
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical and photochemical DNA “gears” reversibly control stiffness, shape-memory, self-healing and controlled release properties of polyacrylamide hydrogels†
Xia
Liu‡
a,
Junji
Zhang‡
 b,
Michael
Fadeev
a,
Ziyuan
Li
b,
Verena
Wulf
a,
He
Tian
b and
Itamar
Willner
*a
b,
Michael
Fadeev
a,
Ziyuan
Li
b,
Verena
Wulf
a,
He
Tian
b and
Itamar
Willner
*a
aInstitute of Chemistry, The Center for Nanoscience and Nanotechnology, The Hebrew University of Jerusalem, Jerusalem, 91904, Israel. E-mail: willnea@vms.huji.ac.il; Fax: +972-2-6527715; Tel: +972-2-6585272
bKey Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, School of Chemistry and Molecular Engineering, East China University of Science & Technology, 130 Meilong Road, Shanghai 200237, China
First published on 31st October 2018
Abstract
A new class of stimuli-responsive DNA-based polyacrylamide hydrogels is described. They consist of glucosamine–boronate ester-crosslinked polyacrylamide chains being cooperatively bridged by stimuli-responsive nucleic acids. The triggered closure and dissociation of the stimuli-responsive units lead to switchable stiffness properties of the hydrogel. One hydrogel includes glucosamine–boronate esters and K+-ion-stabilized G-quadruplex units as cooperative crosslinkers. The hydrogel bridged by the two motifs reveals high stiffness, whereas the separation of the G-quadruplex bridges by 18-crown-6-ether yields a low stiffness hydrogel. By cyclic treatment of the hydrogel with K+-ions and 18-crown-6-ether, it is reversibly cycled between high and low stiffness states. The second system involves a photo-responsive hydrogel that reveals light-induced switchable stiffness functions. The polyacrylamide chains are cooperatively crosslinked by glucosamine–boronate esters and duplex nucleic acid bridges stabilized by trans-azobenzene intercalator units. The resulting hydrogel reveals high stiffness. Photoisomerization of the trans-azobenzene units to the cis-azobenzene states results in the separation of the duplex nucleic acid bridges and the formation of a low stiffness hydrogel. The control over the stiffness properties of the hydrogel matrices by means of K+-ions/crown ether or photoisomerizable trans-azobenzene/cis-azobenzene units is used to develop shape-memory, self-healing, and controlled drug-release hydrogel materials.
Introduction
Stimuli-responsive hydrogels attract growing interest as functional materials for sensing,1 controlled drug release,2 tissue engineering,3 self-healing of hydrogels,4 and switchable or robotic applications.5,6 Different triggers, such as temperature,7–10 pH,11–14 chemical agents,15 light,16 electrical fields,17 magnetic fields18,19 and ultrasound irradiation20,21 were used to reversibly switch between hydrogel and solid or hydrogel and liquid phases. For example, trans-azobenzene-modified polyacrylamide chains and β-cyclodextrin-functionalized polyacrylamide chains form a hydrogel by the crosslinking of the polymers by trans-azobenzene/β-cyclodextrin supramolecular complexes.22 Photoisomerization of the trans-azobenzene units to cis-azobenzene units, that lack binding affinity toward the β-cyclodextrin receptor sites, leads to the separation of the crosslinking units and to the transition of the hydrogel into a polymer solution phase. Within the family of stimuli-responsive polymers, nucleic acid-based hydrogels represent an important class of materials.23,24 The reversible reconfiguration of nucleic acid structures by a variety of triggers/counter-triggers, such as the separation of nucleic acid duplexes by fuel strands and the reformation of the duplexes by anti-fuel strands, using the strand displacement process,25 the pH-stimulated formation or dissociation of i-motif12,26,27 or triplex DNA nanostructures,28 the reversible K+-ions-stimulated formation of G-quadruplexes and their separation in the presence of crown ether,29 the bridging of duplex nucleic acids by C–Ag+–C or T–Hg2+–T bridges and their separation by metal-ion-binding ligands, e.g., cysteine,30 and the switchable stabilization of duplex nucleic acids by trans-azobenzene units and their separation upon photoisomerization of the intercalated trans-azobenzene units to cis-azobenzene,31 provide a rich “tool-box” of structural motifs to construct stimuli-responsive DNA-based hydrogels. Indeed, stimuli-responsive nucleic acid-based hydrogels undergoing reversible gel-to-solution transitions using pH,32 metal-ion/ligand,26 K+-stabilized G-quadruplex/crown ether29 and the photoisomerization of azobenzene units were reported.31,33–36 Different applications of nucleic acid-based hydrogels in solution and on surfaces were suggested,37,38 including controlled drug release,28 switchable transport through pores,39 switchable catalysis and sensing.38–41In addition, the constructions of signal-triggered DNA-based hydrogels that reveal controlled stiffness functions were demonstrated. In these systems the hydrogel is cooperatively stabilized by nucleic acid duplex and reconfigurable crosslinking units, e.g., duplex nucleic acids,42 pH-responsive bridges26 or G-quadruplex crosslinkers.42 The triggered separation of the pH-responsive crosslinkers or G-quadruplex bridging units led to a hydrogel of lower stiffness. Such hydrogels were suggested as stimuli-responsive membranes for the release of drugs from nanoparticle carriers.43,44
One interesting application of stimuli-responsive DNA-based hydrogels involves the development of shape-memory hydrogels. Shape-memory polymers represent an interesting class of “smart materials” that undergo, in the presence of an auxiliary trigger, a shape transition into a temporary shape that includes a “code” (memory) to restore the original shape of the material upon applying a counter trigger.45–48 The ability to control the stiffness of DNA-based hydrogels by the cooperative stabilization of the hydrogel with duplex nucleic acid bridges and stimuli-responsive reconfigurable crosslinking units provide a versatile means to assemble shape-memory DNA-based hydrogels. In these systems, the shaped and high-stiffness hydrogel crosslinked by the duplex nucleic acid bridges and the signal-responsive reconfigurable crosslinking units is subjected to a trigger that dissociates the latter bridging units. This results in a shapeless hydrogel of quasi-liquid and low-stiffness properties. The remaining duplex crosslinkers provide, however, a permanent code (of chain entanglement) for the restoration of the original shape, in the presence of the appropriate counter-trigger. Indeed, DNA hydrogels bridged by duplex nucleic acids and pH-responsive crosslinking units32 or duplex nucleic acid bridges and reconfigurable G-quadruplex linkers demonstrated shape-memory functions.42 Different applications of shape-memory hydrogels were suggested, including their use for the encryption and storage of information, the modulation of bilayer DNA hydrogel structures by stress interactions,49 and their use as sensing materials.
The self-healing of hydrogels is a further possible pathway to use stimuli-responsive soft materials. Particularly, hydrogels crosslinked by cooperative bridges may lead to “healed” hydrogels by the conjugation of quasi-liquid soft matrices crosslinked by one type of bridging units, followed by the triggered rigidification of the matrix by the second bridging element.50 Different triggers were used to induce self-healing of polymer matrices including supramolecular complexes,51 H-bonds,52 light53 and more. Different uses of the self-healing process of hydrogels in tissue engineering and tissue repair were suggested.54 The use of stimuli-responsive shape-memory hydrogel matrices consisting of two types of nucleic acid bridges, e.g., duplex nucleic acids and signal-reconfigurable crosslinking units represents, however, a disadvantage. The duplex nucleic acid crosslinking units that act as “memory” elements that entangle the polymer chains in the quasi-liquid reveal limited stability. The thermally equilibrated opening of the duplex nucleic acid bridges is a “memory destructive” path that limits the switchable recovery of the hydrogel shapes. This calls for the development of other hybrid DNA hydrogels, where chemical crosslinkers, other than duplex nucleic acids, act as memory elements.
In the present study, we introduce a new class of hybrid nucleic acid-based polyacrylamide hydrogels that reveal shape-memory and self-healing functionalities. Two hydrogels are presented in the study: one hydrogel is stabilized by cooperative glucosamine–boronate ester55,56 crosslinking units and reconfigurable K+-ion-stabilized G-quadruplex bridges. The second hydrogel is cooperatively stabilized by the glucosamine–boronate ester bridges and photoresponsive nucleic acid units that undergo light-induced formation or separation of duplex units (DNA “gear”). The study paves the way to apply the glucosamine–boronate ester bridging units and other reconfigurable nucleic acid crosslinkers to design hydrogels of controlled stiffness that reveal shape-memory and self-healing functions. In addition, the control over the stiffness properties of the hydrogel is used to develop signal-triggered matrices for the controlled release of drugs.
Results and discussion
The first hybrid hydrogel system is depicted in Fig. 1. The polyacrylamide chains PA and PB were modified with the phenylboronic acid ligand and glucosamine, respectively, and with the guanosine-rich nucleic acid tethers (1), where the tethers (1) are subunits that, under appropriate conditions, self-assemble into a G-quadruplex. The average molecular weight and loading of the polymer chains PA with the boronic acid ligand and tether (1) were evaluated by NMR and absorption spectroscopy to be MW ∼ 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, loading degree of the boronic acid ligand/acrylamide units = 1:40 and nucleotide tether (1)/acrylamide units = 1:44 (see ESI, Fig. S1† and the accompanying discussion). Similarly, the molecular weight of polymer PB was evaluated to be MW ∼ 137000 Da and the loading of the glucosamine units and of the tether (1) corresponded to 1:12 and 1:12, with respect to the acrylamide monomers associated with the polymer PB (see ESI, Fig. S2† and the accompanying discussion). Mixing of the two polymers leads to the formation of a hydrogel due to the crosslinking of the polymer chains by glucosamine–boronate ester bridges (Fig. 1A). Rheometry characterization of the resulting hydrogel indicated the formation of a hydrogel of low stiffness (G′ ≈ 27 Pa, curve (a), and G′′ ≈ 1 Pa, curve (a′), in Fig. 1B). Treatment of the resulting hydrogel with K+-ions resulted in the additional crosslinking of the polymer chains by K+-ion-stabilized G-quadruplexes. The cooperative crosslinking of the hydrogel by the glucosamine–boronate ester units and the K+-ion-stabilized G-quadruplexes yields a hydrogel of enhanced stiffness (G′ ≈ 72 Pa, curve (b), and G′′ ≈ 16 Pa, curve (b′), Fig. 1B). Subjecting the resulting hydrogel to reaction with crown ether resulted in the elimination of the K+-ions from the G-quadruplex bridges, and the separation of the G-quadruplex bridging units. The separation of the G-quadruplex crosslinking units leads to a hydrogel of low stiffness (G′ ≈ 34 Pa, curve (c) and G′′ ≈ 3 Pa, curve (c′), Fig. 1B). By the cyclic treatment of the hydrogel with K+-ions and crown ether, the hydrogel is reversibly switched between high and low stiffness properties, Fig. 1C. The control over the stiffness properties of the glucosamine–boronate ester cooperatively crosslinked by the K+-ion-stabilized G-quadruplex units, and by the crown ether-stimulated separation of the G-quadruplex units was further supported by micro-indentation experiments. The G-quadruplex-stabilized hydrogel revealed an effective Young's modulus of 14.9 ± 0.1 kPa, and the crown ether-separated G-quadruplex showed an effective Young's modulus of 6.4 ± 0.1 kPa. Scanning electron microscopy (SEM) images further supported the switchable stiffness properties of the hydrogel.57Fig. 1D, panel (i), shows the SEM image of the quasi-liquid hydrogel crosslinked by the boronate ester bridges. The hydrogel includes large pores, suggesting the low degree of crosslinking. Fig. 1D, panels (ii) and (iii) show the SEM images upon the reversible transition of the hydrogel between the stiff state crosslinked by the boronate ester units and the K+-ion-stabilized G-quadruplexes and the quasi-liquid hydrogel crosslinked by the boronate ester crosslinkers. While the stiff hydrogel consists of a dense small-pore matrix consistent with a high degree of crosslinking, panel (ii), the crown ether-stimulated dissociation of the G-quadruplexes yields the quasi-liquid state that reveals large pores, implying low stiffness, panel (iii).
000 Da, loading degree of the boronic acid ligand/acrylamide units = 1:40 and nucleotide tether (1)/acrylamide units = 1:44 (see ESI, Fig. S1† and the accompanying discussion). Similarly, the molecular weight of polymer PB was evaluated to be MW ∼ 137000 Da and the loading of the glucosamine units and of the tether (1) corresponded to 1:12 and 1:12, with respect to the acrylamide monomers associated with the polymer PB (see ESI, Fig. S2† and the accompanying discussion). Mixing of the two polymers leads to the formation of a hydrogel due to the crosslinking of the polymer chains by glucosamine–boronate ester bridges (Fig. 1A). Rheometry characterization of the resulting hydrogel indicated the formation of a hydrogel of low stiffness (G′ ≈ 27 Pa, curve (a), and G′′ ≈ 1 Pa, curve (a′), in Fig. 1B). Treatment of the resulting hydrogel with K+-ions resulted in the additional crosslinking of the polymer chains by K+-ion-stabilized G-quadruplexes. The cooperative crosslinking of the hydrogel by the glucosamine–boronate ester units and the K+-ion-stabilized G-quadruplexes yields a hydrogel of enhanced stiffness (G′ ≈ 72 Pa, curve (b), and G′′ ≈ 16 Pa, curve (b′), Fig. 1B). Subjecting the resulting hydrogel to reaction with crown ether resulted in the elimination of the K+-ions from the G-quadruplex bridges, and the separation of the G-quadruplex bridging units. The separation of the G-quadruplex crosslinking units leads to a hydrogel of low stiffness (G′ ≈ 34 Pa, curve (c) and G′′ ≈ 3 Pa, curve (c′), Fig. 1B). By the cyclic treatment of the hydrogel with K+-ions and crown ether, the hydrogel is reversibly switched between high and low stiffness properties, Fig. 1C. The control over the stiffness properties of the glucosamine–boronate ester cooperatively crosslinked by the K+-ion-stabilized G-quadruplex units, and by the crown ether-stimulated separation of the G-quadruplex units was further supported by micro-indentation experiments. The G-quadruplex-stabilized hydrogel revealed an effective Young's modulus of 14.9 ± 0.1 kPa, and the crown ether-separated G-quadruplex showed an effective Young's modulus of 6.4 ± 0.1 kPa. Scanning electron microscopy (SEM) images further supported the switchable stiffness properties of the hydrogel.57Fig. 1D, panel (i), shows the SEM image of the quasi-liquid hydrogel crosslinked by the boronate ester bridges. The hydrogel includes large pores, suggesting the low degree of crosslinking. Fig. 1D, panels (ii) and (iii) show the SEM images upon the reversible transition of the hydrogel between the stiff state crosslinked by the boronate ester units and the K+-ion-stabilized G-quadruplexes and the quasi-liquid hydrogel crosslinked by the boronate ester crosslinkers. While the stiff hydrogel consists of a dense small-pore matrix consistent with a high degree of crosslinking, panel (ii), the crown ether-stimulated dissociation of the G-quadruplexes yields the quasi-liquid state that reveals large pores, implying low stiffness, panel (iii).
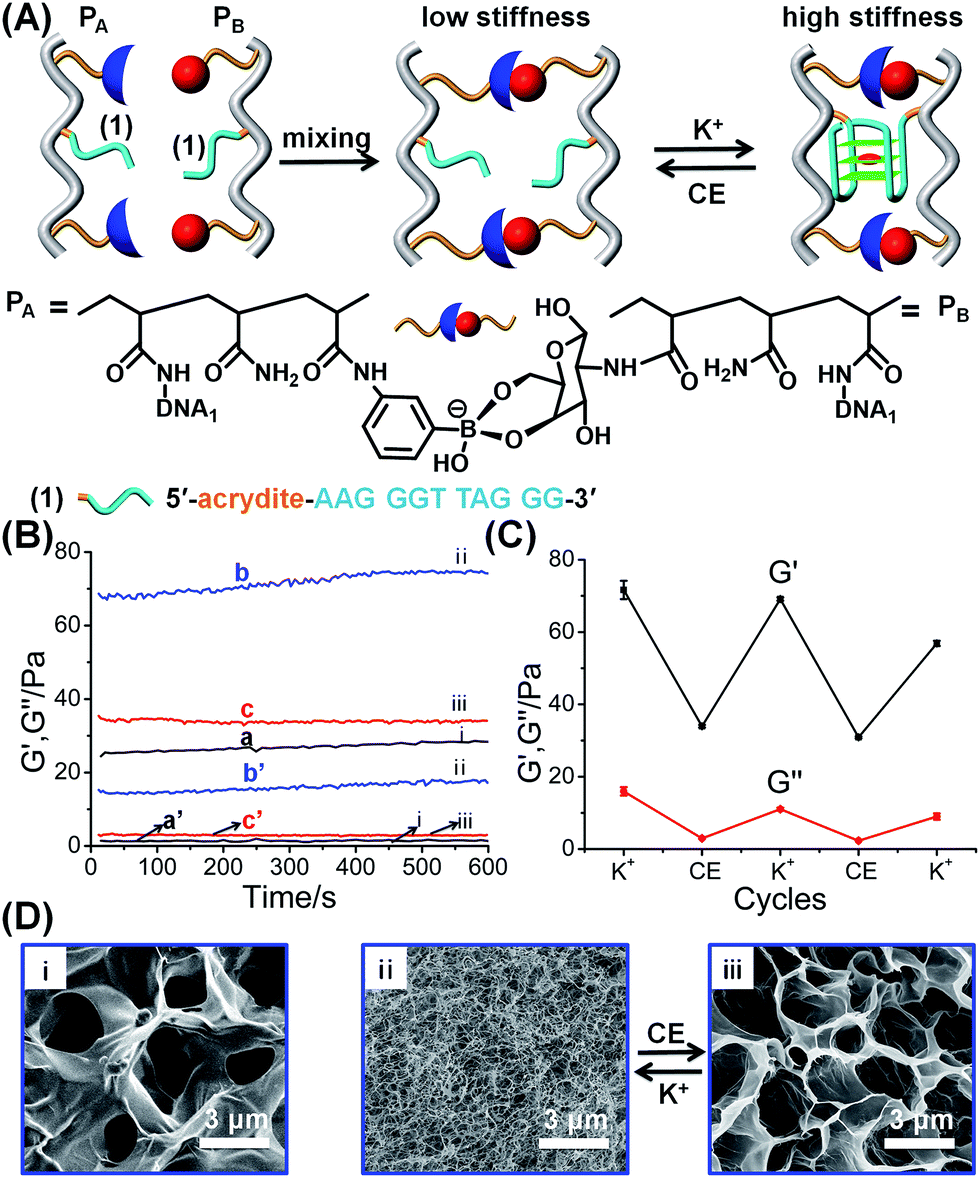 | ||
| Fig. 1 (A) Schematic synthesis of polyacrylamide hydrogel crosslinked by glucosamine–boronate esters and K+-ion-stabilized G-quadruplexes as crosslinking units. The cyclic and reversible separation of the G-quadruplex by 18-crown-6-ether and the reformation of the K+-ion-stabilized G-quadruplex shifts the hydrogel between low stiffness and high stiffness states, respectively. (B) Rheometry features of the K+-ion/crown ether-triggered hydrogels: (a) and (a′) correspond to the storage modulus, G′, and loss modulus, G′′, of the as-prepared glucosamine–boronate ester crosslinked hydrogel. (b) and (b′) correspond to the G′ and G′′ values of the stiff hydrogel crosslinked by the glucosamine–boronate ester and K+-ion-stabilized G-quadruplex as crosslinkers. (c) and (c′) correspond to the G′ and G′′ values of the low-stiffness hydrogel generated by the treatment of the stiff hydrogel with 18-crown-6-ether (CE) that separates the G-quadruplex crosslinkers. (C) Cyclic and reversible G′/G′′ values of the high-stiffness and low-stiffness hydrogels, generated in the presence of K+-ion and CE, respectively. (D) SEM images corresponding to: panel (i) – the low stiffness hydrogel crosslinked by the glucosamine–boronate ester bridges only. Panels (ii) and (iii) – typical SEM images of the hydrogel shifted between the high stiffness, in the presence of K+-ions, and low stiffness, in the presence of CE, respectively. | ||
The K+-ion/crown ether-stimulated stiffness properties of the hydrogel were then used to demonstrate shape-memory and self-healing functions of the hydrogel. The shape-memory properties of the hydrogel, are shown in Fig. 2A. The hydrogel in its high-stiffness state, cooperatively stabilized by the glucosamine–boronate ester groups and by the K+-stabilized G-quadruplex crosslinkers, was prepared in a triangle-shaped Teflon mold. The extruded triangle-shaped hydrogel, state “I”, was then subjected to cyclic interactions with crown ether and K+-ions. In the presence of crown ether, the triangle-shaped hydrogel was converted into a shapeless and quasi-liquid state, state “II”. The glucosamine–boronate ester bridges provide, however, an “internal permanent memory” to restore the triangle-shape. The glucosamine–boronate ester bridges entangle the polymer chains, and retain the tethers (1) in spatial orientations to regenerate the stiff, triangle-shaped hydrogel, upon the addition of K+-ions. By the cyclic addition of K+-ions and crown ether, the hydrogel was reversibly switched between the triangle-shaped hydrogel and the quasi-liquid hydrogel that lacks a defined shape. It should be noted that the spatial organization of the nucleic acid tethers by the glucosamine–boronate ester bridges, acting as permanent memory by the entanglement of the crosslinked polymer chains, is supported by the effect of temperature on the shape-memory properties of the hydrogel. The hydrogel with switchable and reversible shape recovery properties can be recycled at room-temperature (25 °C) for at least eight cycles. Nonetheless, heating the hydrogel to 35–38 °C adversely affects the shape-memory properties of the hydrogel, and the addition of K+-ions to the low-stiffness hydrogel restores only partially the triangle-shaped structure after two K+/crown ether cycles, with constant degradation of the shape-recovery properties upon increasing the number of shape regeneration cycles. That is, the thermally induced Brownian motion of the polymer chains in the low stiffness hydrogel perturbs the memory code embedded in the entangled chains, leading to the perturbation of the shape recovery properties of the hydrogel. In addition, the cooperative stabilization of the hydrogel by the glucosamine–boronate ester and G-quadruplex bridges was used to develop a self-healing mechanism of the hydrogel, Fig. 2B. The hydrogel stabilized by the two crosslinking motifs was prepared as a square-shaped hydrogel, and the extruded hydrogel was cut into two pieces. The two pieces were physically attached and subjected to reaction with crown ether to dissociate the G-quadruplex and form the quasi-liquid hydrogel parts that include the glucosamine–boronate ester ligand as the permanent crosslinking element. Subsequently, the interconnected hydrogel matrix was treated with K+-ions for a time-interval of 1 hour. The hydrogel recovered to the non-separable, square-shaped hydrogel, Fig. 2B, indicating that the cooperative formation of the G-quadruplex provided a self-healing path. In a control experiment, the physically interconnected hydrogel pieces remained in the quasi-liquid state for a time-interval of 1 hour, yet the process did not yield a healed hydrogel, and the two hydrogel pieces were separated upon gentle shaking, Fig. S3.† Furthermore, the shaking of the two quasi-liquid hydrogel pieces led to the labile interconnection of the pieces, and these were subsequently healed in the presence of K+-ions. Nonetheless, the time-interval to reach the interconnected pieces varied from sample-to-sample.
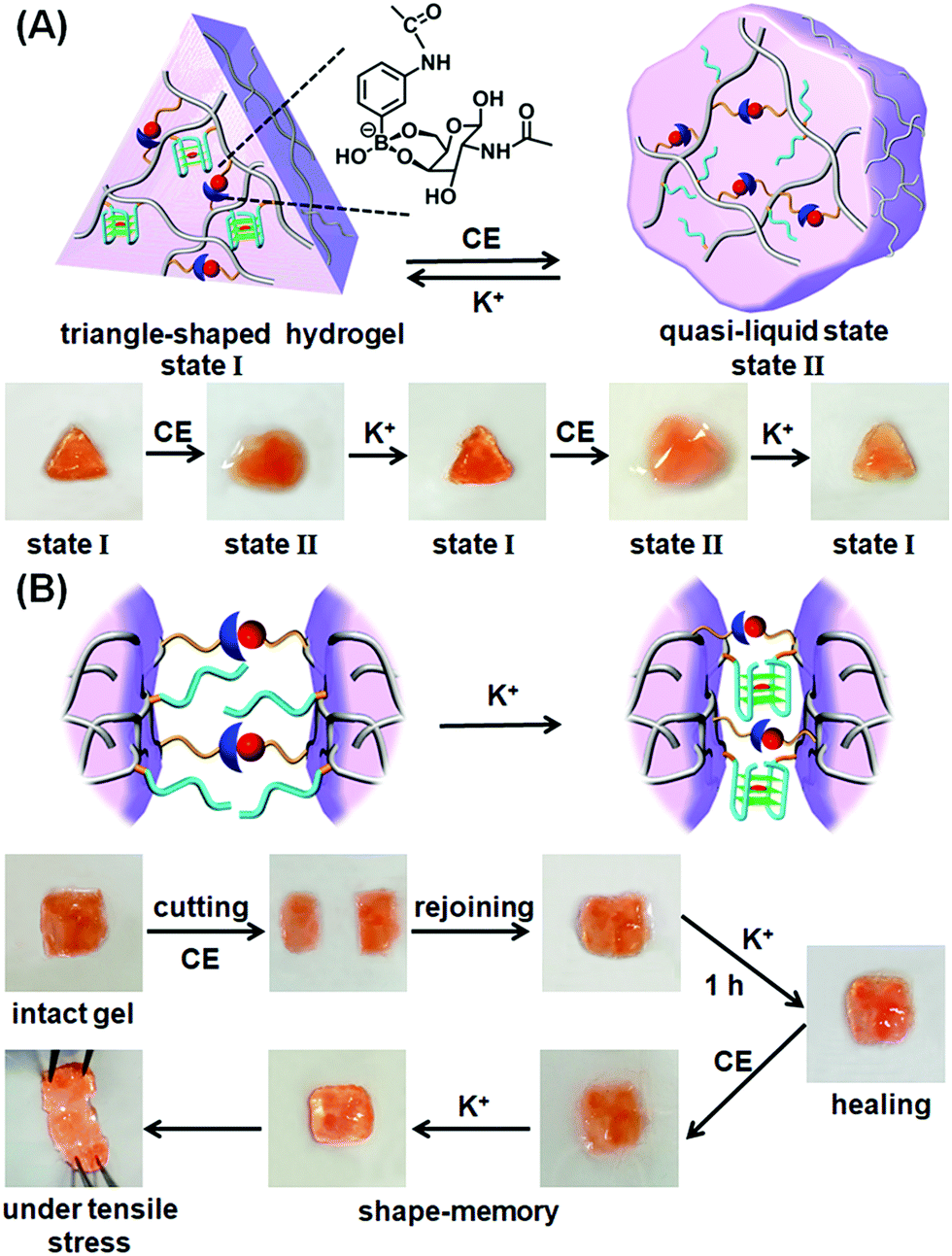 | ||
| Fig. 2 (A) Schematic shape-memory transitions between the triangle-shaped stiff hydrogel crosslinked by the glucosamine–boronate esters and the K+-stabilized G-quadruplexes and the crown ether-induced transition of the shaped hydrogel to a low-stiffness, quasi-liquid, shapeless hydrogel that includes the glucosamine–boronate ester as the permanent bridging memory-code. Images at the bottom show the reversible shape transitions between the stiff shaped hydrogel, state “I”, and the low-stiffness, shapeless hydrogel, state “II”. (B) Schematic self-healing of two low-stiffness hydrogel pieces into an integrated and non-separable hydrogel crosslinked by the glucosamine–boronate ester and the K+-stabilized G-quadruplex crosslinkers. Bottom part shows the images of the stepwise self-healing process. | ||
The second class of stimuli-responsive boronate ester-crosslinked hydrogels has involved the use of light as the stiffness controlling trigger. The synthesis and characterization of the photoresponsive boronate ester bridged hydrogel are presented in Fig. 3, S4 and S5.† Two polyacrylamide chains PC and PD were functionalized with the phenylboronic acid ligand and glucosamine, respectively. The polymer chain PC was modified with the nucleic acid tether (2), and the polymer chain PD was functionalized with the nucleic acid tether (3). The average molecular weights of polymers PC and PD were evaluated by NMR spectroscopy, and these corresponded to 140000 Da and 200000 Da, respectively (see ESI, Fig. S4 and S5†). The loading of the boronic acid ligand and tether (2) was evaluated by NMR and absorption spectroscopy to be 1:46 and 1:81 relative to the acrylamide monomer residues. Also, the loading of glucosamine and of tether (3), associated with polymer PD, corresponded to 1:13 and 1:98 relative to the acrylamide residues (see ESI, Fig. S4 and S5†). The nucleic acid tethers (2) and (3) linked to the polymer chains PC and PD, respectively, were further hybridized with the single strand (4) and the trans-azobenzene-functionalized strand (5), respectively. The strands (4) and (5) include complementary single-strand domains, where the domain “GGT AAC AA” of (4) hybridizes with the azobenzene-modified single strand domain, where the eight-base (relatively unstable) duplex is cooperatively stabilized by the trans-azobenzene units that intercalate into the (4)/(5) duplex domain. The crosslinking of the tethers (3) and (2)–(4) by the photo-responsive strand t-azo (5) results in the cooperative stabilization of a stiff hydrogel bridged by the glucosamine–boronate ester bridges and the photo-responsive (2)–(4)/(3)-t-azo (5) DNA units. The mechanism for the photo-stimulated control of the stiffness of the hydrogel is shown in Fig. 3A. The photoisomerization of the trans-azobenzene units to the cis-azobenzene units (λ = 365 nm), that lack affinity toward the duplex nucleic acids, results in the dissociation of the duplex region between the photoactive domain associated with c-azo (5) and the domain “GGT AAC AA” being a part of tether (4). The separation of the duplex leads to a hydrogel of lower stiffness that is crosslinked by the glucosamine–boronate ester bridges only. Photoisomerization of the cis-azobenzene unit to the trans-azobenzene state (λ > 420 nm) restores the stiff hydrogel that is cooperatively stabilized by the two crosslinking motifs. By the reversible photoisomerization of the azobenzene photoactive groups between the trans and cis states, the photoactive bridges are switched between a “locked” and an “unlocked” configuration, leading to the formation of a hydrogel of higher and lower stiffness, respectively. Fig. 3B shows the rheometry features of the hydrogel. The hydrogel that is cooperatively stabilized by the glucosamine–boronate ester bridges and the trans-azobenzene (2)–(4)/(3)-t-azo (5) units reveals high stiffness (G′ ≈ 65 Pa, curve (a), and G′′ ≈ 11 Pa, curve (a′), in Fig. 3B). Photoisomerization of the trans-azobenzene (t-azo) (5) to the cis-azobenzene (c-azo) (5) (λ = 365 nm) results in hydrogel of lower stiffness due to the unlocking of the (3)-c-azo (5) and (2)–(4) crosslinking duplex, G′ ≈ 26 Pa, curve (b), and G′′ ≈ 2 Pa, curve (b′), in Fig. 3B. By the cyclic photoisomerization of the azobenzene modified (5) between the trans and cis states, the hydrogel undergoes reversible light-induced transitions between high and low stiffness states, respectively, Fig. 3C. The light-stimulated stiffness transitions of the hydrogel were further supported by micro-indentation experiments. The effective Young's modulus of the cooperatively crosslinked glucosamine–boronate ester bridges and the trans-azobenzene bridged duplexes (2)–(4)/(3)-t-azo (5) corresponded to 13.8 ± 0.2 kPa. In turn, the hydrogel crosslinked by the glucosamine–boronate ester units, in the presence of the cis-azobenzene (3)-c-azo (5) and (2)–(4) non-hybridized bridges, revealed an effective Young's modulus of only 5.4 ± 0.2 kPa. The photoinduced control over the stiffness properties of the hydrogel was further supported by SEM imaging of the hydrogel in the different states, Fig. 3D. The SEM image of the hydrogel bridged by the two crosslinking motifs, e.g., the glucosamine–boronate ester supramolecular complex and the trans-azobenzene-stabilized duplexes (4)/t-azo (5) is shown in panel (I). A matrix consisting of dense small pores is observed, consistent with a high degree of crosslinking. Panel (II) shows the SEM image of the hydrogel of lower stiffness that is formed upon the photoisomerization of the trans-azobenzene units to the cis-azobenzene units (λ = 365 nm) that leads to the separation of the (4)/(5) duplexes and to a hydrogel crosslinked only by the glucosamine–boronate ester bridges. A matrix with large pores is observed, consistent with the formation of a hydrogel of lower stiffness.
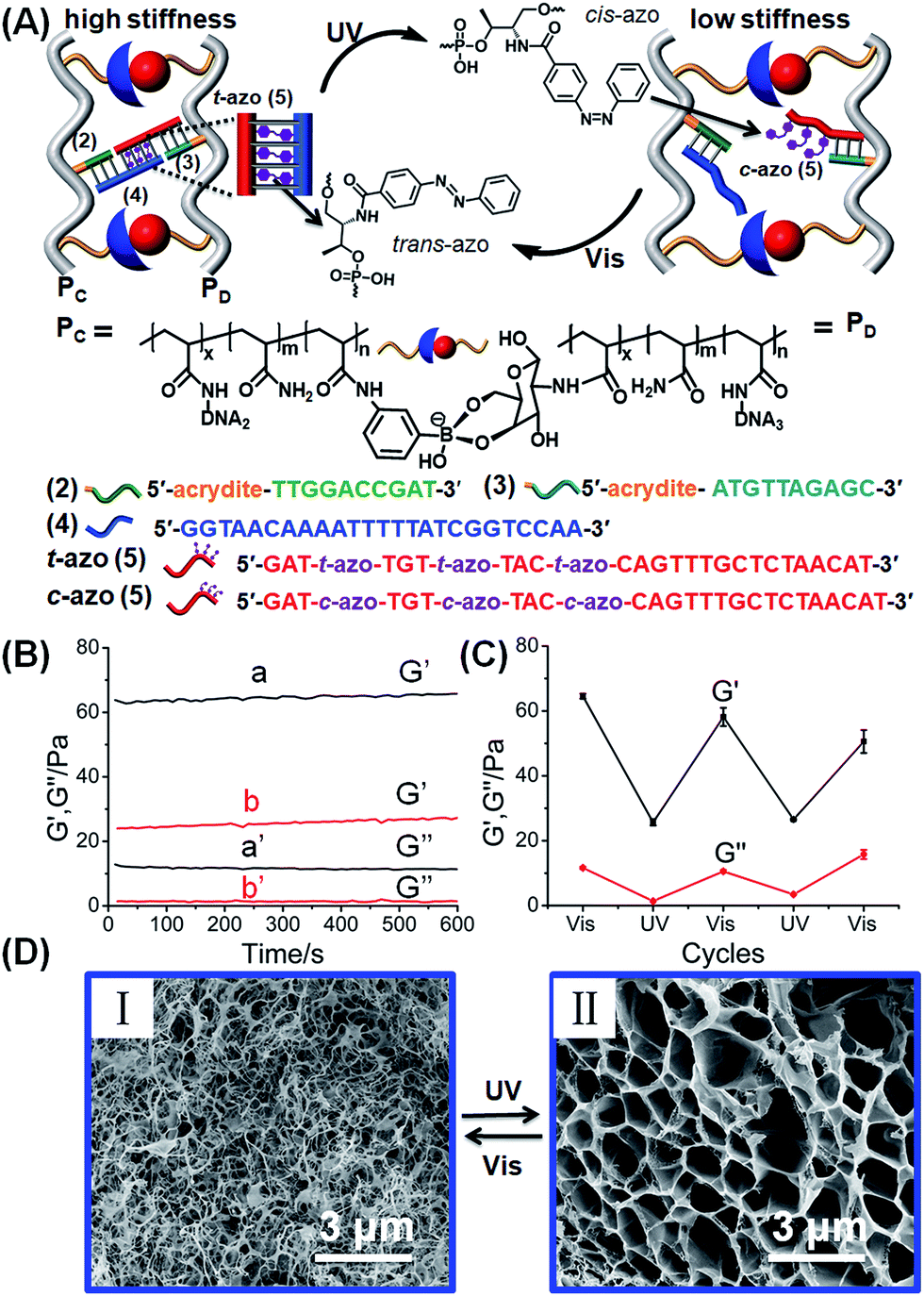 | ||
| Fig. 3 (A) Schematic design of a photoresponsive hydrogel that undergoes cyclic and reversible photoinduced transitions between a hydrogel of high stiffness and low stiffness, respectively. The stiff hydrogel is crosslinked by glucosamine–boronate ester bridges and duplex nucleic acids composed of the (2)–(4) and (3)-trans-azobenzene (5) (t-azo (5)). Photoisomerization of the t-azo (5) units to c-azo (5) (λ = 365 nm) leads to the separation of the nucleic acid duplexes, and to the formation of a low-stiffness hydrogel. The reverse photoisomerization of c-azo (5) (λ > 420 nm) restores the high-stiffness hydrogel. (B) Rheometry experiments that follow the photochemically generated hydrogels of high and low stiffness: (a) and (a′) correspond to the G′ and G′′ values of the stiff hydrogel crosslinked by the glucosamine–boronate esters and the (2)–(4)/(3)-t-azo (5) duplexes. (b) and (b′) correspond to the G′ and G′′ values of the photogenerated low-stiffness hydrogel crosslinked by the glucosamine–boronate ester only and unlocked (2)–(4) and (3)-c-azo (5). (C) Cyclic and reversible photoinduced G′/G′′ values of the stiff hydrogel crosslinked by the glucosamine–boronate ester bridges and (2)–(4) and (3)-t-azo (5) crosslinkers, and the low-stiffness hydrogel crosslinked by only the glucosamine–boronate ester crosslinking units. (D) SEM images corresponding to the photo-induced reversible transitions of the stiff hydrogel crosslinked by the glucosamine–boronate ester bridges and (2)–(4) and (3)-t-azo (5), crosslinkers, panel (I), and the low stiffness hydrogel crosslinked by the glucosamine–boronate ester only and unlocked (2)–(4) and (3)-c-azo (5), panel (II). | ||
The photoresponsive hydrogel shown in Fig. 3A was then applied to develop shape-memory and self-healing soft material matrices. The stiff hydrogel crosslinked by the glucosamine–boronate ester and the trans-azobenzene (2)–(4)/(3)-t-azo (5) was prepared in a Teflon mold in the form of a triangle-shaped structure. The extruded, triangle-shaped hydrogel, state “A”, was subjected to the light-induced isomerization of the trans-azobenzene units to cis-azobenzene (5) state (λ = 365 nm). Unlocking of the (2)–(4)/(3)-t-azo (5) bridges resulted in the formation of the low stiffness, quasi-liquid hydrogel that lost the triangle-shape, Fig. 4A, state “B”. Irradiation of the shapeless hydrogel, state “B”, with visible light (λ > 420 nm) regenerated trans-azobenzene (5) units that cooperatively locked the trans-azobenzene (2)–(4)/(3)-t-azo (5) bridges. This process restored the triangle-shaped hydrogel, state “A”, demonstrating that the glucosamine–boronate ester bridges provide an internal permanent memory for re-shaping the hydrogel. That is, the entanglement of the polymer by means of the glucosamine–boronate ester supramolecular bridges confined the unlocked photo-responsive units to a spatial configuration that allowed, upon irradiation, the locking of the trans-azobenzene (2)–(4)/(3)-t-azo (5) units and the re-shaping of the hydrogel.
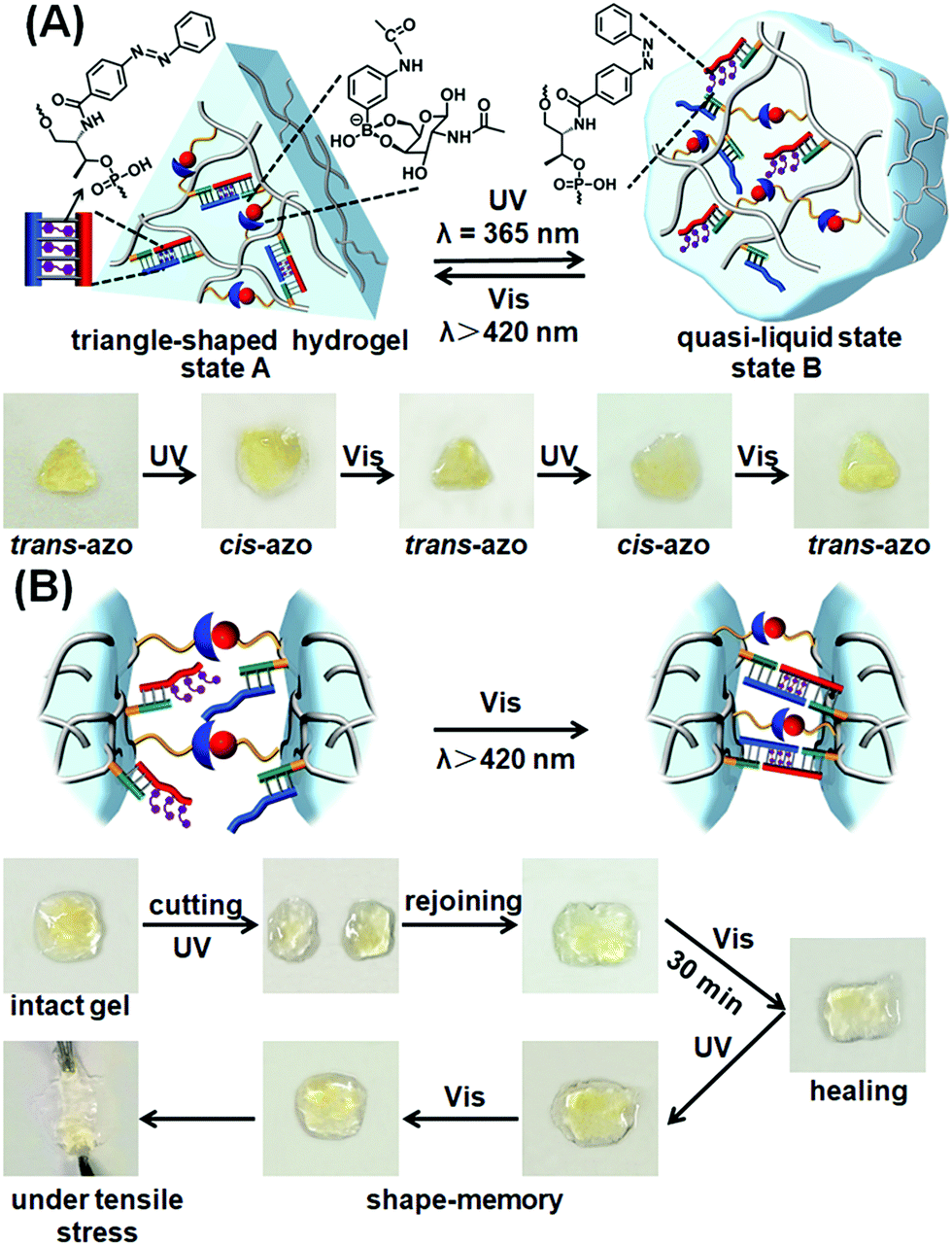 | ||
| Fig. 4 (A) Schematic light-induced shape-memory transitions between the triangle-shaped, high-stiffness hydrogel crosslinked by glucosamine–boronate ester bridges and trans-azobenzene stabilized duplex crosslinkers, and the low-stiffness hydrogel crosslinked by the glucosamine–boronate esters only (the duplex bridges are separated through the photoisomerization of the trans-azobenzene units to the cis-azobenzene states). The glucosamine–boronate ester bridges provide the memory code to regenerate the high-stiffness hydrogel, by the photoisomerization of the cis-azobenzene units to the trans-azobenzene states. Bottom: images corresponding to the reversible and cyclic transitions between the triangle-shaped, stiff hydrogel in state “A” and the quasi-liquid, lower-stiffness, shapeless hydrogel in state “B” upon the photoinduced cyclic transitions between the trans-azobenzene and cis-azobenzene tethers conjugated to the polymer chains. (B) Schematic self-healing of two hydrogel pieces composed of the soft, low-stiffness hydrogel as a result of the photochemically triggered self-healing of the hydrogel pieces by the light-induced transformation of the cis-azobenzene units to the trans-azobenzene units, and the crosslinking of the hydrogel by the glucosamine–boronate esters and the trans-azobenzene units associated with the duplex units. Bottom: images corresponding to the steps involved in the self-healing process. | ||
The photoresponsive hybrid hydrogel composed of the glucosamine–boronate ester supramolecular crosslinker and the trans-azobenzene-stabilized duplexes (4)/t-azo (5) was applied to develop a light-induced self-healing hydrogel matrix, Fig. 4B. The stiff hydrogel crosslinked by the glucosamine–boronate ester and the trans-azobenzene-stabilized duplex bridges (4)/t-azo (5) was cut into two pieces, and the pieces were transformed into the quasi-liquid hydrogel state stabilized by the glucosamine–boronate ester bridges only, by the light-induced isomerization of the t-azo (5) units to the c-azo (5) tethers (λ = 365 nm). The two quasi-liquid hydrogel pieces were combined and subjected to visible light irradiation (λ > 420 nm). Transforming the c-azo (5) tethers to the t-azo (5) states activates the crosslinking of the hydrogel pieces by the two crosslinking motifs, resulting in the healed intact hydrogel, Fig. 4B. It should be noted that the gentle shaking of the physically coupled quasi-liquid hydrogel pieces, that were not illuminated by visible light, resulted in their separation in Fig. S6.†
The control over the stiffness properties of the glucosamine–boronate ester-crosslinked acrylamide hydrogel by the stimuli-responsive G-quadruplex bridging units or the photochemically triggered azobenzene crosslinking units suggests that in addition to the shape-memory and self-healing properties of the hydrogels, these soft-materials could be applied as functional matrices for controlled drug delivery.
Fig. 5 shows the controlled release of the doxorubicin anti-cancer drug from the hydrogel crosslinked by the glucosamine–boronate esters and the K+-ion-stabilized G-quadruplex units (cf.Fig. 1). The doxorubicin drug was encapsulated in the hydrogel. Fig. 5A, inset, shows the fluorescence spectra of the released doxorubicin upon subjecting the doxorubicin-loaded hydrogel to reaction with 18-crown-6-ether at different time intervals. As the release process is prolonged the fluorescence of the released drug is intensified. Fig. 5A, curve (a) shows the time-dependent fluorescence changes originating from the release of doxorubicin from the crosslinked hydrogel, upon treatment with 18-crown-6-ether. Evidently, the release of the drug is fast, and after a time-interval of ca. 90 minutes the fluorescence of the released drug reaches saturation, implying that the release of the anti-cancer drug was completed. From the fluorescence saturation value and using an appropriate calibration curve we estimated that ca. 7.23 nmol of doxorubicin was released from 800 μg of the hydrogel. For comparison, Fig. 5A, curve (b) shows the time-dependent release of doxorubicin from the stiff hydrogel that was not treated with 18-crown-6-ether. Very inefficient release of doxorubicin is observed, and this release process reaches a saturation value after ca. 90 minutes. This release process is attributed to the leakage of doxorubicin from large-pore “defective” domains in the stiff hydrogel and suggests that no leakage proceeds from the small-pores, highly crosslinked, hydrogel matrix. Fig. 5B shows the switchable controlled release of doxorubicin from the glucosamine–boronate ester/G-quadruplex crosslinked hydrogel. Treatment of the hydrogel with the crown ether results in the hydrogel exhibiting low stiffness, resulting in the release of the drug. The subsequent treatment of the hydrogel with K+-ion crosslinks the hydrogel by the G-quadruplex units, resulting in the stiff hydrogel that reveals inefficient release properties. By the cyclic treatment of the hydrogel with 18-crown-6-ether and K+-ions the release of doxorubicin is switched between “ON” and “OFF” states, respectively.
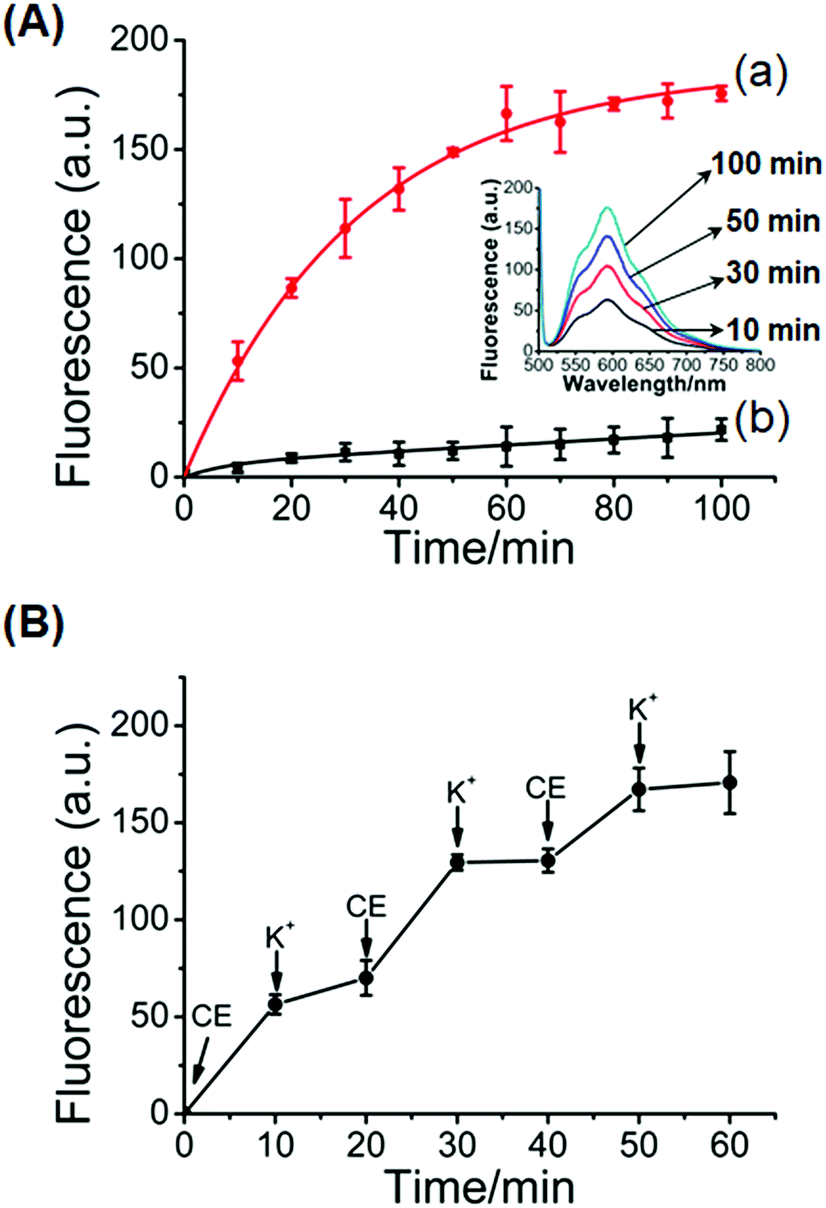 | ||
| Fig. 5 (A) Controlled release of doxorubicin from the glucosamine–boronate ester and K+-stabilized G-quadruplex crosslinked hydrogel: (a) time dependent release of doxorubicin upon treatment of the stiff glucosamine–boronate ester and K+-ion-stabilized hydrogel with 18-crown-6-ether. (b) Release of the doxorubicin from the stiff hydrogel in the absence of crown ether. Inset: fluorescence spectra of the released doxorubicin from the stiff hydrogel treated with crown ether at different time-intervals. (B) Switchable “ON” and “OFF” time-dependent release of doxorubicin from the glucosamine–boronate ester and K+-stabilized G-quadruplex crosslinked hydrogel upon its cyclic treatment with crown ether (CE) and K+-ions. | ||
In addition, the stiff photoresponsive hydrogel crosslinked by glucosamine–boronate ester complexes and the (3)-t-azo (5)/(2)–(4) duplexes was loaded with the doxorubicin drug and subjected to the light-induced release of the drug. The photo-induced isomerization of the t-azo (5) units to the c-azo (5), λ = 365 nm, results in the separation of the (2)–(4) and c-azo (5) (cf.Fig. 3) and the formation of the low stiffness hydrogel crosslinked by the glucosamine–boronate ester complexes. The low-stiffness, quasi-liquid hydrogel matrix allowed then the release of the encapsulated drug. Fig. 6A, inset, shows the fluorescence spectra of the released doxorubicin at different time-intervals. As the release time is prolonged the amount of the released drug is higher. Fig. 6A, curve (a), shows the time-dependent release of doxorubicin from the low-stiffness hydrogel that is crosslinked by the glucosamine–boronate ester complexes only. After a time-interval of ca. 200 minutes the release process reaches saturation, implying that the release of the encapsulated drug in the hydrogel was completed. Using an appropriate calibration, we estimated that ca. 4.4 nmol of doxorubicin was loaded per 1 mg of hydrogel. For comparison, Fig. 6A, curve (b), shows the release of doxorubicin from the stiff hydrogel crosslinked by the glucosamine–boronate ester and the (2)–(4)/(3)-t-azo (5) duplexes as crosslinkers. Very inefficient release of doxorubicin, that reaches saturation after ca. 100 minutes, is observed. This residual release of doxorubicin is attributed to the leakage of the drug from large pores that are still accompanying the stiff hydrogel. Fig. 6B shows the cyclic “ON” and “OFF” light-induced release of the drug from the hydrogel, upon photoisomerization of the azobenzene units across the c-azo (5) and t-azo (5), respectively. It should be noted that the resulting unloaded glucosamine–boronate ester/G-quadruplex (or photoresponsive (2)–(4)/(3)-t-azo (5) duplexes) crosslinked hydrogels could be reloaded with the drug to yield new drug-loaded matrices (for a detailed discussion and experimental results, see Fig. S7 and S8†).
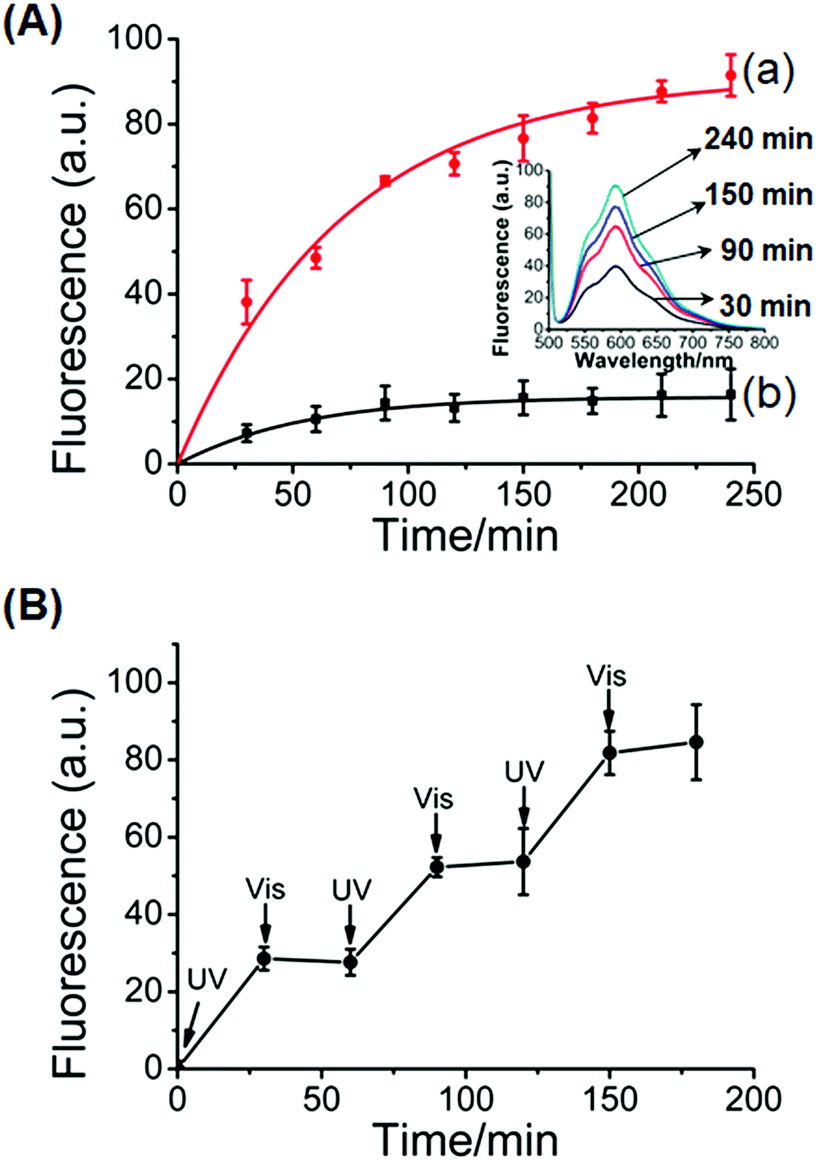 | ||
| Fig. 6 (A) Controlled release of doxorubicin from the glucosamine–boronate ester and trans-azobenzene stabilized duplex crosslinked hydrogel: (a) time-dependent release of doxorubicin upon the UV light-induced transition (λ = 365 nm) of the stiff glucosamine–boronate ester and trans-azobenzene stabilized duplex crosslinked hydrogel, into the low-stiffness hydrogel composed of glucosamine–boronate ester bridges only, and free cis-azobenzene tethers. (b) Release of the doxorubicin from the stiff hydrogel in the absence of UV light irradiation. Inset: fluorescence spectra corresponding to the released doxorubicin from the low-stiffness hydrogel crosslinked by the glucosamine–boronate ester bridges only at different time-intervals. (B) Switchable light-induced “ON” and “OFF” release of doxorubicin upon the cyclic photoisomerization of the hydrogel between the low-stiffness hydrogel crosslinked by the glucosamine–boronate ester bridges only (free cis-azobenzene tethers), λ = 365 nm (switch “ON”), and the high-stiffness hydrogel crosslinked by glucosamine–boronate ester bridges and the trans-azobenzene-stabilized duplexes as crosslinkers, λ > 420 nm (switch “OFF”). | ||
Conclusions
The present study has expanded the area of stimuli-responsive DNA-based hydrogels by synthesizing hydrogels that are crosslinked by covalent glucosamine–boronate ester bridges and stiff stimuli-responsive DNA bridges. In one system the hydrogel was synthesized by the cooperative crosslinking of the matrix by glucosamine–boronate ester bridges and by K+-ion-stabilized G-quadruplex units. The dissociation of the K+-stabilized G-quadruplex crosslinkers in the presence of 18-crown-6-ether led to the transition of the hydrogel into a soft, quasi-liquid matrix. The reversible triggered transitions of the hydrogel between stiff and quasi-liquid states allowed the development of a chemically stimulated shape-memory hydrogel and a self-healing matrix, as well as a stimuli-responsive material for the switchable and controlled drug release. In the second system, a light-responsive stiff hydrogel was synthesized by the cooperative crosslinking of the hydrogel with the glucosamine–boronate ester bridges and trans-azobenzene-stabilized duplex nucleic acid crosslinkers. By the photoisomerization of the azobenzene photoactive intercalator units between the trans and cis states the nucleic acid tethers could be reversibly switched between duplex bridged and open tether configurations, respectively. This allowed the development of hydrogels undergoing light-induced transitions between stiff and quasi-liquid states. These switchable functions of the light-responsive hydrogel were used to develop shape-memory hydrogels and self-healing hydrogel matrices. In addition, the light-induced control of the stiffness of the hydrogel led to the development of a functional material for the switchable controlled release of the doxorubicin drug. The ability to control the stiffness of hydrogels by chemical or light stimuli paves the way to apply these hydrogel matrices for controlled drug delivery43 or for the preparation of “mechanical” hydrogels acting as actuators.49 At present, among the many photoisomerization compounds, only azobenzene and arylazopyrazole58 are known to interact with DNA and to control the stability of duplex nucleic acid structure. The development of new photoisomerizable switches interacting with nucleic acids (particularly switchable operating in the visible/IR regions) is a future challenge.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Israel Science Foundation (ISF 1613/16), the Minerva Center for Biohybrid Complex Systems, and the National Natural Science Foundation of China (21420102004), Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX03) and the international cooperation program of Shanghai Science and Technology Committee (17520750100).Notes and references
- J. H. Holtz and S. A. Asher, Nature, 1997, 389, 829–832 CrossRef CAS PubMed.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS PubMed.
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS PubMed.
- A. J. Amaral and G. Pasparakis, Polym. Chem., 2017, 8, 6464–6484 RSC.
- P. Calvert, Adv. Mater., 2009, 21, 743–756 CrossRef CAS.
- C. Majidi, Soft Robot., 2014, 1, 5–11 CrossRef.
- S. Jiang, F. Liu, A. Lerch, L. Ionov and S. Agarwal, Adv. Mater., 2015, 27, 4865–4870 CrossRef CAS PubMed.
- T. a. Asoh, M. Matsusaki, T. Kaneko and M. Akashi, Adv. Mater., 2008, 20, 2080–2083 CrossRef CAS.
- J. Kim, J. A. Hanna, R. C. Hayward and C. D. Santangelo, Soft Matter, 2012, 8, 2375–2381 RSC.
- Y. S. Kim, M. Liu, Y. Ishida, Y. Ebina, M. Osada, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nat. Mater., 2015, 14, 1002–1007 CrossRef CAS PubMed.
- M. Motornov, R. Sheparovych, R. Lupitskyy, E. MacWilliams, O. Hoy, I. Luzinov and S. Minko, Adv. Funct. Mater., 2007, 17, 2307–2314 CrossRef CAS.
- E. Cheng, Y. Xing, P. Chen, Y. Yang, Y. Sun, D. Zhou, L. Xu, Q. Fan and D. Liu, Angew. Chem., Int. Ed., 2009, 48, 7660–7663 CrossRef CAS PubMed.
- R. Marcombe, S. Cai, W. Hong, X. Zhao, Y. Lapusta and Z. Suo, Soft Matter, 2010, 6, 784–793 RSC.
- S. L. Zhou, S. Matsumoto, H. D. Tian, H. Yamane, A. Ojida, S. Kiyonaka and I. Hamachi, Chem.–Eur. J., 2005, 11, 1130–1136 CrossRef CAS PubMed.
- D. P. Holmes, M. Roché, T. Sinha and H. A. Stone, Soft Matter, 2011, 7, 5188–5193 RSC.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed.
- A. Guiseppi-Elie, Biomaterials, 2010, 31, 2701–2716 CrossRef CAS PubMed.
- T.-Y. Liu, S.-H. Hu, K.-H. Liu, D.-M. Liu and S.-Y. Chen, J. Controlled Release, 2008, 126, 228–236 CrossRef CAS PubMed.
- Y. Y. Liang, L. M. Zhang, W. Jiang and W. Li, ChemPhysChem, 2007, 8, 2367–2372 CrossRef CAS PubMed.
- R. Ebrahimi and M. Ebrahimi, J. Polym. Eng., 2014, 34, 625–632 CAS.
- B. Rokita, J. M. Rosiak and P. Ulanski, Macromolecules, 2009, 42, 3269–3274 CrossRef CAS.
- A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128–2140 CrossRef CAS PubMed.
- J. Li, L. Mo, C.-H. Lu, T. Fu, H.-H. Yang and W. Tan, Chem. Soc. Rev., 2016, 45, 1410–1431 RSC.
- J. S. Kahn, Y. Hu and I. Willner, Acc. Chem. Res., 2017, 50, 680–690 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- W. Guo, C. H. Lu, X. J. Qi, R. Orbach, M. Fadeev, H. H. Yang and I. Willner, Angew. Chem., Int. Ed., 2014, 53, 10134–10138 CrossRef CAS PubMed.
- L. Heinen, T. Heuser, A. Steinschulte and A. Walther, Nano Lett., 2017, 17, 4989–4995 CrossRef CAS PubMed.
- J. Ren, Y. Hu, C.-H. Lu, W. Guo, M. A. Aleman-Garcia, F. Ricci and I. Willner, Chem. Sci., 2015, 6, 4190–4195 RSC.
- C.-H. Lu, X.-J. Qi, R. Orbach, H.-H. Yang, I. Mironi-Harpaz, D. Seliktar and I. Willner, Nano Lett., 2013, 13, 1298–1302 CrossRef CAS PubMed.
- W. Guo, X.-J. Qi, R. Orbach, C.-H. Lu, L. Freage, I. Mironi-Harpaz, D. Seliktar, H.-H. Yang and I. Willner, Chem. Commun., 2014, 50, 4065–4068 RSC.
- L. Peng, M. You, Q. Yuan, C. Wu, D. Han, Y. Chen, Z. Zhong, J. Xue and W. Tan, J. Am. Chem. Soc., 2012, 134, 12302–12307 CrossRef CAS PubMed.
- W. Guo, C. H. Lu, R. Orbach, F. Wang, X. J. Qi, A. Cecconello, D. Seliktar and I. Willner, Adv. Mater., 2015, 27, 73–78 CrossRef CAS PubMed.
- Y. Maeda, T. Nakamura and I. Ikeda, Macromolecules, 2001, 34, 1391–1399 CrossRef CAS.
- A. Desponds and R. Freitag, Langmuir, 2003, 19, 6261–6270 CrossRef CAS.
- T. Ueki, Y. Nakamura, A. Yamaguchi, K. Niitsuma, T. P. Lodge and M. Watanabe, Macromolecules, 2011, 44, 6908–6914 CrossRef CAS.
- Y. Shiraishi, R. Miyamoto and T. Hirai, Org. Lett., 2009, 11, 1571–1574 CrossRef CAS PubMed.
- J. Wang, J. Chao, H. Liu, S. Su, L. Wang, W. Huang, I. Willner and C. Fan, Angew. Chem., Int. Ed., 2017, 56, 2171–2175 CrossRef CAS PubMed.
- J. S. Kahn, A. Trifonov, A. Cecconello, W. Guo, C. Fan and I. Willner, Nano Lett., 2015, 15, 7773–7778 CrossRef CAS PubMed.
- Y. Wu, D. Wang, I. Willner, Y. Tian and L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 7790–7794 CrossRef CAS PubMed.
- H. Yang, H. Liu, H. Kang and W. Tan, J. Am. Chem. Soc., 2008, 130, 6320–6321 CrossRef CAS PubMed.
- X. Mao, G. Chen, Z. Wang, Y. Zhang, X. Zhu and G. Li, Chem. Sci., 2018, 9, 811–818 RSC.
- C.-H. Lu, W. Guo, Y. Hu, X.-J. Qi and I. Willner, J. Am. Chem. Soc., 2015, 137, 15723–15731 CrossRef CAS PubMed.
- W.-C. Liao, S. Lilienthal, J. S. Kahn, M. Riutin, Y. S. Sohn, R. Nechushtai and I. Willner, Chem. Sci., 2017, 8, 3362–3373 RSC.
- W. H. Chen, W. C. Liao, Y. S. Sohn, M. Fadeev, A. Cecconello, R. Nechushtai and I. Willner, Adv. Funct. Mater., 2018, 28, 1705137 CrossRef.
- M. Yoshida, R. Langer, A. Lendlein and J. Lahann, J. Macromol. Sci., Polym. Rev., 2006, 46, 347–375 CrossRef CAS.
- H. Meng and G. Li, Polymer, 2013, 54, 2199–2221 CrossRef CAS.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- K. K. Julich-Gruner, C. Löwenberg, A. T. Neffe, M. Behl and A. Lendlein, Macromol. Chem. Phys., 2013, 214, 527–536 CrossRef CAS.
- Y. Hu, J. S. Kahn, W. Guo, F. Huang, M. Fadeev, D. Harries and I. Willner, J. Am. Chem. Soc., 2016, 138, 16112–16119 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS PubMed.
- J. Cui and A. del Campo, Chem. Commun., 2012, 48, 9302–9304 RSC.
- W. M. Xu, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2016, 4, 10683–10690 RSC.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- J. Zhang, Y. Liu, J. Lv and G. Li, Nano Res., 2015, 8, 920–930 CrossRef CAS.
- J. Zhang, J. Lv, X. Wang, D. Li, Z. Wang and G. Li, Nano Res., 2015, 8, 3853–3863 CrossRef CAS.
- Z. Zhang, L. Chen, C. Zhao, Y. Bai, M. Deng, H. Shan, X. Zhuang, X. Chen and X. Jing, Polymer, 2011, 52, 676–682 CrossRef CAS.
- V. Adam, D. K. Prusty, M. Centola, M. Škugor, J. S. Hannam, J. Valero, B. Klöckner and M. Famulok, Chem.–Eur. J., 2018, 24, 1062–1066 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04292f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |