
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling magnetism of Au133(TBBT)52 nanoclusters at single electron level and implication for nonmetal to metal transition†
Chenjie
Zeng‡§
 ,
Andrew
Weitz‡
,
Gayathri
Withers
,
Tatsuya
Higaki
,
Shuo
Zhao
,
Yuxiang
Chen
,
Roberto R.
Gil
,
Michael
Hendrich
* and
Rongchao
Jin
*
,
Andrew
Weitz‡
,
Gayathri
Withers
,
Tatsuya
Higaki
,
Shuo
Zhao
,
Yuxiang
Chen
,
Roberto R.
Gil
,
Michael
Hendrich
* and
Rongchao
Jin
*
Department of Chemistry, Carnegie Mellon University, 4400 Fifth Ave, Pittsburgh, PA, USA. E-mail: hendrich@andrew.cmu.edu; rongchao@andrew.cmu.edu
First published on 4th September 2019
Abstract
The transition from the discrete, excitonic state to the continuous, metallic state in thiolate-protected gold nanoclusters is of fundamental interest and has attracted significant efforts in recent research. Compared with optical and electronic transition behavior, the transition in magnetism from the atomic gold paramagnetism (Au 6s1) to the band behavior is less studied. In this work, the magnetic properties of 1.7 nm [Au133(TBBT)52]0 nanoclusters (where TBBT = 4-tert-butylbenzenethiolate) with 81 nominal “valence electrons” are investigated by electron paramagnetic resonance (EPR) spectroscopy. Quantitative EPR analysis shows that each cluster possesses one unpaired electron (spin), indicating that the electrons fill into discrete orbitals instead of a continuous band, for that one electron in the band would give a much smaller magnetic moment. Therefore, [Au133(TBBT)52]0 possesses a nonmetallic electronic structure. Furthermore, we demonstrate that the unpaired spin can be removed by oxidizing [Au133(TBBT)52]0 to [Au133(TBBT)52]+ and the nanocluster transforms from paramagnetism to diamagnetism accordingly. The UV-vis absorption spectra remain the same in the process of single-electron loss or addition. Nuclear magnetic resonance (NMR) is applied to probe the charge and magnetic states of Au133(TBBT)52, and the chemical shifts of 52 surface TBBT ligands are found to be affected by the spin in the gold core. The NMR spectrum of Au133(TBBT)52 shows a 13-fold splitting with 4-fold degeneracy of 52 TBBT ligands, which are correlated to the quasi-D2 symmetry of the ligand shell. Overall, this work provides important insights into the electronic structure of Au133(TBBT)52 by combining EPR, optical and NMR studies, which will pave the way for further understanding of the transition behavior in metal nanoclusters.
Introduction
Metal nanoclusters represent an important stage in the evolution from discrete atoms to regular nanoparticles as many transitions in terms of structure and properties are expected to occur over the nanocluster regime.1 Recent advances in solution-phase synthesis of atomically precise metal nanoclusters and their total structure determination by X-ray crystallography have opened up exciting opportunities for exploring the precise structure–property correlations.1–5 Significant progress has been achieved in controlling the size and structure of gold,6–12 silver,13–21 copper,22–24 and alloy nanoclusters.25–30 Such new materials hold potential in a wide range of applications, such as catalysis, chemical and biological detection, drug delivery, to name a few.31–36With respect to the optical transition, recent work has mapped out that the transition from excitonic to plasmonic state occurs between Au246(SR)80 to Au279(SR)84 for quasi-spherical gold nanoclusters.37–39 In contrast, the magnetic transition of Aun(SR)m nanoclusters remains largely elusive.40,41 It is known that discrete gold atoms are paramagnetic due to an unpaired electron in the 6s orbital (6s1), while bulk gold is diamagnetic because the diamagnetic contribution from paired core-electrons outweigh the weak Pauli paramagnetism from itinerant electrons in the conduction band.42–44 Therefore, it is important to investigate the transition from atomic paramagnetism to bulk diamagnetism via atomically precise Aun(SR)m nanoclusters. However, this remains a challenge because of major difficulties in controlling nanoclusters at the single-electron level.45–48 For example, oxidation treatment of the 8-electron Au23(SR)16− unfortunately led to size conversion,49,50 rather than the desired sole change of charge state as in Au25(SR)18−.46–48
Despite the challenges in single-electron level control of metal nanoclusters,51 impressive progress has recently been achieved.52–57 Maran and coworkers have done a series of work on the magnetism of nanoclusters.41 Using Au25(SR)18 as an example, controlling its charge states has led to the observation of paramagnetism (e.g. neutral Au25) and diamagnetism (e.g. ±1 states of Au25) probed by electron paramagnetic resonance (EPR) and nuclear magnetic resonance (NMR) measurements. In a simplified picture, neutral [Au25(SR)18]0 has an electron count of 7e (i.e. 25 − 18 = 7); note that each monovalent thiolate covalently localizes one 6s1 electron from gold. The seven delocalized Au 6s1 electrons are distributed in the superatomic orbitals (1s21p5) of the Au25.40 The odd number of valence electron count leads to one unpaired electron (i.e. spin), which indeed explains the origin of the observed paramagnetism in [Au25(SR)18]0. When the spin is removed by oxidation to [Au25(SR)18]+ (6e) or is paired again with an electron via chemical reduction (i.e. [Au25(SR)18]−, 8e), the nanocluster becomes diamagnetic.40,47,48,52 For Pd or Pt doped [Au24M(SR)18]0 (M = Pd or Pt), Wu and coworkers reported the absence of paramagnetism despite the same charge state as [Au25(SR)18]0.55 Knappenberger et al. probed the spin-conversion dynamics in [Au25(SC8H9)18]0 and revealed multiple spin-polarized transient signals.45 Solid-state magnetic properties of Au25 have also been probed by various groups.58,59 Maran et al. has recently provided an excellent summary on the Au25 magnetic properties.41 While much work has been done on the Au25 system, there has been no extension yet to larger sizes of Aun(SR)m; the large-size regime (with hundreds of gold atoms) is desirable as transitions are expected to occur.
In this work we report our effort in controlling magnetism of a 1.7 nm Au133(TBBT)52 nanocluster (where TBBT represents 4-tert-butylbenzenethiolate) at the single-electron level. Unlike the vast majority of nanoclusters ranging from small sizes (e.g. 4e Au20(SR)16,60 8e Au28(SR)20,61,62 14e Au38(SR)24 (ref. 63)) to large ones (e.g. 80e Au130(SR)50,64 84e Au144(SR)60,8 166e Au246(SR)80 (ref. 6)), where all the clusters have even counts on valence electrons, Au133(TBBT)52 is unique in that it possesses an odd count (81e).65 Significantly, in this work we find that Au133(TBBT)52 exhibits interesting paramagnetism, with one spin per particle. This result is surprising given its large size and seemly metallic state.66 We have further investigated the spin effects on the optical properties and ligand's chemical shifts, as well as comparisons with the classical Au25 system.
Results and discussions
The Au133(TBBT)52 nanocluster was synthesized by the LEIST method50,65 (where, LEIST = ligand-exchange induced size/structure transformation reaction). Briefly, Au144(SC2H4Ph)60 nanoclusters were first made and then reacted with excess TBBT thiols at 80 °C under ambient conditions. After 4 days, Au144(SC2H4Ph)60 were completely transformed into Au133(TBBT)52 (see ESI† for details). To ensure the highest purity and quality, Au133(TBBT)52 was crystallized prior to EPR, NMR and optical measurements. The total structure of Au133(TBBT)52 was reported by Zeng et al.;65 of note, the partial structure without the surface ligand patterns was reported by Dass et al.66 Both Au133 and Au25 possess a common icosahedral core,65–67 with Au25 being built from a one-shelled icosahedron containing 13 atoms (Au13@[Au2(SR)3]6, Scheme 1A),67 whereas Au133 from a two-shell icosahedron containing 55 atoms (i.e. Au13@Au42@Au52@[Au(SR)2]26,65,66Scheme 1B).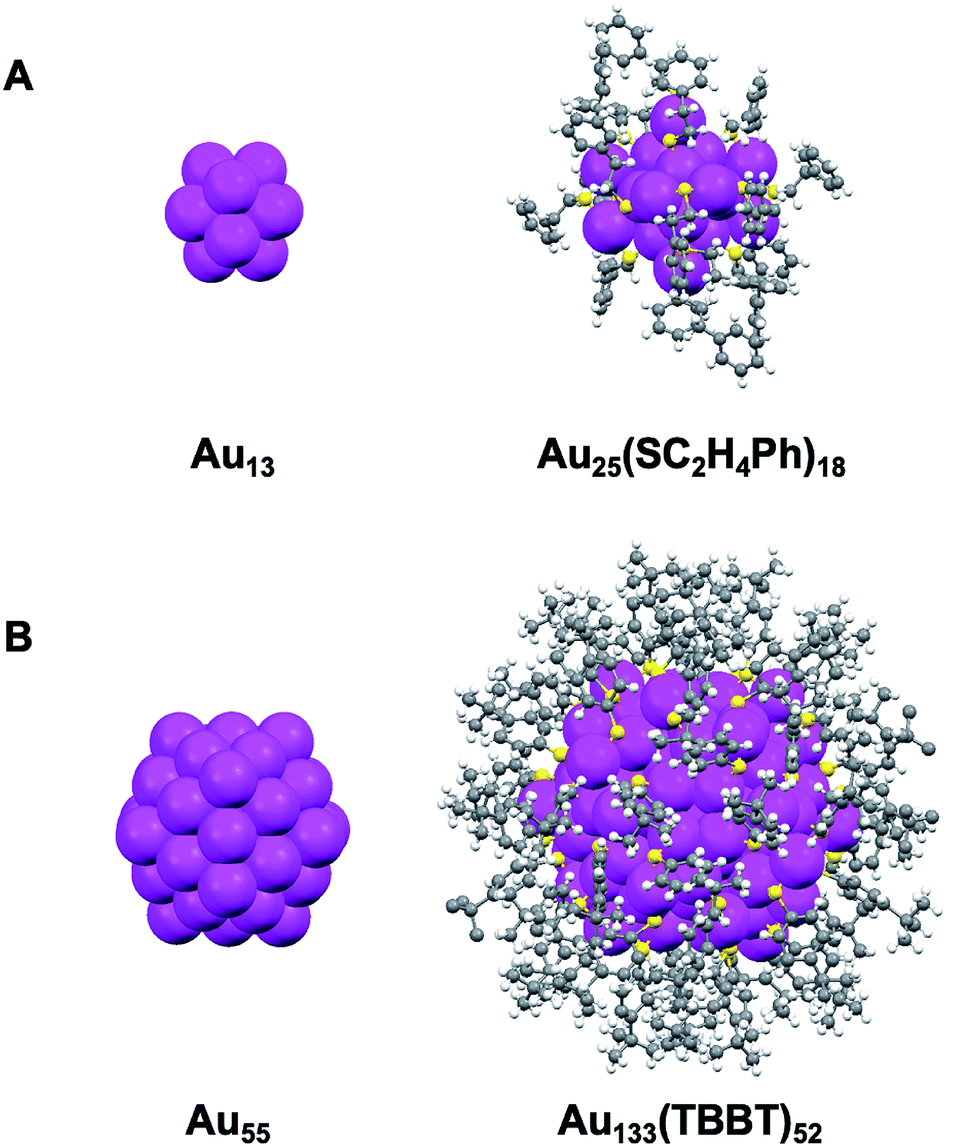 | ||
| Scheme 1 Comparison between the X-ray structures of (A) Au25(SC2H4Ph)18 and (B) Au133(TBBT)52 (TBBT = S-Ph-p-tBu). Redrawn from ref. 65 and 67. | ||
Spin properties of Au133(TBBT)52
To measure the EPR spectra, [Au133(TBBT)52]0 crystals65 were dissolved in a mixed solvent of toluene/CH2Cl2 (volumetric ratio of 1/1, which forms a clear glass at cryogenic temperatures). Fig. 1 shows S-band (3.480 GHz) and X-band (9.645 GHz) EPR spectra of [Au133(TBBT)52]0. The spectra are displayed on different magnetic field scales that equate the g-value scales for both microwave frequencies. Both S- and X-band spectra display a signal with g‖ = 2.47 and g⊥ = 1.69. The signal at g = 2.03 (S-band only) is from a minor extraneous copper impurity. The temperature dependence of the signal intensity from 2 to 25 K was observed to obey the Curie law (intensity vs. 1/T), which indicate the behavior of molecular magnetism as opposed to the electron-conduction-band Pauli paramagnetism (i.e., temperature independent).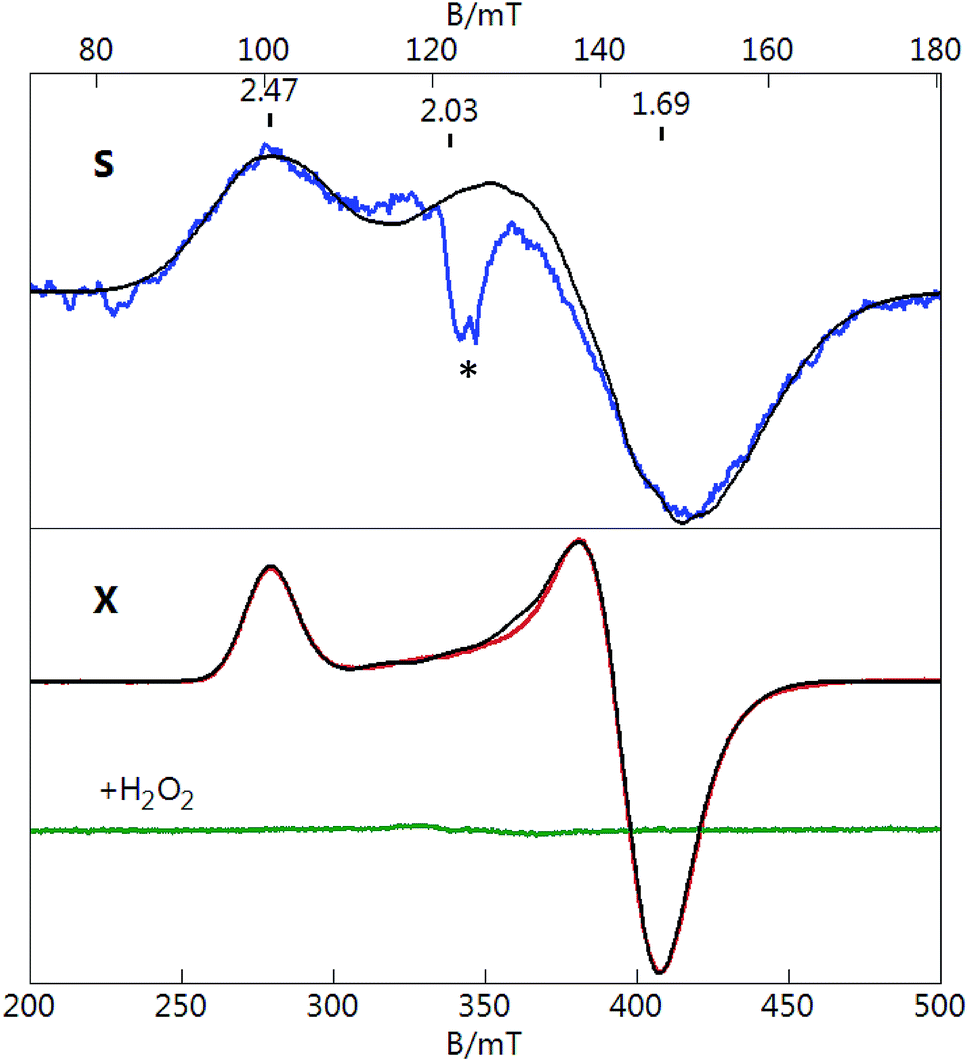 | ||
Fig. 1 S-band (3.480 GHz) and X-band (9.645 GHz) EPR spectra of [Au133(TBBT)52]0 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2:toluene at T = 10 K (S-band), 6 K (X-band). Microwave power: 0.3 mW (S-band), 2 mW (X-band), see text for parameters of simulations (black lines). The green trace shows loss of signal after H2O2 addition. The g = 2.03 signal (indicated by *) is from the extraneous copper impurity. :1 CH2Cl2:toluene at T = 10 K (S-band), 6 K (X-band). Microwave power: 0.3 mW (S-band), 2 mW (X-band), see text for parameters of simulations (black lines). The green trace shows loss of signal after H2O2 addition. The g = 2.03 signal (indicated by *) is from the extraneous copper impurity. | ||
EPR quantification reveals a S = 1/2 spin concentration that agrees with the molar concentration of the prepared [Au133(TBBT)52]0 solution. Thus, each [Au133(TBBT)52]0 nanocluster hosts a total of one unpaired spin, which results from the odd 81 electrons. The observation of an unpaired spin in [Au133(TBBT)52]0 indicates that the 6s1 electrons from Au atoms are filled onto discrete energy levels of the nanocluster, which infers that the continuous band structure has not formed in Au133(TBBT)52 despite its 1.7 nm size. Similar to the case of [Au25(SR)18]0, the unpaired spin can be removed by oxidation to [Au133(TBBT)52]+, which has an electron count of 80. As shown in Fig. 1 (green line), when reacting with H2O2, the sample showed total loss of the paramagnetic signal, indicating that [Au133(TBBT)52]+ is diamagnetic.
To gain a better understanding of the unpaired spin and the EPR spectrum of Au133, we first consider the same effects from the previously characterized [Au25(SR)18]0 nanoclusters. Fig. 2 displays S- and X-band EPR spectra of [Au25(SR)18]0 in 1:1 DCM:toluene solution at cryogenic temperatures. The X-band spectrum reproduces our previous work.40 Both spectra display a signal with a g-tensor of (2.61, 2.34, 1.81), which in contrast to [Au133(TBBT)52]0 is not axial. If the larger two g-values of Au25 (2.61 and 2.34) are averaged and associated with an axial tensor to give g‖ = 1.81 and g⊥ = 2.48, then the values are roughly similar to Au133 but have swapped directions, indicating a change in symmetry for the orbital of the unpaired electron. If we follow the superatomic orbital sequence of 1S 1P 1D 2S 1F 2P 1G 2D 1H, we obtain 1H orbital (angular momentum quantum number l = 5, degeneracy 2l + 1 = 11, 13e filled) as the HOMO of [Au133(TBBT)52]0 (cf. 1P5 as the HOMO of [Au25(SR)18]0); apparently, if the last electron is indeed filled into the 1H orbital, then the 1H orbital (a set of 11) should be split, with the topmost orbital being non-degenerate. This is reminiscent of the case of [Au25(SR)18]0, in which the HOMO (1P set) is split into non-degenerate orbitals.52 Future theoretical simulations40,52 may reveal more insight into the cluster/molecular orbitals of Au133.
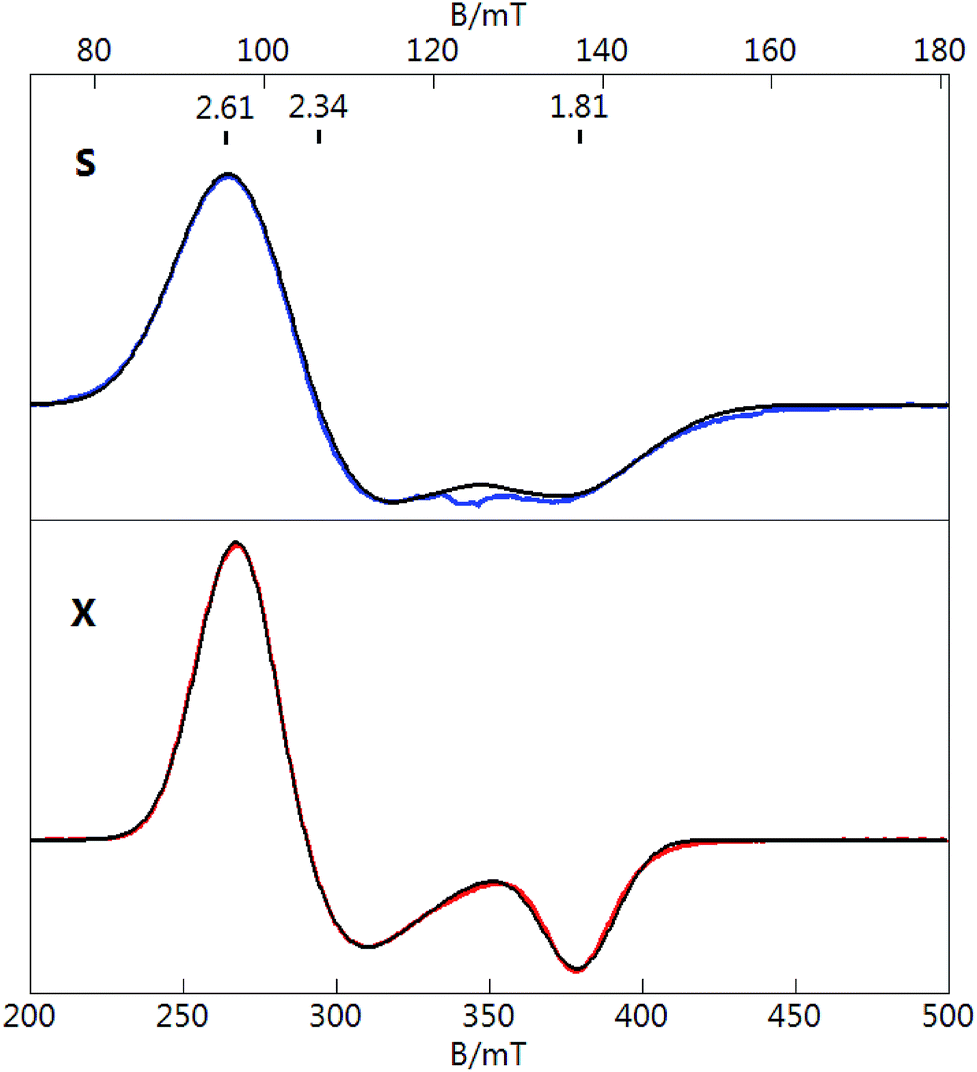 | ||
| Fig. 2 S-band (3.489 GHz) and X-band (9.644 GHz) EPR spectra of [Au25(SR)18]0 in 1:1 CH2Cl2:toluene at T = 10 K (S), 6 K (X). Microwave power: 0.3 mW (S), 2 mW (X). See text for parameters of simulations (black lines). | ||
The S-band spectrum is broader that the X-band spectrum due to unresolved Au hyperfine structure of [Au25(SR)18]0 (note: 197Au nuclear spin I = 3/2, natural abundance 100%). The black traces overlaid on both spectra (Fig. 2) are simultaneous least-squares fits to the data for S = 1/2 and 12 equivalent Au nuclei with an isotropic A-tensor of Aiso = 48 MHz. The value of Aiso agrees with that determined from ENDOR spectroscopy for the icosahedral core of inner Au nuclei.53 One unpaired electron equally delocalized over 12 Au nuclei would equate to a value of Aiso ≈ 600 MHz for an unpaired electron localized at a single Au atom. This value can be compared to the value of Aiso for a free Au atom of 3050 MHz. The reduction of Aiso for the Au nanoclusters is consistent with the significant sp hybridization of the Au centers as described previously.40
The simulations displayed in Fig. 1 for [Au133(TBBT)52]0 are least-squares fits for S = 1/2 and 12 equivalent Au (I = 3/2) nuclei with an isotropic A-tensor of Aiso = 50 MHz. These values are comparable to that of [Au25(SR)18]0. We considered that the unpaired electron for [Au133(TBBT)52]0 could be distributed over a significantly higher number of Au centers; for example, the inner 55 Au centers (Scheme 1) that are all bonded only to neighboring Au center. Simulations with 55 equivalent Au centers required Aiso = 26 MHz. This value would give an Aiso for an unpaired electron localized at a single Au center of 1500 MHz. The higher values would require a significant change in the hybridization of unpaired orbital, which is unlikely. Thus, although the number of Au centers is significantly greater for [Au133(TBBT)52]0, the delocalization of the wavefunction of the unpaired electron is similar to that of [Au25(SR)18]0. Therefore, the EPR analysis implies that the 1H orbital should be in the inner-most icosahedron.
The observation of a single unpaired electron in the [Au133(TBBT)52]0 infers that the Au133 cluster has similar discrete electronic orbitals as in the Au25 to hold the unpaired spin. In other words, Au133 has a non-metallic nature; otherwise, an itinerant metallic-like conduction band would be formed and the cluster would show Pauli paramagnetism (much weaker than the observed molecular paramagnetism). The observation of molecular magnetism is important for understanding of the transition between molecular and metallic-state behaviors in Au nanoclusters/nanoparticles, and Au nanomagnetism in general. From the EPR study, the conclusion of Au133 being nonmetallic is consistent with the conclusion provided by the optical absorption of Au133, which shows multiple peaks at 336, 421, 503 and 712 nm (Fig. 3), as well as the femtosecond transient signal which showed a laser-power independent decay rate of photoexcited electron,65 hence, nonmetallic or non-plasmonic nature of the Au133 cluster. Our results disagree with the previous conclusion that Au133(SR)52 was metallic.66
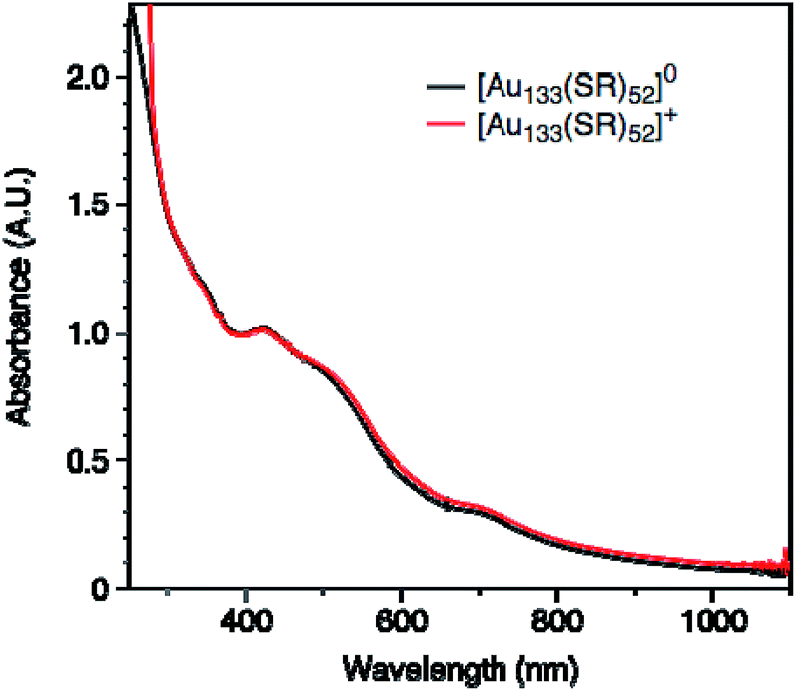 | ||
| Fig. 3 UV-vis absorption spectra of [Au133(TBBT)52]0 and [Au133(TBBT)52]+. | ||
Optical absorption properties of [Au133(TBBT)52]0 and [Au133(TBBT)52]+
We further measured the UV-vis absorption spectra of two charge states, which showed no distinguishable difference between [Au133(TBBT)52]0 and [Au133(TBBT)52]+ (Fig. 3). Therefore, it is difficult to differentiate the charge states of Au133(TBBT)52 by its absorption spectrum. This is different from the case of Au25(SR)18,46 in which the change of the oxidation state from [Au25(SR)18]− to [Au25(SR)18]0 resulted in a blue shift of the ∼670 nm peak as well as an increase in the ∼400 nm peak intensity. The similarity between the absorption spectra of [Au133(TBBT)52]0 and [Au133(TBBT)52]+ may be ascribed to the much less perturbation of one electron loss in Au133 (1 out 81e) than Au25 (1 out of 8e) as well as possibly less structural distortion in Au133 than Au25 when switching the charge state.46,56,57We also performed electrochemical oxidation of [Au133(TBBT)52]0 by applying positive voltages and monitored the optical spectra in situ,68,69 but no change in optical spectra was found, even after applying a 1 V bias for 90 min (Fig. S1†). This further indicates that the optical spectrum of Au133 is insensitive to the change of oxidation state. In comparison, the absorption spectra of [Au25(SR)18]− changed at 0.6 V (for 60 min) due to the formation of [Au25(SR)18]0 (Fig. S2†).
Spin effects on the surface ligands probed by NMR
NMR analysis on ligand-protected nanoclusters can provide important information on the symmetry of surface ligand shell and the charge state of nanoclusters, as demonstrated previously.41,64,70–72 Unlike the free ligand in solution, the ligands on the surface of nanoclusters are confined to different chemical environments. Therefore, the same type of ligands can show different chemical shifts and split into different groups. When the charge state and the corresponding magnetic state of the nanocluster change, the NMR spectrum also changes accordingly. Here we employed NMR to first analyze the symmetry of ligand shell and then probe the redox properties of Au133(TBBT)52. In the smaller Au25(SCH2CH2Ph)18 case, it was demonstrated that the NMR peaks shifted significantly when going from diamagnetic (q = −1) to paramagnetic (q = 0), especially for the α-carbon hydrogen atoms (–SCαH2CβH2Ph) since they are close to the gold core.47,48 Interestingly, we found that, despite Au133(TBBT)52 being two shells larger than Au25, the NMR spectrum of Au133(TBBT)52 can still be affected by the magnetic state of the core. In addition, not only the closer aromatic 1H but also the further tert-butyl 1H can be influenced by the change in magnetism.We first analyze the NMR spectra of free TBBT thiols and [Au133(TBBT)52]0 nanocluster. For the free TBBT thiol, HS–C6H4–C(CH3)3, the nine identical hydrogens in the tert-butyl group give rise to an intense singlet peak at 1.30 ppm, and the four hydrogens in the aromatic part give rise to a pair of symmetrical doublets at 7.22/7.24 and 7.26/7.29 ppm (Fig. 4A and S3†). Such simple, symmetric and intense NMR signals of TBBT thiols greatly facilitate the further analysis of complex ligand patterns on the Au133 surface. When confined on the Au133 surface, the 52 TBBT thiolates are split into 13 groups, with each group containing 4 ligands. This is mostly evidenced in the tert-butyl 1H; the 1.30 ppm peak in free thiol is split into 13 peaks ranging from ∼0.72 to ∼1.39 ppm (Fig. 4B and S4†). A similar splitting pattern can also be identified in the aromatic region, but not as distinct as the singlet tert-butyl 1H (i.e. 2H vs. 9H). As shown in the 2D correlation spectrum (1H–1H COSY) in the aromatic region (Fig. 4C), 10 pairs of aromatic doublets from the TBBT ligands can be clearly identified and the detailed peak positions are listed in Table S1.† It is difficult to identify the remaining 3 pairs, probably due to the overlap of signals or broadening of peaks when protons are very close to the gold core.
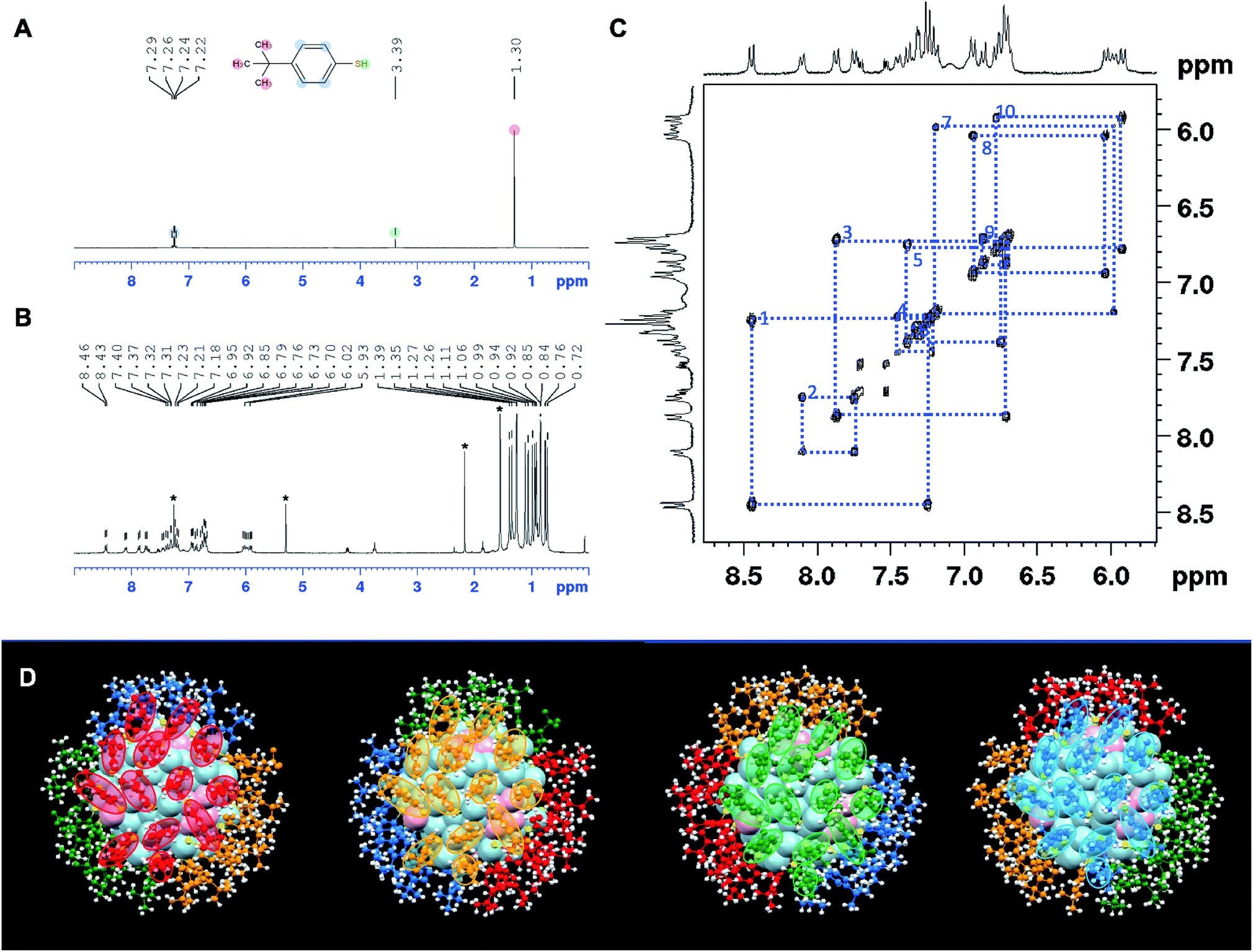 | ||
| Fig. 4 Correlation between the NMR spectra and ligand shell symmetry of [Au133(TBBT)52]0. (A) NMR of free TBBT thiol. (B) NMR of [Au133(TBBT)52]0 nanoclusters, the stars indicate solvent peaks: chloroform (7.26), CH2Cl2 (5.30), acetone (2.17) and H2O (1.56). (C) 1H–1H COSY of the aromatic region of [Au133(TBBT)52]0 for the correlation of doublet pairs. (D) Symmetry of ligand shells. The four symmetrically identical surface ligand patches are highlighted in red, orange, green and blue colors, corresponding to the 4-fold degeneracy of 52 TBBT ligands. The 13 TBBT ligands in each patch correlate to the 13-fold splitting of NMR pattern. Au: pink and light blue; S: yellow; C: red, orange, green and blue; and H: white. | ||
The 13-fold splitting and 4-fold degeneracy of the 52 TBBT ligands are the reflection of the symmetry of the ligand shell in Au133(TBBT)52. Since all the thiolates are assembled into monomeric staple motifs (i.e. 26 of –S–Au–S–) in Au133(TBBT)52, there is no symmetry breaking due to formation of different staple motifs. Therefore, the 4-fold degeneracy indicates that there are four identical ligand patches on Au133 surface, and the 13-fold splitting infers that in each patch, there are 13 chemically different TBBT ligands. Indeed, this NMR splitting pattern can be correlated with the symmetry of surface ligand shell. As shown in Fig. 4D, the 52 TBBT ligands can be divided into four symmetrically identical patches as labeled in red, orange, green and blue colors. Each surface patch contains 13 TBBT ligands. Therefore, the ligand shell has a quasi-D2 symmetry. Such splitting of NMR signal of ligands according to the symmetry of the nanoclusters have also been inferred in Au130(SR)50, which exhibits a 10-fold degeneracy and 5-fold splitting in its NMR spectrum, reflecting its quasi D5 symmetry.64 We note that it is possible to qualitatively correlates some NMR peaks with ligands location based on the electron density around the ligands, but a quantitative correlation requires in-depth theoretical calculations given that 13 correlation needs to be made. Nonetheless, the detailed correlation of the complex NMR pattern with the symmetry of Au133(TBBT)52 proves the potential of using NMR to characterize the structural symmetry of atomically precise nanoclusters, similar to the case of the protein structure determination. It is worth noting that Maran et al. recently probed the Au144(SR)60 structure by NMR.72
Redox properties of [Au133(TBBT)52]0 and [Au133(TBBT)52]+
As aforementioned, optical spectra of two charge states showed no discernable difference, but NMR can indeed be useful. We probed the aliphatic region (0–2 ppm) in the NMR spectra to further analyze the redox behavior of Au133(TBBT)52 since the tert-butyl 1H gives stronger signals than the phenyl 1H. Fig. 5 shows that when paramagnetic [Au133(TBBT)52]0 is oxidized by H2O2 to diamagnetic [Au133(TBBT)52]+, the NMR spectrum changes, with shifts of peaks to both lower and higher fields. The differences in NMR spectra between [Au133(TBBT)52]0 and [Au133(TBBT)52]+ should result from the change of magnetic state and charge state of the gold core. The protons in tert-butyl group, about 1 nm away from the gold surface, can still sense the change of magnetic states of the gold core; while the slight shifts and similar splitting ranges indicates that magnetic field disturbance in Au133 is not as significant as in the smaller Au25 nanocluster. The shifts of the proton peaks may also imply that the superatomic orbital that hold the unpaired electron spin may have none-spherical shape which cause inhomogeneous distribution of magnetic field on the ligand shell, therefore leading to both up and down shifts.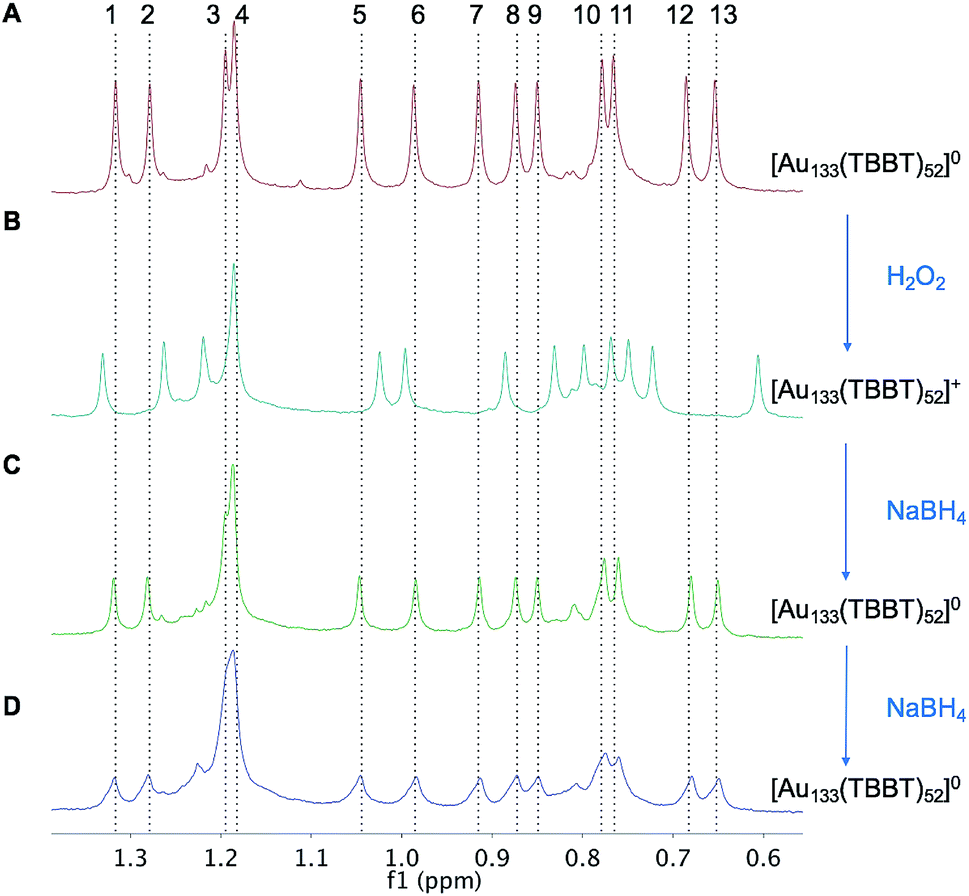 | ||
| Fig. 5 Redox property of Au133(TBBT)52 probed by NMR. (A) NMR of tert-butyl–H in [Au133(TBBT)52]0. (B) Oxidation of [Au133(TBBT)52]0 to [Au133(TBBT)52]+ by H2O2. (C) Reduction of [Au133(TBBT)52]+ to [Au133(TBBT)52]0 by NaBH4. (D) Further reaction of [Au133(TBBT)52]0 with NaBH4. | ||
The [Au133(TBBT)52]+ can be reduced to [Au133(TBBT)52]0 by NaBH4, as evidenced in the recovery of the NMR spectrum to that of [Au133(TBBT)52]0 (Fig. 5C). Increasing the reaction time as well as adding more NaBH4 cannot further reduce [Au133(TBBT)52]0 to [Au133(TBBT)52]−, based on the identical peak positions in NMR spectra after adding more NaBH4 (Fig. 5D). Instead, Au133 gradually decomposed in the presence of large excess NaBH4, as evidenced in the decreasing intensity and broadening of peaks in NMR. This may be due to the stripping of surface TBBT ligands by excess NaBH4 and then the aggregation of nanoclusters.
Conclusion
In summary, paramagnetism is discovered in the 1.7 nm Au133(TBBT)52 nanocluster and reversible switch between paramagnetism and diamagnetism is demonstrated. Through EPR measurements, we proved that [Au133(TBBT)52]0 (an 81-electron nanocluster) hosts one unpaired electron. The observation of one spin per cluster in Au133 implies that Au133 is non-metallic, with its electrons being distributed in discrete orbitals instead of filling a continuous band. EPR simulations suggest that the unpaired spin is mainly delocalized in the inner 13-atom icosahedral core, which is similar to the case of paramagnetic [Au25(SR)18]0 (7e). NMR measurements show a 13-fold splitting and 4-fold degeneracy of the 52 TBBT ligands on the Au133 surface, which is correlated to the D2 symmetry of the ligand shell. When [Au133(TBBT)52]0 is oxidized to [Au133(TBBT)52]+, the nanocluster changes to diamagnetism, as evidenced in the disappearance of EPR signal as well as the shift of NMR peaks. [Au133(TBBT)52]+ can be reduced back to [Au133(TBBT)52]0 and the NMR peaks are restored, hence, a reversible process. Further reduction of Au133(0) by NaBH4 lead to degradation of nanoclusters instead of forming [Au133(TBBT)52]−. In future work, the spin effects on the ultrafast electron dynamics and catalytic reactivity deserve to be pursued.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Science Foundation (DMR-1808675). Funding for the EPR spectrometer was from National Science Foundation grant CHE1126268.References
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346 CrossRef CAS PubMed.
- S. Takano, S. Hasegawa, M. Suyama and T. Tsukuda, Acc. Chem. Res., 2018, 51, 3074 CrossRef CAS PubMed.
- Y. Negishi, T. Nakazaki, S. Malola, S. Takano, Y. Niihori, W. Kurashige, S. Yamazoe, T. Tsukuda and H. Häkkinen, J. Am. Chem. Soc., 2015, 137, 1206 CrossRef CAS PubMed.
- Q. Yao, X. Yuan, T. Chen, D. T. Leong and J. Xie, Adv. Mater., 2018, 30, 1802751 CrossRef PubMed.
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580 CrossRef CAS PubMed.
- Z. Lei, X. K. Wan, S. F. Yuan, Z. J. Guan and Q. M. Wang, Acc. Chem. Res., 2018, 51, 2465 CrossRef CAS PubMed.
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin, R. Jin and Z. Wu, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS PubMed.
- K. Konishi, M. Iwasaki and Y. Shichibu, Acc. Chem. Res., 2018, 51, 3125 CrossRef CAS PubMed.
- N. A. Sakthivel and A. Dass, Acc. Chem. Res., 2018, 51, 1774 CrossRef CAS PubMed.
- S. Takano, H. Hirai, S. Muramatsu and T. Tsukuda, J. Am. Chem. Soc., 2018, 140, 8380 CrossRef CAS PubMed.
- Y. Z. Li, R. Ganguly, K. Y. Hong, Y. Li, M. E. Tessensohn, R. Webster and W. K. Leong, Chem. Sci., 2018, 9, 8723 RSC.
- B. Bhattarai, Y. Zaker, A. Atnagulov, B. Yoon, U. Landman and T. P. Bigioni, Acc. Chem. Res., 2018, 51, 3104 CrossRef CAS PubMed.
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084 CrossRef CAS PubMed.
- M. J. Alhilaly, M. S. Bootharaju, C. P. Joshi, T. M. Besong, A.-H. Emwas, R. Juarez-Mosqueda, S. Kaappa, S. Malola, K. Adil, A. Shkurenko, H. Häkkinen, M. Eddaoudi and O. M. Bakr, J. Am. Chem. Soc., 2016, 138, 14727 CrossRef CAS PubMed.
- S. M. Aly, L. G. AbdulHalim, T. M. D. Besong, G. Soldan, O. M. Bakr and O. F. Mohammed, Nanoscale, 2016, 8, 5412 RSC.
- S. Bestgen, O. Fuhr, B. Breitung, V. S. K. Chakravadhanula, G. Guthausen, F. Hennrich, W. Yu, M. M. Kappes, P. W. Roesky and D. Fenske, Chem. Sci., 2017, 8, 2235 RSC.
- I. Russier-Antoine, F. Bertorelle, R. Hamouda, D. Rayane, P. Dugourd, Ž. Sanader, V. Bonačić-Koutecký, P. F. Brevet and R. Antoine, Nanoscale, 2016, 8, 2892 RSC.
- G. X. Duan, L. Tian, J. B. Wen, L. Y. Li, Y. P. Xie and X. Lu, Nanoscale, 2018, 10, 18915 RSC.
- L. Suber, P. Imperatori, L. Pilloni, D. Caschera, N. Angelini, A. Mezzi, S. Kaciulis, A. Iadecola, B. Joseph and G. Campi, Nanoscale, 2018, 10, 7472 RSC.
- Y. L. Li, Z. Y. Wang, X. H. Ma, P. Luo, C. X. Du and S. Q. Zang, Nanoscale, 2019, 11, 5151 RSC.
- T.-A. D. Nguyen, Z. R. Jones, B. R. Goldsmith, W. R. Buratto, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2015, 137, 13319 CrossRef CAS PubMed.
- A. W. Cook and T. W. Hayton, Acc. Chem. Res., 2018, 51, 2456 CrossRef CAS PubMed.
- K. K. Chakrahari, R. P. B. Silalahi, J. H. Liao, S. Kahlal, Y. C. Liu, J. F. Lee, M. H. Chiang, J. Y. Saillard and C. W. Liu, Chem. Sci., 2018, 9, 6785 RSC.
- L. V. Nair, S. Hossain, S. Takagi, Y. Imai, G. Hu, S. Wakayama, B. Kumar, W. Kurashige, D. Jiang and Y. Negishi, Nanoscale, 2018, 10, 18969 RSC.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114 CrossRef CAS PubMed.
- X. Kang, M. Zhou, S. Wang, S. Jin, G. Sun, M. Zhu and R. Jin, Chem. Sci., 2017, 8, 2581 RSC.
- Z. Wang, R. Senanayake, C. M. Aikens, W.-M. Chen, C.-H. Tung and D. Sun, Nanoscale, 2016, 8, 18905 RSC.
- Y. Song, Y. Lv, M. Zhou, T. Y. Luo, S. Zhao, N. L. Rosi, H. Yu, M. Zhu and R. Jin, Nanoscale, 2018, 10, 12093 RSC.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan and T. Pradeep, Nat. Commun., 2016, 7, 13447 CrossRef CAS PubMed.
- Z. Li, W. Li, H. Abroshan, Q. Ge, G. Li and R. Jin, Nanoscale, 2018, 10, 6558 RSC.
- S. Antonello, M. Hesari, F. Polo and F. Maran, Nanoscale, 2012, 4, 5333 RSC.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12 CrossRef CAS PubMed.
- Q. Li, Y. Pan, T. Chen, Y. Du, H. Ge, B. Zhang, J. Xie, H. Yu and M. Zhu, Nanoscale, 2018, 10, 10166 RSC.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669 CrossRef CAS.
- M. Li, Y. H. Lao, R. L. Mintz, Z. Chen, D. Shao, H. Hu, H. X. Wang, Y. Tao and K. W. Leong, Nanoscale, 2019, 11, 2631 RSC.
- M. Zhou, C. Zeng, Y. Chen, S. Zhao, M. Y. Sfeir, M. Zhu and R. Jin, Nat. Commun., 2016, 7, 13240 CrossRef CAS PubMed.
- M. Zhou, C. Zeng, Y. Song, J. W. Padelford, G. Wang, M. Y. Sfeir, T. Higaki and R. Jin, Angew. Chem., Int. Ed., 2017, 56, 16257 CrossRef CAS PubMed.
- T. Higaki, M. Zhou, K. J. Lambright, K. Kirschbaum, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2018, 140, 5691 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. Qian, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2009, 131, 2490 CrossRef CAS PubMed.
- M. Agrachev, M. Ruzzi, A. Venzo and F. Maran, Acc. Chem. Res., 2019, 52, 44 CrossRef CAS PubMed.
- Z. Wu, J. Chen and R. Jin, Adv. Funct. Mater., 2011, 21, 177 CrossRef CAS.
- G. L. Nealon, B. Donnio, R. Greget, J.-P. Kappler, E. Terazzi and J.-L. Gallani, Nanoscale, 2012, 4, 5244 RSC.
- A. Cirri, A. Silakov and B. J. Lear, Angew. Chem., Int. Ed., 2015, 54, 11750–11753 CrossRef CAS PubMed.
- L. J. Williams, P. J. Herbert, M. A. Tofanelli, C. J. Ackerson and K. L. Knappenberger Jr, J. Chem. Phys., 2019, 150, 101102 CrossRef PubMed.
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221 CrossRef CAS.
- A. Venzo, S. Antonello, J. A. Gascón, I. Guryanov, R. D. Leapman, N. V. Perera, A. Sousa, M. Zamuner, A. Zanella and F. Maran, Anal. Chem., 2011, 83, 6355 CrossRef CAS PubMed.
- Z. Liu, M. Zhu, X. Meng, G. Xu and R. Jin, J. Phys. Chem. Lett., 2011, 2, 2104 CrossRef CAS.
- T. Higaki, C. Liu, Y. Chen, S. Zhao, C. Zeng, R. Jin, S. Wang, N. L. Rosi and R. Jin, J. Phys. Chem. Lett., 2017, 8, 866 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976 CrossRef CAS PubMed.
- Z. Wu, C. Gayathri, R. R. Gil and R. Jin, J. Am. Chem. Soc., 2009, 131, 6535 CrossRef CAS PubMed.
- S. Antonello, N. V. Perera, M. Ruzzi, J. A. Gascón and F. Maran, J. Am. Chem. Soc., 2013, 135, 15585 CrossRef CAS PubMed.
- T. Dainese, S. Antonello, J. A. Gascón, F. Pan, N. V. Perera, M. Ruzzi, A. Venzo, A. Zoleo, K. Rissanen and F. Maran, ACS Nano, 2014, 8, 3904 CrossRef CAS PubMed.
- M. Agrachev, S. Antonello, T. Dainese, J. A. Gascón, F. Pan, K. Rissanen, M. Ruzzi, A. Venzo, A. Zoleo and F. Maran, Chem. Sci., 2016, 7, 6910 RSC.
- S. Tian, L. Liao, J. Yuan, C. Yao, J. Chen, J. Yang and Z. Wu, Chem. Commun., 2016, 52, 9873 RSC.
- S. Antonello, T. Dainese, F. Pan, K. Rissanen and F. Maran, J. Am. Chem. Soc., 2017, 139, 4168 CrossRef CAS PubMed.
- M. A. Tofanelli, K. Salorinne, T. W. Ni, S. Malola, B. Newell, B. Phillips, H. Häkkinen and C. J. Ackerson, Chem. Sci., 2016, 7, 1882 RSC.
- K. S. Krishna, P. Tarakeshwar, V. Mujica and C. S. S. R. Kumar, Small, 2014, 10, 907 CrossRef CAS PubMed.
- M. Agrachev, S. Antonello, T. Dainese, M. Ruzzi, A. Zoleo, E. Aprà, N. Govind, A. Fortunelli, L. Sementa and F. Maran, ACS Omega, 2017, 2, 2607 CrossRef CAS PubMed.
- C. Zeng, C. Liu, Y. Chen, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2014, 136, 11922 CrossRef CAS PubMed.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011 CrossRef CAS PubMed.
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D. E. Jiang, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 1482 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280 CrossRef CAS PubMed.
- Y. Chen, C. Zeng, C. Liu, K. Kirschbaum, C. Gayathri, R. R. Gil, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2015, 137, 10076 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Kirschbaum, K. Appavoo, M. Y. Sfeir and R. Jin, Sci. Adv., 2015, 1, e1500045 CrossRef PubMed.
- A. Dass, S. Theivendran, P. R. Nimmala, C. Kumara, V. R. Jupally, A. Fortunelli, L. Sementa, G. Barcaro, X. Zuo and B. C. Noll, J. Am. Chem. Soc., 2015, 137, 4610 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883 CrossRef CAS PubMed.
- D. Wang, J. W. Padelford, T. Ahuja and G. Wang, ACS Nano, 2015, 9, 8344 CrossRef CAS PubMed.
- J. W. Padelford, M. Zhou, Y. Chen, R. Jin and G. Wang, J. Phys. Chem. C, 2017, 121, 21217 CrossRef CAS.
- H. Qian, M. Zhu, C. Gayathri, R. R. Gil and R. Jin, ACS Nano, 2011, 5, 8935–8942 CrossRef CAS PubMed.
- K. Salorinne, S. Malola, O. A. Wong, C. D. Rithner, X. Chen, C. J. Ackerson and H. Häkkinen, Nat. Commun., 2016, 7, 10401 CrossRef CAS PubMed.
- T. Dainese, M. Agrachev, S. Antonello, D. Badocco, D. M. Black, A. Fortunelli, J. A. Gascón, M. Stener, A. Venzo, R. L. Whetten and F. Maran, Chem. Sci., 2018, 9, 8796 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02736j |
| ‡ These authors contribute equally. |
| § Current address: Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA, 19104. |
| This journal is © The Royal Society of Chemistry 2019 |