DOI:
10.1039/C9SC02711D
(Edge Article)
Chem. Sci., 2019,
10, 9692-9698
Influence of the distal guanidine group on the rate and selectivity of O2 reduction by iron porphyrin†
Received
4th June 2019
, Accepted 28th August 2019
First published on 29th August 2019
Abstract
The O2 reduction reaction (ORR) catalysed by iron porphyrins with covalently attached pendant guanidine groups is reported. The results show a clear enhancement in the rate and selectivity for the 4e−/4H+ ORR. In situ resonance Raman investigations show that the rate determining step (rds) is O2 binding to ferrous porphyrins in contrast to the case of mononuclear iron porphyrins and heme/Cu analogues where the O–O bond cleavage of a heme peroxide is the rds. The selectivity is further enhanced when an axial imidazole ligand is introduced. Thus, the combination of the axial imidazole ligand and pendant guanidine ligand, analogous to the active site of peroxidases, is determined to be very effective in enabling a facile and selective 4e−/4H+ ORR.
Introduction
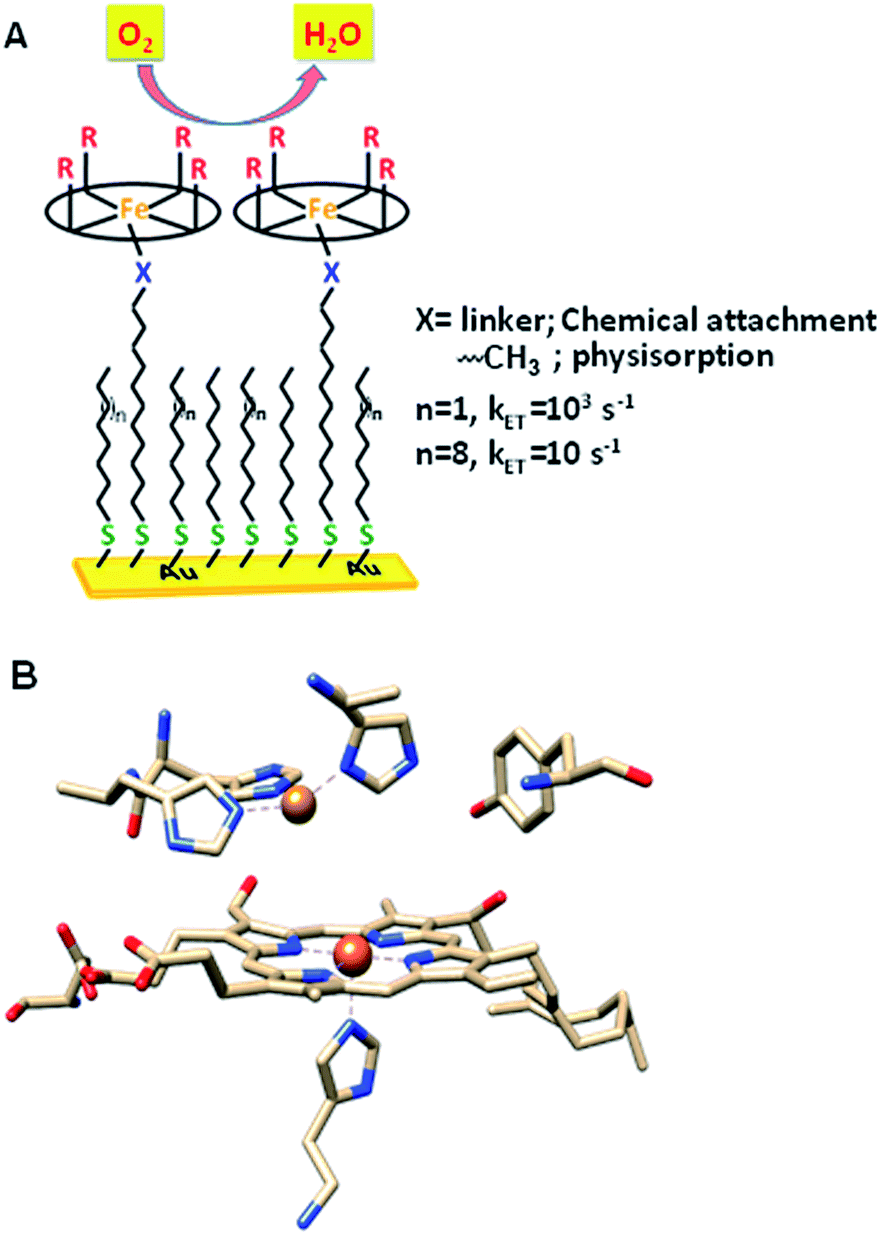
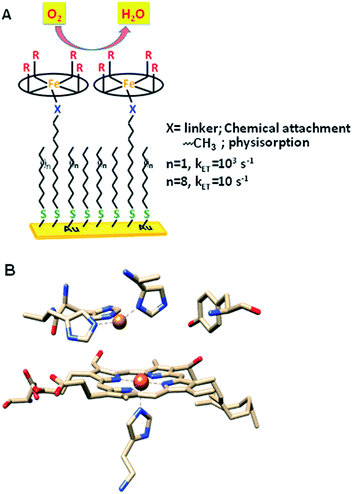
The factors that affect the rate and selectivity of the catalytic oxygen reduction reaction (ORR) are of fundamental interest as it is an essential process for several energy conversion technologies ranging from fuel cells to metal air batteries.1 The ORR can proceed either via a 2e−/2H+ pathway to produce H2O2 or via a 4e−/4H+ pathway to produce water.2 The latter pathway is favoured both from a thermodynamic point of view2 and because it avoids partially reduced oxygen species (PROS) such as H2O2 and O2− which are very reactive and often detrimental to the catalyst.3 Iron porphyrins have been long used to extensively investigate the O2 reduction process in solution as well as under heterogeneous electrochemical conditions by immobilizing them on electrodes (Fig. 1A).4–6 Erstwhile investigations have revealed that these complexes prefer 2e−/2H+ reduction of O2 resulting in H2O2.7
 |
| | Fig. 1 (A) Heterogeneous electrochemical construct (the oval ring represents the porphyrin ligand); (B) binuclear active site of CcO (PDB ID: 1OCC).9 | |
Inspiration for ORR catalyst design can be obtained from nature as the ORR is a key process in cell metabolism. Eukaryotes use molecular oxygen as the terminal electron acceptor in the final stage of the respiration process where cytochrome c oxidase (CcO) catalyses the reduction of O2 to H2O to drive the oxidative phosphorylation activity.8 CcO belongs to the heme–Cu oxidase superfamily of enzymes and its active site comprises a bimetallic heme/Cu core and a post transitionally modified Tyr244 residue (Fig. 1B).9,10 The mechanism of the CcO assisted ORR was extensively investigated using its elaborate structural and functional mimics, to design a suitable molecular electrocatalyst. Unlike mononuclear iron porphyrins, these analogues produce minimal PROS and catalyse the electrochemical 4e−/4H+ ORR at neutral pH under heterogeneous conditions with 2nd order rates of 105 M−1 s−1.4,11–13 The generation of PROS was thought to occur via hydrolysis of dioxygen derived intermediates (FeIII–O2− and/or FeIII–OOH)14 and the extent of PROS generation increases with a decrease in electron flux from the electrode to the catalyst.15 The electron flux can be conveniently tuned between 1 and 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 by utilizing self-assembled monolayer covered Au electrodes where the electron tunnelling rate depends on the chain length of the thiol used.16,17 Recent reports show that similar selectivity and rates can be obtained by using iron porphyrins bearing both hydrogen bonding and redox active moieties such as ferrocene even under slow electron flux from the electrode.18,19
000 s−1 by utilizing self-assembled monolayer covered Au electrodes where the electron tunnelling rate depends on the chain length of the thiol used.16,17 Recent reports show that similar selectivity and rates can be obtained by using iron porphyrins bearing both hydrogen bonding and redox active moieties such as ferrocene even under slow electron flux from the electrode.18,19
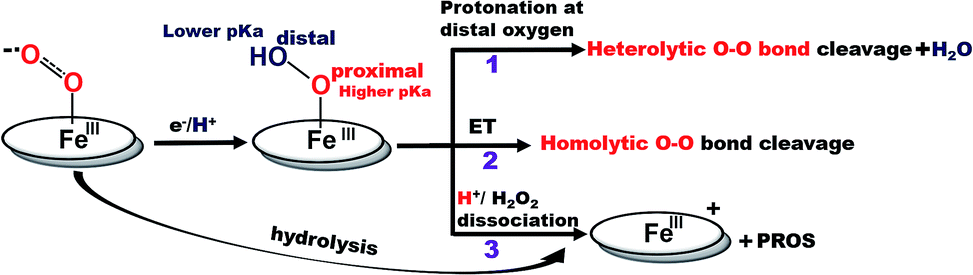
Recently surface enhanced resonance Raman spectroscopy coupled to rotating disk electrochemistry (SERRS-RDE) has been utilised for detection of intermediates accumulated when the electrocatalyst is in a steady state.20 The results reveal that one of the key intermediates produced during the ORR cycle catalysed by iron porphyrins is a low spin FeIII–OOH species.21 The O–O bond of this species is strong and the step leading to its cleavage is the rate determining step (rds) of the electrochemical ORR for several iron porphyrins and heme/Cu systems.13,20,22 The facile cleavage of the O–O bond requires protonation of the distal oxygen of the FeIII–OOH species for the anionic thiolate and phenolate axial ligands or reduction to a FeII–OOH species for neutral axial ligands to form water.14,15,23 The protonation of the distal oxygen (one bound to proton) of a FeIII–OOH species leads to heterolytic O–O bond cleavage and subsequent release of water (Scheme 1, pathway 1). However, the protonation of the proximal oxygen (one bound to the iron) leading to hydrolysis and release of H2O2 is favoured due to a 1.5 unit higher pKa of the proximal oxygen relative to the distal oxygen (Scheme 1, pathway 3).20,24–26 In iron porphyrins with neutral axial ligands the O–O bond cleavage in the subsequent steps requires electron transfer (ET).18,27 Thus, when ET is inhibited, the extent of H2O2 release is increased. However, if the O–O bond can be activated for protonation and stabilized by hydrogen bonding one may be able to enhance the rate and selectivity of the ORR. Accordingly, distal amine groups and heteroatoms that can specifically deliver protons to the distal oxygen have been shown to facilitate the selective protonation of the distal oxygen atom of this FeIII–OOH intermediate resulting in very facile and selective 4e−/4H+ reduction of O2 by mononuclear iron porphyrins.2,26
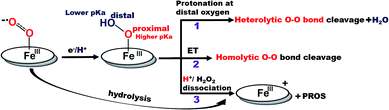 |
| | Scheme 1 Schematic representation of factors determining the 2e−/2H+vs. 4e−/4H+ ORR selectivity of porphyrin complexes. (The oval ring represents the porphyrin ligand.)26 | |
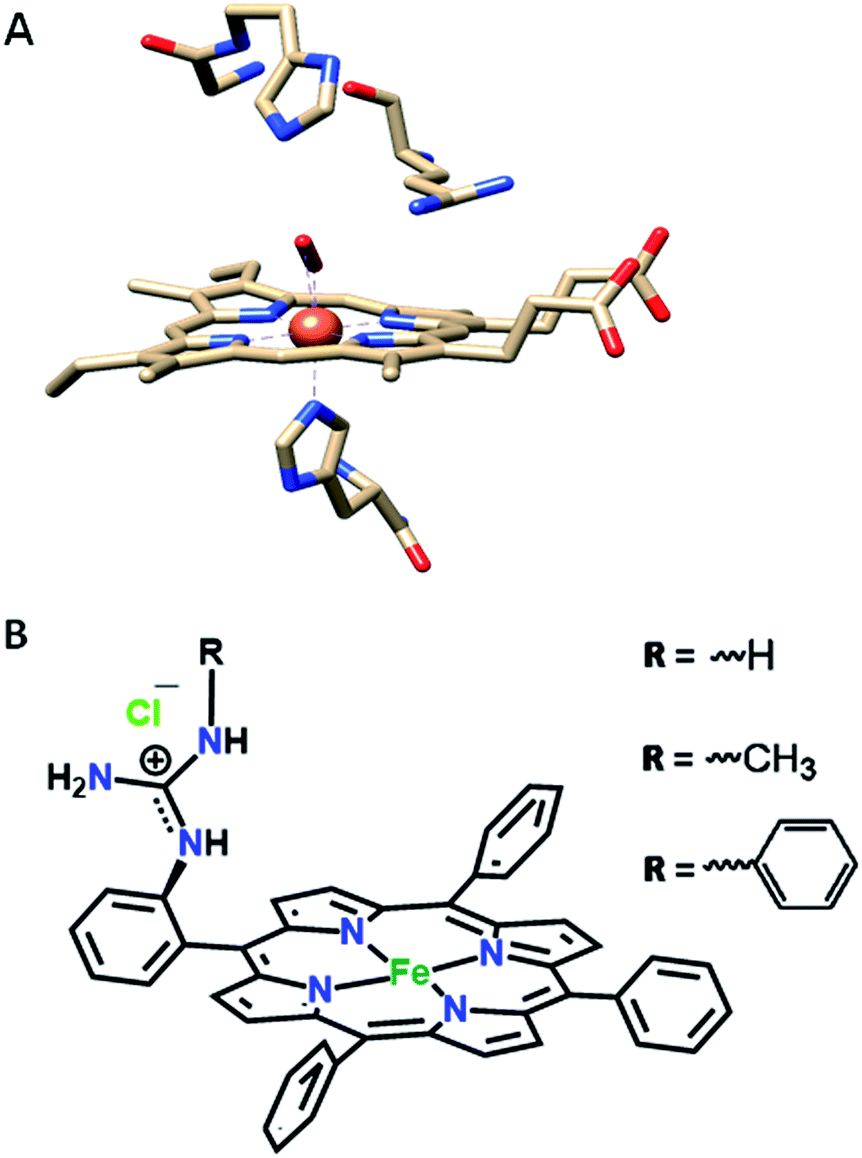
In our continued pursuit of understanding the role of the distal superstructure of the porphyrin active site we are drawn to peroxidases. Heme peroxidases are ubiquitous in nature, where they catalyse the oxidation of organic substrates using H2O2.28 The presence of basic distal residues such as Arg38 and His42 in the distal site (Fig. 2A)29 facilitates the O–O bond cleavage of a LS FeIII–OOH species to form compound I via selective protonation of the distal oxygen atom – the pull effect.30 Recently, a mononuclear iron porphyrin o-monoguanidinotetraphenyliron(III)–porphyrin (FeIIICl–MARG, Fig. 2B, R = H) mimicking the active site of HRP has been developed where a covalently attached guanidium moiety is included in the second sphere mimicking the Arg38 group of HRP. This iron porphyrin is a functional model of HRP and can catalyse the oxidation of most HRP substrates with H2O2. This biomimetic functional model utilizes the ‘ping pong’ mechanism for substrate oxidation akin to HRP with KM and kcat values similar to those of the native enzyme.31
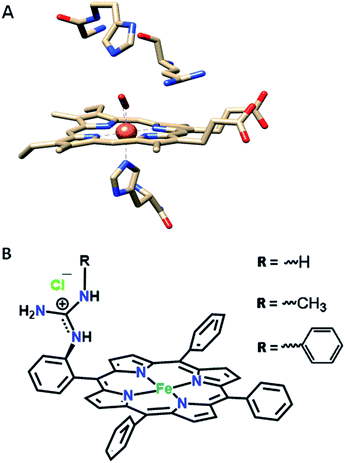 |
| | Fig. 2 (A) The active site structure of HRP (PDB ID: 1H57);32 (B) pictorial representation of all the electrocatalysts used (R = H, FeIIICl–MARG; R = methyl, FeIIICl–MeMARG and R = phenyl, FeIIICl–PhMARG). | |
In this manuscript, the electrochemical oxygen reduction activity of a series of guanidine bearing iron porphyrins is investigated under heterogeneous electrochemical conditions in aqueous solvents, along with their initial spectroscopic and theoretical characterisation. These porphyrins vary in their substitution of the guanidine moiety (Fig. 2B). The results indicate that the inclusion of the guanidine group in the distal environment of a synthetic iron porphyrin enhances the selectivity as well as the rate of the 4e−/4H+ ORR. The rates and selectivity are substantially affected either by inclusion of an axial imidazole ligand or by making the distal pocket more hydrophobic.
Results
The iron porphyrin complexes (Fig. 2) are physiadsorbed over edge plane graphite (EPG) electrodes and alkyl thiol SAM (C8SH/C16SH) covered Au electrodes, and electrochemical data are collected in pH 7 phosphate buffer solution against a Ag/AgCl in saturated KCl reference electrode and a Pt counter electrode. The electron transfer rates vary between 105 and 101 s−1 between EPG and C16SH SAM and this allows evaluation of the role of the pendant guanidine group in stabilizing the intermediates formed during the ORR (Scheme 1) against hydrolysis. The further activation of these species for O–O bond cleavage requires reduction and in the absence of rapid electron flux from the electrode it enhances the release of PROS via hydrolysis. These iron porphyrins can also be chemically attached to the electrode using a mixed thiol SAM combining linker ImdC11SH and diluent C8SH (ImdC11SH SAM) functionalised Au electrode where the imidazole terminal of the SAM acts as an axial ligand (Fig. 1A, ESI†). SERRS data show that the iron porphyrins are in their high spin state bearing an axial –OH ligand with the Fe–OH frequency at 585 cm−1 (Fig. S3, ESI†). The cyclic voltammetry (CV) data of FeIIICl–MARG show a well-developed FeIII/II redox couple for the complex in all the constructs in the absence of O2 at pH 7. For the EPG electrode and C8SH SAM covered Au electrodes, the E1/2 value of the FeIII/II process of Fe–MARG is at −255 mV (Fig. 3A green) and −253 mV (Fig. 3A red), respectively, vs. Ag/AgCl, while that for imidazole bound Fe–MARG is at −221 mV (Fig. 3A blue) vs. Ag/AgCl. Integrating these redox waves, the surface coverage is found to be 0.73 ± 0.60 × 10−12, 1.86 ± 0.40 × 10−12, and 4.84 ± 0.30 × 10−12 mol cm−2 over the EPG electrode, C8SH SAM- and ImdC11SH SAM modified Au electrodes, respectively. In air saturated pH-7 buffer the reversible couple disappears and is replaced by a large irreversible oxygen reduction current indicating the electrocatalytic ORR by FeIIICl–MARG (Fig. 3B). The LSV data show that at the EPG electrode Fe(III)-Cl–MARG reduces O2 at a potential of −240 mV (Fig. 3B green). In contrast, on C8SH SAM and ImdC11SH SAM the complex shows the ORR at a further negative potential of −313 mV (Fig. 3B red) and −368 mV (Fig. 3B blue), respectively, vs. Ag/AgCl compared to the FeIII/II reduction potential indicating that the reduction from FeIII to FeII is not the potential determining step or thermodynamically the most uphill process of the ORR when these complexes are immobilized on SAM.
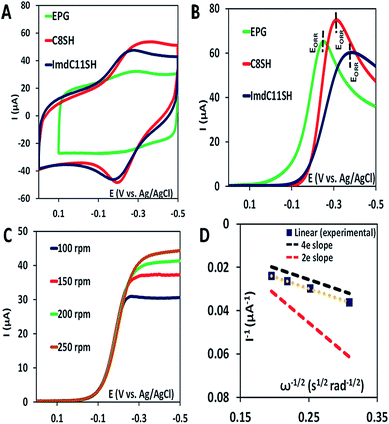 |
| | Fig. 3 (A) CV data of FeIIICl–MARG over the EPG electrode (green), C8SH SAM- (red) and ImdC11SH SAM (blue) modified Au electrodes in the absence of O2 in pH 7 phosphate buffer with KPF6 supporting electrolyte at a scan rate of 1 V s−1; (B) LSV data of the complex in air saturated pH 7 phosphate buffer over EPG (green), C8SH SAM (red) and ImdC11SH SAM (blue) at 0 rpm at a scan rate of 50 mV s−1; (C) RDE plots of the catalyst over the EPG electrode for multiple rotations at a scan rate of 50 mV s−1; (D) K–L plot of the catalyst at a potential of −0.5 V. | |
Rotating disk electrochemistry (RDE) data of FeIIICl–MARG on EPG indicate a normal substrate diffusion limited current for the complex below −240 mV (Fig. 3C). The K–L plot of 1/Icatvs. 1/ω1/2 is linear and the slope is close to the theoretical value for the 4e− reduction process of O2. The 2nd order rate constant for the electrocatalytic ORR by this complex is calculated using K–L analysis. The kcat value obtained in the EPG electrode is 4.2 × 106 M−1 s−1, which exceeds those of synthetic heme/Cu CcO model systems by one order of magnitude.33 Thus, the iron porphyrin having a guanidium moiety in the 2nd sphere, emulating the ‘pull effect’ of HRP, increases the rate and selectivity of the 4e−/4H+ ORR substantially compared to unfunctionalized porphyrins.
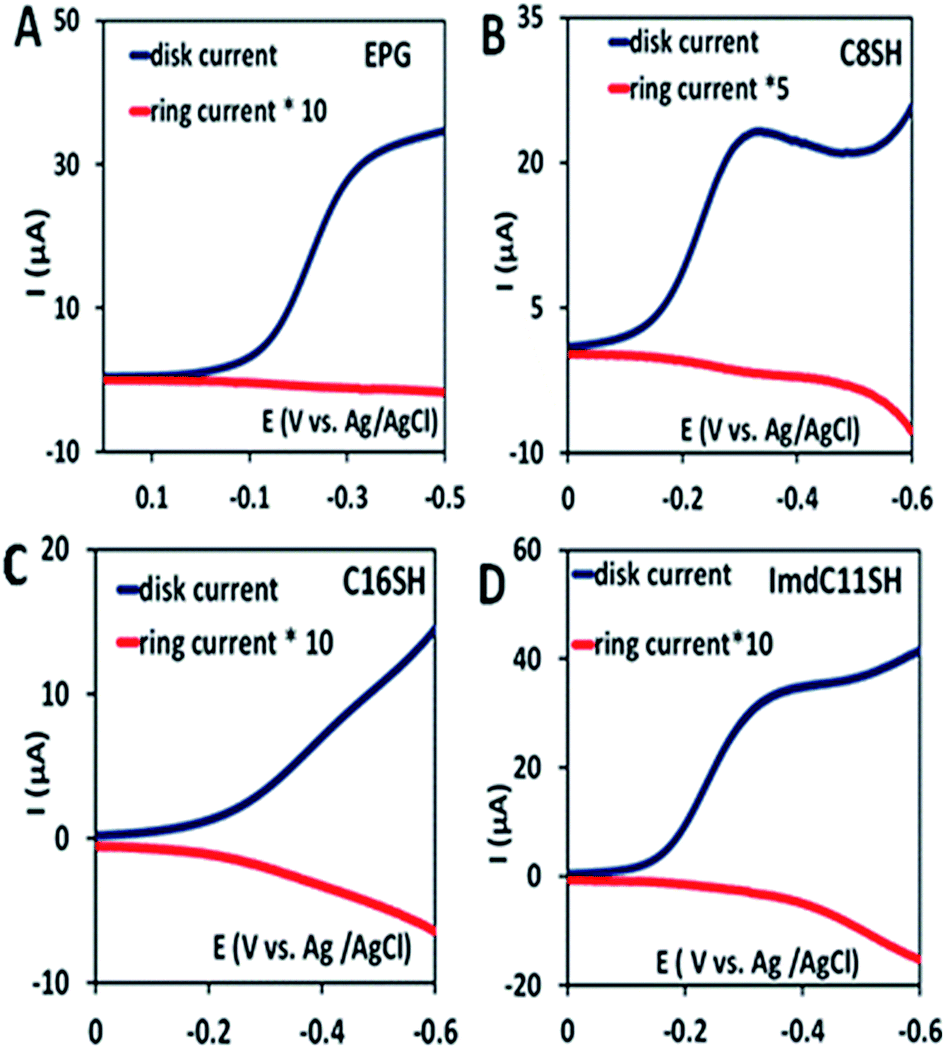
In the rotating ring disk electrochemistry (RRDE) experiment, partially reduced oxygen species (PROS) such as H2O2/O2− produced on the working electrode diffuses to the Pt ring electrode encircling the disk electrode where it can be oxidised back to O2 allowing quantitative detection of PROS (Fig. 4). The FeIIICl–MARG physiadsorbed over the EPG electrode, C8SH SAM covered, and C16SH SAM covered Au electrodes produce 2.54 ± 0.1%, 8.85 ± 0.1% and 18 ± 0.2% PROS (Table 1), respectively. The rate of electron transfer (ET) from the electrode to the catalyst is >105 s−1, 103 s−1, and 10 s−1 for EPG, C8SH and C16SH SAM, respectively.34 Thus, with a decrease in the electron transfer rate from the electrode, i.e. with the increase in the chain length of the SAM, the rate of hydrolysis of the hydroperoxide intermediate species (Scheme 1) and PROS formation increases. FeIIICl–MARG produces a maximum of 18% PROS in contrast to iron porphyrins with hydrogen bonding triazole residues in the distal pocket which produce ∼100% PROS.19 Although the distal guanidine can enhance the selectivity towards 4e−/4H+ reduction of oxygen, it is not as efficient as the recently reported mono-nuclear porphyrins with pendant primary amines or pyridine.26
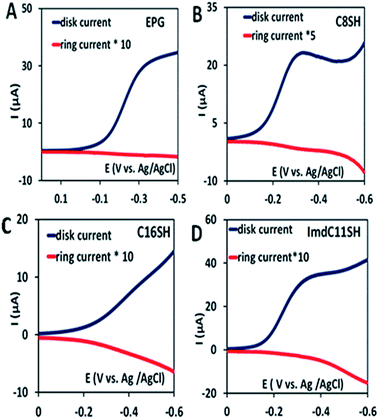 |
| | Fig. 4 RRDE data of the complex on the EPG electrode (A), C8SH SAM modified Au electrode (B), C16SH SAM modified Au electrode (C), and ImdC11SH SAM modified Au electrode (D) at a scan rate of 10 mV s−1 at 300 rpm using a Pt counter electrode and Ag/AgCl (sat. KCl) as a reference electrode. | |
Table 1 Electrochemical ORR data
| Catalyst |
PROS analysis (%) |
| EPGa (106 s−1) |
C8SHa (103 s−1) |
C16SHa (10 s−1) |
ImdC11SHa (103 s−1) |
|
Standard rate of electron transfer.
|
| FeIIICl–MARG |
2.54 ± 0.1 |
8.85 ± 0.1 |
18.0 ± 0.2 |
4.95 ± 0.4 |
| Fe–MARG–N-MeImd |
1.43 ± 0.2 |
3.10 ± 0.5 |
7.00 ± 0.1 |
NA |
| FeIIICl–MeMARG |
2.40 ± 0.3 |
5.43 ± 0.5 |
7.9 ± 0.1 |
2.64 ± 0.1 |
| FeIIICl–PhMARG |
1.80 ± 0.1 |
4.73 ± 0.5 |
9.20 ± 0.4 |
1.32 ± 0.1 |
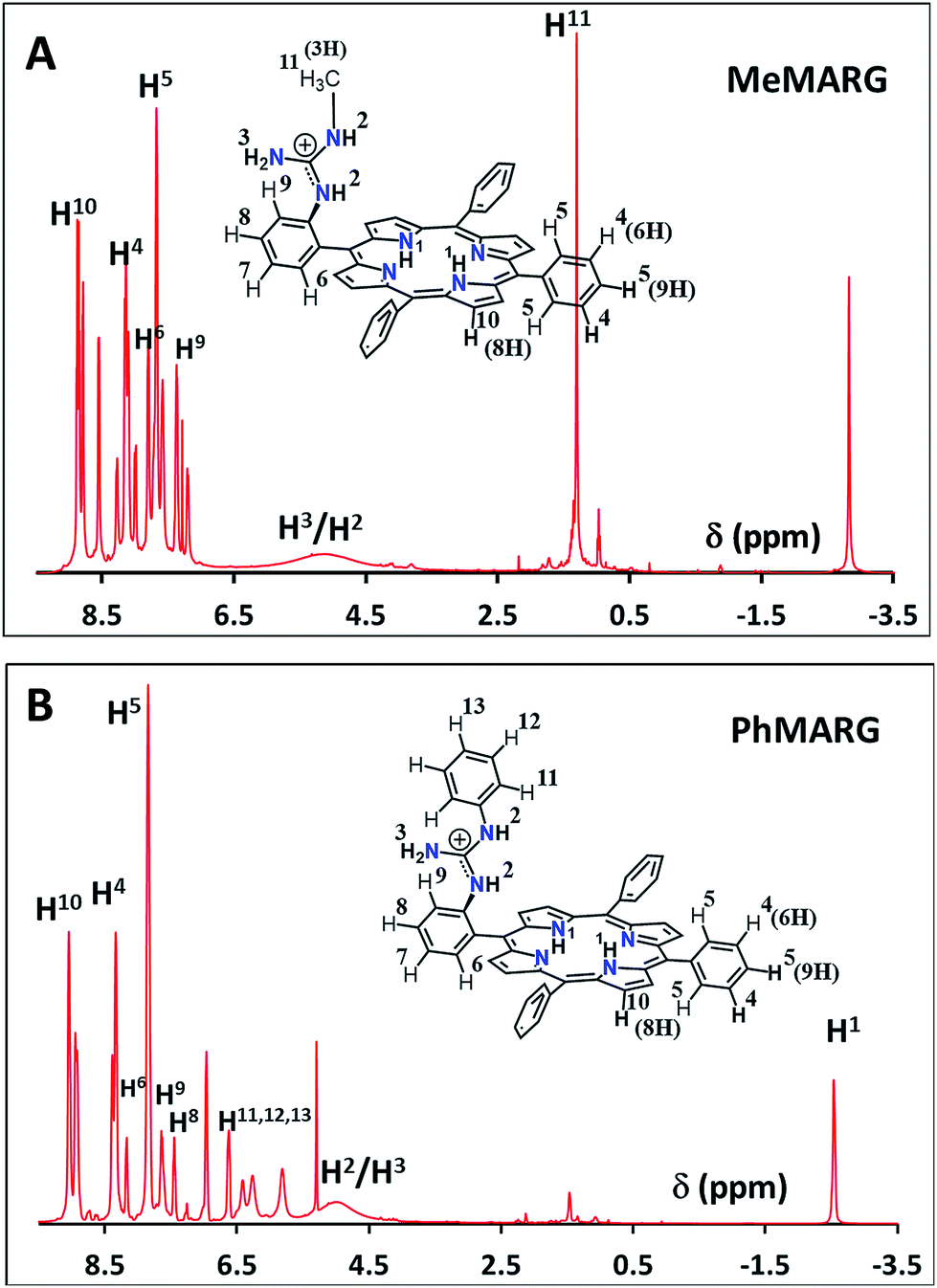
Two structural variants of FeIIICl–MARG are evaluated as well, namely o-monomethylguanidinotetraphenyliron(III)–porphyrin (FeIIICl–MeMARG) and o-monophenylguanidinotetraphenyliron(III)–porphyrin (FeIIICl–PhMARG) (Fig. 2). The 1H NMR data of the free ligand of MeMARG and PhMARG (details in the ESI†) in CDCl3 show that the resonances for the methyl CHs of the guanidine group (Fig. 2, R = methyl) are at 1.35 ppm (Fig. 5A) and the resonances for the aromatic CHs of the guanidine Ph group (Fig. 2, R = phenyl) are at 5.8, 6.2, and 6.4 ppm (Fig. 5B). The 1H resonances of the guanidine substituents indicate that both methyl and phenyl protons are shielded by the aromatic porphyrin group implying that these groups are poised on top of the porphyrin ring. This ought to make the environment of the distal site of the iron porphyrin more hydrophobic. The iron complexes of these ligands show a clear FeIII/II redox process under a N2 atmosphere in pH 7 phosphate buffer (Fig. S7 and S8, ESI†). In the presence of air saturated pH 7 phosphate buffer both FeIIICl–MeMARG and FeIIICl–PhMARG show catalytic ORR currents. Using RRDE the amounts of PROS produced for FeIIICl–MeMARG are determined to be 2.40 ± 0.3%, 5.43 ± 0.5%, 7.9 ± 0.1% for the ORR when it is physiadsorbed over EPG, C8SH and C16SH SAM, respectively (Fig. S9, ESI†). In the case of FeIIICl–PhMARG the same trend is followed with a maximum of 9.20 ± 0.4% PROS on C16SH SAM and only 1.80 ± 0.1% PROS on EPG (Fig. S10, ESI,†Table 1). These data clearly indicate that the % PROS released during the ORR decreases as more hydrophobic residues are included in the pendant guanidine group, enhancing the selectivity towards the 4e−/4H+ ORR. The presence of methyl and phenyl groups just on top of the porphyrin ring likely creates a hydrophobic environment, which slows the hydrolysis of the hydroperoxide intermediate species (Scheme 1).
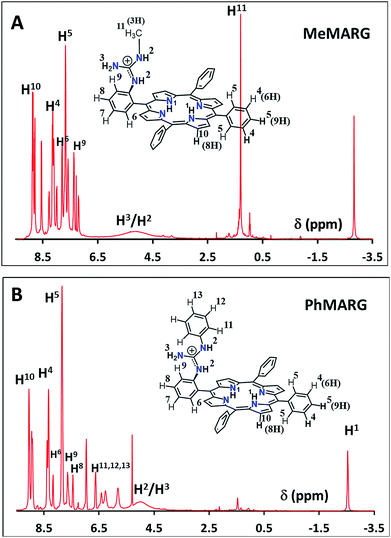 |
| | Fig. 5
1H NMR spectra of (A) MeMARG and (B) PhMARG in CDCl3. | |
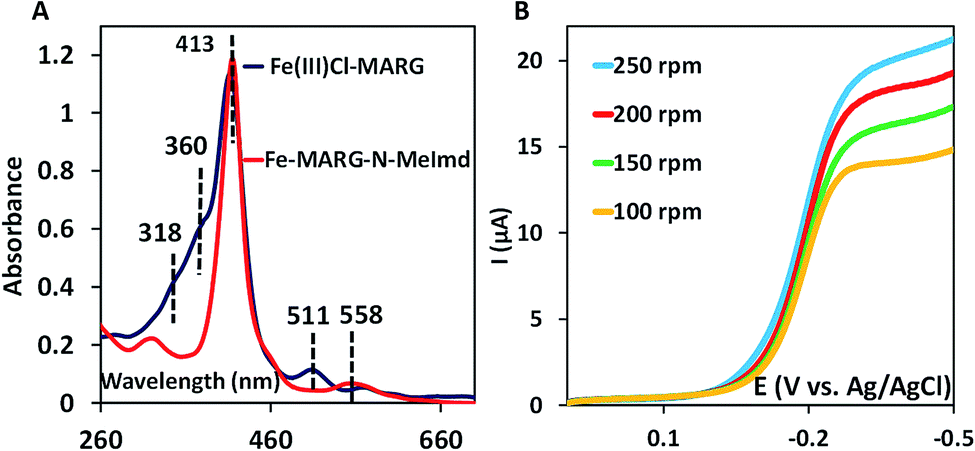
In nature, the heme in the active site of HRP is bound to the protein via an axial histidine ligand (Fig. 1B) which has an imidazole head group. The axial imidazole ligand can be introduced in FeIIICl–MARG either via using an imidazole group terminated SAM or by chemically binding an imidazole to Fe–MARG before its immobilization on the electrode. First, when FeIIICl–MARG is chemically attached to ImdC11SH SAM, the RRDE experiments show that it produces only 4.95 ± 0.4% PROS (Fig. 4D and Table 1) during the ORR, which is almost half of the PROS produced when FeIIICl–MARG is physiadsorbed atop C8SH. Similarly, FeIIICl–MeMARG and FeIIICl–PhMARG produce 2.64 ± 0.3% and 1.32 ± 0.1% PROS when attached to imidazole SAM (Fig. S9 and S10, ESI†). Alternatively, an N-methyl imidazole bound FeIIICl–MARG (Fe–MARG–N-MeImd) complex is obtained by addition of 5 eq. N-methylimidazole to the solution of the complex and confirmed through the UV titration technique (details in the ESI†). In the absorption spectra, Fe–MARG–N-MeImd shows a Soret band at 413 nm and Q bands at 558 nm and 605 nm with the disappearance of characteristic Q bands (511 nm, 580 nm and 690 nm) of FeIII-Cl–MARG (Fig. 6A). Fe–MARG–N-MeImd is physiadsorbed over the EPG electrode, C8SH- and C16SH SAM-covered Au electrodes and its heterogeneous electrochemical ORR is evaluated (Fig. S11, ESI†). The RRDE experiments show only 1.43 ± 0.2, 3.0 ± 0.5% and 7.0 ± 0.05% (Fig. S12, ESI,†Table 1) PROS over the EPG electrode, C8SH- and C16SH SAM-covered Au electrodes, respectively. The K–L analysis of this Fe–MARG–N-MeImd complex adsorbed on the EPG electrode (Fig. 6B) yields a 2nd order rate constant for the 4e−/4H+ ORR of 1.76 × 106 M−1 s−1. These data clearly indicate that the combination of “push” of imidazole and “pull” of guanidine is very effective in allowing a facile and selective ORR for mononuclear iron porphyrins.
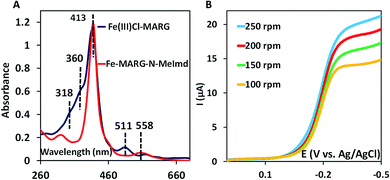 |
| | Fig. 6 (A) Absorption spectra of FeIIICl–MARG (blue) and Fe–MARG–N-MeImd (red); (B) RDE plots of Fe–MARG–N-MeImd over EPG at a scan rate of 50 mV s−1 for multiple rotations. | |
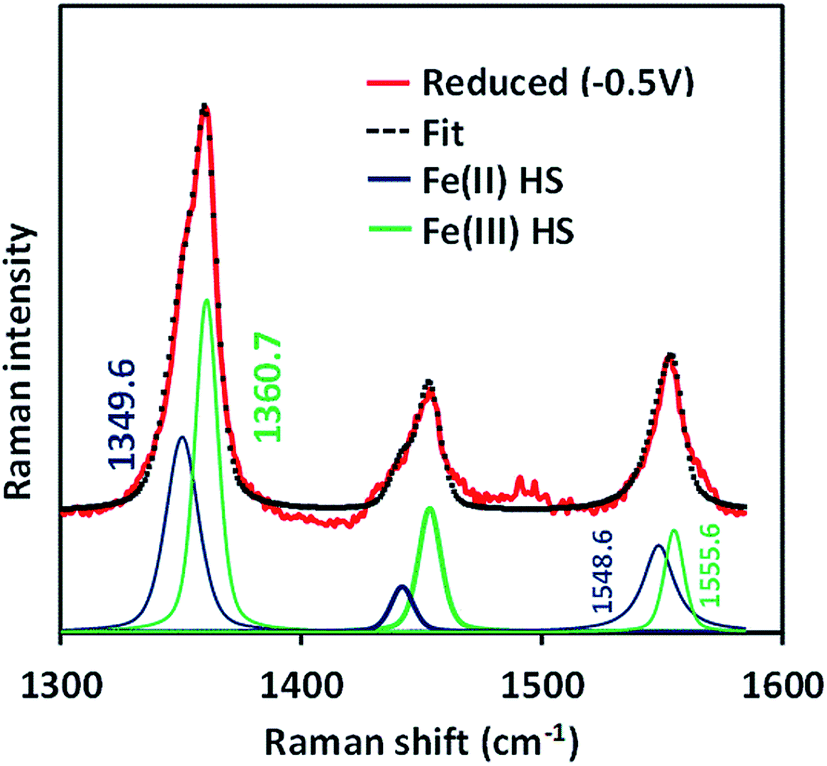
The SERRS-RDE data of FeIIICl–MARG adsorbed on the C8SH SAM modified Ag electrode at −0.5 V vs. Ag/AgCl (sat. KCl) do not show any characteristic oxidation or spin state marker band corresponding to a LS FeIII species as observed for simple iron porphyrins and synthetic models of CcO, rather unreacted HS FeII species is observed with ν4 and ν2 at 1349 cm−1 and 1548 cm−1, respectively (Fig. 7 and S4†). The presence of the reactant HS FeII state in a catalytic steady state implies that O2 binding to the iron is the rds of catalysis commensurate with the proposal that the O–O cleavage is no longer the rds for the iron porphyrin with pendant guanidine groups.
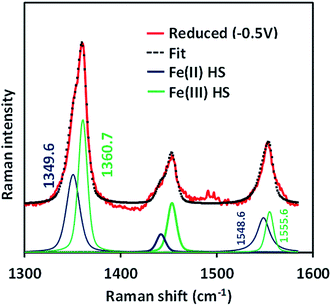 |
| | Fig. 7 SERRS-RDE data of FeIIICl–MARG physiadsorbed on a C8SH SAM modified Ag disc, at reducing potential (−0.5 V, red) in pH 7 phosphate buffer under aerobic conditions at a constant rotation rate of 200 rpm along with its Lorentzian fit showing different components. | |
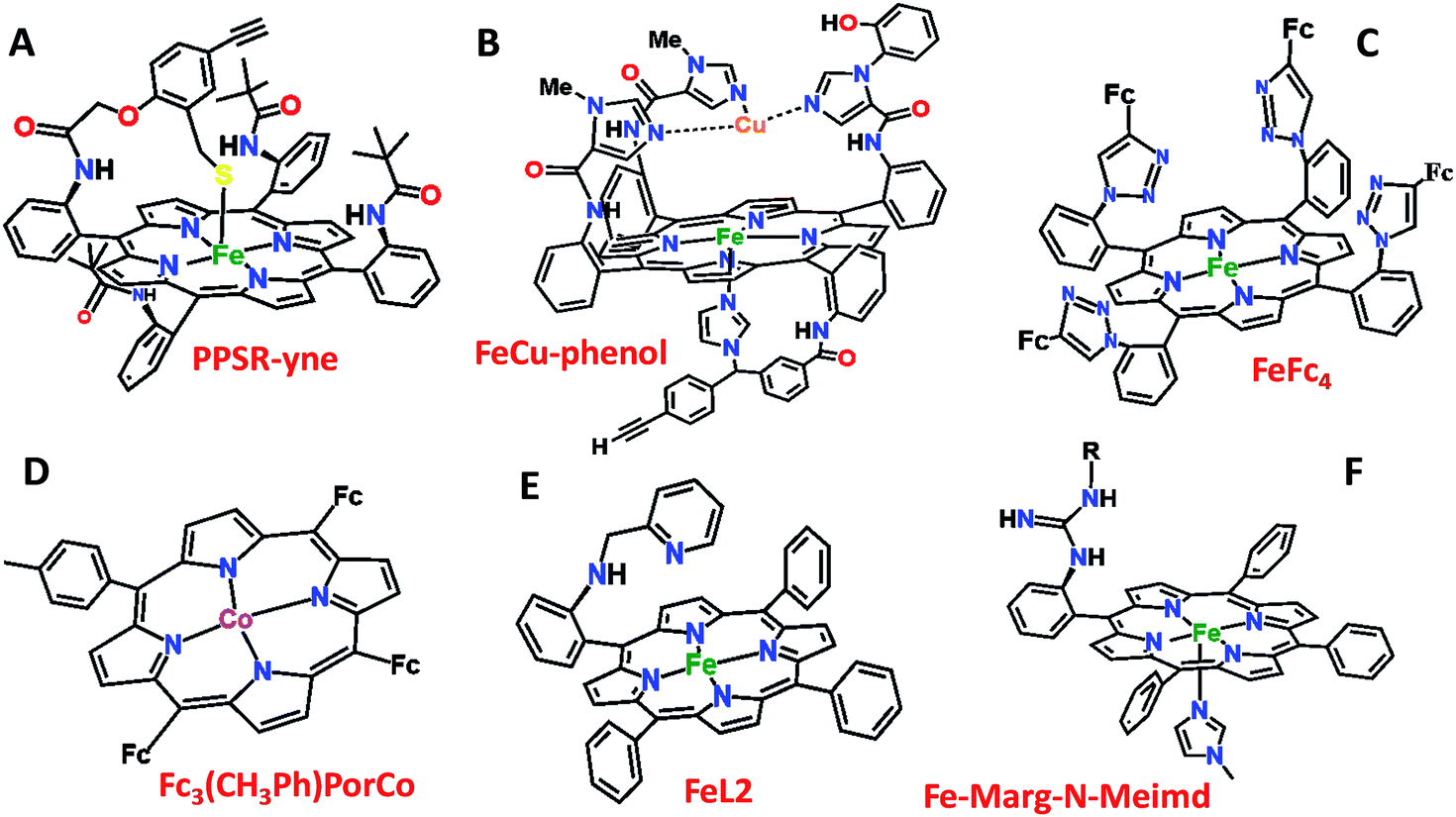
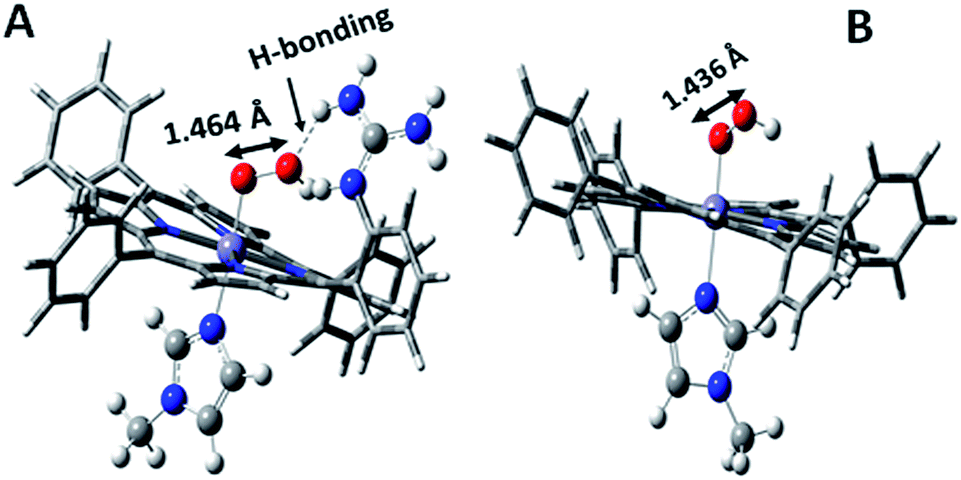
Over the last few years we have explored several factors that affect the selective and facile 4e−/4H+ ORR using iron porphyrins. These include the “push” effect of thiolate, presence of 2nd sphere electron transfer and hydrogen bonding effects.15,26,35,36 Previously the best results were obtained with synthetic mimics of CcO which includes a redox active Cu and phenol (Fig. 8B and Table 2 row 2). The direct in situ SERRS-RDE results for the system clearly indicated that the rate is determined by the rate of O–O bond cleavage of a LS FeIII–OOH species while the selectivity is determined by the site of protonation i.e. on the distal/proximal oxygen atom of the LS FeIII–OOH intermediate.20,21 Although the push effect of a thiolate enhances the pKa of the bound hydroperoxide making the protonation of this species efficient resulting in faster O–O bond cleavage, the protonation is not selective in the distal oxygen atom and substantial H2O2 is released (Fig. 8A and Table 2, row 1).15 In contrast, inclusion of additional electron donor groups does not help the selectivity as the peroxide intermediate is prone to hydrolysis compromising the selectivity (Fig. 8D and Table 2, row 4).37,38 However, when stabilisation of this intermediate via H-bonding or bonding with a distal metal atom is included along with the ET site (e.g. FeFc4 and FeCu–phenol) there is a distinct improvement in selectivity (Fig. 8C and Table 2, row 3; Fig. 8B and Table 2, row 2).11,35 The rate, on the other hand, depends solely on the extent of activation of the O–O bond of the bound peroxide. The activation of the O–O bond can be achieved by a distal metal like Cu (Fig. 8B). The O–O bond cleavage in heme/Cu systems can be further enhanced by H-bonding to a bridging Fe–O–O–Cu peroxide.39 The same is most conveniently achieved in mononuclear porphyrins by introducing protonated basic groups that can form H-bonds as well as translocate protons to the distal oxygen of the bound peroxide (Fig. 8E and Table 2, row 5).26 The iron porphyrins discussed here are designed to do the same. Geometry optimized DFT calculations of the putative imidazole bound LS FeIII–OOH species show that the O–O bond in FeIII–MARG is elongated to 1.46 Å relative to the 1.43 Å in FeIII–TPP (Fig. 9). This weakening of the O–O bond is brought about by the H-bonding with the protonated guanidine group (1.45 Å in neutral guanidine, Fig. S13, ESI†). No accumulation of FeIII–OOH species during the ORR as shown by the SERRS-RDE data is consistent with a fast O–O bond cleavage step. Note that the H-bonding to the guanidine results in 16 kcal mol−1 stronger O2 binding on this site of the porphyrin relative to the open site, suggesting that the ORR is indeed likely to proceed on the side bearing the guanidine (Fig. S14, ESI†). Thus, the H-bonding from the protonated guanidine activates the O–O bond for cleavage which results in enhancement in both the rate and selectivity of the ORR.
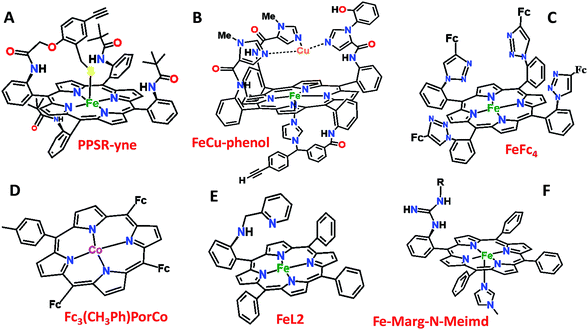 |
| | Fig. 8 Pictorial representation of all the electrocatalysts used in Table 2. Fc: ferrocene. | |
Table 2 Comparison of rates and % PROS values of different porphyrins
| Catalysts |
Effect |
Rate (Kcat) (M−1 S−1) |
PROS (%) C16SH SAM |
Ref. |
| PPSR-yne (Fig. 8A) |
Only ‘push’ from the axial site |
1.11 × 107 |
22 |
15
|
| FeCu–phenol (Fig. 8B) |
Redox species in the 2nd sphere |
1.2 × 105 |
11 |
11
|
| FeFc4 (Fig. 8C) |
Redox species + H-bond (2nd sphere) |
5 × 104 |
10 |
35
|
| Fc3(CH3Ph)–PorCo (Fig. 8D) |
Electron donor groups in the 2nd sphere |
|
100 (EEPG) |
33
|
| FeL2 (Fig. 8E) |
H-bond + protonated basic grp. (2nd sphere) |
1.80 × 107 |
5 |
26
|
| FeIIICl–MARG (Fig. 2) |
H-bond + protonated basic grp. (2nd sphere) |
4.2 × 106 |
18 |
|
| Fe–MARG–N-MeImd (Fig. 8F) |
H-bond + protonated basic grp. (2nd sphere) + push effect |
1.76 × 106 |
7 |
|
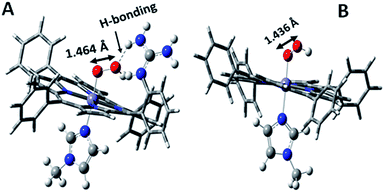 |
| | Fig. 9 DFT optimized structures of N-methylimidazole bound (A) protonated FeIII–MARG hydroperoxide and (B) FeIII–TPP–hydroperoxide. | |
Conclusion
In summary, inclusion of pendant guanidine groups in the iron porphyrin architecture, inspired by the Arg38 residue in the distal site of HRP, increases both the rate and selectivity of the electrocatalytic 4e−/4H+ oxygen reduction reaction in an aqueous environment. Inclusion of the axial imidazole ligand further enhances the selectivity of the oxygen reduction process producing minimal PROS. Thus, the inclusion of push and pull effects of the HRP active site in synthetic iron porphyrins enhances the rate as well as the selectivity of oxygen reduction substantially. The hydrogen bonding from the pendant guanidine group activates the O–O bond of hydroperoxide species and thus is responsible for the selectivity and rate enhancement observed here. In situ spectroscopy indicates the oxygen binding to iron step as the rds of the ORR process.
Materials and methods
Details of the experimental procedure and materials are included in the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by Department of Science and Technology Grant SERM/EMR/008063 and Council of Scientific and Industrial Research Grant (CSIR) No. 01(2874)/17/EMR-II. A. G. and S. B. thank UGC SRF and CSIR SPMF, respectively, and S. B. acknowledges IACS integrated Ph.D.
Notes and references
- Y.-H. Wang, Z. K. Goldsmith, P. E. Schneider, C. W. Anson, J. B. Gerken, S. Ghosh, S. Hammes-Schiffer and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 10890–10899 CrossRef CAS
 .
.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS .
- J. P. Collman, R. A. Decréau, H. Lin, A. Hosseini, Y. Yang, A. Dey and T. A. Eberspacher, Proc. Natl. Acad. Sci., 2009, 106, 7320 CrossRef CAS .
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS .
- S. M. Adam, G. B. Wijeratne, P. J. Rogler, D. E. Diaz, D. A. Quist, J. J. Liu and K. D. Karlin, Chem. Rev., 2018, 118, 10840–11022 CrossRef CAS .
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 0098 CrossRef CAS .
- M. L. Rigsby, D. J. Wasylenko, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2015, 137, 4296–4299 CrossRef CAS .
- S. Ferguson-Miller and G. T. Babcock, Chem. Rev., 1996, 96, 2889–2908 CrossRef CAS .
- T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono and S. Yoshikawa, Science, 1996, 272, 1136 CrossRef CAS .
- M. Svensson-Ek, J. Abramson, G. Larsson, S. Tornroth, P. Brzezinski and S. Iwata, J. Mol. Biol., 2002, 321, 329–339 CrossRef CAS .
- J. P. Collman, N. K. Devaraj, R. A. Decréau, Y. Yang, Y.-L. Yan, W. Ebina, T. A. Eberspacher and C. E. D. Chidsey, Science, 2007, 315, 1565–1568 CrossRef CAS .
- S. Hematian, I. Garcia-Bosch and K. D. Karlin, Acc. Chem. Res., 2015, 48, 2462–2474 CrossRef CAS .
- S. Chatterjee, K. Sengupta, S. Hematian, K. D. Karlin and A. Dey, J. Am. Chem. Soc., 2015, 137, 12897–12905 CrossRef CAS .
- S. Chatterjee, K. Sengupta, B. Mondal, S. Dey and A. Dey, Acc. Chem. Res., 2017, 50, 1744–1753 CrossRef CAS .
- S. Chatterjee, K. Sengupta, S. Samanta, P. K. Das and A. Dey, Inorg. Chem., 2015, 54, 2383–2392 CrossRef CAS .
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS .
- E. D. C. Christopher, Science, 1991, 251, 919–922 CrossRef .
- K. Mittra, S. Chatterjee, S. Samanta and A. Dey, Inorg. Chem., 2013, 52, 14317–14325 CrossRef CAS .
- S. Samanta, K. Sengupta, K. Mittra, S. Bandyopadhyay and A. Dey, Chem. Commun., 2012, 48, 7631–7633 RSC .
- K. Sengupta, S. Chatterjee, S. Samanta and A. Dey, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8431 CrossRef CAS .
- K. Sengupta, S. Chatterjee and A. Dey, ACS Catal., 2016, 6, 6838–6852 CrossRef CAS .
- S. Mukherjee, M. Mukherjee, A. Mukherjee, A. Bhagi-Damodaran, Y. Lu and A. Dey, ACS Catal., 2018, 8, 8915–8924 CrossRef CAS .
- K. Sengupta, S. Chatterjee and A. Dey, ACS Catal., 2016, 6, 1382–1388 CrossRef CAS .
- S. Samanta, P. K. Das, S. Chatterjee, K. Sengupta, B. Mondal and A. Dey, Inorg. Chem., 2013, 52, 12963–12971 CrossRef CAS .
- K. Sengupta, S. Chatterjee, S. Mukherjee, S. G. Dey and A. Dey, Chem. Commun., 2014, 50, 3806–3809 RSC .
- S. Bhunia, A. Rana, P. Roy, D. J. Martin, M. L. Pegis, B. Roy and A. Dey, J. Am. Chem. Soc., 2018, 140, 9444–9457 CrossRef CAS .
- S. Chatterjee, K. Sengupta, S. Samanta, P. K. Das and A. Dey, Inorg. Chem., 2013, 52, 9897–9907 CrossRef CAS .
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS .
- M. Gajhede, D. J. Schuller, A. Henriksen, A. T. Smith and T. L. Poulos, Nat. Struct. Biol., 1997, 4, 1032–1038 CrossRef CAS .
- E. Derat and S. Shaik, J. Phys. Chem. B, 2006, 110, 10526–10533 CrossRef CAS .
- S. Bhakta, A. Nayek, B. Roy and A. Dey, Inorg. Chem., 2019, 58, 2954–2964 CrossRef CAS .
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, Nature, 2002, 417, 463–468 CrossRef CAS .
- R. Boulatov, J. P. Collman, I. M. Shiryaeva and C. J. Sunderland, J. Am. Chem. Soc., 2002, 124, 11923–11935 CrossRef CAS .
- C. E. D. Chidsey, Science, 1991, 251, 919 CrossRef CAS .
- S. Samanta, K. Mittra, K. Sengupta, S. Chatterjee and A. Dey, Inorg. Chem., 2013, 52, 1443–1453 CrossRef CAS .
- K. Sengupta, S. Chatterjee, S. Samanta, S. Bandyopadhyay and A. Dey, Inorg. Chem., 2013, 52, 2000–2014 CrossRef CAS .
- L. Ye, Y. Fang, Z. Ou, S. Xue and K. M. Kadish, Inorg. Chem., 2017, 56, 13613–13626 CrossRef CAS .
- B. Sun, Z. Ou, D. Meng, Y. Fang, Y. Song, W. Zhu, P. V. Solntsev, V. N. Nemykin and K. M. Kadish, Inorg. Chem., 2014, 53, 8600–8609 CrossRef CAS .
- A. W. Schaefer, M. T. Kieber-Emmons, S. M. Adam, K. D. Karlin and E. I. Solomon, J. Am. Chem. Soc., 2017, 139, 7958–7973 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, the synthetic procedure, construction of electrodes, spectral data, electrochemical data and DFT optimized structures. See DOI: 10.1039/c9sc02711d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*