
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorogenic atom transfer radical polymerization in aqueous media as a strategy for detection†
Zachary T.
Allen
,
Jemima R.
Sackey-Addo
,
Madeline P.
Hopps
,
Danyal
Tahseen
,
Joseph T.
Anderson
,
Tyler A.
Graf
and
Christina B.
Cooley
 *
*
Department of Chemistry, Trinity University, One Trinity Place, San Antonio, TX 78212, USA. E-mail: ccooley@trinity.edu
First published on 8th November 2018
Abstract
The development of novel approaches to signal amplification in aqueous media could enable new diagnostic platforms for the detection of water-soluble analytes, including biomolecules. This paper describes a fluorogenic polymerization approach to amplify initiator signal by the detection of visible fluorescence upon polymerization in real-time. Fluorogenic monomers were synthesized and co-polymerized by atom transfer radical polymerization (ATRP) in water to reveal increasing polymer fluorescence as a function of both reaction time and initiator concentration. Optimization of the fluorogenic ATRP reaction conditions allowed for the quantitative detection of a small-molecule initiator as a model analyte over a broad linear concentration range (pM to mM). Raising the reaction temperature from 30 °C to 60 °C facilitated sensitive initiator detection at sub-picomolar concentrations in as little as 1 h of polymerization. This method was then applied to the detection of streptavidin as a model biological analyte by fluorogenic polymerization from a designed biotinylated ATRP initiator. Taken together, these studies represent the first example of a fluorogenic ATRP reaction and establish fluorogenic polymerization as a promising approach for the direct detection of aqueous analytes and biomolecular recognition events.
Introduction
Signal amplification is a key step of any sensitive detection assay. Traditional approaches have relied on biological enzymes for signal amplification; however, there is growing interest in the development of sensitive and economical chemical amplification methods as alternatives to enzymatic detection.1–3 Polymerization amplification is a relatively new approach that takes advantage of the growth of a long polymer chain from one initiation event. If that event is coupled to the detection of a desired analyte, the formation of detectable polymer signals analyte presence. Radical polymerization techniques are particularly attractive platforms for signal amplification as the reactions are robust and sensitive to very low radical concentrations.2Radical polymerization amplification methods can sensitively detect biomolecules from DNA to proteins, with polymer formation detected by methods such as polymer film or hydrogel formation on a sensor surface,3–9 electrochemical analysis,10–12 nanoparticle aggregation,13 fluorophore conjugation14 or enzymatic amplification.15 However, these approaches rely on either pre-polymerization derivatization with macroinitiating polymers or post-polymerization elaboration or experimentation, and are not direct readouts of polymerization progress.
We sought to develop a simple, real-time fluorescence method for signal amplification by fluorogenic polymerization, in which non-fluorescent, “dark” monomers become visibly fluorescent upon incorporation into the growing polymer chain. We postulated that a controlled, living polymerization approach16 could facilitate quantitative analyte detection as the linear chain growth should allow for polymer length to correlate with initiator concentration.
Atom transfer radical polymerization (ATRP)17–19 was explored for the initial development of this fluorogenic polymerization detection method as it exhibits controlled, living character, robust turnover and kinetics, broad functional group tolerance, bioorthogonal initiation, and is viable in aqueous solvents.20–22 Recent advances in aqueous ATRP by the activators regenerated by electron transfer (ARGET-ATRP) method using a reducing agent to regenerate active catalyst in situ has allowed for aqueous polymerizations with low catalyst loading and high control over the resultant polymer's molecular weights and polydispersity.23,24
Here, we describe a new fluorogenic ATRP reaction by synthesis of polyaromatic hydrocarbon probe monomers and their co-polymerization with PEG methacrylate to generate visibly fluorescent polymers in water. Real-time monitoring of polymer fluorescence tracks with early polymerization progress and with initiator concentration, enabling both qualitative (visual) and quantitative detection of a broad range of initiator concentrations by fluorescence spectroscopy. Optimization for detection applications allows for sub-picomolar initiator sensitivity in 1 h of reaction time. Extension of this method to the detection of streptavidin as a model biological analyte explores the potential utility of fluorogenic polymerization as a new strategy for biomolecular detection.
Results and discussion
Fluorogenic monomer synthesis
Our initial targets were fluorogenic monomers that are “dark” in monomer form, but reveal fluorescence once incorporated into a growing polymer chain. These probes are methacrylamide derivatives of polycyclic aromatic hydrocarbon (PAH) probes such as pyrene,25 anthracene26 and acridine (Fig. 1). Upon polymerization, they exhibit UV excitation (337–431 nm) and visible emission (∼400–500 nm), which allows for visual fluorescence detection following UV irradiation. While pyrene, anthracene and acridine are fluorescent, the methacrylamide monomers 1–3 are non-fluorescent due to quenching by the covalently attached α,β-unsaturated amide.25–27 As the fluorogenic monomer polymerizes, the carbon–carbon double bond becomes saturated, and fluorescence from the probes is observed. | ||
| Fig. 1 Structures of synthesized fluorogenic monomers based on pyrene (Py 1), anthracene (An 2) and acridine (Ac 3) fluorophores. | ||
Pyrene and anthracene methacrylamide monomers Py 1 and An 2 have been utilized as fluorescent probes to monitor the kinetics of radiation-induced radical polymerizations. The fluorescence emission intensity was found to be proportional to early conversion of monomer into polymer.25,26,28 Acridine methacrylamide monomer Ac 3 was designed in this study to improve the fluorogenic probe features for aqueous detection, as acridine has a higher quantum yield of fluorescence and slightly improved water solubility relative to pyrene and anthracene.29,30 Fluorogenic monomers 1–3 were synthesized by reacting their respective commercially available amino-probe precursers with freshly distilled methacryloyl chloride and triethylamine in DCM or THF to achieve monomers 1–3 in good yields (see ESI†).
Fluorogenic aqueous ATRP
Fluorogenic ATRP polymerization was initially investigated by reaction of the anthracene fluorogenic monomer An 2 with a water-soluble co-monomer such as PEG methacrylate 5 under modified aqueous ARGET-ATRP conditions (Scheme 1).23 We hypothesized that inclusion of a co-monomer would be necessary to generate water-soluble polymers that exhibit bright fluorescence by spacing out the fluorogenic monomers and avoiding self-quenching by pi-stacking or excimer formation.31,32 The ATRP reaction was initiated by 2-hydroxyethyl-2-bromoisobutyrate 4, a water-soluble initiator that serves as a model detectable analyte. Other reaction components included the CuBr2 catalyst, tris(2-pyridylmethyl)-amine ligand (TPMA), NaCl and ascorbic acid as the reducing agent to generate active catalyst for ARGET ATRP.23 Sodium dodecyl sulfate (SDS) was also added as a surfactant to ensure solubility of the fluorogenic monomer in water (Scheme 1). Upon heating the reaction to 30 °C, fluorescence emission spectra of the solution were monitored at various time points and showed that anthracene methacrylamide monomer An 2 exhibited no fluorescence before initiation of polymerization (t = 0). However, fluorescence from random co-polymer 6 was clearly detected as early as five h into the reaction. Notably, fluorescence increased with reaction time until 24 h, enabling a real-time monitoring of polymerization progress (Fig. 2). | ||
| Scheme 1 Fluorogenic ATRP polymerization in aqueous media with anthracene monomer An 2. | ||
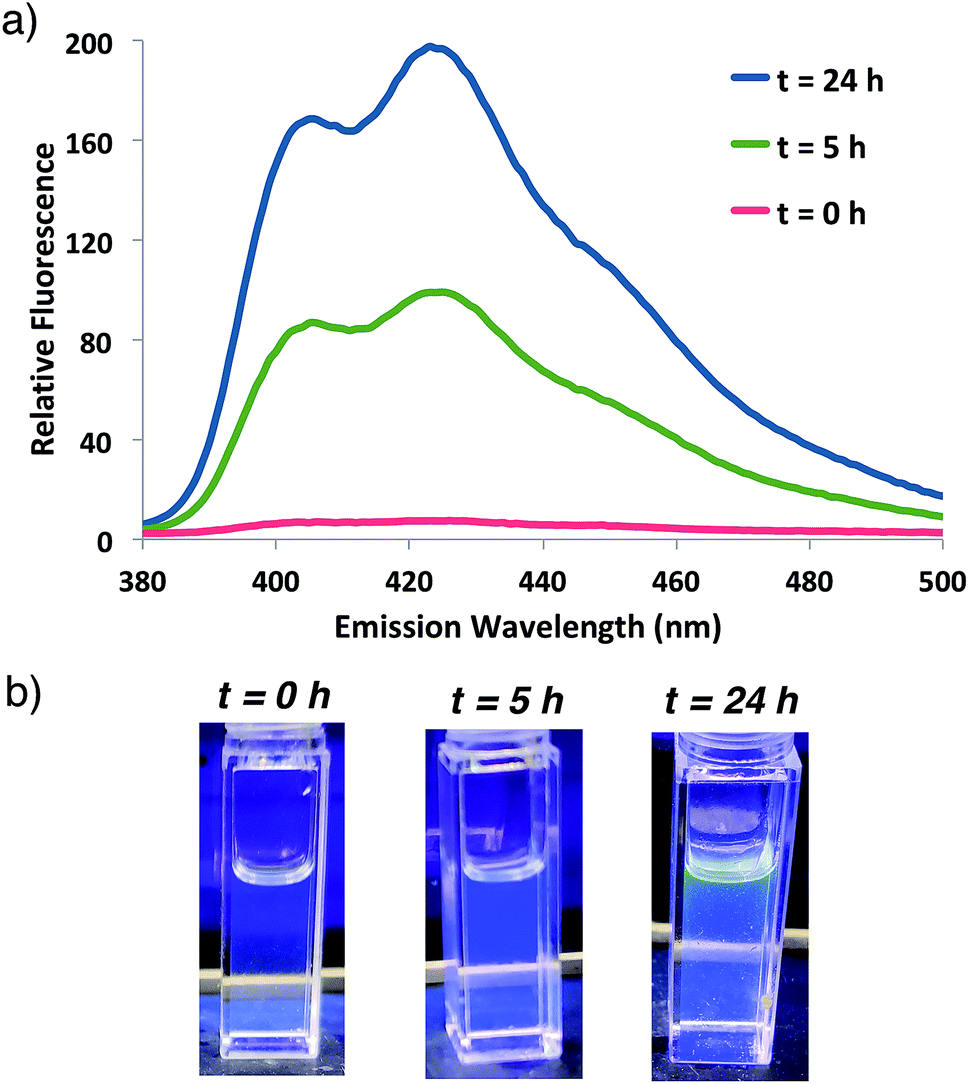 | ||
| Fig. 2 (a) Fluorescence emission spectrum for ATRP reaction shown in Scheme 1 at indicated times following 337 nm excitation. (b) Photographs of the reaction at indicated times illuminated by a hand-held UV light (365 nm). | ||
The fluorogenic polymerization reaction was also monitored by 1H NMR by examining the loss of monomer vinyl peaks over time (Fig. S1†). Monomer consumption was complete after 5 days of reaction at 30 °C. Concomitant fluorescence analysis demonstrates that fluorescence monitoring of polymerization best tracks with early monomer conversion (Fig. S1b†), consistent with previous reports.33 Following complete monomer conversion, the visibly fluorescent polymer was isolated by precipitation from THF in 90% yield. Gel permeation chromatography (GPC) analysis was performed on the isolated fluorescent polymer to confirm a narrow molecular weight distribution of 1.197, which is consistent with a living polymerization mechanism (Mw/Mn < 1.3). Incorporation of the fluorogenic monomer was further verified by trace nitrogen detection via elemental analysis (0.1% N, see ESI†), as only the fluorogenic monomers contain nitrogen atoms.
Monomer ratio optimization
Having demonstrated that fluorogenic, aqueous ATRP reactions are indeed feasible, we sought to increase the incorporation of fluorescent probe(s) into the polymers to optimize the signal intensity and kinetics. Initial attempts to increase fluorogenic monomer incorporation did not improve fluorescence due to a concomitant increase in SDS concentrations (>100 mM) that interfered with signal detection. Experiments to reduce the SDS concentration determined that a 3-fold reduction in SDS from the original reaction conditions retained adequate monomer solubilization with improved fluorescence levels (Fig. S2†). After lowering the SDS concentration, 10-fold additional fluorogenic anthracene monomer An 2 (from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9300 anthracene:PEG molar ratio to 1:930) dramatically increased the fluorescence signal and polymer kinetics. The 10× greater anthracene-content polymer exhibited a higher fluorescence signal after 1 h of reaction compared to the original conditions at 22 h (Fig. 3). Further attempts to incorporate additional fluorogenic monomer did not lead to a fluorescence increase (Fig. S3†), presumably due to the requirement for more SDS to maintain An 2 solubility.
:9300 anthracene:PEG molar ratio to 1:930) dramatically increased the fluorescence signal and polymer kinetics. The 10× greater anthracene-content polymer exhibited a higher fluorescence signal after 1 h of reaction compared to the original conditions at 22 h (Fig. 3). Further attempts to incorporate additional fluorogenic monomer did not lead to a fluorescence increase (Fig. S3†), presumably due to the requirement for more SDS to maintain An 2 solubility.
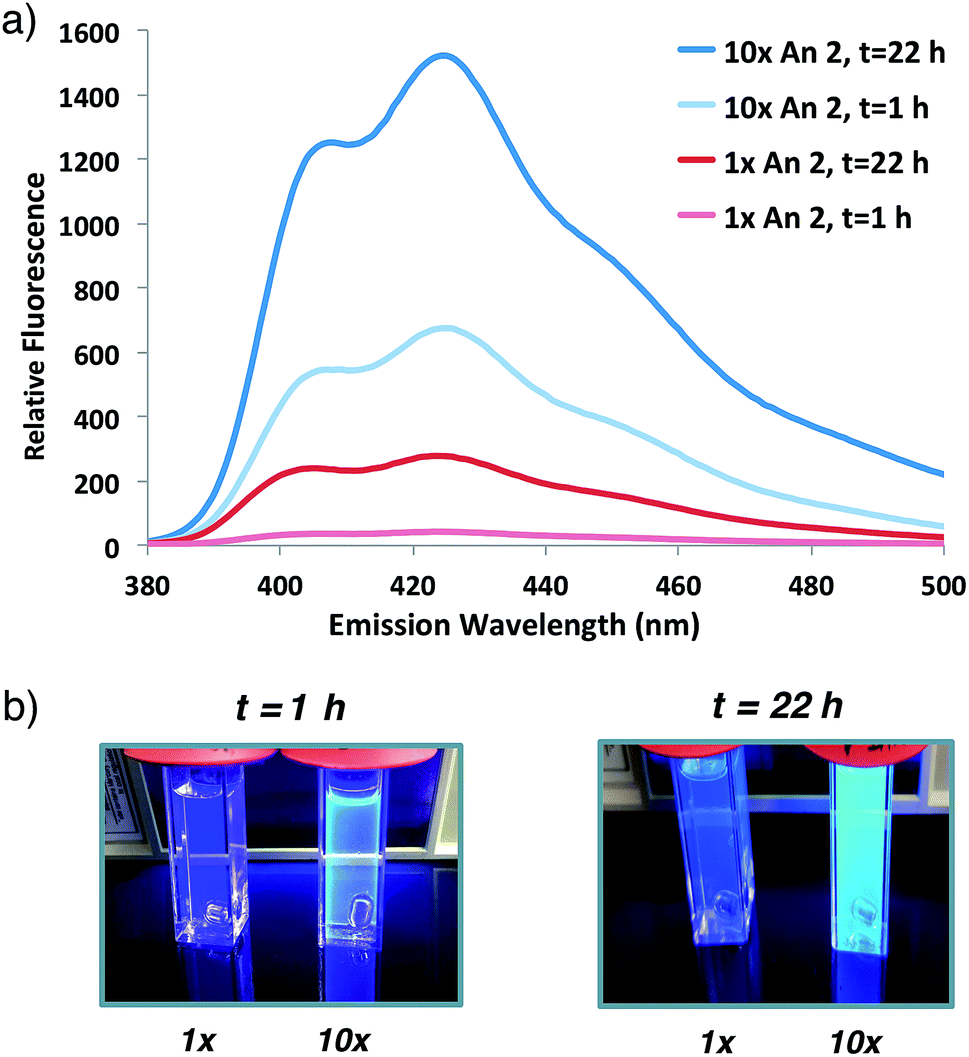 | ||
| Fig. 3 (a) Fluorescence emission spectrum of initial anthracene:PEG 2:5 molar ratio (1×, 1:9300) or 10-fold increase in An 2 (10×, 1:930) co-polymerization at indicated times following 337 nm excitation. (b) Photographs of sample illuminated by hand-held UV light (365 nm). | ||
Fluorogenic monomer comparison
The improved fluorescent signal obtained with 10-fold additional anthracene incorporation was sufficient to move forward to compare the relative fluorescence characteristics of fluorogenic methacrylamide monomers 1–3. Following confirmation that newly designed acridine monomer Ac 3 exhibited similar fluorogenic characteristics as the other probe monomers (Fig. S4†), side-by-side ATRP polymerization reactions were conducted as shown in Scheme 1. In these experiments, fluorogenic monomer An 2 was replaced with either the pyrene- or acridine-containing monomers Py 1 and Ac 3, respectively. Monitoring fluorescence emission at 2 h and 24 h following the start of polymerization (Fig. 4) showed that as the emission spectra shift further into the visible range, the signal intensity progressively increases. Polymerization with acridine monomer Ac 3 exhibited the brightest signal as predicted due to its increased quantum yield.29,30 Significant pyrene fluorescence was not observed until 24 h, which indicates that either polymerization of the pyrene monomer occurs at a slower rate relative to the other two monomers, or that the fluorescence increase for pyrene is less efficient than anthracene and acridine at similar levels of monomer conversion. Both effects likely contribute to the observed kinetic delay in pyrene fluorescence, as Py 1 exhibits a slight reduction in polymerization reactivity compared to An 225,26 and the excited state of pyrene is significantly less stable relative to the other probes used in this study.26,29
 | ||
| Fig. 4 Emission spectra and photographs of pyrene, anthracene and acridine co-polymerization reactions following excitation at 337, 371 or 431 nm, respectively, at 1 h (a) or 24 h (b). | ||
Sensitivity to initiator concentration
In order to investigate the potential for this fluorogenic ATRP reaction to be utilized as a diagnostic assay, we sought to assess fluorescence variance with initiator concentration. If small-molecule initiator 4 represents a model detectable analyte or is coupled to a detectable analyte, then the lowest detectable initiator concentration would provide a measure of assay sensitivity. Whereas the previous polymerizations were performed with a standard initiator concentration (2 mM), we performed anthracene ATRP co-polymerization experiments either in the absence of initiator or with concentrations of initiator 4 from 2 pM to 2 mM (Fig. 5). Fluorescence measurements at t = 24 h showed that 0 initiator gives negligible background fluorescence, indicating no detectable polymer formation (Fig. 5a). Further, fluorescence tracks with initiator concentration over this broad concentration range, spanning 9 orders of magnitude (Fig. 5b). The lower limit of detection under these conditions by fluorescence measurement was in the upper fM to low pM range, while qualitative analysis of fluorescence by eye allowed for reproducible discrimination between 0 and 2 pM concentrations of initiator and above. However, the relatively slow kinetics of fluorescence onset (∼24 h), especially at the lower range of initiator concentrations, is a drawback of this approach for detection applications. | ||
| Fig. 5 (a) Emission spectra and photograph of anthracene ATRP co-polymerization at 30 °C with indicated concentrations of initiator 4 at 24 h of reaction time following excitation at 371 nm. (b) Plot of relative fluorescence intensity at 426 nm versus initiator concentration from the reaction in (a). Error bars represent standard error of n = 4 separate trials. | ||
To speed up the reaction kinetics and the corresponding appearance of a detectable fluorescence signal, we investigated the effects of reaction temperature. Increasing the reaction temperature to 60 °C allowed for faster polymerization kinetics and higher fluorescence readings at shorter times (1–3 h), although at longer time points (24 h) significant background polymerization was observed (Fig. S5†). While ATRP reactions are known to proceed with control and living character at higher temperatures,34,35 we postulated that in the relatively unstudied case in the absence of initiator, the diradical nature of trace oxygen in solution could initiate polymerization and lead to the observed background fluorescence. Fluorogenic ATRP experiments conducted at 30 °C without rigorous degassing showed that trace oxygen is capable of initiating polymerization and increases background fluorescence in the absence of initiator (Fig. S6†). This higher background fluorescence is likely exacerbated when the reaction is performed at higher temperature, and can be observed at 60 °C even with rigorous degassing efforts. However, analysis of early reaction times prior to significant background fluorescence accumulation at 60 °C allowed for detection down to 200 fM of initiator 4 after only 1 h of reaction time (Fig. 6). Continued optimization experiments to reduce the detection limit and time to detectable fluorescence are ongoing; particularly to explore alternative oxygen scavenging methods such as chemical or enzymatic approaches. However, for this proof-of-concept study, fM initiator concentrations detectable by 1–24 h was sufficient to move forward with detection of a model biological analyte.
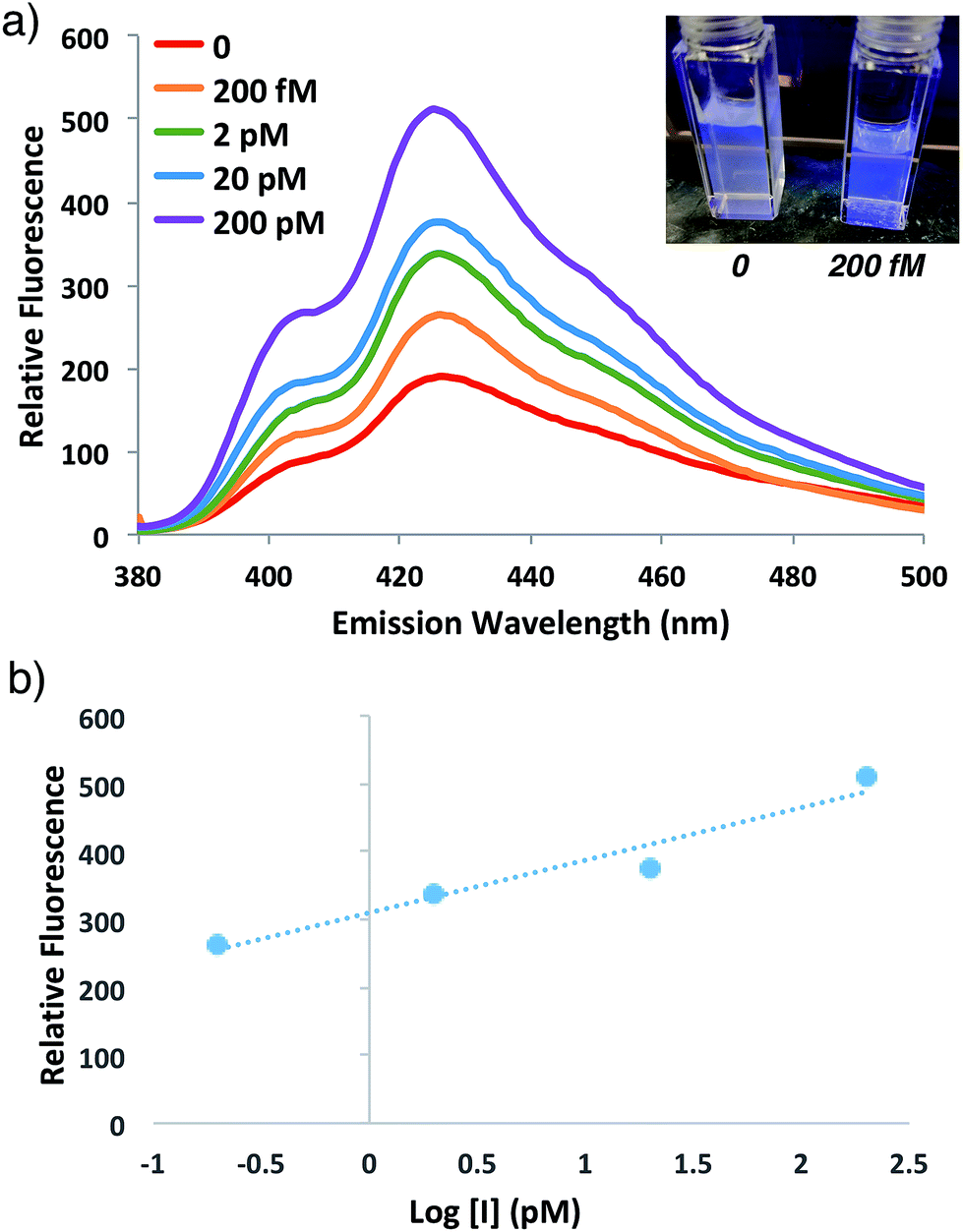 | ||
| Fig. 6 (a) Emission spectra and photograph of anthracene ATRP co-polymerization at 60 °C with indicated concentrations of initiator 4 at 1 h of reaction time following excitation at 371 nm. (b) Plot of relative fluorescence intensity at 426 nm versus of initiator concentration from the reaction in (a). | ||
Streptavidin detection by fluorogenic polymerization
To demonstrate the potential of this new fluorogenic ATRP polymerization technique, we applied this new reaction to the detection of a model biomolecule, streptavidin. Streptavidin was chosen as it is a well-known, tetrameric protein that specifically binds four equivalents of the small-molecule biotin (one per subunit) with extremely high affinity (Kd = 10−15 M).36,37 In order to detect streptavidin, we designed a dual-functionality small-molecule with an isobutyryl bromide head group capable of initiating ATRP and a biotin moiety for streptavidin binding (Scheme 2). Esterification of biotin with 2-hydroxyethyl-2-bromoisobutyrate 4 in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 4-(dimethylamino)pyridine (DMAP) facilitated the formation of biotinylated initiator 7 in good yield.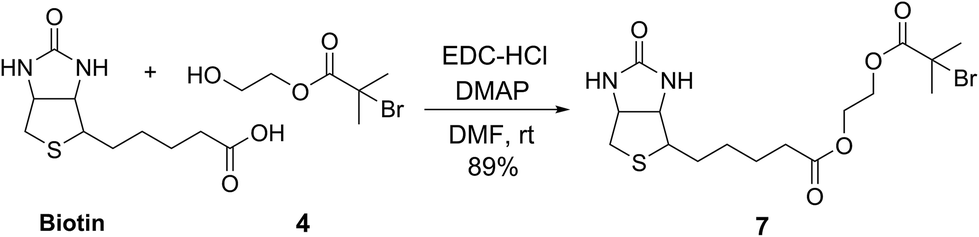 | ||
| Scheme 2 Synthesis of biotinylated initiator 7. | ||
Following confirmation that 7 initiates fluorogenic ATRP reactions in a similar fashion to unmodified initiator 4 (Fig. S7†), the ability of 7 to bind and detect streptavidin by fluorogenic ATRP polymerization was explored as shown in Fig. 7. Varying amounts of streptavidin-coated magnetic beads were washed and incubated with excess biotinylated initiator 7, followed by repeated washes to remove all unbound species (Fig. 7a). Bound biontinylated initiator 7 was then eluted by heating in water to 75 °C. These relatively mild conditions are known to gently elute bound biotin–streptavidin species.38 The supernatant containing biotinylated initiator 7 was then reacted with anthracene methacrylamide monomer An 2 and PEG methacrylate 5 under standard fluorogenic ATRP reaction conditions at 30 °C (Scheme 1). Fluorescence analysis at 24 h indicated increasing fluorescence signal as a function of increasing streptavidin concentrations (Fig. 7b). Further, streptavidin concentrations in the pM range are easily detectible by this fluorogenic ATRP amplification approach, both quantitatively by fluorescence spectroscopy and qualitatively by eye.
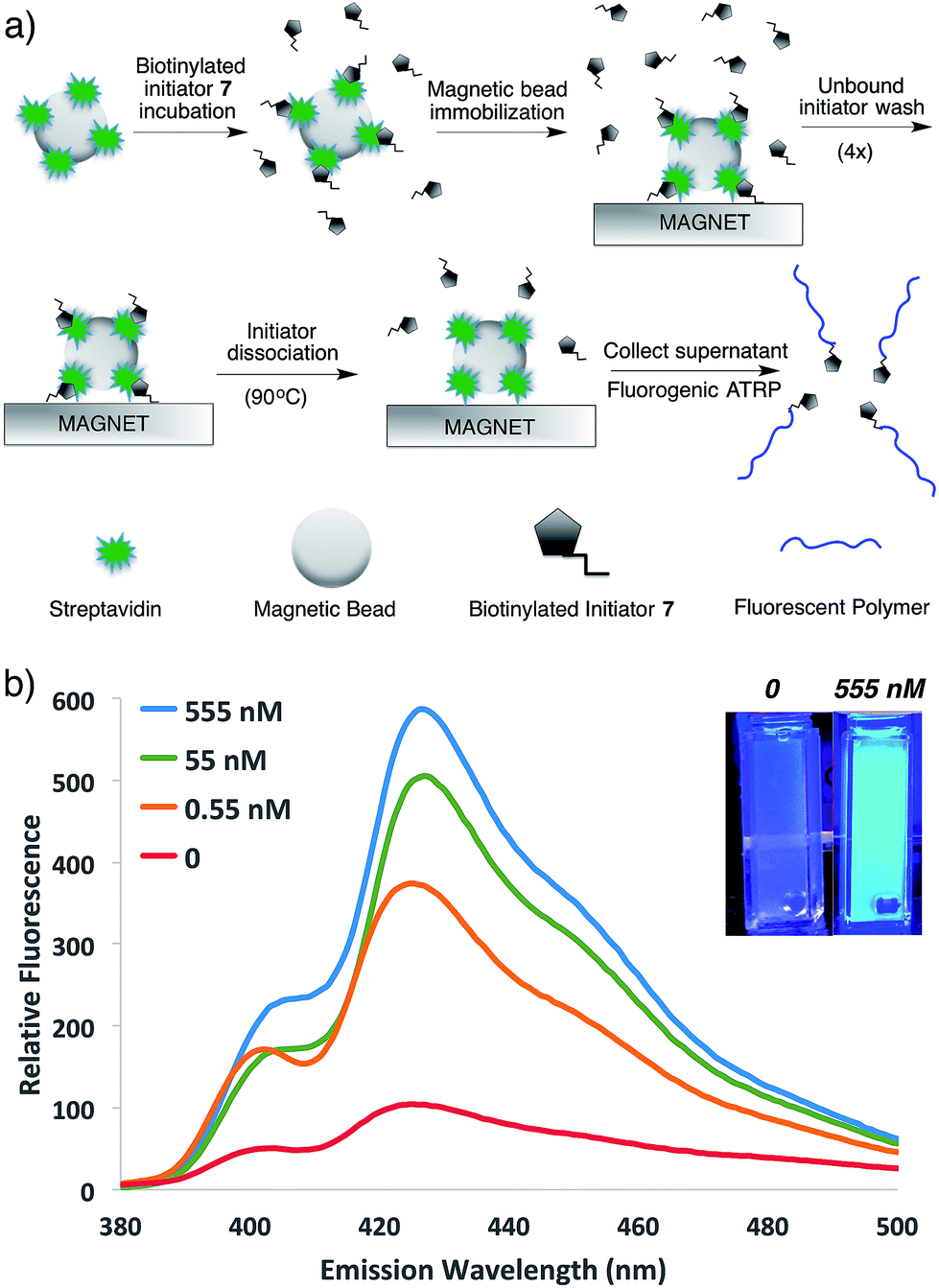 | ||
| Fig. 7 (a) Schematic for the streptavidin detection assay. (b) Emission spectra and photographs of anthracene ATRP co-polymerization initiated from 7 following incubation with various concentrations of streptavidin by the protocol shown in (a). The spectra and photographs for various conditions were taken after 24 h of reaction time following excitation at 371 nm or by a hand-held UV lamp (365 nm). | ||
Conclusions
We have developed the first fluorogenic ATRP reaction and applied it to the detection of a water-soluble, small-molecule analyte and a biologically relevant protein–ligand interaction. This was achieved by the design and synthesis of fluorogenic methacrylamide probe monomers and the optimization of aqueous ATRP co-polymerization conditions to provide a visible fluorescent signal as a function of monomer conversion and initiator concentration. Notably, the reaction occurs in aqueous media and allows for the detection of biological analytes in solution, which is currently under investigation by our laboratory and will be reported in due course. Further, the relatively low cost and spectral properties (UV excitation, visible emission) of the PAH probes used in this study affords unique advantages in low-resource settings as polymerization can be detected without specialized equipment by the naked eye. The ability to sensitively amplify initiator signals directly by fluorescence in real-time could have many unique applications ranging from basic studies of polymerization kinetics and mechanism to the development of new and economical diagnostic assay platforms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ned Bowden and Chad Gilmer at The University of Iowa for assistance with polymer polydispersity and molecular weight measurements. Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for support of this research. We gratefully acknowledge further support from the Welch Foundation (department grant W-0031), the Arnold and Mabel Beckman Foundation, the National Science Foundation (CHE-1726441), the San Antonio Area Foundation, and Trinity University.Notes and references
- M. Urdea, L. A. Penny, S. S. Olmsted, M. Y. Giovanni, P. Kaspar, A. Shepherd, P. Wilson, C. A. Dahl, S. Buchsbaum, G. Moeller and D. C. Hay Burgess, Nature, 2006, 444, 73 CrossRef PubMed
.
- K. Kaastrup and H. D. Sikes, Chem. Soc. Rev., 2016, 45, 532 RSC
- H. Qian and L. He, Sens. Actuators, B, 2010, 150(2), 594 CrossRef CAS
- H. D. Sikes, R. R. Hansen, L. M. Johnson, R. Jenison, J. W. Birks, K. L. Rowlen and C. N. Bowman, Nat. Mater., 2007, 7(1), 52 CrossRef PubMed
- R. R. Hansen, H. D. Sikes and C. N. Bowman, Biomacromolecules, 2008, 9(1), 355 CrossRef CAS PubMed
- H. D. Sikes, R. Jenison and C. N. Bowman, Lab Chip, 2009, 9(5), 653 RSC
- Y. Liu, Y. Dong, J. Jauw, M. J. Linman and Q. Cheng, Anal. Chem., 2010, 82(9), 3679 CrossRef CAS PubMed
- A. K. Badu-Tawiah, S. Lathwal, K. Kaastrup, M. Al-Sayah, D. C. Christodouleas, B. S. Smith, G. M. Whitesides and H. D. Sikes, Lab Chip, 2015, 15, 655 RSC
- K. Kaastrup and H. D. Sikes, Lab Chip, 2012, 12(20), 4055 RSC
- Y. Wu, S. Liu and L. He, Anal. Chem., 2009, 81(16), 7015 CrossRef CAS PubMed
- Y. Wu, W. Wei and S. Liu, Acc. Chem. Res., 2012, 45(9), 1441 CrossRef CAS PubMed
- Y. Wu, P. Xue, K. M. Hui and Y. Kang, Biosens. Bioelectron., 2014, 52(C), 180 CrossRef CAS PubMed
- A. J. Gormley, R. Chapman and M. M. Stevens, Nano Lett., 2014, 14(11), 6368 CrossRef CAS PubMed
- L. Zhang, W. Zhao, X. Liu, G. Wang, Y. Wang, D. Li, L. Xie, Y. Gao, H. Deng and W. Gao, Biomaterials, 2015, 64(C), 2 CrossRef CAS PubMed
- B. J. Berron, L. M. Johnson, X. Ba, J. D. McCall, N. J. Alvey, K. S. Anseth and C. N. Bowman, Biotechnol. Bioeng., 2011, 108(7), 1521 CrossRef CAS PubMed
- O. W. Webster, Science, 1991, 251(4996), 887 CrossRef CAS PubMed
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed
- T. E. Patten, J. Xia, T. Abernathy and K. Matyjaszewski, Science, 1996, 272(5263), 866 CrossRef CAS PubMed
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015 CrossRef CAS
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136(18), 6513 CrossRef CAS PubMed
- C. Boyer, N. A. Corrigan, K. Jung, D. Nguyen, T.-K. Nguyen, N. N. M. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116(4), 1803 CrossRef CAS PubMed
- A. Simakova, S. E. Averick, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45(16), 6371 CrossRef CAS
- S. Averick, A. Simakova, S. Park, D. Konkolewicz, A. J. D. Magenau, R. A. Mehl and K. Matyjaszewski, ACS Macro Lett., 2012, 1(1), 6 CrossRef CAS
- M. S. Frahn, R. D. Abellon, W. F. Jager, L. H. Luthjens and J. M. Warman, Nucl. Instrum. Methods Phys. Res., Sect. B, 2001, 185, 241 CrossRef CAS
- M. S. Frahn, L. H. Luthjens and J. M. Warman, Polymer, 2003, 44(26), 7933 CrossRef CAS
- M. R. Eftink, T. J. Selva and Z. Wasylewski, Photochem. Photobiol., 1987, 46(1), 23 CrossRef CAS
- M. S. Frahn, R. D. Abellon, L. H. Luthjens, M. J. W. Vermeulen and J. M. Warman, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 208, 405 CrossRef CAS
- F. P. Schwarz and S. P. Wasik, Anal. Chem., 1976, 48(3), 524 CrossRef CAS PubMed
- A. Kellmann, J. Phys. Chem., 1977, 81(12), 1195 CrossRef CAS
- S. P. Kozel, Y. Y. Gotlib, G. I. Lashkov and N. S. Shelekhov, Polym. Sci. U.S.S.R., 1982, 24(7), 1578 CrossRef
- T. Seko, K. Ogura, Y. Kawakami, H. Sugino, H. Toyotama and J. Tanaka, Chem. Phys. Lett., 1998, 291, 438 CrossRef CAS
- H. Feng, Y. Dan and Y. Zhao, Can. J. Chem., 2010, 88(3), 185 CrossRef CAS
- F. Seeliger and K. Matyjaszewski, Macromolecules, 2009, 42(16), 6050 CrossRef CAS
- A. Mohammad Rabea and S. Zhu, Ind. Eng. Chem. Res., 2014, 53(9), 3472 CrossRef CAS
- P. C. Weber, D. H. Ohlendorf, J. J. Wendoloski and F. R. Salemme, Science, 1989, 243, 85 CrossRef CAS PubMed
- A. Chilkoti and P. S. Stayton, J. Am. Chem. Soc., 1995, 117(43), 10622 CrossRef CAS
- A. Holmberg, A. Blomstergren, O. Nord, M. Lukacs, J. Lundeberg and M. Uhlén, Electrophoresis, 2005, 26(3), 501 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, including spectroscopic and analytical data and supplemental figures are provided. See DOI: 10.1039/c8sc03938k |
| This journal is © The Royal Society of Chemistry 2019 |