DOI:
10.1039/C8SC04279A
(Edge Article)
Chem. Sci., 2019,
10, 1780-1785
Ni-catalysed reductive arylalkylation of unactivated alkenes†
Received
26th September 2018
, Accepted 3rd December 2018
First published on 4th December 2018
Abstract
In this protocol Ni-catalysed reductive arylalkylation of unactivated alkenes tethered to aryl bromides with primary alkyl bromides has been accomplished, providing a new path to construct diverse benzene-fused carbo- and heterocyclic cores including indanes, tetrahydroisoquinolines, indolines and isochromanes. Notably, this new method circumvents the pregeneration of organometallics and demonstrates high tolerance to a wide range of functional groups. The preliminary mechanistic investigations suggest a reaction pathway with an intermediate reduction.
Introduction
In recent years, transition-metal catalyzed dicarbo-functionalisation of unactivated alkenes has gained increasing interest in the organic community, because simple olefin precursors can be converted into structurally more complex molecules in one single step with the formation of two C–C bonds. Significant progress has been achieved in this area using both redox-neutral1–3 and reductive strategies.4,5 For instance, aryl organometallics containing a pendant olefinic unit were successfully reacted with diverse alkyl or aryl halides as electrophiles under redox-neutral conditions to construct a series of carbo- and heterocyclic cores, such as indanes, dihydrobenzofurans and indolines (Scheme 1A).2b–d However, the use of pregenerated organometallics is less desirable from the viewpoint of step economy and functional group tolerance. In contrast, through reductive dicarbofunctionalisation two different alkyl or aryl electrophiles can be directly installed across the C–C double bonds under mild reaction conditions. Although a few examples of reductive dicarbofunctionalisation have been reported,4,5 application of this reductive strategy in a two-component reaction to prepare benzene-fused cyclic compounds is still elusive. In this protocol we report Ni-catalyzed reductive arylalkylation of tethered olefins with various primary alkyl bromides providing a path for benzene-fused cyclic compounds, such as indanes, tetrahydro-isoquinolines, indolines and isochromanes, which are characteristic motifs in numerous biologically active compounds6 (Scheme 1B).
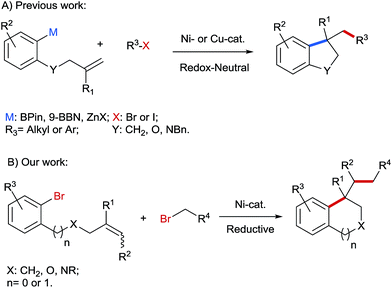 |
| | Scheme 1 Redox-neutral (A) and reductive (B) arylalkylation of tethered alkenes for the synthesis of benzene-fused cyclic compounds. | |
Results and discussion
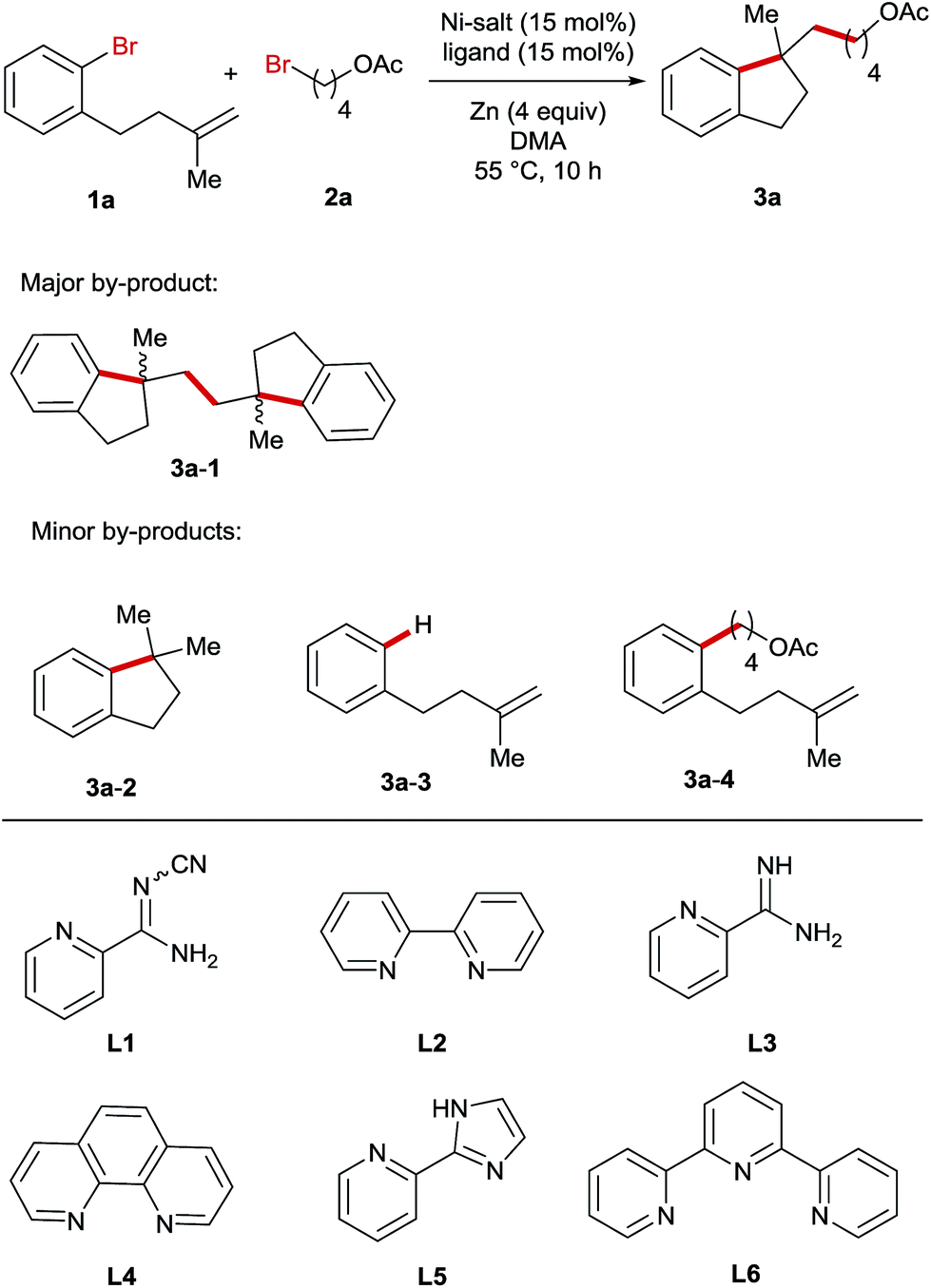
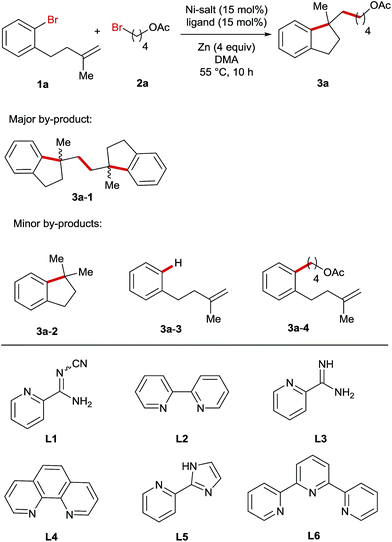
For the optimization of reaction conditions, we used bromobenzene (1a) tethering a terminal olefinic unit and 4-bromobutyl acetate (2a) as the standard substrate (Table 1). Systematic screening of the reaction parameters provided the optimum conditions using NiBr2 as a catalyst, L1 as a ligand, DMA as a solvent and Zn as a reducing agent at 55 °C (entry 1). Generally, all the reactions delivered a dimer compound 3a-1 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio as the major by-product and its yields are also shown in the table (entries 2–23). Furthermore, the formation of a low amount of reductive Heck product 3a-2, debromination product 3a-3 and cross-coupling product 3a-4 was also observed. In comparison, replacing either or both of these bromide precursors with their iodo-analogues gave rise to increasing amounts of the reductive Heck product 3a-2 in the product mixtures (entries 2–4). The use of other pyridine-based ligands L2–L6 resulted in lower reaction efficiency (entries 5–9). Ni-salt screening indicated that the studied reactions could be promoted by both Ni(II)- and Ni(0)-catalysts (entries 10–13). Performing the reaction in other polar solvents including DMF and NMP led to decreased yields (entries 14 and 15), whereas the reaction was completely shut down when using THF or MeCN as the solvent (entries 16 and 17). The reaction employing Mn instead of Zn as a reducing agent yielded the product in a reduced yield (entry 18). In addition, the temperature impact on this reaction was also investigated and both raising and lowering the reaction temperature had a detrimental effect on the reaction efficiency (entries 19 and 20). Moreover, reducing the amount of alkyl bromide 4, NiBr2 or Zn-powder all resulted in lower yields (entries 21–23).
:1 diastereomeric ratio as the major by-product and its yields are also shown in the table (entries 2–23). Furthermore, the formation of a low amount of reductive Heck product 3a-2, debromination product 3a-3 and cross-coupling product 3a-4 was also observed. In comparison, replacing either or both of these bromide precursors with their iodo-analogues gave rise to increasing amounts of the reductive Heck product 3a-2 in the product mixtures (entries 2–4). The use of other pyridine-based ligands L2–L6 resulted in lower reaction efficiency (entries 5–9). Ni-salt screening indicated that the studied reactions could be promoted by both Ni(II)- and Ni(0)-catalysts (entries 10–13). Performing the reaction in other polar solvents including DMF and NMP led to decreased yields (entries 14 and 15), whereas the reaction was completely shut down when using THF or MeCN as the solvent (entries 16 and 17). The reaction employing Mn instead of Zn as a reducing agent yielded the product in a reduced yield (entry 18). In addition, the temperature impact on this reaction was also investigated and both raising and lowering the reaction temperature had a detrimental effect on the reaction efficiency (entries 19 and 20). Moreover, reducing the amount of alkyl bromide 4, NiBr2 or Zn-powder all resulted in lower yields (entries 21–23).
Table 1 Variation of the reaction parameters for the Ni-catalysed reductive arylalkylation reactiona
|
|
| Entry |
Variation from the optimum conditions |
Yield 3ab (%) |
Yield: 3a-1b (%) |
|
Unless otherwise specified, reactions were performed on a 0.2 mmol scale of aryl bromide 1a with 2 equiv. bromobutyl acetate (2a), 15 mol% NiBr2, 15 mol% ligand L1 and 4 equiv. Zn as the reductant in 0.5 mL DMA at 55 °C for 10 h.
GC-yields using n-dodecane as an internal standard.
Yield of the isolated product.
Yield of 3a-2.
|
| 1 |
None |
69 (67c) |
9 |
| 2 |
Iodo-analogue of 1a used |
32 |
19 (8d) |
| 3 |
Iodo-analogue of 2a used |
10 |
18 (31d) |
| 4 |
Both iodo-analogues of 1a and 2a used |
17 |
10 (30d) |
| 5 |
L2 instead of L1 |
30 |
49 |
| 6 |
L3 instead of L1 |
45 |
2 |
| 7 |
L4 instead of L1 |
54 |
13 |
| 8 |
L5 instead of L1 |
32 |
50 |
| 9 |
L6 instead of L1 |
0 |
0 |
| 10 |
NiI2 instead of NiBr2 |
61 |
8 |
| 11 |
Ni(OTf)2 instead of NiBr2 |
32 |
4 |
| 12 |
NiBr2·glyme instead of NiBr2 |
63 |
8 |
| 13 |
Ni(COD)2 instead of NiBr2 |
63 |
7 |
| 14 |
DMF instead of DMA |
43 |
3 |
| 15 |
NMP |
68 |
8 |
| 16 |
THF instead of DMA |
0 |
0 |
| 17 |
MeCN instead of DMA |
0 |
0 |
| 18 |
Mn instead of Zn |
51 |
17 |
| 19 |
75 °C instead of 55 °C |
62 |
8 |
| 20 |
35 °C instead of 55 °C |
27 |
2 |
| 21 |
1.2 equiv. 2a used |
50 |
13 |
| 22 |
10 mol% NiBr2 used |
63 |
13 |
| 23 |
2 equiv. Zn used |
50 |
7 |
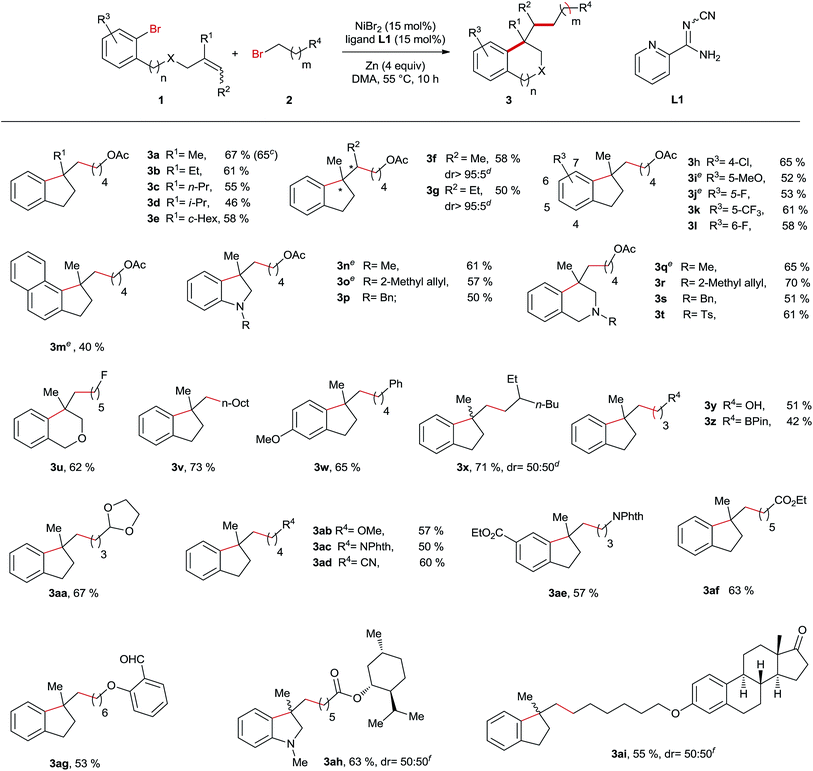
After establishing the best reaction conditions, we started to evaluate the substrate spectrum of this Ni-catalyzed reductive arylalkylation reaction by varying the structure of both pendant alkenes 1 and alkyl bromides 2 (Table 2). First, we studied the influence of the alkene substitution pattern on the outcome of this reaction. In the case of disubstituted terminal olefins all the reactions provided the products 3a–e in moderately good yields. Remarkably, the reactions employing 1,1,2-trisubstituted alkenes also proceeded smoothly under the optimum reaction conditions yielding the products 3f and 3g in excellent diastereoselectivities, although the E/Z ratios of the alkene precursors are nearly 1:1. The high diastereocontrol indicates that this Ni-catalyzed reaction is probably not initiated by the radical addition of the alkyl group to the C–C double bond. When 1,2-disubstituted and monosubstituted alkenes were used as substrates, no desired products were obtained due to the high tendency to undergo the Heck reaction. Next, the examination of the substituent effect on the phenyl ring was undertaken. To our delight, all the substrates bearing electron-withdrawing or donating groups turned out to be suitable substrates providing the corresponding products 3h–m, 3w and 3ae in moderate to good yields. Furthermore, our method is not limited to the synthesis of indane derivatives. A series of indolines 3n–p and 3ah and tetrahydroisoquinolines 3q–t and an isochromane 3u were also successfully prepared through this Ni-catalysed reaction. Subsequently, diverse primary alkyl bromides were reacted with various pendant olefins. Of note is that this Ni-catalysed reaction demonstrates the high compatibility of a wide range of functional moieties including alcohol (3y), boronate (3z), acetal (3aa), imide (3ac and 3ae), nitrile (3ad), ester (3af and 3ah), aldehyde (3ag) and ketone (3ai). Moreover, the reaction using 1a and 2a was performed on a one-gram scale still furnishing the product 3a in a 65% yield with 5 mol% catalyst loading. A limitation of this method was observed in the case of secondary and tertiary bromides, which failed to yield the arylalkylation products.
Table 2 Evaluation of the substrate scope of the Ni-catalysed reductive arylalkylation reactiona,b
|
Unless otherwise specified, reactions were performed on a 0.4 mmol scale of aryl bromides 1 with 2 equiv. alkyl bromides 2, 15 mol% NiBr2, 15 mol% ligand L1 and 4 equiv. Zn as the reductant in 1.0 mL DMA at 55 °C for 10 h.
Yields of the isolated products.
Reaction was performed on a 1 g scale using 5 mol% NiBr2 and 5 mol% ligand L1 at 65 °C for 12 h.
Determined by 13C-NMR-spectroscopy.
Reactions were performed at 70 °C.
Determined by HPLC-analysis.
|
|
A series of control experiments were carried out to explore the mechanism of this Ni-catalyzed ring opening reaction. First, we reacted Zn-powder with both bromide precursors under the standard reaction conditions and the results indicated that no organozincs were formed in the reaction mixture (Scheme 2). Consequently, the Negishi coupling reaction pathway is less likely for the studied reaction.
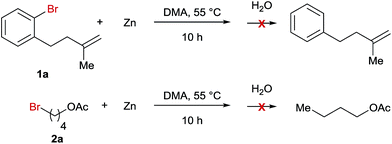 |
| | Scheme 2 Stoichiometric reactions of bromides 1a and 2a with Zn-powder. | |
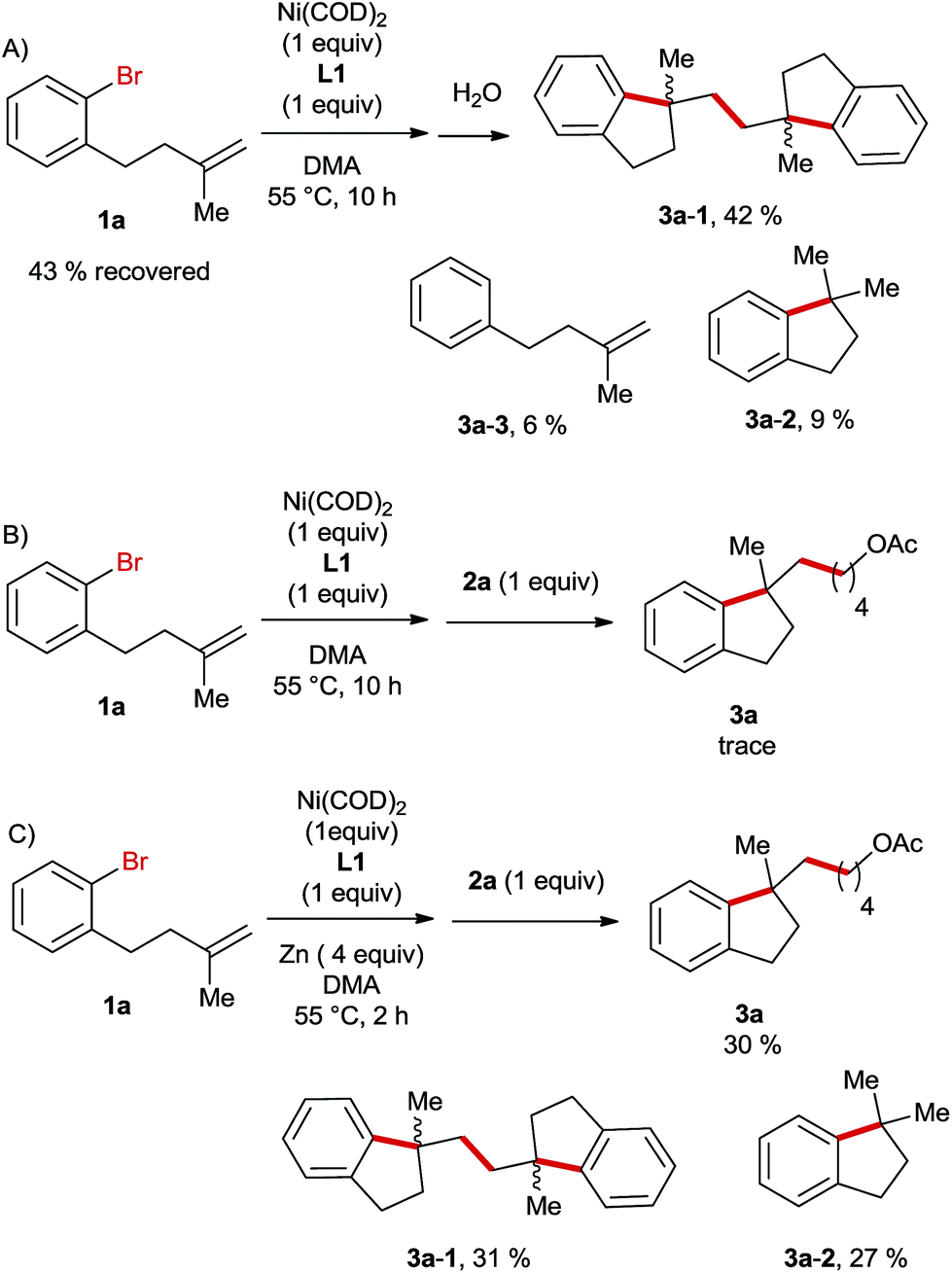
Next, we carried out a stoichiometric reaction between Ni(COD)2 and the aryl bromide 1a. After 10 h the reaction was quenched with water and it turned out that nearly half of the starting material was recovered and the major product was the dimer 3a-1 (Scheme 3A). If the alkyl bromide 2a was added to the reaction mixture instead of water, only traces of the desired product 3a were formed in this case (Scheme 3B). In contrast, the sequential stoichiometric reactions of Ni(COD)2 with 1a and 2a in the presence of Zn furnished the product 3a in a 30% yield (Scheme 3C). These results suggest that Zn is required in a step of an intermediate reduction instead of serving as a terminal reductant in this Ni-catalysed reaction, which is likely initiated by the oxidative addition of aryl bromide to a Ni(0)-species followed by an intramolecular migratory insertion. A similar process was reported very recently by Kong et al. in Ni-catalysed reductive diarylation of activated olefins.5
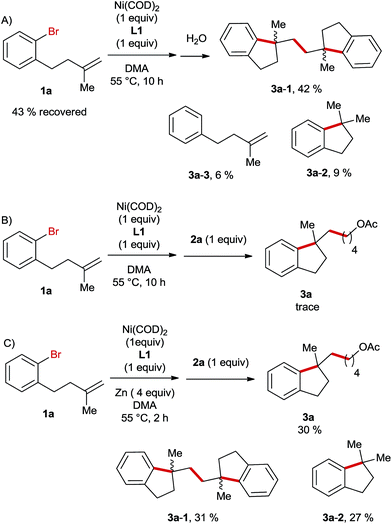 |
| | Scheme 3 Stoichiometric control experiments for the Ni-catalyzed reductive arylalkylation. | |
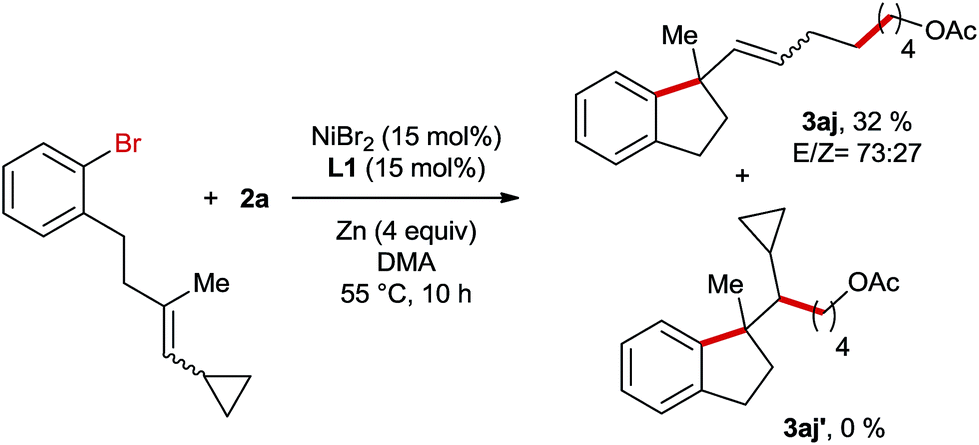
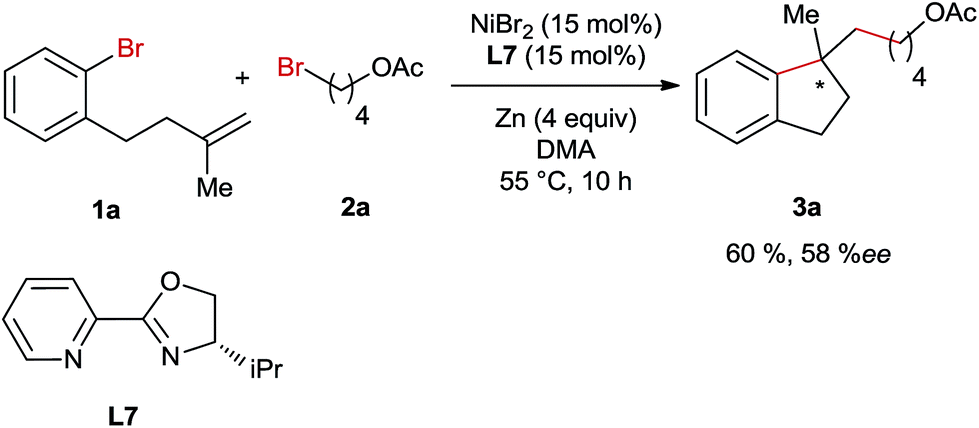
Subsequently, we conducted the reaction using TEMPO as a radical scavenger and in this case the reaction was completely shut down revealing that a radical species might be involved in the key step of this reaction. This is not surprising since alkyl bromide can easily form radicals in Ni-catalyzed reductive coupling reactions according to numerous reports in the literature.7,8 However, it is unknown whether the migratory insertion of Ni–Ar into the C–C double bond proceeds through a radical pathway or not under reductive conditions. In order to gain more insights into the mechanism of this reaction, a radical clock experiment employing a cyclopropyl-substituted alkene as a substrate was conducted (Scheme 4). In this case a ring opening product 3aj was obtained in 32% yield, whereas the formation of unrearranged product 3aj′ was not observed. This result could be rationalized by cyclisation involving an aryl radical, which is generated through the interaction of aryl bromide with the Ni-catalyst. Alternatively, the initial migratory insertion could be non-radical, but the resultant Ni-alkyl species after cyclisation might undergo homolytic Ni–alkyl bond cleavage, affording the same C-centered radical as the one generated in the aryl-radical-mediated ring closure. Moreover, the high diastereoselectivities of 3f and 3g also support the formation of this alkyl radical; otherwise similar diastereomeric ratios would be obtained to their trisubstituted alkene-precursors. To differentiate the two possible pathways mentioned above we conducted Ni-catalysed reductive arylalkylation employing a chiral oxazoline ligand (Scheme 5). In this case the product 3a was obtained with a moderate enantioselectivity. This report clearly excludes the possibility of radical-mediated cyclisation, which is supposed to provide the product as a racemic mixture.
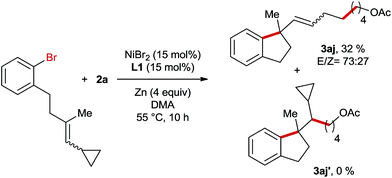 |
| | Scheme 4 Radical clock experiment for the Ni-catalyzed reductive arylalkylation. | |
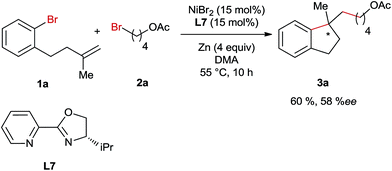 |
| | Scheme 5 Ni-catalysed arylalkylation employing a chiral oxazoline ligand. | |
Based on the aforementioned experimental results we proposed the following plausible mechanism for this Ni-catalyzed reaction (Scheme 6). Initially, under the reductive reaction conditions a Ni(0)-species I is generated, which undergoes oxidative addition with aryl bromides 1 to afford a Ni(II) complex II. Next, the ring closure is accomplished via an intramolecular non-radical migratory insertion. The generated Ni(II) species III stays in equilibrium with an alkyl radical IV and Ni(I)LnBr before Zn-mediated reduction to the Ni(I) species V. The subsequent oxidative addition of alkyl bromides 2 involves the formation of a cage VI and the following recombination provides a Ni(III) intermediate VII. Finally, the reductive elimination of the Ni(III) complex VII furnishes the products 3 and the Ni(I) species VIII, which is subsequently reduced to the Ni(0)-species I for the next catalytic cycle.
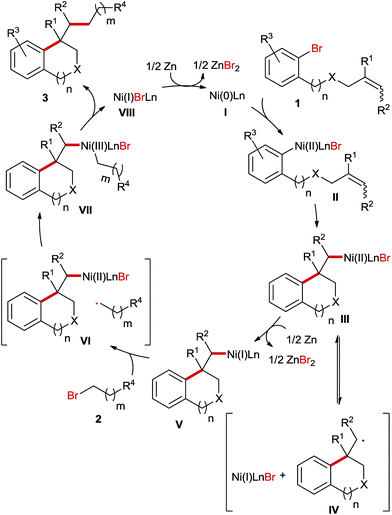 |
| | Scheme 6 Proposed mechanism of the Ni-catalyzed reductive arylalkylation. | |
Conclusions
In conclusion, we have developed Ni-catalysed reductive arylalkylation of unactivated alkenes tethered to aryl bromides with an array of primary alkyl bromides, providing a new path to synthesize benzene-fused cyclic compounds such as indanes, tetrahydroisoquinolines, indolines and isochromanes with an all-carbon-stereogenic center. This new method is distinguished by avoidance of the use of pregenerated organometallics, high tolerance of a broad range of functional moieties and base-free reaction conditions. The preliminary mechanistic studies indicate that this Ni-catalysed reaction proceeds in a reaction pathway with an intermediate reduction. Further investigations into the asymmetric version of this reaction are in progress and will be published in due course.
Conflicts of interest
There is no conflict to declare.
Acknowledgements
This work is supported by the “1000-Youth Talents Plan” starting up funding, National Natural Science Foundation of China (Grant No. 21772183), the Fundamental Research Funds for the Central Universities (WK2060190086), and the University of Science and Technology of China.
Notes and references
- For a review on redox-neutral dicarbofunctionalisation of unactivated alkenes, see: S. KC and R. Giri, J. Org. Chem., 2018, 83, 3013 CrossRef PubMed.
- For selected examples on two-component redox-neutral dicarbofunctionalisation of unactivated alkenes see:
(a) V. B. Phapale, E. Buñuel, M. García-Iglesias and D. J. Cárdenas, Angew. Chem., Int. Ed., 2007, 46, 8790 CrossRef CAS PubMed;
(b) H. Cong and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 3788 CrossRef CAS PubMed;
(c) W. You and M. K. Brown, J. Am. Chem. Soc., 2014, 136, 14730 CrossRef CAS PubMed;
(d) S. Thapa, P. Basnet and R. Giri, J. Am. Chem. Soc., 2017, 139, 5700 CrossRef CAS PubMed;
(e) S. KC, P. Basnet, S. Thapa, B. Shrestha and R. Giri, J. Org. Chem., 2018, 83, 2920 CrossRef CAS PubMed.
- For selected examples on three-component redox-neutral dicarbofunctionalisation of unactivated alkenes see:
(a) K. Mizutani, H. Shinokubo and K. Oshima, Org. Lett., 2003, 5, 3959 CrossRef CAS PubMed;
(b) J. Terao, Y. Kato and N. Kambe, Chem.–Asian J., 2008, 3, 1472 CrossRef CAS PubMed;
(c) B. Shrestha, P. Basnet, R. K. Dhungana, S. KC, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653 CrossRef CAS PubMed;
(d) J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657 CrossRef CAS PubMed;
(e) J. Derosa, V. A. van der Puy, V. T. Tran, M. Liu and K. M. Engle, Chem. Sci., 2018, 9, 5278 RSC;
(f) S. KC, R. K. Dhungana, B. Shrestha, S. Thapa, N. Khanal, P. Basnet, R. W. Lebrun and R. Giri, J. Am. Chem. Soc., 2018, 140, 9801 CrossRef CAS PubMed;
(g) P. Gao, L.-A. Chen and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 10653 CrossRef CAS PubMed;
(h) W. Li, J. W. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600 RSC.
-
(a) Y. Peng, C.-S. Yan, X.-B. Xu and Y.-W. Wang, Chem.–Eur. J., 2012, 18, 6039 CrossRef PubMed;
(b) Y. Peng, X.-B. Xu, J. Xiao and Y.-W. Wang, Chem. Commun., 2014, 50, 472 RSC;
(c) Y. Peng, J. Xiao, X.-B. Xu, S.-M. Duan, L. Ren, Y.-L. Shao and Y.-W. Wang, Org. Lett., 2016, 18, 5170 CrossRef CAS PubMed;
(d) Y.-L. Kuang, X.-F. Wang, D. Anthony and T.-N. Diao, Chem. Commun., 2018, 54, 2558 RSC;
(e) J. Xiao, X.-W. Cong, G.-Z. Yang, Y.-W. Wang and Y. Peng, Org. Lett., 2018, 20, 1651 CrossRef CAS PubMed;
(f) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835 CrossRef PubMed.
- Very recently, Kong et al. reported Ni-catalysed reductive diarylation of activated alkenes: K. Wang, Z. Ding, Z. Zhou and W. Kong, J. Am. Chem. Soc., 2018, 140, 12364 CrossRef CAS PubMed.
-
(a) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Chem. Biol., 2010, 14, 347 CAS;
(b) J. Lee, H. Kim, T. G. Lee, I. Yang, D. H. Won, H. Choi, S.-J. Nam and H. Kang, J. Nat. Prod., 2014, 77, 1528 CrossRef CAS PubMed;
(c) V. Vecchietti, G. D. Clark, R. Colle, G. Giardina, G. Petrone and S. Sbacchi, J. Med. Chem., 1991, 34, 2624 CrossRef CAS PubMed;
(d) P. G. Baraldi, R. Makaeva, M. G. Pavani, M. d. C. Nunez, G. Spalluto, S. Moro, S. Falzoni, F. Di Virgilio and R. Romagnoli, Arzneim. Forsch., 2002, 52, 273 CAS;
(e) S. Zeeli, T. Weill, E. Finkin-Groner, C. Bejar, M. Melamed, S. Furman, M. Zhenin, A. Nudelman and M. Weinstock, J. Med. Chem., 2018, 61, 4004 CrossRef CAS PubMed;
(f) M. Yamamoto, K. Hashigaki, K. Hiromatsu and K. Tasaka, Chem. Pharm. Bull., 1983, 31, 521 CrossRef.
- For detailed mechanistic studies on cross-electrophile coupling involving aryl halides and alkyl halides, see: S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192 CrossRef CAS PubMed.
- For reviews on cross-electrophile coupling, see:
(a) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793 CrossRef CAS PubMed;
(b) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411 RSC;
(c) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767 CrossRef CAS PubMed;
(d) E. Richmond and J. Moran, Synthesis, 2018, 50, 499 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectral data, NMR-data, and HPLC-data. See DOI: 10.1039/c8sc04279a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*