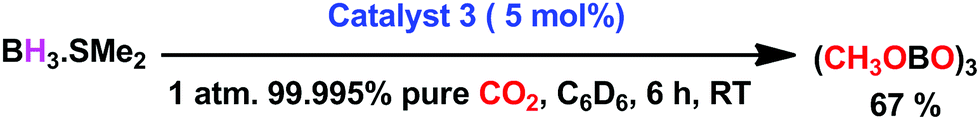DOI:
10.1039/C8SC03581D
(Edge Article)
Chem. Sci., 2019,
10, 1879-1884
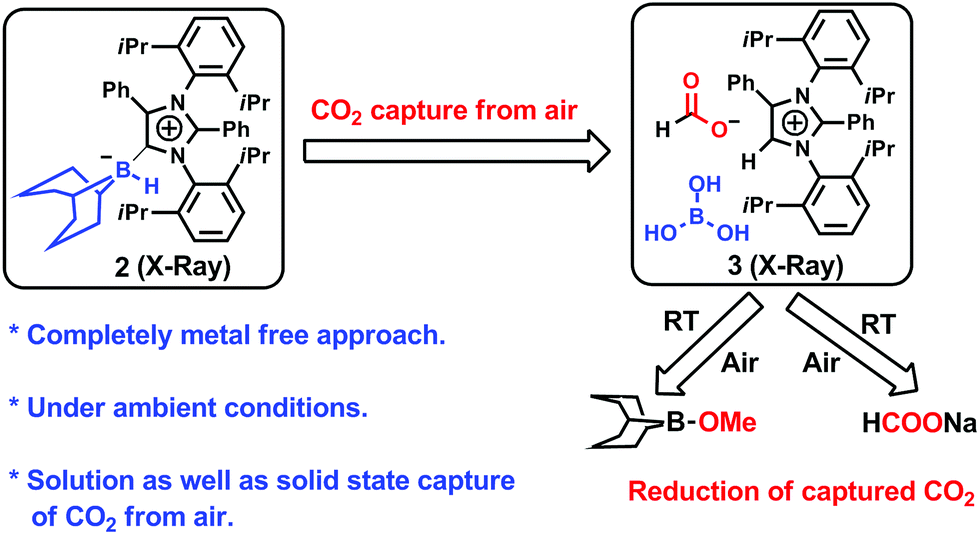
Transforming atmospheric CO2 into alternative fuels: a metal-free approach under ambient conditions†
Received
12th August 2018
, Accepted 29th November 2018
First published on 30th November 2018
Abstract
This work demonstrates the first-ever completely metal-free approach to the capture of CO2 from air followed by reduction to methoxyborane (which produces methanol on hydrolysis) or sodium formate (which produces formic acid on hydrolysis) under ambient conditions. This was accomplished using an abnormal N-heterocyclic carbene (aNHC)–borane adduct. The intermediate involved in CO2 capture (aNHC-H, HCOO, B(OH)3) was structurally characterized by single-crystal X-ray diffraction. Interestingly, the captured CO2 can be released by heating the intermediate, or by passing this compound through an ion-exchange resin. The capture of CO2 from air can even proceed in the solid state via the formation of a bicarbonate complex (aNHC-H, HCO3, B(OH)3), which was also structurally characterized. A detailed mechanism for this process is proposed based on tandem density functional theory calculations and experiments.
Introduction
Carbon dioxide is an attractive, economical, and renewable C1 source for the production of value added chemicals and fuels.1–5 Many fuel-related C1 products, such as methane (CH4), methanol (CH3OH) and formic acid (HCOOH), can be obtained by treating CO2 with a reducing agent as a hydride source.6 Among the various possible CO2 hydrogenation products, CH3OH and formic acid are the most attractive candidates. Methanol can be used as a direct replacement for gasoline in internal combustion engines and also in direct methanol fuel cells.7 In addition, formic acid has applications in the textile industry in dyeing processes, as a food preservative and as a hydrogen storage material.8–10 During the last 20 years, numerous reports have described the reduction of CO2 by various transition metal based compounds.11–17 The reduction was also accomplished catalytically without a transition metal based compound18 or by direct reduction with NaBH4 without requiring any catalyst.19 The reduction of CO2 has been achieved even with metal-free catalysts.20–25 These reports utilized commercially available pure CO2 gas from a cylinder. However, it remains difficult to capture CO2 present in the ambient atmosphere at extremely low concentrations and to perform reduction reactions to obtain fuels under ambient conditions. Only a very few compounds26–32 have demonstrated selective CO2 absorption from air, but these compounds are not capable of reducing the captured CO2 to generate methanol. Recently, Olah and coworkers combined two compounds with the aim of capturing CO2 from air followed by reduction into methanol. However, this process required the use of a rare and expensive Ru-based species at 155 °C under harsh reaction conditions (50 bar H2 pressure).33 At present, there are no methods for the capture of CO2 from the atmosphere and the consecutive reduction of this CO2 to produce methoxyborane (which can be hydrolyzed to methanol) under ambient conditions using a single chemical system without any metal-based compounds. Moreover, there is currently only a very limited understanding of the mechanistic pathways for such key chemical transformations based on isolating and characterizing various intermediates. Density functional theory (DFT) calculations along with several analyses, including structure determination by single-crystal X-ray diffraction (SC-XRD), were employed in this work to gain better insights into the mechanistic details of the conversion. The present study describes metal-free capture of CO2 from air and its reduction under ambient conditions (Fig. 1) using an abnormal N-heterocyclic carbene.
 |
| | Fig. 1 Metal-free CO2 fixation from air followed by reduction to methoxyborane or sodium formate under ambient conditions. | |
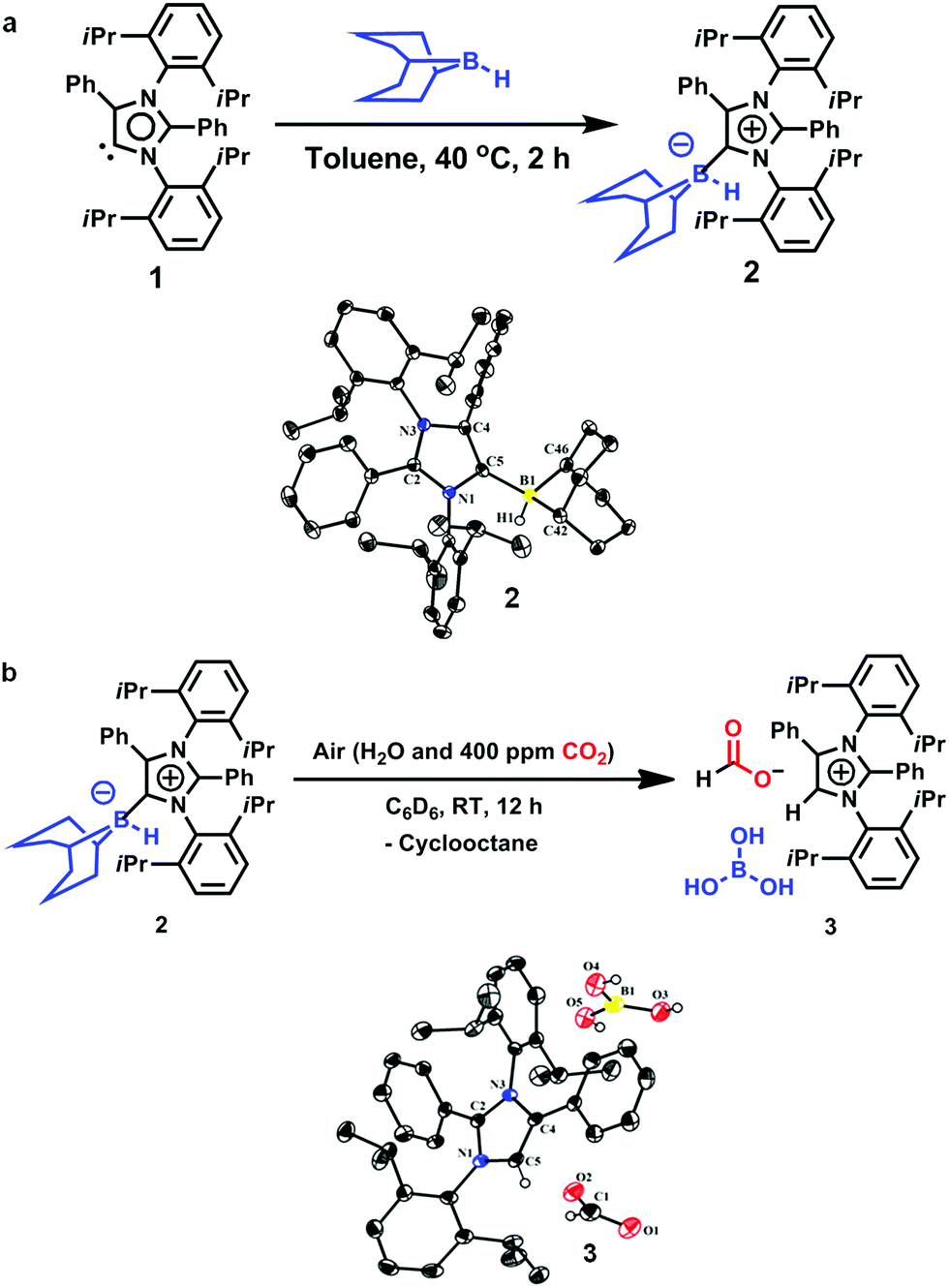
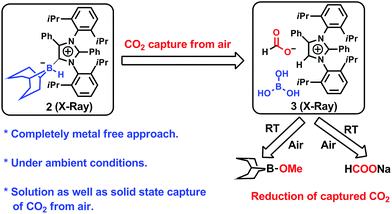
An abnormal N-heterocyclic carbene (aNHC; 1) was prepared according to a literature procedure,34 following which the aNHC–9BBN adduct, 2, was synthesized in a 75% isolated yield by reacting 1 and 9-BBN in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equivalent ratio in toluene at 40 °C for 2 h (Fig. 2a). Because a higher yield and crystals suitable for XRD analysis were obtained, this reaction protocol represents an improvement over the previously reported procedure by Crudden and coworkers using an aNHC salt, in which the XRD structural analysis of the adduct 2 was not carried out.35 Crystals of 2 suitable for XRD analysis were successfully grown from a toluene/hexane mixture under an inert atmosphere and the resulting structure is presented in Fig. 2a. When a solution of 2 in deuterated benzene was left exposed to the ambient air overnight, a distinct color change from light yellow to green was observed. The formation of a new product (3, Fig. 2b) was also confirmed by 1H NMR spectroscopy. The conversion of the original compound during this process was 66% and the isolated crystal yield of 3 was 40%. The 1H NMR spectrum of 3 in deuterated chloroform showed two singlets at δ = 8.53 and 8.55 ppm with a 1:1 signal intensity ratio, attributed to the formate anion36 and C5–H of the azolium cation,37 respectively. The presence of formate anions indicated that CO2 in the ambient air may have been fixed during the transformation to provide these species. The 13C NMR spectrum of 3 contains signals at δ = 169.2 ppm (assigned to the C
:1 equivalent ratio in toluene at 40 °C for 2 h (Fig. 2a). Because a higher yield and crystals suitable for XRD analysis were obtained, this reaction protocol represents an improvement over the previously reported procedure by Crudden and coworkers using an aNHC salt, in which the XRD structural analysis of the adduct 2 was not carried out.35 Crystals of 2 suitable for XRD analysis were successfully grown from a toluene/hexane mixture under an inert atmosphere and the resulting structure is presented in Fig. 2a. When a solution of 2 in deuterated benzene was left exposed to the ambient air overnight, a distinct color change from light yellow to green was observed. The formation of a new product (3, Fig. 2b) was also confirmed by 1H NMR spectroscopy. The conversion of the original compound during this process was 66% and the isolated crystal yield of 3 was 40%. The 1H NMR spectrum of 3 in deuterated chloroform showed two singlets at δ = 8.53 and 8.55 ppm with a 1:1 signal intensity ratio, attributed to the formate anion36 and C5–H of the azolium cation,37 respectively. The presence of formate anions indicated that CO2 in the ambient air may have been fixed during the transformation to provide these species. The 13C NMR spectrum of 3 contains signals at δ = 169.2 ppm (assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of formate) and 125.4 ppm (the C5 of azolium), which further substantiates the incorporation of a formate anion in 3.24,36 The fate of the B atom in the 9-BBN backbone of 3 during this transformation was determined using 11B NMR spectroscopy, which showed a singlet at δ = 20.8 ppm, attributed to free boric acid {B(OH)3}. Based on these data, a structure for 3 was developed (Fig. 2b).This compound was successfully crystallized into colorless crystals from a benzene/n-hexane mixture in open atmosphere. Single-crystal XRD analysis confirmed the proposed structure (Fig. 2b), in which a CO2 molecule is fixed as a formate anion by reaction with the aNHC–borane adduct 2.
O of formate) and 125.4 ppm (the C5 of azolium), which further substantiates the incorporation of a formate anion in 3.24,36 The fate of the B atom in the 9-BBN backbone of 3 during this transformation was determined using 11B NMR spectroscopy, which showed a singlet at δ = 20.8 ppm, attributed to free boric acid {B(OH)3}. Based on these data, a structure for 3 was developed (Fig. 2b).This compound was successfully crystallized into colorless crystals from a benzene/n-hexane mixture in open atmosphere. Single-crystal XRD analysis confirmed the proposed structure (Fig. 2b), in which a CO2 molecule is fixed as a formate anion by reaction with the aNHC–borane adduct 2.
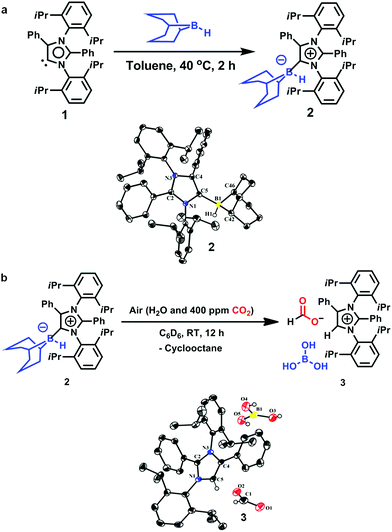 |
| | Fig. 2 Fixation of CO2 in air. (a) Synthesis of an abnormal N-heterocyclic carbene–9-borabicyclo(3.3.1)nonane adduct (aNHC–9BBN, 2) and an ORTEP drawing of the molecular structure of 2. (b) Fixation of CO2 from air with the formation of 3 and an ORTEP drawing of 3. | |
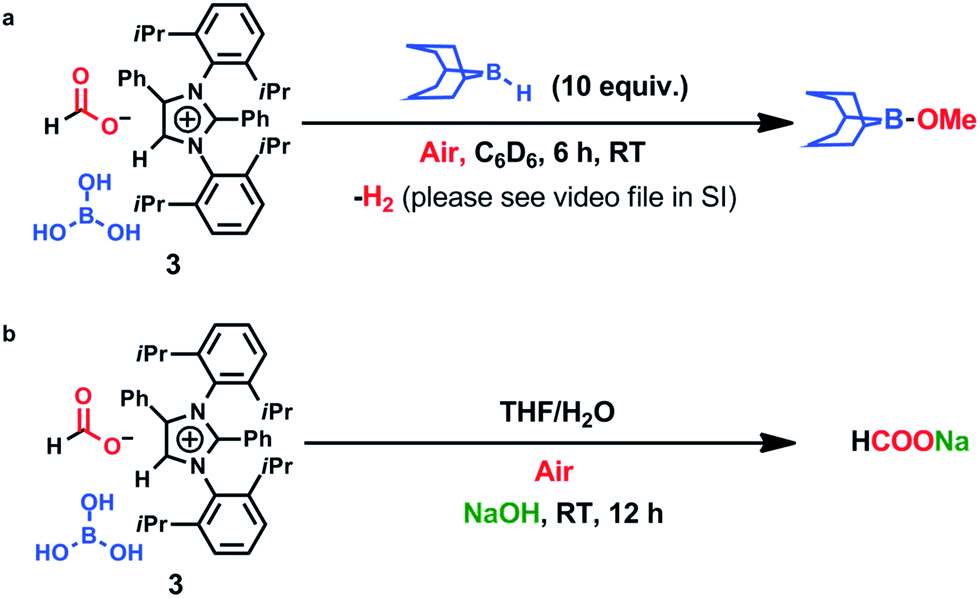
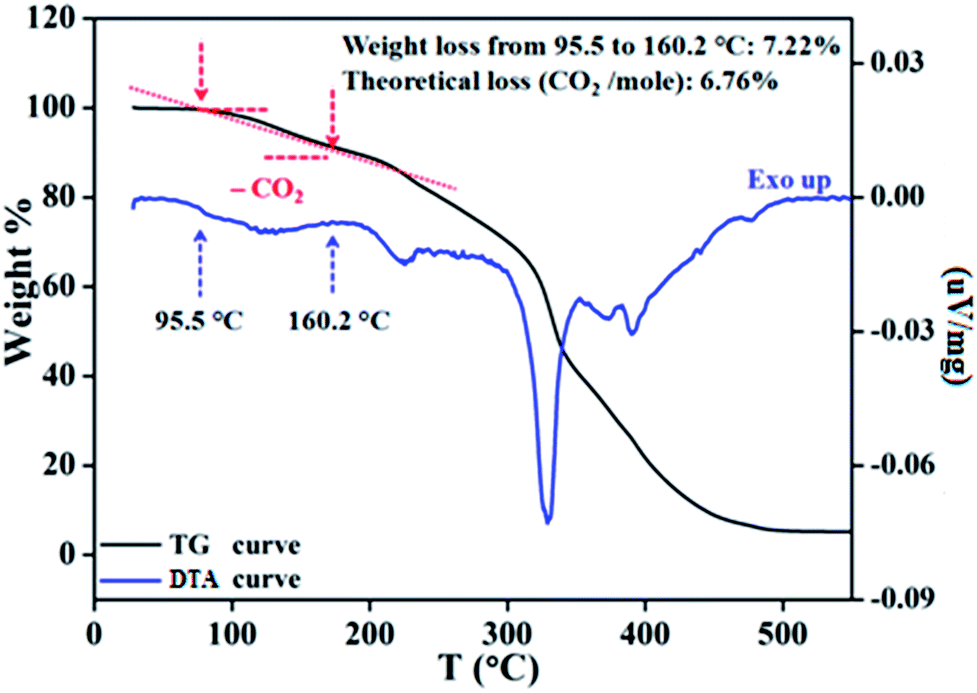
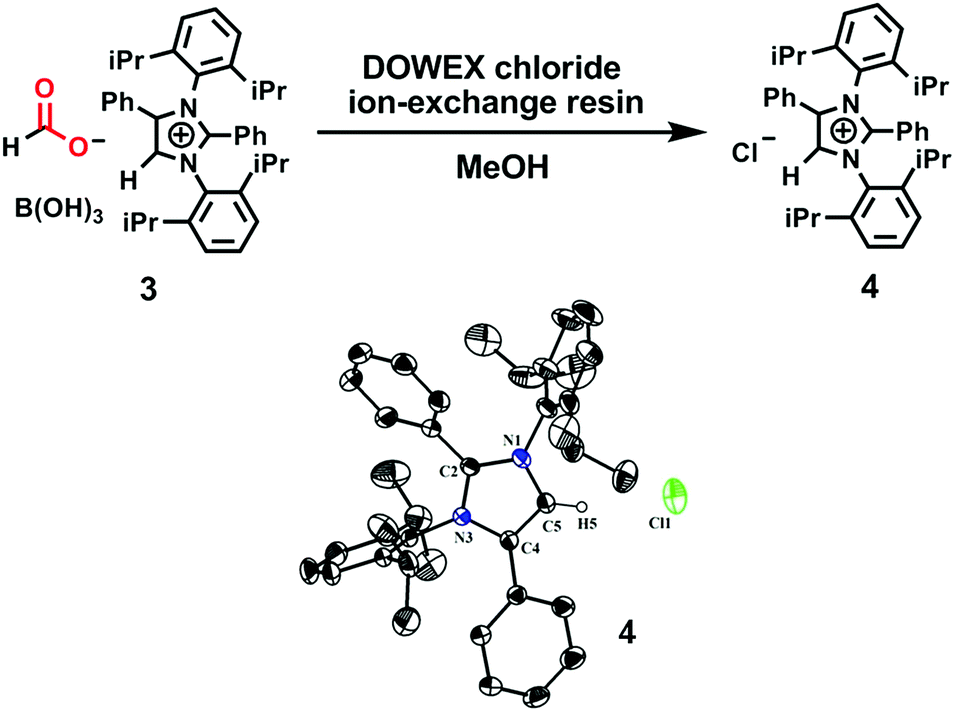
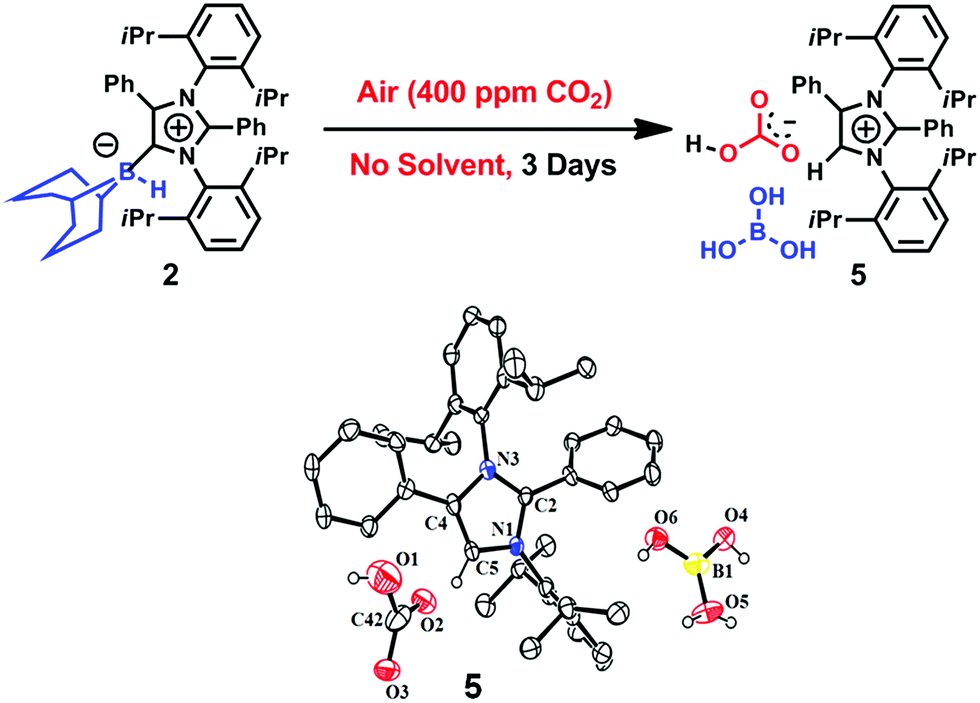
To ascertain the reactivity of the captured CO2, we treated 3 with a boron hydride. Upon reaction of 3 with 10 equiv. of the hydroborane 9-BBN in the presence of air, the corresponding methoxyborane product (CH3OBBN) was obtained with complete consumption of 3 within 6 h in deuterated benzene (Scheme 1a, methoxyborane). The formation of CH3OBBN was confirmed by 1H NMR (δ = 3.44 ppm) and 11B NMR (δ = 57 ppm) spectroscopy in deuterated benzene and by comparing our results to previous literature data.20 During the course of the reaction, gas evolution was observed (see the video file in the ESI†), which was confirmed to be H2 by 1H NMR spectroscopy (δ = 4.47 ppm).38 Additionally, upon introducing dry CO2 from a cylinder (99.995% pure) and performing the same reaction under moisture-free pure CO2 instead of air, 3 exhibited catalytic activity, which may be explained based on a mechanism previously proposed by our group.24 Under these conditions, CO2 was catalytically converted to the corresponding trimethoxyboroxine21,39 with 67% conversion, using a low cost borane (BH3·SMe2) as the hydride source (Scheme 2). The feasibility of applying 3 to the synthesis of formic acid via the formation of sodium formate was also assessed. Adding 3 to 5 mL of a 2 M sodium hydroxide solution and 0.5 mL tetrahydrofuran at room temperature in air resulted in complete consumption of the compound (Scheme 1b, sodium formate). The formation of sodium formate (HCOONa) was confirmed by 1H NMR spectroscopy (δ = 8.38 ppm in deuterated water) based on previous literature data.11 We subsequently attempted to remove the CO2 captured in 3 by heating. Raising the temperature of 3 to 150 °C for 12 h in the solid state was found to promote the loss of the formate anion, leading to an abrupt color change from colorless to brown, and forming a new product as determined by 1H NMR spectroscopy.40,41 As noted previously, 3 showed two singlets in its 1H NMR spectrum at δ = 8.53 and 8.55 ppm with a 1:1 intensity ratio in deuterated chloroform. After heating 3 at 150 °C for 12 h, the 1H NMR spectrum of the resulting compound contained only a new singlet (δ = 8.35 ppm) while the other original singlet was absent. This observation indicates that the formate anion might have been lost during the heating process. To further support this assumption, a 13C NMR spectroscopic experiment was performed. Following heating, the singlet in the downfield region of the 13C NMR spectrum acquired in deuterated chloroform (at δ = 169.2 ppm) disappeared completely, confirming the loss of the formate counter anion. Thermogravimetric-differential thermal analysis (TG-DTA) analyses were also performed by heating 3 from 27 to 300 °C at a rate of 4 °C min−1. The TG data in Fig. 3 (black line) demonstrate mass loss beginning at 95.5 °C and continuing up to 160.2 °C, for a total loss of 7.22% (going from a molecular mass of 651.4 to 603.9). This may be attributed to the loss of a CO2 molecule from 3 on heating (the theoretical mass loss is 6.76%). The DTA data in Fig. 3 (blue line) exhibit an exotherm in agreement with the loss of CO2. Based on the NMR evidence and the TG-DTA analysis,30,42–44 we may conclude that heating 3 at 150 °C releases the captured CO2 that is present as a formate anion. It was also found possible to replace the formate anion in 3 with a chloride ion simply by passing it through a Dowex ion-exchange resin. This procedure generated compound 4 with a 50% isolated yield (Fig. 4).45 The formation of 4 was confirmed by NMR analysis and single-crystal XRD. The 1H NMR spectrum of the resulting compound in deuterated chloroform contained only one singlet at δ = 8.84 ppm, while the 13C NMR spectrum did not display any signals beyond 145.1 ppm, confirming the absence of formate ions in 4. Finally, 4 was obtained as colorless crystals from methanol, and single-crystal XRD confirmed the expected structure (Fig. 4), in which the formate anion was replaced by a chloride ion.45 Based on the above ion exchange chromatographic experiment, it was possible to regenerate the aNHC salt (4). Interestingly, when 2 was exposed to air in the solid state for three days as a fine powder in a Petri dish, the formation of a new product was observed, as evidenced by 1H NMR spectroscopy (Fig. 5).26,30,46
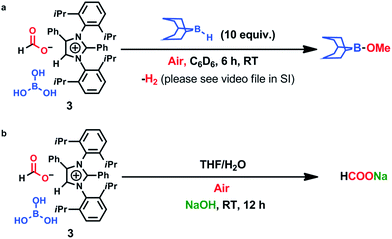 |
| | Scheme 1 Reduction of CO2 to methoxyborane and sodium formate in air. | |
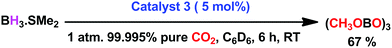 |
| | Scheme 2 Metal-free catalytic reduction of CO2 under ambient conditions. | |
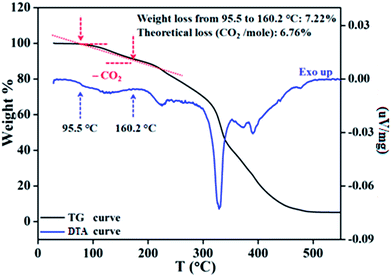 |
| | Fig. 3 Removal of captured CO2 in solid state, as monitored by thermogravimetric-differential thermal analysis of compound 3. | |
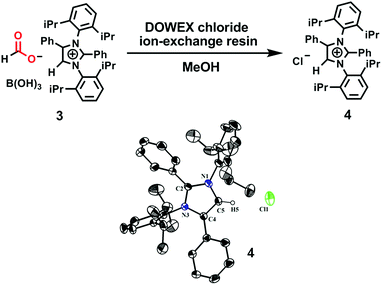 |
| | Fig. 4 Substitution of a formate anion with a chloride ion upon passing 3 through an ion-exchange resin, and an ORTEP drawing of the molecular structure of 4. | |
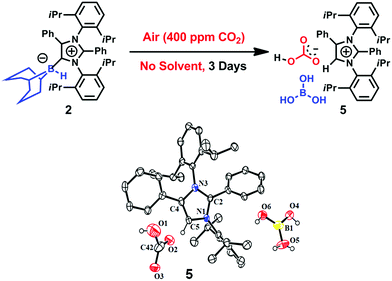 |
| | Fig. 5 Capture of CO2 from air by 2 in the solid state with the formation of the bicarbonate 5, and its ORTEP drawing. | |
The 1H NMR spectrum of this new compound in deuterated chloroform at room temperature contained two singlets at δ = 8.58 and 8.77 ppm with a 1:1 intensity ratio. These two signals (which are different from those produced by 3) were attributed to a bicarbonate anion30 and an imidazolium cation,24 respectively. These assignments were further substantiated by the corresponding 13C NMR spectroscopic signals at δ = 169 ppm (CO of bicarbonate) and 125.4 ppm (C5 of azolium). Furthermore, the 11B NMR spectrum of 5 exhibits a singlet at δ = 21 ppm that can be ascribed to free boric acid. Based on these data, it is evident that 5 was obtained (Fig. 5). Colorless crystals of 5 were grown from a benzene/n-hexane mixture with a 50% yield and the single-crystal XRD analysis of compound 5 confirmed the expected structure (Fig. 5).
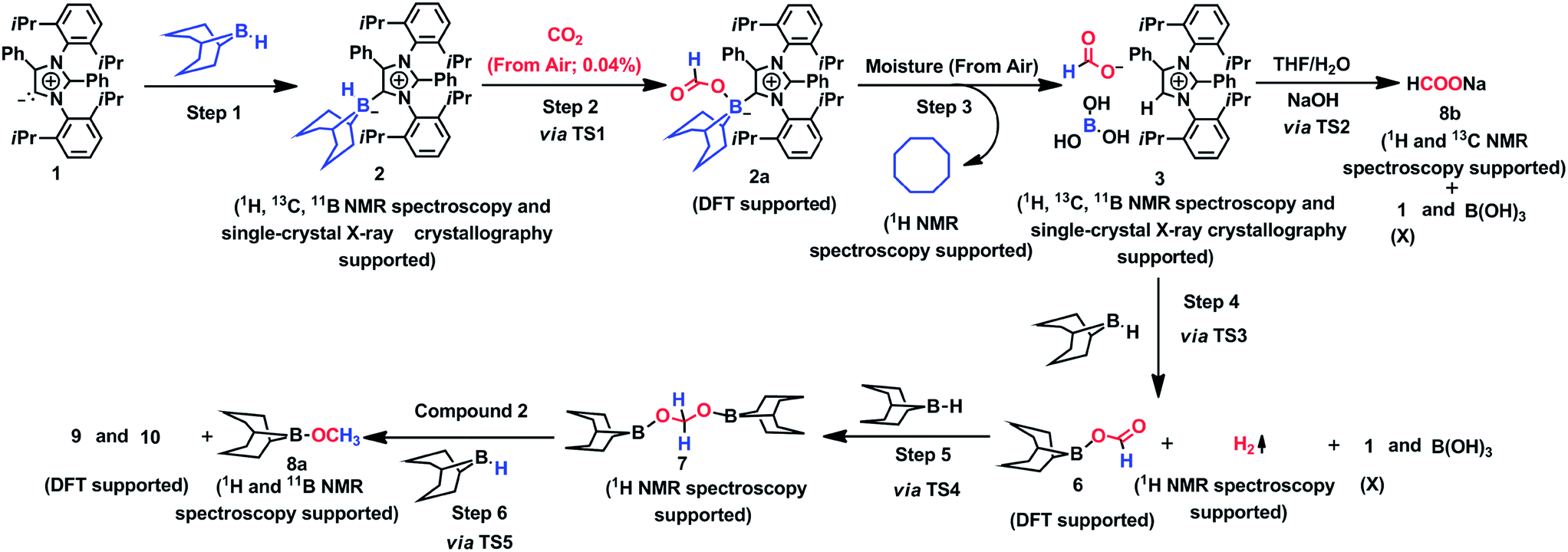
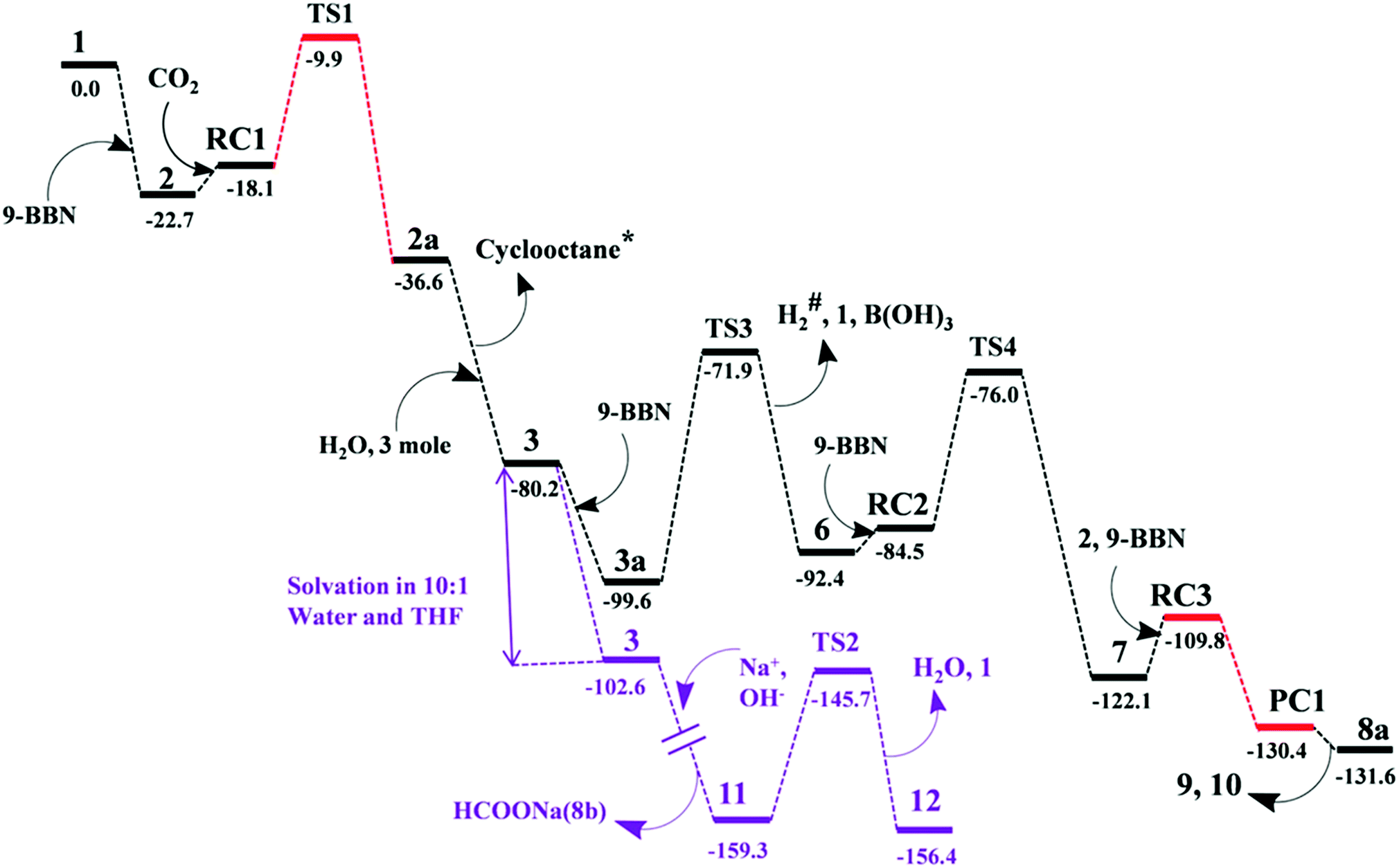
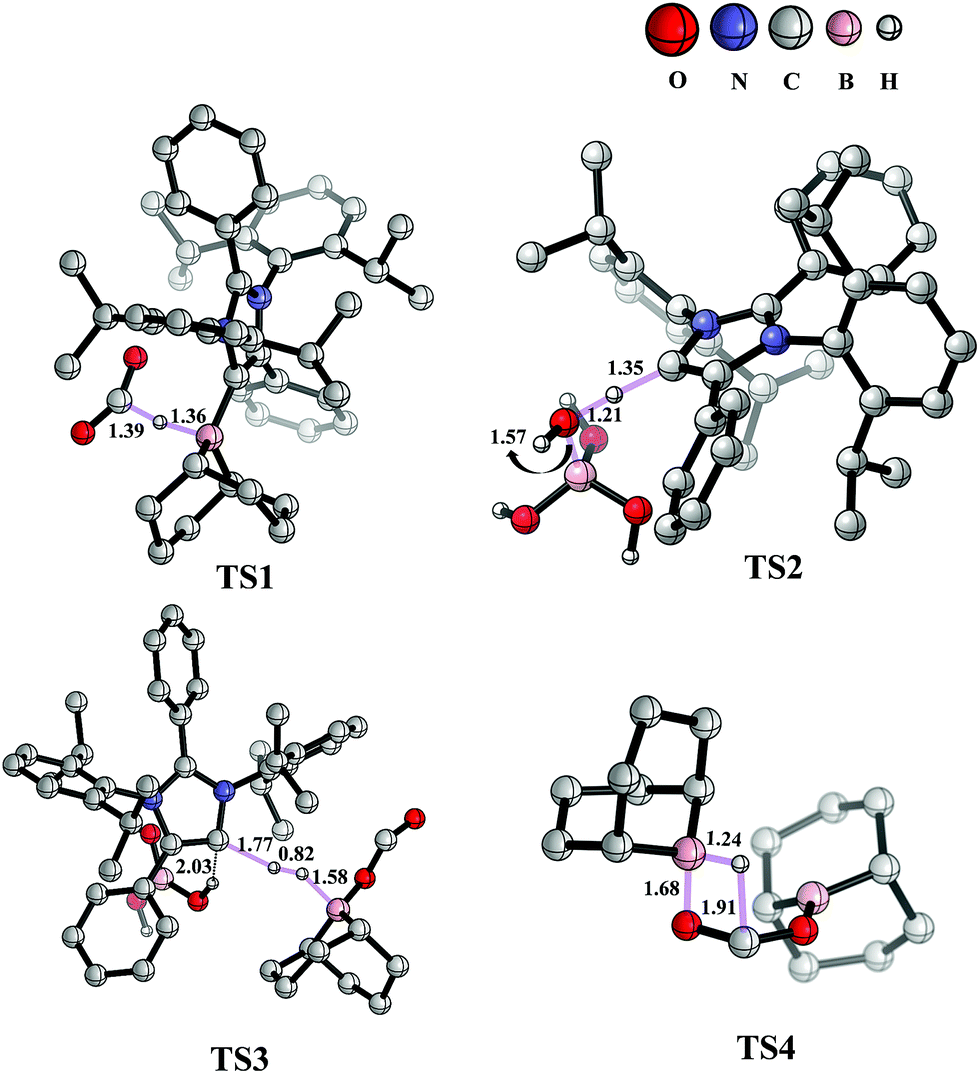
The mechanism associated with the reduction of the captured CO2 was examined by performing several stoichiometric reactions in addition to high level DFT calculations. On the basis of the experimental results, a full mechanistic path (Fig. 6) is proposed together with the energy profile diagram shown in Fig. 7. The optimized transition states for the mechanistic path are presented in Fig. 8. The combination of 1 with 9-BBN in an equimolar amount in toluene at 40 °C furnishes 2,35 as confirmed by NMR and XRD analysis. The electrostatic potential (ESP) surface and the highest occupied molecular orbital of 2 were also determined (Fig. S1, ESI†). In the ESP surface diagram, the red region around the B–H bond of intermediate 2 indicates higher electron density around that area. Thus, 2 could potentially serve as a hydride donor during the course of the reaction. This can be explained by the activation of the B–H bond of 2 as a result of its proximity to the strongly σ-donating aNHC moiety. In the presence of air, a CO2 molecule is selectively incorporated into the B–H bond36,47 of 2 to form 2avia reactant complex RC1 (Fig. S3†) and transition state TS1 (Fig. 8). During the reaction of 2 with dry air, we observed the capture of CO2 as formate ions36 as confirmed by the corresponding 13C NMR signal at δ = 168.2 ppm (Fig. S25†). The activation barrier for this elementary step is much lower (ΔG‡ = 8.2 kcal mol−1). The formation of 2a is thermodynamically favorable, as this particular step involves a heat release of 18.5 kcal mol−1. As a result of atmospheric moisture, 2a is hydrolyzed to 3, which stabilizes the compound as a zwitterion.47 During this process, the 9-BBN fragment is hydrolyzed to boric acid with the elimination of a cyclooctane molecule, as confirmed by 1H NMR analysis (δ = 1.55 ppm in deuterated benzene) of the reaction mixture.48 Compound 3 was thoroughly characterized by XRD, NMR spectroscopy and elemental analysis. This compound was also found to react with sodium hydroxide solution to furnish sodium formate (8b), as confirmed through NMR analysis.11
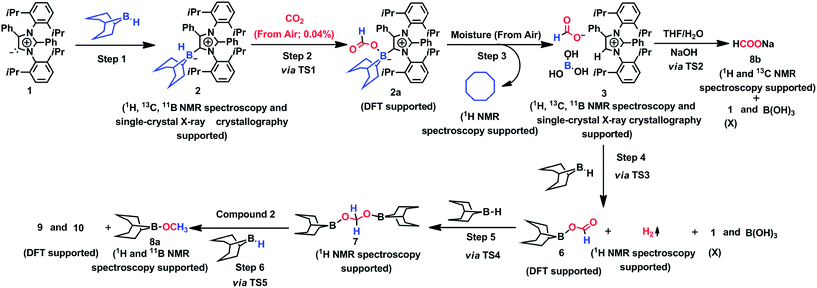 |
| | Fig. 6 Mechanistic scheme for the reduction of CO2 to methoxyborane and sodium formate with 2 in air. For drawings of 9 and 10 see Fig. S3.† | |
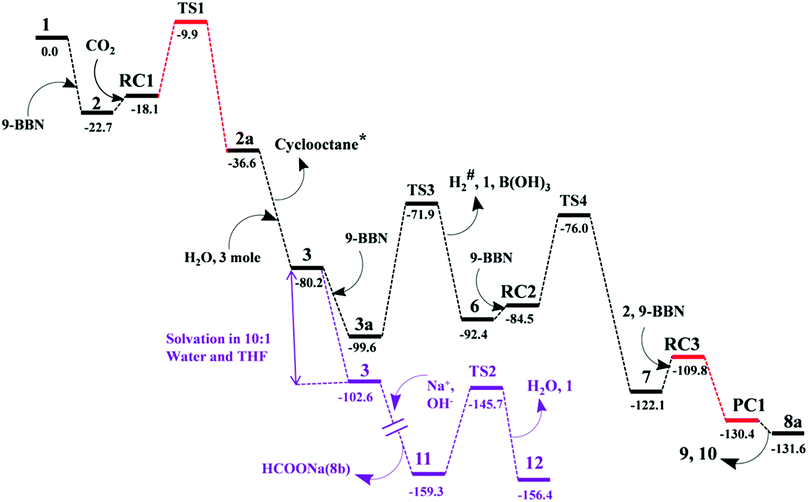 |
| | Fig. 7 Computed Gibbs free energy profiles at 25 °C for the conversion of CO2 to methoxyborane or sodium formate with 2 in air. The relative free energies (in kcal mol−1) obtained in solvent were calculated with respect to the energy of the separate reactants {aNHC (1) and 9-BBN}. *Formation of cyclooctane was confirmed by 1H NMR spectroscopy. #H2 gas evolution was confirmed by 1H NMR spectroscopy and visual observation (see the video file in the ESI†). For drawings of all compounds see Fig. S3.† | |
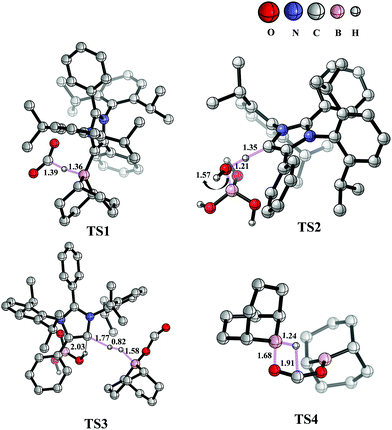 |
| | Fig. 8 Optimized structures of the transition states involved in the potential energy surface of CO2 reduction to methoxyborane and sodium formate with 2 in air. Important bond distances (in Å) are shown. Only relevant H atoms are provided in the structures. | |
When sodium hydroxide was added to a solution of 3 in water/tetrahydrofuran, sodium formate was released with the formation of intermediate 11 (Fig. S3†). The structure of 11, as shown in Fig. S3,† consists of two fragments (viz. B(OH)4− and aNHC-H+) that subsequently release a water molecule to form boronic acid (12) and free aNHC (1) (Fig. S3†) viaTS2 (Fig. 8). The free energy of activation for water elimination was calculated to be 13.6 kcal mol−1. In addition, another 9-BBN molecule reacts with 3 to furnish 3a (Fig. S3†), thus regenerating free aNHC (1) with the formation of boron formate20 (6) via hydrogen gas evolution38 (see the video file in the ESI†) through TS3 (Fig. 8). The spatial orientation of TS3 is shown in Fig. 8, in which the distance between the two hydrogen atoms is 0.82 Å. The free energy barrier for this hydrogen evolution reaction was calculated to be 27.7 kcal mol−1. In addition, 6 can be reduced to its acetal form (H2C(OBBN)2; 7) in the presence of 9-BBN viaTS4 (Fig. 8), through reactant complex RC2 (Fig. S3†).20,22 The free energy barrier for this hydroboration is 8.5 kcal mol−1 and this particular step is highly exothermic (ΔG = − 37.6 kcal mol−1). The formation of 7 was confirmed from its characteristic chemical shift (δ = 5.34 ppm in deuterated benzene) upon analysis of the reaction mixture by 1H NMR spectroscopy.20 Finally, 7 is reduced to the methoxide derivative 8a in the presence of 2 and 9-BBN (Fig. 7), through reactant complex RC3 (Fig. S3†) and product complex PC1 (Fig. S2†).20 The formation of 8a is associated with the elimination of 9 and 10 (Fig. S3†), and the free energy change (ΔG) for this exothermic elementary step was determined to be 20.6 kcal mol−1.
In summary, in this work we synthesized an abnormal N-heterocyclic carbene (aNHC) supported 9-BBN adduct (2) for CO2 capture from air with subsequent conversion to methoxyborane or sodium formate under ambient conditions. The intermediate (aNHC-H, HCOO, B(OH)3; 3) was structurally characterized to unequivocally establish the capture of atmospheric CO2. It was further shown that CO2 present in low concentrations (such as in ambient air) could be captured by this compound in the solid state. This represents the first-ever demonstration of the possibility of CO2 capture from air followed by its reduction to methanol without employing any metal under very mild conditions. A detailed mechanistic pathway for this fascinating transformation was proposed based on tandem experimental and computational studies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. K. M. thanks the IISER Kolkata for financial support. S. C. S. thanks the UGC for a research fellowship and Invictus Oncology, Delhi, for providing a research scientist position. P. K. V. and G. V. K. thank the UGC, New Delhi, for a research fellowship. P. K. H. thanks the IISER Kolkata for a research fellowship. R. G. thanks the SERB for a National Postdoctoral Fellowship. R. B. thanks the CSIR India for a research fellowship, and A. D. thanks the INSA, DST and BRNS for partial funding and providing access to a CRAY supercomputer. The authors gratefully acknowledge Dr Tapan Kanti Paine, Dr Bikash Shaw and Mr Dinesh Mullangi for their help in acquiring DTA data and Dr Soumik Mandal for his assistance during XRD work.
References
- Y.-N. Li, R. Ma, L.-N. He and Z.-F. Diao, Catal. Sci. Technol., 2014, 4, 1498–1512 RSC.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef PubMed.
- H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453–456 CrossRef PubMed.
- A. Goeppert, M. Czaun, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC.
- D. W. Keith, Science, 2009, 325, 1654–1655 CrossRef PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef PubMed.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef PubMed.
- S. Enthaler, J. V. Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 RSC.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- R. Langer, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 9948–9952 CrossRef PubMed.
- M. S. Jeletic, M. T. Mock, A. M. Appel and J. C. Linehan, J. Am. Chem. Soc., 2013, 135, 11533–11536 CrossRef PubMed.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef PubMed.
- C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef.
- A. Paparo, J. S. Silvia, C. E. Kefalidis, T. P. Spaniol, L. Maron, J. Okuda and C. C. Cummins, Angew. Chem., Int. Ed., 2015, 54, 9115–9119 CrossRef.
- F.-G. Fontaine, M.-A. Courtemanche and M.-A. Légaré, Chem.–Eur. J., 2014, 20, 2990–2996 CrossRef.
- I. Knopf and C. C. Cummins, Organometallics, 2015, 34, 1601–1603 CrossRef.
- C. D. N. Gomes, E. Blondiaux, P. Thuéry and T. Cantat, Chem.–Eur. J., 2014, 20, 7098–7106 CrossRef.
- M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2013, 135, 9326–9329 CrossRef PubMed.
- T. Wang and D. W. Stephan, Chem.–Eur. J., 2014, 20, 3036–3039 CrossRef.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef.
- S. C. Sau, R. Bhattacharjee, P. K. Vardhanapu, G. Vijaykumar, A. Datta and S. K. Mandal, Angew. Chem., Int. Ed., 2016, 55, 15147–15151 CrossRef.
- T. Wanga and D. W. Stephan, Chem. Commun., 2014, 50, 7007–7010 RSC.
- M. Yamashita, K. Goto and T. Kawashima, J. Am. Chem. Soc., 2005, 127, 7294–7295 CrossRef PubMed.
- U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Nat. Commun., 2014, 5, 5883 CrossRef.
- C. Liu, B. C. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 1210–1213 CrossRef.
- F. Inagaki, C. Matsumoto, T. Iwata and C. Mukai, J. Am. Chem. Soc., 2017, 139, 4639–4642 CrossRef.
- C. A. Seipp, N. J. Williams, M. K. Kidder and R. Custelcean, Angew. Chem., Int. Ed., 2017, 56, 1042–1045 CrossRef.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef.
- P. M. Bhatt, Y. Belmabkhout, A. Cadiau, K. Adil, O. Shekhah, A. Shkurenko, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2016, 138, 9301–9307 CrossRef CAS.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2016, 138, 778–781 CrossRef CAS.
- E. Aldeco-Perez, A. J. Rosenthal, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Science, 2009, 326, 556–559 CrossRef CAS.
- P. Eisenberger, B. P. Bestvater, E. C. Keske and C. M. Crudden, Angew. Chem., Int. Ed., 2015, 54, 2467–2471 CrossRef CAS.
- C. Chauvier, A. Tlili, C. D. N. Gomes, P. Thuéry and T. Cantat, Chem. Sci., 2015, 6, 2938–2942 RSC.
- P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433–7437 CrossRef CAS.
- M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2014, 136, 10708–10717 CrossRef CAS.
- K. Fujiwara, S. Yasuda and T. Mizuta, Organometallics, 2014, 33, 6692–6695 CrossRef CAS.
- F. S. Pereira, D. L. d. S. Agostini, R. D. d. E. Santo, E. R. deAzevedo, T. J. Bonagamba, A. E. Joba and E. R. P. Gonzalez, Green Chem., 2011, 13, 2146–2153 RSC.
- C. M. Crudden, J. H. Horton, M. R. Narouz, Z. Li, C. A. Smith, K. Munro, C. J. Baddeley, C. R. Larrea, B. Drevniok, B. Thanabalasingam, A. B. McLean, O. V. Zenkina, I. I. Ebralidze, Z. She, H. B. Kraatz, N. J. Mosey, L. N. Saunders and A. Yag, Nat. Commun., 2016, 7, 12654 CrossRef CAS.
- H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 112–113 RSC.
- A. Tudose, A. Demonceau and L. Delaude, J. Organomet. Chem., 2006, 691, 5356–5365 CrossRef CAS.
- B. R. V. Ausdall, J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, J. Org. Chem., 2009, 74, 7935–7942 CrossRef PubMed.
- D. H. Brown and B. W. Skelton, Dalton Trans., 2011, 40, 8849–8858 RSC.
- M. Vogt, J. E. Bennett, Y. Huang, C. Wu, W. F. Schneider, J. F. Brennecke and B. L. Ashfeld, Chem.–Eur. J., 2013, 19, 11134–11138 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS.
-
https://www.sigmaaldrich.com/spectra/fnmr/FNMR009622.PDF
.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR data, a video clip, computational details, XRD details with CIF files and NMR images. CCDC 1583077–1583080. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03581d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Pradip Kumar
Hota
b,
Pavan K.
Vardhanapu
b,
Gonela
Vijaykumar
a,
Pradip Kumar
Hota
b,
Pavan K.
Vardhanapu
b,
Gonela
Vijaykumar