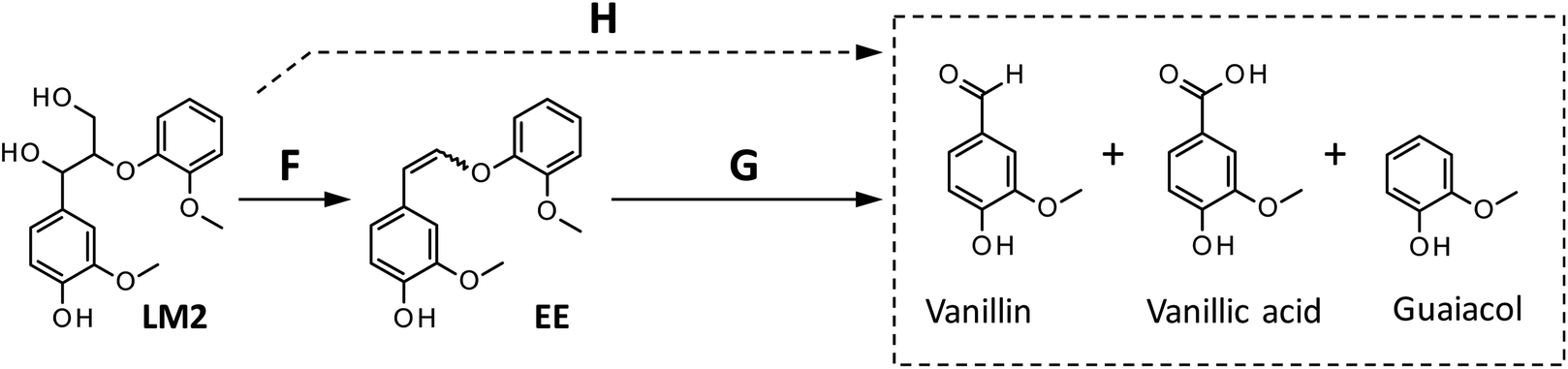 | ||
| Scheme 3 Reaction pathways from LM2 to the products detected in this study. The formation of vanillic acid via pathway H is less significant than that via pathway G. | ||
We also carried out degradation of cis/trans mixture of isolated EE (3.0 mg) in the Bu4NOH aq. (1.0 mL) under air. As summarized in Table 5, the degradation for 4 h resulted in the formation of 43.6, 22.2, and 41.4 mol% of vanillin, vanillic acid, and guaiacol, respectively. Thus, the yields of vanillin and guaiacol from EE are moderately smaller than those from LM2 obtained under the same reaction conditions (vanillin: 59.5 mol% and guaiacol: 61.9 mol%, see Fig. 5 and Table 5). These results mildly suggest that there is a reaction pathway forming vanillin and guaiacol directly from LM2 (pathway H in Scheme 3). For vanillic acid, on the other hand, the yield from EE (22.2 mol%, Table 5) was greater than that from LM2 (12.7 mol%, Fig. 5 and Table 5), which suggests that vanillic acid is formed mostly via EE in the degradation of LM2.
The degradation of LM2 in the NaOH aq. was then carried out under the same reaction conditions. As shown in Fig. 5B, we observed reaction behaviors qualitatively the same as those observed in the case of the Bu4NOH aq. This indicates that the degradation follows the pathways in Scheme 3 both in the Bu4NOH and the NaOH aq. Through quantitative comparison of the reaction behaviors between the Bu4NOH and the NaOH aq. (Fig. 5), the degradation in the NaOH aq. exhibited (1) slower degradation of LM2, (2) lower yields and faster degradation of EE, (3) larger yields of vanillin and guaiacol, and (4) lower yields of vanillic acid. We will discuss these four differences below.
The relatively slow degradation of the starting material in the NaOH aq., the first difference mentioned above, can be explained again with the stronger basicity of Bu4OH over NaOH. The reactions in the pathway G (Scheme 3) requires the deprotonation of the phenolic OH of LM2 and Cγ–OH of the intermediate quinone methide. These deprotonation steps are considered to be less feasible in the NaOH aq., ending up the slow degradation of LM2.
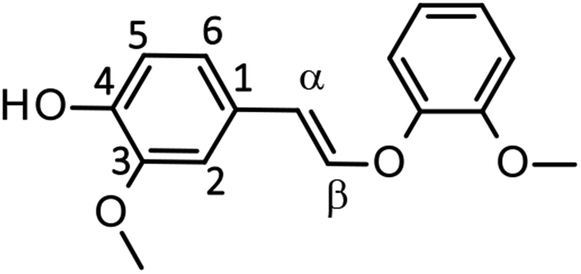
The second and third differences are closely linked together, as the faster degradation of EE is the greater the yields of the products (vanillin and guaiacol) should become. It is possible that, as an analogy to the stabilization of veratraldehyde by Bu4N+ (see previous section), EE is stabilized by the presence of Bu4N+. The larger recovery of EE (29.2%) after the degradation for 4 h in the Bu4NOH aq. than that in the NaOH aq. (17.7%, Table 5) also supports this idea. As summarized in Table 6, 1H NMR analyses of trans-EE in the 1.25 mol L−1 NaOD/D2O with and without the addition of Bu4NCl indicated that C2–H and C6–H on the benzene ring and the protons on the CαCβ group significantly shifted to upper magnetic fields when Bu4NCl was added to the NaOD/D2O solution, whereas the shift observed for C5–H was relatively small. The addition of the same molar amount of NaCl again did not cause those shifts. These results suggest that Bu4N+ is located mainly around the CαCβ bond of EE. It has been reported that the initial step of the aerobic oxidation of EE to vanillin is addition of O2 to the CαCβ group.26,28 Binding of the CαCβ group to Bu4N+ will render the O2 addition more demanding, by which the degradation of EE to vanillin is inhibited. Note that the changes in the chemical shifts of EE by the addition of Bu4N+ (Table 6) were opposite from those observed for the veratraldehyde–NaOD/D2O system (Table 4), that is, upper-magnetic field shift and downer one were observed in the EE and in the veratraldehyde solutions, respectively. However, the reasons for this difference are not clear at the moment.
|
|||||
|---|---|---|---|---|---|
| C2–H (d) | C5–H (d) | C6–H (dd) | Cα–H (s) | Cβ–H (s) | |
| a The results only for the trans-isomer are shown as most of the cis-isomer signals were overlapped with those of the trans-isomer.b Bu4NCl and NaCl were added to the NaOD solution with the concentration of the additives being set at 1.25 mol L−1.c Changes in the chemical shift (Hz) caused by the additive were shown in the parenthesis. | |||||
| 1.25 mol L−1 NaOD/D2O | 6.65 | 6.33 | 6.54 | 6.83 | 6.00 |
| 1.25 mol L−1 NaOD/D2O + Bu4NCl | 6.46 (−76)c | 6.27 (−24) | 6.38 (−64) | 6.71 (−48) | 5.89 (−44) |
| 1.25 mol L−1 NaOD/D2O + NaCl | 6.65 (±0) | 6.36 (+12) | 6.56 (+8) | 6.85 (+8) | 6.02 (+8) |
The reasons for the low yield of vanillic acid in the NaOH aq. (the fourth difference between the Bu4NOH aq. and the NaOH aq. mentioned above) are not clear at the moment. As presented in Table 5, this vanillic acid-oriented degradation behavior was re-produced also in the degradation of EE in the Bu4NOH aq.: EE formed vanillic acid in 22.2 mol% on the basis of degraded starting material in the Bu4NOH aq., whereas the yield of the compound was much smaller in the NaOH aq. (6.5 mol%). This suggests that the degradation of EE (pathway G in Scheme 3) is modified by the presence of Bu4N+ to be more vanillic acid-selective. The interaction between EE and Bu4N+ discussed above would play some roles in this selectivity change, but more systematic investigation is necessary to elucidate the underlying mechanism.
Effects of Bu4N+ on aerobic oxidation of lignin polymer
We have so far discussed the degradation behavior of the lignin model compounds processing β-O-4 linkages, which consist around 50% of the inter-unit linkages of native lignin polymer. In this section we wish to discuss aerobic oxidation of native lignin polymer focusing mainly on vanillin production, on the basis of the results obtained above. In the discussion below, we assume that the non-phenolic model LM1 represents the reactivity of the monomeric unit in the middle of the polymer and the phenolic model LM2 corresponds to that of the phenolic end of the polymer.The initial step of the degradation of lignin polymer is simple alkaline degradation of the phenolic end, which is the fastest reaction observed in this study (Fig. 5). When the phenolic end has a β-O-4 linkage, the degradation results in the formation of an enol ether end group. The enol ether end formed – if it is in contact with O2 – is further oxidized into vanillin or vanillic acid and forms a new phenolic end in the adjacent monomeric unit. When these stepwise reactions continued, lignin polymers could degrade completely into the low molecular weight compounds. However, this type of “unzipping” reaction seems not to play major roles in the degradation. The reaction time required for completing the degradation of LM2 was ∼8 h, which was far from sufficient for vanillin production from the wood flour under the same conditions: at least 43 h is necessary to fully produce vanillin from the wood flour at 120 °C under air,8 although the introduction of pure O2 shortens the reaction time (see above). On one hand, the unzipping vanillin/vanillic acid production may be stopped when the reaction “bumps” into a less reactive linkage such as β–5, β–β, 4-O-5 and 5–5′ linkages. On the other hand, other types of reactions such as condensations are possible to prevent the vanillin/vanillic acid production from the phenolic end.
Our results obtained for LM2 revealed that Bu4N+ did no good for the vanillin production after all: Bu4N+ stabilizes the enol ether to make the vanillin formation slow and changes the selectivity to vanillic acid-oriented. However, these negative effects of Bu4N+ are not very serious in the vanillin production from native lignin in Bu4NOH, since the vanillin production from the phenolic end is fundamentally of little importance in entire vanillin production reactions.
Besides a series of the reactions on the phenolic end, alkaline degradation of non-phenolic middle units gradually takes place. As shown in Scheme 4, the initial step of the degradation on a non-phenolic β-O-4 type middle unit is alkaline-catalyzed ether link cleavage to form a glycerol end group, which corresponds to the pathway A in Scheme 2. This step is significantly enhanced in Bu4NOH due to its strong basicity and this is one of the benefits in the utilization of Bu4NOH instead of NaOH.
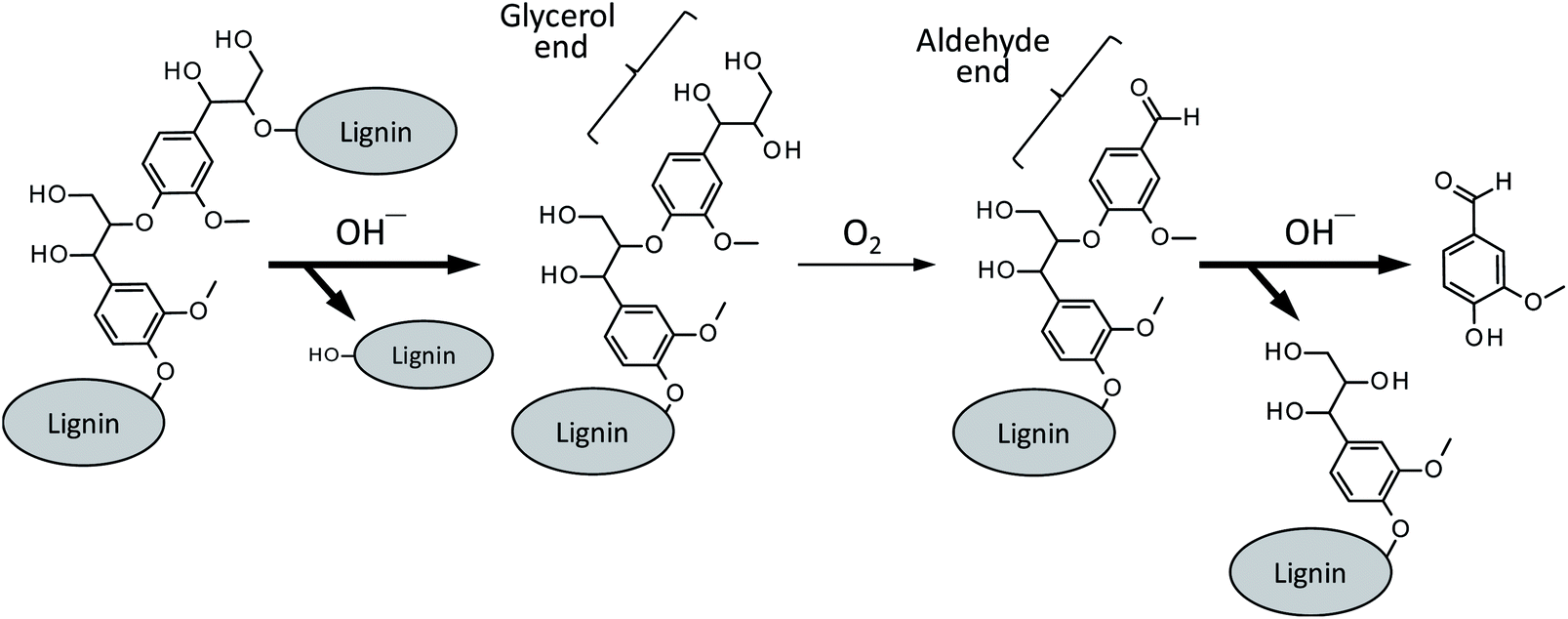 | ||
| Scheme 4 Vanillin formation from a β-O-4 middle unit of lignin polymer. The pathways with bold arrows are positively influenced by Bu4N+. | ||
The oxidation of the glycerol group by O2, pathway B in Scheme 2 in the case of the model compound LM1, then occurs to form a benzaldehyde end group (vanillin end group when the lignin sample is derived from soft wood lignin). There are two possibilities for the fate of the benzaldehyde end group. One is the free benzaldehyde formation by the cleavage of the ether link connecting the benzaldehyde end to the adjacent monomeric unit, which corresponds to the pathway C in Scheme 2. The other is the disproportionation of the aldehyde group resulting in the formation of benzoic acid and benzyl alcohol ends (pathway D in Scheme 2). The degradation in Bu4NOH facilitates the former reaction pathway to increase the benzaldehyde yield.
These proposed reactions suggest that the reaction mixture from the wood flour should involve some amount of vanillyl alcohol. However, the compound was not detected in the degradation of the wood flour under any conditions, although vanillic acid was detected as a major product (Fig. 1). This is probably because vanillyl alcohol end group is unstable due to the presence of benzyl hydroxy group which is readily subjected to condensation reactions.22 It is also noted that vanillic acid, unlike vanillyl alcohol, can also be formed from the oxidation of the enol ether end, as indicated from Fig. 5, and some of the origins of vanillic acid produced from the wood flour is considered to be phenolic end groups of lignin polymer.
Conclusions
The degradation of Japanese cedar wood flour under O2 produced vanillin and vanillic acid in 23.2 and 1.2% yields on the basis of the Klason lignin amount, respectively, at 120 °C for 4 h in 1.25 mol L−1 Bu4NOH aq., where the OH− concentration was enhanced to 3.75 mol L−1 by the addition of NaOH. These yields have been significantly improved, compared to those obtained under air reported in our previous studies.8 Also, the reaction time required for achieving the maximum vanillin and vanillic acid yields was drastically shortened to 4 h by the O2 introduction. It should be noted that the yields of the compounds were comparable to those obtained in the AN oxidation, which is a benchmark for evaluating selectivity of lignin depolymerization methods.The mechanical investigation with the lignin model compounds suggested that the major vanillin formation pathway involved the alkaline-catalyzed β-ether cleavage on the non-phenolic middle units of lignin polymer to produce the glycerol end group, followed by the oxidation of the end group by O2 to aldehyde and subsequent releasement of free vanillin. The positive effect of Bu4NOH over NaOH with regard to the vanillin formation was partially attributed to the basicity of Bu4NOH stronger than that of NaOH. More fundamentally, Bu4N+ significantly suppressed the disproportionation of the aldehyde end so that the releasement of vanillin became to play more roles in entire degradation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Technologies for Creating Next-Generation Agriculture, Forestry, and Fisheries under the Cross-Ministerial Strategic Innovation Promotion Program (SIP) administered by the Council for Science, Technology, and Innovation (CSTI), Japan, and a Grant-in-Aid for Young Scientists (B) (No. 17K18008) from the Japan Society for the Promotion of Science. The GC/MS analysis was carried out with the apparatus in Kyoto Research Park Corp.References
- H.-M. Chang, Oxidation, in Lignins—Occurrence, Formation, Structure and Reactions, ed. K. V. Sarkanen and C. H. Ludwig, John Wiley and Sons, NY, 1971, pp. 433–485 Search PubMed.
- T. P. Schultz and M. C. Templeton, Proposed Mechanism for the Nitrobenzene Oxidation of Lignin, Holzforschung, 1986, 40, 93–97 CrossRef CAS.
- (a) M. B. Hocking, Vanillin: Synthetic Flavoring from Spent Sulfite Liquor, J. Chem. Educ., 1997, 74, 1055–1059 CrossRef CAS; (b) D. Havkin-Frenkel, Vanillin, Kirk-Othmer Encyclopedia of Chemical Technology, 2018, pp. 1–12 Search PubMed.
- (a) J. R. Silverman, A. M. Danby and B. Subramaniam, Intensified ozonolysis of lignins in a spray reactor: insights into product yields and lignin structure, React. Chem. Eng., 2019, 4, 1421–1430 RSC; (b) A. Gavriilidis, A. Constantinou, K. Hellgardt, K.-K. Hii, G. J. Hutchings, G. L. Brett, S. Kuhn and S. P. Marsden, Aerobic oxidations in flow: opportunities for the fine chemicals and pharmaceuticals industries, React. Chem. Eng., 2016, 1, 595–612 RSC.
- M. Maeda, T. Hosoya, K. Yoshioka, H. Miyafuji, H. Ohno and T. Yamada, Vanillin Production from Native Softwood Lignin in the Presence of Tetrabutylammonium Ion, J. Wood Sci., 2018, 64, 810–815 CrossRef CAS.
- M. Fache, B. Boutevin and S. Caillol, Vanillin Production from Lignin and Its Use as a Renewable Chemical, ACS Sustain. Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- S. G. Santos, A. P. Marques, D. L. D. Lima, D. V. Evtuguin and V. I. Esteves, Kinetics of Eucalypt Lignosulfonate Oxidation to Aromatic Aldehydes by Oxygen in Alkaline Medium, Ind. Eng. Chem. Res., 2011, 50, 291–298 CrossRef CAS.
- K. Yamamoto, T. Hosoya, K. Yoshioka, H. Miyafuji, H. Ohno and T. Yamada, Tetrabutylammonium Hydroxide 30-Hydrate as Novel Reaction Medium for Lignin Conversion, ACS Sustain. Chem. Eng., 2017, 5, 10111–10115 CrossRef CAS.
- P. C. Rodrigues Pinto, E. A. Borges da Silva and A. E. Rodrigues, Insights into Oxidative Conversion of Lignin to High-Added-Value Phenolic Aldehydes, Ind. Eng. Chem. Res., 2011, 50, 741–748 CrossRef CAS.
- J. D. P. Araújo, C. A. Grande and A. E. Rodrigues, Vanillin Production from Lignin Oxidation in a Batch Reactor, Chem. Eng. Res. Des., 2010, 88, 1024–1032 CrossRef.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Formic-acid-induced Depolymerization of Oxidized Lignin to Aromatics, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- T. Rinesch, J. Mottweiler, M. Puche, P. Concepción, A. Corma and C. Bolm, Mechanistic Investigation of the Catalyzed Cleavage for the Lignin β-O-4 Linkage: Implications for Vanillin and Vanillic Acid Formation, ACS Sustain. Chem. Eng., 2017, 5, 9818–9825 CrossRef CAS.
- S. Gharehkhani, Y. Zhang and P. Fatehi, Lignin-derived Platform Molecules through TEMPO Catalytic Oxidation Strategies, Prog. Energy Combust. Sci., 2019, 72, 59–89 CrossRef.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Catalytic Transformation of Lignin for the Production of Chemicals and Fuels, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- H. Kawamoto, Lignin Pyrolysis Reactions, J. Wood Sci., 2017, 63, 117–132 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, From Lignin-derived Aromatic Compounds to Novel Biobased Polymers, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed.
- H. Zhang, X. Yong, J. Zhou, J. Deng and Y. Wu, Biomass Vanillin-Derived Polymeric Microspheres Containing Functional Aldehyde Groups: Preparation, Characterization, and Application as Adsorbent, ACS Appl. Mater. Interfaces, 2016, 8, 2753–2763 CrossRef CAS PubMed.
- G. H. Tomlinson and H. Hibbert, Studies on Lignin and Related Compounds. XXIV. The Formation of Vanillin from Waste Sulfite Liquor, J. Am. Chem. Soc., 1936, 58, 345–348 CrossRef CAS.
- G. H. Tomlinson and H. Hibbert, Studies on Lignin and Related Compounds. XXV. Mechanism of Vanillin Formation from Spruce Lignin Sulfonic Acids in Relation to Lignin Structure, J. Am. Chem. Soc., 1936, 58, 348–353 CrossRef CAS.
- P. Ding, M. Garrett, Ø. Loe, A. W. Nienow and A. W. Pacek, Generation of Hydrogen Gas during the Catalytic Oxidation of Sodium Lignosulfonate to Vanillin: Initial Results, Ind. Eng. Chem. Res., 2012, 51, 184–188 CrossRef CAS.
- J. Gierer, Chemistry of Delignification Part 1: General Concept and Reactions during Pulping, Wood Sci. Technol., 1985, 19, 289–312 CAS.
- D. R. Dimmel and L. F. Schuller, Structural/Reactivity Studies (I): Soda Reactions of Lignin Model Compounds, J. Wood Chem. Technol., 1986, 6, 535–564 CrossRef CAS.
- T. F. Hubbard, T. P. Schultz and T. H. Fisher, Alkaline Hydrolysis of Nonphenolic β-O-4 Lignin Model Dimers: Substituent Effects on the Leaving Phenoxide in Neighboring Group vs. Direct Nucleophilic Attack, Holzforschung, 1992, 46, 315–320 CrossRef CAS.
- W. E. Collier, T. H. Fisher, L. L. Ingram Jr, A. L. Harris and T. P. Schultz, Alkaline Hydrolysis of Nonphenolic β-O-4 Lignin Model Dimers: Further Studies of the Substituent Effect on the Leaving Phenoxide, Holzforschung, 1996, 50, 420–424 CrossRef CAS.
- J. Gierer, Chemistry of Delignification Part 2: Reactions of Lignins during Bleaching, Wood Sci. Technol., 1986, 20, 1–33 CrossRef CAS.
- E. Johansson and S. Ljunggren, The Kinetics of Lignin Reactions During Oxygen Bleaching. IV. The Reactivities of Different Lignin Model Compounds and the Influence of Metal Ions on the Rate of Degradation, J. Wood Chem. Technol., 1994, 14, 507–525 CrossRef CAS.
- J. Gierer, T. Reitberger, E. Yang and B.-H. Yoon, Formation and Involvement of Radicals in Oxygen Delignification Studied by the Autoxidation of Lignin and Carbohydrate Model Compounds, J. Wood Chem. Technol., 2001, 21, 313–341 CrossRef CAS.
- S. Kuitunen, A. Kalliola, V. Tarvo, T. Tamminen, S. Rovio, T. Liitiä, T. Ohra-aho, T. Lehtimaa, T. Vuorinen and V. Alopaeus, Lignin Oxidation Mechanisms under Oxygen Delignification Conditions. Part 3. Reaction Pathways and Modeling, Holzforschung, 2011, 65, 587–599 CAS.
- S. Ohmura, T. Yokoyama and Y. Matsumoto, Progress of Oxidation of Non-phenolic Lignin Moiety in an Oxygen Bleaching Process via the Conversion of Non-phenolic into Phenolic Lignin Moiety, J. Wood Sci., 2012, 58, 243–250 CrossRef CAS.
- D. R. Dimmel and L. F. Bovee, Pulping Reactions of Vinyl Ethers, J. Wood Chem. Technol., 1993, 13, 583–592 CrossRef CAS.
- S. Kubo, K. Hashida, S. Hishiyama, T. Yamada and S. Hosoya, Possibilities of the Formation of Enol-Ethers in Lignin by Soda Pulping, J. Wood Chem. Technol., 2015, 35, 62–72 CrossRef.
- K. Lundquist and S. v. Unge, Stability of Arylglycerols During Alkaline Cooking, Holzforschung, 2004, 58, 330–333 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Total ion chromatogram in GC/MS analysis of the acetylated reaction mixture obtained after the degradation of LM1. Values in Fig. 1, 3, and 5. See DOI: 10.1039/d0ra03420g |
| This journal is © The Royal Society of Chemistry 2020 |