The challenges of characterising nanoparticulate catalysts: general discussion
Rosa
Arrigo
,
Kassim
Badmus
,
Francesca
Baletto
,
Maurits
Boeije
,
Michael
Bowker
,
Katharina
Brinkert
,
Aram
Bugaev
 ,
Valerii
Bukhtiyarov
,
Michele
Carosso
,
Richard
Catlow
,
Revana
Chanerika
,
Philip R.
Davies
,
Wilke
Dononelli
,
Hans-Joachim
Freund
,
Cynthia
Friend
,
Simone
Gallarati
,
Bruce
Gates
,
Alexander
Genest
,
Emma K.
Gibson
,
Justin
Hargreaves
,
Stig
Helveg
,
Haoliang
Huang
,
Graham
Hutchings
,
Nicola
Irvine
,
Roy
Johnston
,
Stanley
Lai
,
Carlo
Lamberti
,
Joseph
Macginley
,
David
Marchant
,
Toru
Murayama
,
Rene
Nome
,
Yaroslav
Odarchenko
,
Jonathan
Quinson
,
Scott
Rogers
,
Andrea
Russell
,
Said
Said
,
Paul
Sermon
,
Parag
Shah
,
Sabrina
Simoncelli
,
Katerina
Soulantica
,
Federico
Spolaore
,
Bob
Tooze
,
Laura
Torrente-Murciano
,
Annette
Trunschke
,
David
Willock
and
Jiaguang
Zhang
,
Valerii
Bukhtiyarov
,
Michele
Carosso
,
Richard
Catlow
,
Revana
Chanerika
,
Philip R.
Davies
,
Wilke
Dononelli
,
Hans-Joachim
Freund
,
Cynthia
Friend
,
Simone
Gallarati
,
Bruce
Gates
,
Alexander
Genest
,
Emma K.
Gibson
,
Justin
Hargreaves
,
Stig
Helveg
,
Haoliang
Huang
,
Graham
Hutchings
,
Nicola
Irvine
,
Roy
Johnston
,
Stanley
Lai
,
Carlo
Lamberti
,
Joseph
Macginley
,
David
Marchant
,
Toru
Murayama
,
Rene
Nome
,
Yaroslav
Odarchenko
,
Jonathan
Quinson
,
Scott
Rogers
,
Andrea
Russell
,
Said
Said
,
Paul
Sermon
,
Parag
Shah
,
Sabrina
Simoncelli
,
Katerina
Soulantica
,
Federico
Spolaore
,
Bob
Tooze
,
Laura
Torrente-Murciano
,
Annette
Trunschke
,
David
Willock
and
Jiaguang
Zhang
First published on 10th August 2018
Haoliang Huang opened discussion of the paper by Carlo Lamberti: My question is about carbon species in "palladium carbide", since when the Pd was exposed to acetylene, the changes were observed in XANES but not in XRD. Is it possible that such carbon species just the acetylene, or its fragment, absorbed on Pd, or Pd-C solid solution, rather than forming a palladium carbide phase. Have you used XPS to confirm the oxidation states of carbon? It may be more applicable for alumina supported or unsupported Pd.Carlo Lamberti answered: When the Pd/C system was exposed to C2H2 at 100 °C we observed changes in all the three techniques (XRPD, EXAFS and XANES: data in the 0–40 and 100–140 min t-intervals in Fig. 5 (DOI: 10.1039/c7fd00211d)). Conversely, when, after a long exposure to C2H2 at 100 °C, we exposed the Pd/C catalyst to H2 (data at t = 80 min in Fig. 5 (DOI: 10.1039/c7fd00211d)), we observed a change in the Pd K-edge XANES (orange triangle) but no changes in the XRD and Pd K-edge EXAFS (gray circle and black square, respectively). This has been interpreted in terms of the removal of C species from the surface of the Pd nanoparticle, while C atoms in the bulk of the NPs were unaffected by H2. We are unfortunately unable to discriminate between physisorbed C2H2 and carbonaceous fragments at the NP surface. We recently performed an in-depth C 1s XPS study of different carbon supports and Pd/C catalysts1,2 but not under reaction conditions. Our XPS studies were aimed at the support characterization only. Unfortunately, I do not believe that it would be possible to discriminate the C 1s signal of the C atoms forming the surface and bulk Pd-carbide to that coming from the vast majority of C atoms of the support. As you suggested such in situ C 1s XPS study would be more interesting on a carbon-free support as γ-Al2O3. In the recent past we performed a combined XRPD/XAS study on alumina supported Pd NPs;3 in that case the quality of the XRPD data was not as good as in the present case because of the presence of the broad, but intense, diffraction peak of γ-Al2O3. Your suggestion to use this system to further investigate the Pd carbide phase with C 1s XPS is worth trying.
1 A. Lazzarini, A. Piovano, R. Pellegrini, G. Leofanti, G. Agostini, S. Rudic, M. R. Chierotti, R. Gobetto, A. Battiato, G. Spoto, A. Zecchina, C. Lamberti and E. Groppo, Catal. Sci. Technol., 2016, 6, 4910–4922.
2 A. Lazzarini, R. Pellegrini, A. Piovano, S. Rudic, C. Castan-Guerrero, P. Torelli, M. R. Chierotti, R. Gobetto, C. Lamberti and E. Groppo, Catal. Sci. Technol., 2017, 7, 4162–4172.
3 A. L. Bugaev, A. A. Guda, K. A. Lomachenko, V. V. Srabionyan, L. A. Bugaev, A. V. Soldatov, C. Lamberti, V. P. Dmitriev and J. A. van Bokhoven, J. Phys. Chem. C, 2014, 118, 10416–10423.
Paul Sermon asked: I was intrigued by the long periodicity (3 min) structural and catalytic oscillations that you see. IR thermography (IRT) reveals intriguing synchronous exothermic oscillations with a 20 s frequency in growth-defined biological systems over a wide area (see Fig. 1a).1–4 Do you have an explanation for the long 3 min periodicity that you find? Could you elaborate on the gradual transition of lattice parameters in your Fig. 2a, because when crystallite size effects in the PdHx system were investigated for unsupported Pd blacks some years ago,5 in the α↔β and β↔α plateau transitions, as one varied p(H2) (and hence x), there coexistence of α and β domains, as shown in Fig. 1b (but also seen by others).6
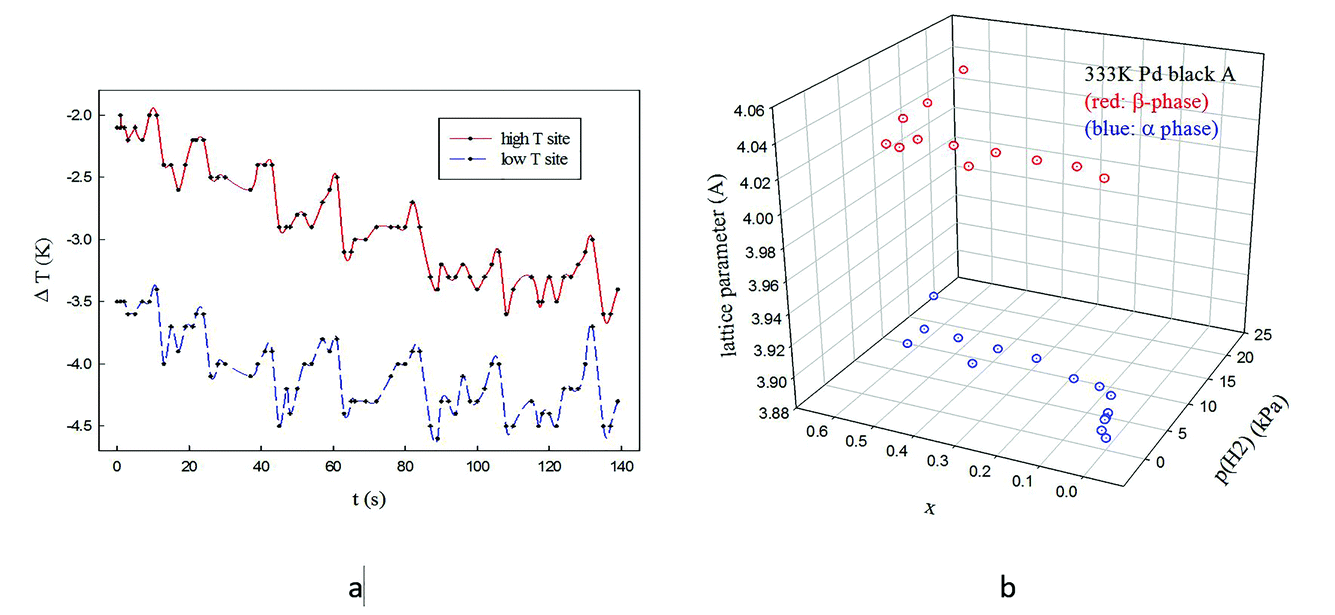 | ||
| Fig. 1 (a) Exothermic synchronised oscillations seen by IRT over macroscopic distances (cm) when Penicillium grows on a moist, naturally-colonised surface (multigrain bread). (b) Plots of pH2-x in PdHx-fcc lattice parameter at 333 K for 86 nm unsupported Pd black (where the Δx width of the hysteresis loop was larger than for a 10 nm sample).5 | ||
1 J. Buck and F. Buck, Science, 1968, 159, 1319–1327.
2 S. Dano, M. F. Madsen and P. G. Sorensen, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12732–12736.
3 A. L. Koch, Crit. Rev. Microbiol., 2001, 27, 223–237.
4 D. Sauvageau, Z. Storms and D. G. Cooper, J. Biotechnol., 2010, 149, 67–73.
5 D. H. Everett and P. A. Sermon, Z. Phys. Chem., 1979, 114, 109–122.
6 Z. L. Vert, I. A. Mosevich and I. R. Tverdovskii, Russ. J. Phys. Chem., 1965, 39, 566.
Carlo Lamberti replied: Concerning the periodicity, it is dependent on several factors: the catalyst itself, the amount of catalyst in the capillary, the flow and the feed. In this regard, very recently, Rupprechter and coworkers1 observed multifrequential oscillations during hydrogen oxidation over rhodium, imaged in situ by using photoemission electron microscopy. Depending on the observed Rh(h,k,l) surface investigated, the observed periodicity ranged from few to several minutes. Concerning the second part of your question, we never claimed to be the first one observing the α↔β transition upon interaction of Pd nanoparticles (NPs) with H2. We believe that our study, combining almost simultaneous XRPD and EXAFS data collections, coupled with independent volumetry measurements (Fig. 2a–c) is a complete and consistent set of data and that (to the best of our knowledge) we have been the first to interpret the different behavior of the XRPD and EXAFS isotherms in terms of a crystalline/amorphous core/shell model of the Pd NPs (DOI: 10.1039/c7fd00211d).2
1 Y. Suchorski, M. Datler, I. Bespalov, J. Zeininger, M. Stoger-Pollach, J. Bernardi, H. Gronbeck and G. Rupprechter, Nat. Commun., 2018, 9, 600, and references therein.
2 A. L. Bugaev, A. A. Guda, K. A. Lomachenko, V. V. Shapovalov, A. Lazzarini, J. G. Vitillo, L. A. Bugaev, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, J. Phys. Chem. C, 2017, 121, 18202–18213.
Andrea Russell enquired: Have you considered using the Pd L-edge to further explore the effects of hydride formation? See for example the paper by van Bokhoven’s group.1
1 M. W. Tew, J. T. Miller and J. A. van Bokhoven, J. Phys. Chem. C, 2009, 113, 15140–15147.
Carlo Lamberti responded: This is indeed an excellent suggestion. Pd L3- and L2-edges XANES spectra would be more informative on the unoccupied d-density-of-states (DOS) of Pd than the K-edge one, which mainly probes the p-DOS. Obviously, such measurements will be much more demanding on the experimental ground because of the much lower penetration depth of 3 keV photons with respect to 24 keV ones; moreover, they could not be coupled with XRPD data collection and will not allow the extraction of an EXAFS signal. Notwithstanding these limitations, they can nicely complement the present study.
Andrea Russell asked: I have also studied the formation of hydrides in Pd nanoparticles using XRD and EXAFS, but from an electrochemical perspective.1,2 Whilst we saw an effect in the electrochemistry of a moving boundary between the alpha and beta phases of the hydride, we didn’t interpret these data in terms of a core–shell argument as you have done. Do you have any additional information to support the core shell model you’re proposing?
1 A. Rose, S. Maniguet, R. J. Mathew, C. Slater, J. Yao and A. E. Russell, Phys. Chem. Chem. Phys., 2003, 5, 3220.
2 A. Rose, O. South, S. Diaz-Moreno, J. R. Owen and A. E. Russell, Phys. Chem. Chem. Phys., 2005, 7, 366.
Carlo Lamberti answered: Many thanks for having pointed out two very relevant studies in this topic.1,2 Our interpretation in terms of a core–shell structure of the nanoparticles (NPs)3 has been further supported by the analysis of the higher shells in the EXAFS signal in the frame of the multiple scattering approach, which is mandatory in the case of fcc NPs (DOI: 10.1039/c7fd00211d). Upon increasing the shell order, the response of the EXAFS analysis become closer and closer to that of XRPD, which reflects the order of the NPs at the long-range scale. Now, the relative elongation of the first (blue triangles) and third (red circles) shell along the 22 °C-isotherm of Pd-hydrate formation in a bulk sample (Pd black) matches, within the experimental incertitude, that obtained from XRPD (gray circles), see Fig. 2a. Conversely, for Pd NPs (Fig. 2b), the relative variation of the first shell clearly behaves differently from that of the third shell, which in turns is very close to the curve obtained from XRPD data.
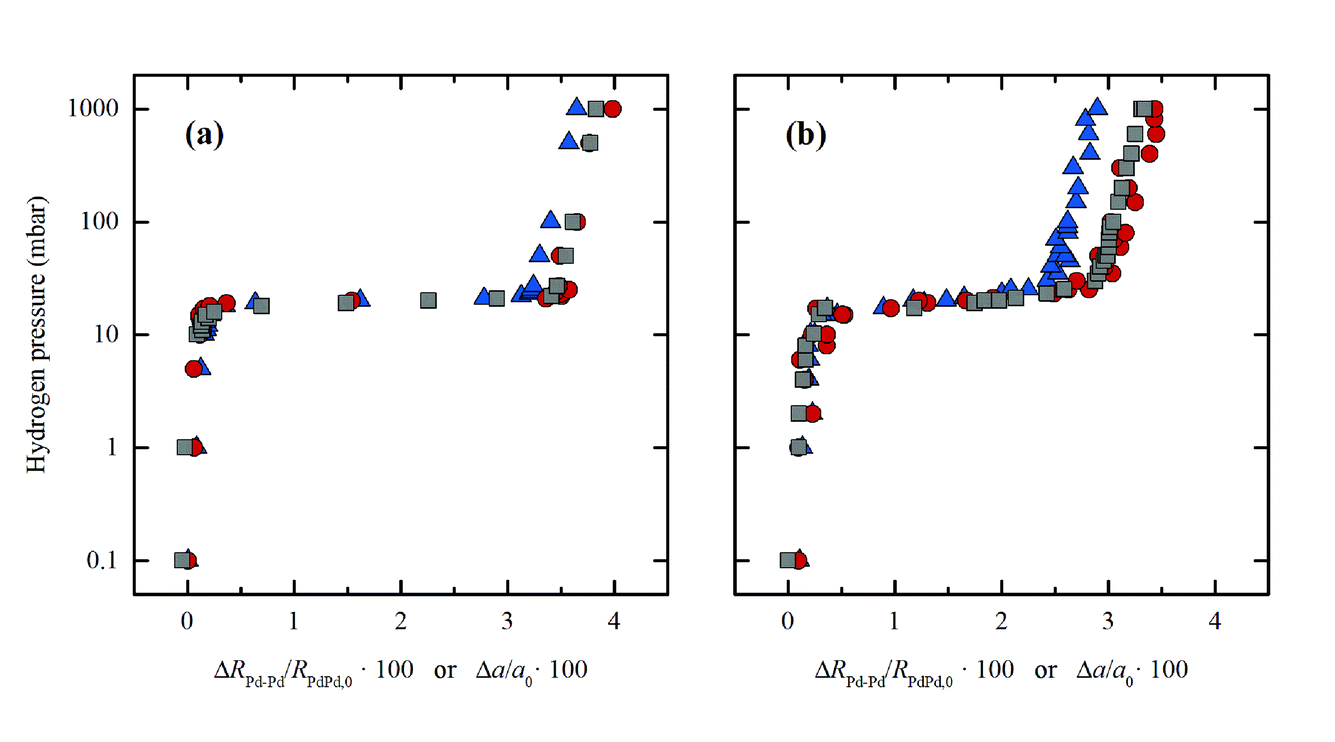 | ||
| Fig. 2 Evolution of the first (blue triangles) and third (red circles) shells determined by EXAFS and lattice parameter determined by XRPD (grey squares) for Pd black (a) and Pd NPs (b) during hydride phase formation at 22 °C. For a direct comparison, that data were reported as relative variation: ΔRPd–Pd/RPd–Pd,0 and Δa/a0 for EXAFS and XRPD, respectively. Previously unpublished figure, reporting data published in ref. 3. | ||
1 A. Rose, S. Maniguet, R. J. Mathew, C. Slater, J. Yao and A. E. Russell, Phys. Chem. Chem. Phys., 2003, 5, 3220–3225.
2 A. Rose, O. South, I. Harvey, S. Diaz-Moreno, J. R. Owen and A. E. Russell, Phys. Chem. Chem. Phys., 2005, 7, 366–372.
3 A. L. Bugaev, A. A. Guda, K. A. Lomachenko, V. V. Shapovalov, A. Lazzarini, J. G. Vitillo, L. A. Bugaev, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, J. Phys. Chem. C, 2017, 121, 18202–18213.
Andrea Russell enquired: You have interpreted your results in terms of a core-shell model, with a hydride phase in the shell, to account for the differences observed in the slope of the plateau regions in the plots in Fig. 2 obtained for EXAFS compared to the other methods. Have you considered the effects of the particle size distribution of your catalyst nanoparticles? XRD and EXAFS each have a different inherent bias, with XRD being much more sensitive to the larger particles and EXAFS being a per-atom technique, which means that even the smallest particles will contribute. As XRD effectively weights the data by volume, it only takes a few of the larger particles to emphasise the different perspectives of the two techniques.
Carlo Lamberti replied: With your question you have stimulated discussion on a very important point in the field of nanoparticle (NP) characterization, which is the intrinsic sensitivity of the different techniques. Besides XRD and EXAFS, I will include also TEM in my answer, but first a comment is deserved. There are several studies on NPs where the authors compare EXAFS and TEM analyses. In a non-negligible fraction of them, the authors use the mean value of the particle size distribution <D > obtained in the TEM study to predict the average coordination number of metal atoms in a particle of diameter <D >: N (<D >). Finally, they compare the N (<D >) value obtained from the TEM analysis with the average first shell co-ordination number obtained from the EXAFS analysis. This approach can be correct only in the ideal case of a NP having a null standard deviation in the particle size distribution. In all the realistic cases it is wrong because TEM distributions weights by particles while, as correctly indicated by Andrea Russell, EXAFS weights by atoms, i.e. by particle volume. If the NP size distribution is sharp (like the case shown in Fig. 3a–c) the systematic error is small, and if it is large (like the case shown in Fig. 3g–i) the error can be macroscopic (compare the blue and green distributions). The correct approach in the comparison between TEM and EXAFS data is to calculate the volume-weighted co-ordination number, using the whole NP size distribution obtained from TEM and not just its mean value. This concept has been well described in several publications.1–5
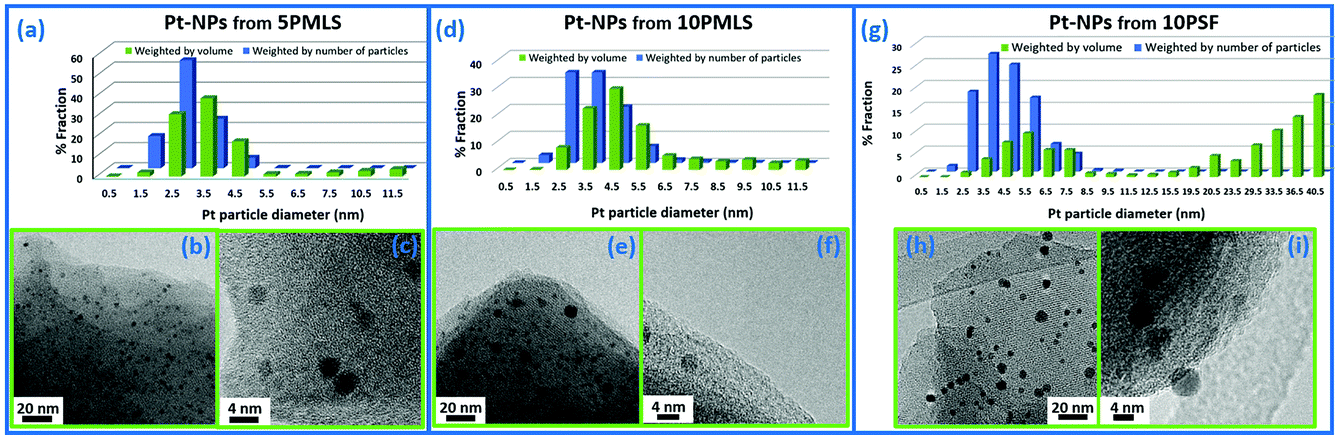 | ||
| Fig. 3 Particle size distribution weighted by number (blue, from a TEM study) and by volume (green, to be used to estimate the average coordination number N obtained by the EXAFS first shell analysis) in three different examples of Pt NPs formed inside a Pt-functionalized UiO-67 MOF. Adapted by permission of the Royal Society of Chemistry (copyright 2017).5 | ||
Now my answer. First, the TEM study (performed over more than 500 independent NPs) on the Chimet catalyst used in this study (DOI: 10.1039/c7fd00211d) has shown a very sharp particle size distribution: <D > = 26 ± 4 Å.6 The N (<D >) value obtained from the TEM analysis, taking into account the whole particle size distribution (as described above), matches with the value obtained in our EXAFS analysis, meaning that we do not have a bimodal particle size distribution with very few (that unfortunately would have escaped the TEM sampling) and very big NPs. Second, both X-ray absorption and X-ray scattering are volume-weighted processes, meaning that the contribution of a single NP to the total absorption or scattering process is proportional to its volume in both cases. Things change when with XRD we analyze the Bragg fraction only of the total X-ray scattering (as we did in our work), because XRD probes only the fraction of Pd atoms characterized by long-range order (the complementary fraction of disordered, or amorphous, Pd atoms will contribute to the diffuse scattering around the Bragg peaks). On this basis, I totally agree with your statement that XRD is much more sensitive to the larger NPs then EXAFS, but the lower contribution of the small NPs to the Pd(h,k,l) reflections (compared to their contribution to EXAFS) is not because they are small, but because they are partially (or totally) disordered. Our model foresees that the disordered (amorphous) part corresponds to the external shell of the NP. On this basis, I believe that the discrepancy in the XRD and EXAFS H2-adsorption isotherms reported in Fig. 2a and 2b, (DOI: 10.1039/c7fd00211d) respectively, should not be related to the presence of small and big Pd NPs, but to the different order ranges (long- vs. short-) that co-exist in different proportions in the overall Pd phase. In this regard, the simplest assumption is to imagine, for each NP, a crystalline core, probed by both EXAFS and XRD, and an amorphous shell, probed by EXAFS only. Obviously, for each NP, the fraction of Pd atoms belonging to the crystalline or to the amorphous phase is size dependent, the former dominating for large NPs, the latter prevailing in small NPs, being the only one present for NPs smaller than about 10 Å in diameter.
1 G. Agostini, R. Pellegrini, G. Leofanti, L. Bertinetti, S. Bertarione, E. Groppo, A. Zecchina and C. Lamberti, J. Phys. Chem. C, 2009, 113, 10485–10492.
2 G. Agostini, A. Piovano, L. Bertinetti, R. Pellegrini, G. Leofanti, E. Groppo and C. Lamberti, J. Phys. Chem. C, 2014, 118, 4085–4094.
3 G. Agostini, C. Lamberti, R. Pellegrini, G. Leofanti, F. Giannici, A. Longo and E. Groppo, ACS Catal., 2014, 4, 187–194.
4 L. Braglia, E. Borfecchia, K. A. Lomachenko, A. L. Bugaev, A. A. Guda, A. V. Soldatov, B. T. L. Bleken, S. Oien-Odegaard, U. Olsbye, K. P. Lillerud, S. Bordiga, G. Agostini, M. Manzoli and C. Lamberti, Faraday Discuss., 2017, 201, 277–298.
5 L. Braglia, E. Borfecchia, A. Martini, A. L. Bugaev, A. V. Soldatov, S. Oien-Odegaard, B. T. Lonstad-Bleken, U. Olsbye, K. P. Lillerud, K. A. Lomachenko, G. Agostini, M. Manzoli and C. Lamberti, Phys. Chem. Chem. Phys., 2017, 19, 27489–27507.
6 A. L. Bugaev, A. A. Guda, K. A. Lomachenko, V. V. Shapovalov, A. Lazzarini, J. G. Vitillo, L. A. Bugaev, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, J. Phys. Chem. C, 2017, 121, 18202–18213.
Justin Hargreaves asked: How amenable is the diffraction you report to detailed line profile analysis and could this be a useful supplementary approach?
Carlo Lamberti responded: Your question deals with a very important methodological aspect in nanoparticle (NP) characterization, which is the full extraction of all information available from an X-ray scattering experiment. Indeed, it is exactly the diffuse scattering around the Pd(h,k,l) Bragg peaks that contains the structural information on the amorphous shell of the NPs. Unfortunately, the large majority of the literature papers that use XRD to characterize NPs limit the data analysis to the Bragg part of the pattern, and so did we in this study. There are two major difficulties in extending the data analysis to the diffuse scattering. The first is methodological and it is related to the fact that the simple Bragg equation does not hold any more and that the much more complex Debye equation must be used.1,2 The latter has not a direct solution but requires the construction of a structural model that must be iteratively refined with a comparison between experimental and predicted scattering profile. The second difficulty is intrinsically related to the high dilution of metal NPs that are relevant in catalysis. The system investigated in this study is a 5 wt% Pd supported on carbon catalyst, which means that for each Pd atom we have about 168 C atoms. As a consequence, notwithstanding the much higher Z value of Pd with respect to C (46 vs. 6), the electrons belonging to Pd are only 4% of the total number of electrons in the sample. Therefore, the non-Bragg scattering profile from a 5 wt% Pd/C catalyst will be dominated by the diffuse scattering from the amorphously carbon support. I do not see any reliable way to separate, in a standard XRD experiment, the diffuse scattering from the shell of the NPs from the much more intense scattering from the substrate and advanced experiments must be foreseen. A first way to overcome this difficulty is to perform an anomalous XRD experiment, which allows the technique to become element selective.2–4 The method consists of collecting three independent XRD patterns using three different λ across the Pd K-edge: in this way only the atomic form factor f′ of Pd will change appreciably in the three data collections and there may be a chance to discriminate the Pd from the C contribution. A second way can be to collect, with a very short λ, a diffraction pattern up to very high Q values (Q = 4π sin(θ)/λ ∼ 30 Å−1) and to analyze the data in the total scattering approach, that treats Bragg and diffuse scattering on an equal basis, allowing us to see beyond the crystal structure and reveal nanoscale features.4,5 As the atomic scattering factor of carbons goes rapidly to zero for Q >5 Å−1, the use of the scattering profile of a Pd/C catalyst in the 5–30 Å−1 region will contain the contribution of Pd atoms mainly. Summarizing it is clear that an accurate analysis of the diffuse scattering from the metal NPs is a very difficult task, however, if properly done it will significantly improve the structural level of knowledge in the field of NP characterization.
1 E. Borfecchia, D. Gianolio, G. Agostini, S. Bordiga and C. Lamberti, Metal Organic Frameworks as Heterogeneous Catalysts, Royal Society of Chemistry, Cambridge, UK, 2013, ch. 5, pp. 143–208.
2 C. Garino, E. Borfecchia, R. Gobetto, J. A. van Bokhoven and C. Lamberti, Coord. Chem. Rev., 2014, 277, 130–186.
3 J. L. Hodeau, V. Favre-Nicolin, S. Bos, H. Renevier, E. Lorenzo and J. F. Berar, Chem. Rev., 2001, 101, 1843–1867.
4 C. Lamberti, E. Borfecchia, J. A. van Bokhoven and M. Fernández-García, in X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, ed. J. A. van Bokhoven and C. Lamberti, John Wiley and Sons, Chichester, UK, 2016, pp. 303–350.
5 E. S. Bozin, P. Juhas and S. J. L. Billinge, in Characterization of Semiconductor Heterostructures and Nanostructures, ed. C. Lamberti and G. Agostini, Elsevier, Amsterdam, 2013, vol. 2, pp. 229–257.
Katerina Soulantica remarked: Considering that surface and sub-surface PdC is an important factor in Pd catalyzed hydrogenation, a core–shell configuration in which Pd is located on the shell and a carbide forming metal (for example Fe) in the core could be interesting. The core could act as a PdC carbide regulator (C "drain" or a C reservoir). Could such a core–shell configuration be helpful by adding a supplementary probe for operando studies?
Carlo Lamberti responded: As we have demonstrated, Pd is able to form both surface, sub-surface and bulk carbides (DOI: 10.1039/c7fd00211d).1 Thus, the introduction of another type of atom can be made to suppress the formation of carbide either in the core or in the shell of the NPs. This could explain why, in several types of reactions, alloyed catalysts, such as PdAg,2 PdZn,3 or PdPb4 demonstrate higher selectivity. Obviously, the idea suggested by you to have an external carbon source may be interesting but will increase the degree of complexity of the system. A more complex structure of the catalyst (Pd shell and Fe core) will definitely be an additional challenge for the operando studies. In such a case, X-ray absorption spectroscopy should be used at both the Pd K- and Fe K-edges. In most of the synchrotron radiation facilities, due to the huge difference in energy between the two edges (24.35 and 7.11 keV for Pd and Fe, respectively) the two edges cannot be measured with the same experimental set-up but must be done at two separate times. To the best of my knowledge the Rock beamline5 of the SOLEIL synchrotron is the only facility where the two edges could be measured almost simultaneously because the beamline is equipped with two sets of independent optics and two triplets of independent ionization chambers. Both optics are based on oscillating channel-cut monochromators that are foreseen to follow fast kinetics in quick-EXAFS mode with a time resolution in the sub-second range. Obviously, before starting the experiments on the Fe/Pd core/shell NPs, the monometallic iron carbide system must be investigated on both experimental and theoretical grounds. On the X-ray scattering ground, I'm expecting that small Fe/Pd NPs (with an additional fraction of carbide phase), are too disordered to provide analyzable Bragg peaks and only a proper analysis of the diffuse scattering could unravel the structure of such complex FeCz/PdHxCy core/shell NPs.
1 A. L. Bugaev, O. A. Usoltsev, A. A. Guda, K. A. Lomachenko, I. A. Pankin, Yu. V. Rusalev, H. Emerich, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, J. Phys. Chem. C , 2018, 122, 12029–12037
2 W. Huang, W. Pyrz, R. F. Lobo, and J. G. Chen, Appl. Catal. A: Gen., 2007, 333, 254–263.
3 M. W. Tew, H. Emerich and J. A. van Bokhoven, J. Phys. Chem. C, 2011, 115, 8457–8465.
4 J. Rajaram, A. P. S. Narula, H. P. S. Chawla and S. Dev, Tetrahedron, 1983, 39, 2315–2322.
5 V. Briois, C. La Fontaine, S. Belin, L. Barthe, T. Moreno, V. Pinty, A. Carcy, R. Girardot and E. Fonda, J. Phys.: Conf. Ser., 2016, 712, 012149.
Michael Bowker asked: I guess you used a temperature of 100 °C for your measurements since that is around the temperature used for hydrogenation. However, this poses some difficulties with respect to hydride and carbide formation/separation. If you went to higher temperatures could you isolate more carbide in the Pd? A number of reports in the literature suggest that x in PdCx is around 0.17, significantly higher than you have (∼0.05) and maybe higher temperatures would give a higher carbide presence in your experiment.
Carlo Lamberti answered: Yes, you are right, the 5 wt% Pd/C catalyst is product D1190 from the Chimet catalyst library (http://www.chimet.com/) used in this study (DOI: 10.1039/c7fd00211d) and usually operates for hydrogenation reactions of pharmaceutical interest in the 70–90 °C range. At 100 °C the catalyst is stable for a very long time. In few cases it has been successively employed at higher temperatures, where it undergoes a progressive sintering that is small, up to 150 °C but that becomes relevant and relatively fast at temperatures higher than 200 °C, that this catalyst should never reach. For hydrogenation reactions that require higher temperatures, e.g. the purification of terephthalic acid (270–290 °C), granular carbon is used as the support: 0.5 wt% Pd/C, type D3065 catalyst, supplied by Chimet, which can work in an industrial reactor for more than 2 years, if properly treated.1 As a consequence, the experiment has been performed at 100 °C, which represents the temperature where our study has not just an academic but also an industrial relevance. I agree with you that, for academic purposes, it would have been interesting to extend the study to higher temperatures and to reach higher carbon loadings of the palladium carbide phase. We did not do so because we were afraid of sintering: even a small sintering would have modified the XANES features (which are particle size-dependent2) and biased, in an uncontrolled way, our XANES analysis. Besides these practical aspects, there are some reasons that could be responsible for the lowering of the carbon concentration y in the PdCy nanoparticles. The most relevant one is the nanometric dimensions of the particles, which in the case of the palladium hydride phase are known to lead to much lower H/Pd ratios (DOI: 10.1039/c7fd00211d).3,4 This phenomenon can be explained by the fact that in the nanoparticles we have a considerable number of atoms forming an amorphous shell4 which could not host the same amount of C per each Pd atom as in the Pd bulk (DOI: 10.1039/c7fd00211d). As a final comment, our XANES simulations may overestimate or underestimate the effect of carbon on the spectral shape. This means that we can fully rely on the relative changes (i.e. increase or decrease of the C/Pd ratio) but the absolute values may change depending on the theoretical approach for simulation, convolution, etc. In this regard, a calibration with the XANES spectrum collected on a buck PdCy system with a known y stoichiometry would be of great help.
1 R. Pellegrini, G. Agostini, E. Groppo, A. Piovano, G. Leofanti and C. Lamberti, J. Catal., 2011, 280, 150–160.
2 J. Timoshenko, D. Y. Lu, Y. W. Lin and A. I. Frenkel, J. Phys. Chem. Lett., 2017, 8, 5091–5098.
3 A. L. Bugaev, A. A. Guda, K. A. Lomachenko, A. Lazzarini, V. V. Srabionyan, J. G. Vitillo, A. Piovano, E. Groppo, L. A. Bugaev, A. V. Soldatov, V. P. Dmitriev, R. Pellegrini, J. A. van Bokhoven and C. Lamberti, J. Phys.: Conf. Ser., 2016, 712, 012032.
4 A. L. Bugaev, A. A. Guda, K. A. Lomachenko, V. V. Shapovalov, A. Lazzarini, J. G. Vitillo, L. A. Bugaev, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, J. Phys. Chem. C, 2017, 121, 18202–18213.
Roy Johnston enquired: In your paper, the phase diagrams for the PdHx system (2.6 nm particles) show clear separation of the α and β phases, but you state that in a previous study of smaller (1 nm) particles (ref. 9) there is no α-β phase separation. Have you studied any intermediate sizes? If so, have you identified a critical particle size where α-β phase separation is first seen?
Carlo Lamberti responded: I agree with you that it would be very interesting to extend our study to at least two Pd/C systems with an average particle size distribution around 1 and 4 nm to complement the present one (where D = 2.6 ± 0.4 nm) (DOI: 10.1039/c7fd00211d), however such studies would be really meaningful only if the particle size distribution of the two new Pd/C systems would be sufficiently small to minimize the size overlap among the three distributions. So far, we have not performed such studies however, according to the present results (DOI: 10.1039/c7fd00211d), we expect that the distinguishable separation between the α and β phases should exist in particles that have a region with a crystalline fcc structure. In addition, it should be noted that initially the term “phase” was used only for macroscopic systems with a huge number of particles. Interestingly, similar phase separation has been recently observed even for an individual palladium nanoparticle.1
1 S. Syrenova, C. Wadell, F. A. A. Nugroho, T. A. Gschneidtner, Y. A. D. Fernandez, G. Nalin, D. Switlik, F. Westerlund, T. J. Antosiewicz, V. P. Zhdanov, K. Moth-Poulsen and C. Langhammer, Nat. Mater., 2015, 14, 1236–1244.
Katerina Soulantica asked: Could the use of D2 be of interest for your studies?
Carlo Lamberti replied: In all vibrational studies (IR, Raman, INS) the isotopic substitution (e.g.16O2 with 18O 2 or 12CO with 13CO) is indeed a powerful experimental tool to validate the assignment of the observed vibrational bands. Unfortunately, in the specific case of INS applied to diluted samples, such as the Pt/C catalyst investigated by Carosso et al. (DOI: 10.1039/c7fd00214a), the isotopic substitution of H2 with D2 is not applicable because the total bound scattering cross section1 of 2D is 27.6 barn, to be compared with 82.0 barn of 1H. This means that all vibrational modes related to H, would lose a factor 10 in intensity in the experiment performed with D2 and will probably be lost in the noise level.
1 V. F. Sears, Neutron News, 1992, 3, 29.
Federico Spolaore opened general discussion of the paper by Annette Trunschke: In your study you used nitrate salts, adsorbed them and thermally decomposed them to form metal nanoparticles. The average size of the support you used was within the range of ca. 250 and 500 nm. Would you expect that the size of the support itself could affect the size of particles during their formation and, eventually, when these nanoparticles are further treated at high temperatures?
Annette Trunschke responded: The size of the supported metal nanoparticles on average was smaller than 2 nm in all catalysts. In the used catalysts some bigger particles were also observed, but not larger than 20 nm. Therefore, we think that the size of the support particle has no impact on the size of the metal particles.
Graham Hutchings enquired: You introduced a theoretical approach to the design of catalysts in your presentation concerning the conversion of synthesis gas to ethanol and that no single metals are predicted on the basis of their assumptions to make ethanol and hence alloys could be the approach needed. In your case you have a Rh–Fe alloy. Is this an alloy that would have been consistent with the theoretical approach?
Annette Trunschke answered: The particular Rh–Fe alloy was not included in the cited work,1 but the authors discuss that a limited dataset was used and that a broader approach would result in further hits.
1 A. Medford, A. Lausche, F. Abild-Pedersen, B. Temel, N. Schjødt, J. Nørskov and F. Studt, Top. Catal., 2014, 57, 135–142.
Graham Hutchings commented: In scheme 1 you appear to have a composite catalyst with the FeRh alloy and FeMnOx. As FeMnOx can be a precursor to a Fischer Tropsch catalyst and this will lead to hydrocarbons being produced. Perhaps you need to decrease the amount of Fe and Mn in the catalysts and it might improve selectivity.
Annette Trunschke responded: The catalyst composition was optimized in terms of ethanol productivity (maximum approximately 30% yield). In our ongoing research we are trying to synthesize the Mn–Fe sub-oxide in the absence of Rh to analyze the reactivity of this catalyst component separately.
David Marchant asked: Do you have an explanation for why co-impregnation yields a more active catalyst than a sequential impregnation approach, in this particular case?
Annette Trunschke answered: The sequentially prepared catalysts were not analyzed in such detail, but co-impregnation resulted in a more homogeneous distribution of all elements on the surface of the support.
Andrea Russell enquired: I found your use of a laboratory based source for the XANES measurements presented in Fig. 2 very interesting. For the benefit of the audience and readers of this discussion, would you please comment on why you chose to use this source and how long it took to collect the spectra?
Annette Trunschke replied: Beam damage is an issue that complicates the analysis of supported nanoparticles, in particular manganese species. In the laboratory-based X-ray absorption experiment we minimized the exposure of the catalyst to X-rays, however, longer accumulation times of approximately 8 h have to be taken into account.
Carlo Lamberti commented: I confirm that useful XAS experiments can be performed on laboratory instrumentation. As an example, in the Smart Materials Research center of the Southern Federal University (Russia) where I’m the scientific director, we have a laboratory XAS spectrometer.1 It can be used to collect in situ XANES spectra that do not have the energy resolution of those collected at the synchrotron sources, but that are still informative on oxidation and coordination state of the selected element. We have even been able to run an operando electrochemical reaction following the charge and the discharge of a Mn-containing Li-battery.
1 http://nano.sfedu.ru/structure/facilities/rigaku-r-xas/.
Michele Carosso asked: My question refers to the FT-IR spectra of CO adsorbed on the un-promoted Rh/SiO2 catalyst and on the promoted ones (both Rh–Mn/SiO2 and Rh–Mn–Fe/SiO2). You showed that the absorption bands due to CO adsorbed on the metal phase are much more intense for the un-promoted catalyst, concluding that, in this case, the metal surface area available for CO adsorption is larger. While the conclusion sounds likely, it is also known that CO is not an innocent probe towards metal surfaces, and in particular towards Rh surfaces. As a matter of fact, CO may lead to the fragmentation of the Rh nanoparticles via the formation of Rh-carbonyls and to a consequent overestimation of the metal surface area. I'm wondering if you considered this point during the analysis of your FT-IR spectra?
Annette Trunschke answered: Fragmentation of the Rh nanoparticles did essentially not occur on the present catalysts in the experiments performed at 313 K. Only very small contributions of Rh+(CO)2 species to the spectrum of CO adsorbed on Rh/SiO2 were observed at 2095 cm−1 and 2015 cm−1. Geminal dicarbonyl species were not formed on the promoted catalysts. These species were also not present under reaction conditions.
Michael Bowker enquired: You refer to higher alcohol synthesis as a basis for this work, and this is a case where there is a need for the bifunctional nature of the catalyst. It needs to be able to both dissociate CO and to adsorb it molecularly, since ethanol contains both a dissociated and associated CO molecule. You showed IR where the bridge site disappeared, but maybe we do need the bridge site as a dissociation centre? So is it a good idea to look to remove the bridge site?
Annette Trunschke responded: We observed that the very active and selective catalysts do not adsorb CO in a bridged configuration on Rh. Therefore we postulated that the active sites might be located on the promoter sub-oxides or on the interface between Rh and the promoter-suboxides. Perhaps hydrogen-assisted C–O dissociation is involved in the mechanism. Further experiments are necessary to verify this postulation experimentally.
Francesca Baletto asked: We are wondering whether your technological technique can detect the chemical composition at the interface cluster/oxide. Indeed, we have predicted a considerable improvement of the adsorption property of MgO-supported PtNi clusters1 when a few Ni atoms are in contact with the substrate. We would like to see any experimental evidence supporting this finding.
1 Asara et al., ACS Catal., 2016, 6, 4388–4393.
Annette Trunschke answered: This can be studied by electron microscopy, albeit the task is challenging due to the charging of MgO.
Bruce Gates said: Can you comment on the reproducibility of these complex catalysts you have investigated in terms of the characterization and performance data?
Annette Trunschke replied: Characterization and testing exhibited high reproducibility. The catalysts were also tested over long times on stream (> 800 h) and under varying reaction conditions.
Joseph Macginley enquired: What is the importance of the Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn ratio in these catalysts?
:Mn ratio in these catalysts?
Annette Trunschke responded: Our main interest was directed at the influence of the individual promoter elements, Mn and Fe, on the nanostructure of the Rh particles. Therefore, we investigated only optimized catalyst compositions using spectroscopy and microscopy.
Nicola Irvine asked: Do you use any alkali components in your Rh–Fe/Mn catalyst, as alkali dopants have been known to enhance oxygenate formation? If so, how does that affect the selectivity to ethanol or any other oxygenated product? If not, how do you think an alkali component may influence your activity and selectivity in the CO hydrogenation?
Annette Trunschke answered: We did not investigate alkali addition, because this would have made our systems even more complex. A positive effect of alkali on oxygenate formation over Rh catalysts has been reported and some authors argued that the presence of alkali induces a weakening of the strength of CO adsorption, which would be in agreement with our observation that strongly adsorbed CO molecules poison the catalyst.
Andrea Russell opened general discussion of the paper by Michele Carosso: The INS data you have shown for the 5 wt% Pt/C catalyst is really lovely and you’ve clearly benefited from the advances in the spectrometer as you’ve described in your paper. This system has been examined using INS previously, but for more highly loaded catalysts (see the references to papers by Parker, Mitchell, and Ramirez-Cuetsa in the manuscript). Other than being the more typical industrial catalyst, is there any advantage to looking at the lower loaded material? Do you expect the more highly loaded material to show evidence of cooperative effects between adjacent particles and if so, do you see evidence of this when you compare your results to those published previously?
Michele Carosso replied: We selected a low loaded Pt sample of relevance for real industrial applications. From a data analysis point of view, a lower Pt loading implies weaker features for chemisorbed hydrogen species, which can represent a problem in terms of signal-to-noise ratio. However, this was not a problem in our case. Indeed, our spectra have a signal-to-noise ratio comparable (or even better) to those reported decades ago by Parker,1–3 Mitchell4 and Ramirez-Cuesta,5 and all of them collected on samples containing no less than 40 wt% Pt, clearly as a consequence of the improvements in the INS instruments in terms of both neutron fluxes on the sample and detector efficiency. From a chemical perspective, the metal loading would influence the dispersion and the average particle size of the metal NPs, and therefore also the type of hydrides present on the particles. Moreover, for a more loaded Pt/C catalyst under reaction conditions sintering phenomena may occur, especially considering that carbon has a weak interaction with the metal NPs.
1 P. Albers, E. Auer, K. Ruth and S. F. Parker, J. Catal., 2000, 196, 174–179.
2 P. W. Albers, M. Lopez, G. Sextl, G. Jeske and S. F. Parker, J. Catal., 2004, 223, 44–53.
3 S. F. Parker, C. D. Frost, M. Telling, P. Albers, M. Lopez and K. Seitz, Catal. Today, 2006, 114, 418–421.
4 P. C. H. Mitchell, A. J. Ramirez-Cuesta, S. F. Parker and J. Tomkinson, J. Mol. Struct., 2003, 651–653, 781–785.
5 A. J. Ramirez-Cuesta, P. C. H. Mitchell, S. F. Parker, J. Tomkinson and D. Thompsett, Stud. Surf. Sci. Catal., 2001, 138, 55–60.
Andrea Russell asked: In your discussion of Fig. 2 in your paper you conclude that "these data suggest that at least some of the platinum nanoparticles in the Pt/C sample sit at the platelets’ edges, preferentially at the regular edges of the sp3 graphitic domains ...". What are the implications of this observation for those who are attempting to use graphene flakes as a support material for Pt or other metal nanoparticles?
Michele Carosso responded: The location of Pt (or other metals) NPs on the support may have a great influence on their reactivity. For example, the edges of the graphitic domains in activated carbons may contain more reactive sites with respect to the extended faces. These reactive sites may be involved in the catalysis, in strong cooperation with the metal NPs. As an example, unsaturated carbon radical species are known to be present at the edges of the graphitic domains. In the presence of hydrogen, the Pt NPs located at the edges of the carbon platelets may promote the transfer of atomic hydrogen from the Pt surface to the carbon edges (spillover effect).1–8 This is the reason why metal-doped carbonaceous materials are widely studied as promising systems for hydrogen storage.9–17 The same concept has to be applied for metal NPs supported on graphene flakes.
1 R. Bhowmick, S. Rajasekaran, D. Friebel, C. Beasley, L. Jiao, H. Ogasawara, H. Dai, B. Clemens and A. Nilsson, J. Am. Chem. Soc., 2011, 133, 5580–5586.
2 C. I. Contescu, C. M. Brown, Y. Liu, V. V. Bath and N. C. Gallego, J. Phys. Chem. C, 2009, 113, 5886–5890.
3 P. C. H. Mitchell, A. J. Ramirez-Cuesta, S. F. Parker, J. Tomkinson and D. Thompsett, J. Phys. Chem. B, 2003, 107, 6838–6845.
4 A. J. Ramirez-Cuesta, P. C. H. Mitchell, S. F. Parker, J. Tomkinson and D. Thompsett, Stud. Surf. Sci. Catal., 2001, 138, 55–60.
5 C. Tsao, Y. Liu, M. Li, Y. Zhang, J. B. Leao, H. Chang, M. Yu and S. Chen, J. Phys. Chem. Lett., 2010, 1, 1569–1573.
6 C. Tsao, Y. Liu, H. Chuang, H. Tseng, T. Chen, C. Chen, M. Yu, Q. Li, A. Lueking and S. Chen, J. Phys. Chem. Lett., 2011, 2, 2322–2325.
7 R. Zacharia, S. Rather, S. W. Hwang and K. S. Nahm, Chem. Phys. Lett., 2007, 434, 286–291.
8 G. M. Psofogiannakis and G. E. Froudakis, J. Phys. Chem. C, 2009, 113, 14908–14915.
9 D. S. Pyle, E. M. Gray and C. J. Webb, Int. J. Hydrogen Energy, 2016, 41, 19098–19113.
10 P. Benard and R. Chahine, Scr. Mater., 2007, 56, 803–808.
11 A. C. Chien and S. S. C. Chuang, Int. J. Hydrogen Energy, 2011, 36, 6022–6030.
12 D. Giasafaki, G. Charalambopoulou, C. Tampaxis, D. M. Gattia, A. Montone, G. Barucca and T. Steriotis, J. Alloys Compd., 2015, 645, S485–S489.
13 S. M. Lee and Y. H. Lee, Appl. Phys. Lett., 2000, 76, 2877–2879.
14 Y. Li and R. T. Yang, J. Phys. Chem. C, 2007, 111, 11086–11094.
15 S. Park and S. Lee, Int. J. Hydrogen Energy, 2010, 35, 13048–13054.
16 A. L. M. Reddy and S. Ramaprabhu, Int. J. Hydrogen Energy, 2008, 33, 1028–1034.
17 L. Wang and R. T. Yang, J. Phys. Chem. C, 2008, 112, 12486–12494.
Philip R. Davies enquired: Do you have any idea of the proportion of sp2/sp3 carbon in your sample? When you introduce the platinum metal using impregnation, are you introducing any new oxygen functionality? Can you rule out the possibility that it is such new oxygen functional groups that are responsible for the loss of the edge hydrogen atoms you have observed? Do you see any other functional groups in your INS spectra?
Michele Carosso replied: The carbon used as the catalyst support in this work was previously characterized in terms of morphological, structural and surface properties, employing a wide range of techniques.1 In particular, Raman spectroscopy has the potential to reveal the proportion of sp2 and sp3 carbon fractions. Fig. 4a shows the Raman spectrum of the activated carbon, collected with the laser line at 514 nm as the exciting source: two intense bands at 1605 cm−1 and 1350 cm−1 (labelled as G and D, respectively) dominate the spectrum, and both of them are attributed to sp2 domains. The G band refers to bond stretching of pairs of sp2 carbon atoms2–4, while the D band is due to a lattice breathing mode, forbidden in ideal graphitic crystals but activated by structural disorder.2–7 The very weak feature around 1150 cm−1 (labelled as I band) originates from amorphous carbon. The absence of sp3 carbon is confirmed by the UV Raman spectrum (Fig. 4b, laser line at 244 nm) that, exciting both the π and the σ states, is able to probe both the sp2 and the sp3 carbon species. The spectrum shows only sp2 species. Concerning the second question, I agree with you that the changes in the INS spectra might be, at least in part, due to the introduction of oxygen functional groups at the edges of the graphitic domains. To clarify this point a strategy could be to measure, by using INS, the same carbon subjected to the same chemical treatment adopted during Pt impregnation, but without the Pt precursor. Finally, regarding the observation of other functional groups by INS, unluckily (or luckily, it depends on what you want to measure), hydrogen-containing species dominate the whole INS spectrum.8 Hence, only functional groups containing hydrogen can be easily detected. According to DFT calculation, a sharp band at 585 cm−1 in the INS spectra (Fig. 2 in the paper) could be attributed to COOH groups.9
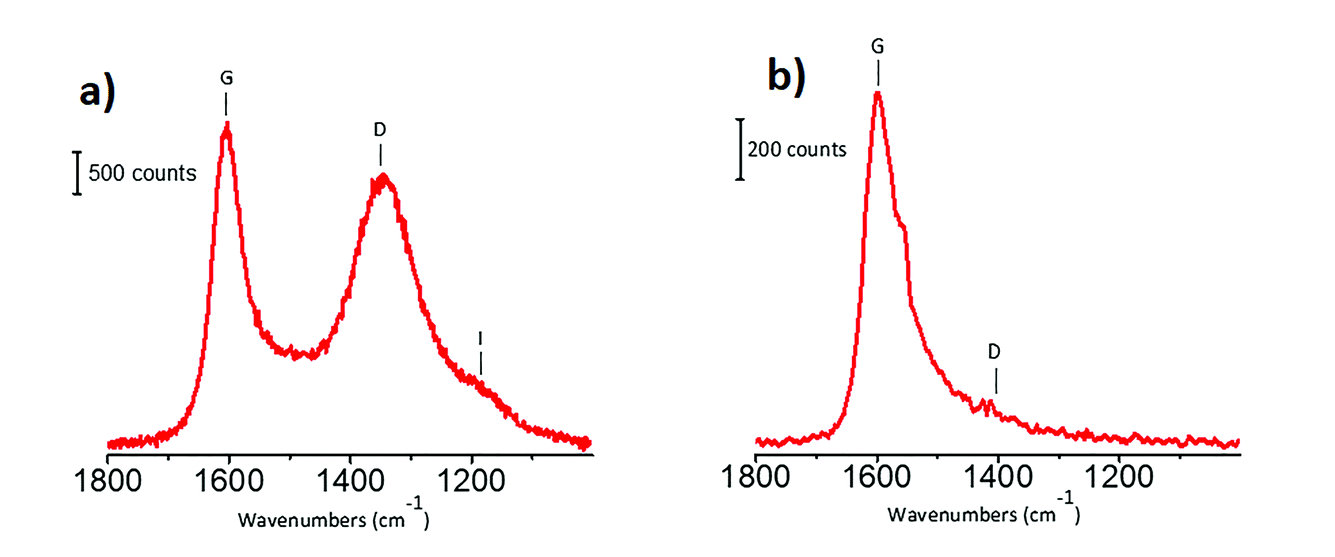 | ||
| Fig. 4 Raman spectra of the carbon support, collected with the laser line at 514 nm (part a) and at 244 nm (part b) as exciting sources. Previously unpublished figure, reporting data published in ref. 1. | ||
1 A. Lazzarini, A. Piovano, R. Pellegrini, G. Leofanti, G. Agostini, S. Rudic, M. R. Chierotti, R. Gobetto, A. Battiato, G. Spoto, A. Zecchina, C. Lamberti and E. Groppo, Catal. Sci. Technol., 2016, 6, 4910–4922.
2 K. D. Henning and H. von Kienle, Ullmann’s Encyclopedia of Industrial Chemistry, 2010.
3 R. Schlogl, Handbook of heterogeneous catalysis, 2008, 1, 357.
4 H. Marsh and F. Rodriguez-Reinoso, Activated carbon, Elsevier Science, Oxford, UK, 2006, p. 13.
5 C. Castiglioni, M. Tommasini and G. Zerbi, Philos. Trans. R. Soc. A, 2004, 362, 2425.
6 M. Tommasini, C. Castiglioni. G. Zerbi, A. Barbon and M. Brustolon, Chem. Phys. Lett., 2011, 516, 220.
7 A. C. Ferrari, Solid State Commun., 2007, 143, 47.
8 V. F. Sears, Methods Exp. Phys., 1986, 23, 521–550.
9 A. Piovano, A. Lazzarini, R. Pellegrini, G. Leofanti, G. Agostini, S. Rudic, A. L. Bugaev, C. Lamberti and E. Groppo, Adv. Condens. Matter Phys., 2015, 2015, 803267.
Michael Bowker said: My question relates to the graphite support and edge sites; you say you treat it at high temperature in air, then cool in air. You might expect a reaction at the C edge sites to adsorb something from the air e.g. water or oxygen/CO2, do you have clear evidence that the edge sites are completely clean?
Michele Carosso answered: I said that the carbon support was steam-treated at high temperature, this relates to the procedure to obtain an activated carbon from the raw material (wood in this case). Concerning the catalyst pre-treatment prior to the INS measurement, it was conducted at 120 °C in dynamic vacuum, in order to eliminate the physisorbed water. The 120 °C treatment was prolonged until a residual pressure of 10−3 mbar was reached. Then the sample was inserted in an Al sample holder by means of an Ar-filled glovebox (Al is an ideal sample holder for INS, owing to its negligible stotal (1.50 barn).1 Considering the pre-treatment and the adopted precautions in order to avoid air contamination, we believe that our sample was clean. This is confirmed by the INS spectra that do not show any water on the sample.
1 V. F. Sears, Methods Exp. Phys., 1986, 23, 521–550.
Jonathan Quinson asked: Are you using the same approach for the investigation of other metals (e.g. Pd), other carbon supports and maybe even other supports? Since the technique is very dependent to the properties of hydrogen, but given the improvement of the technique you mentioned, do you see it being more widely used in the short or long term future, possibly for molecules other than H2?
Michele Carosso responded: Concerning the first question, we have recently employed INS spectroscopy to investigate two different activated carbons and the related Pd-based catalysts.1 Both carbons are of wood-origin, but activated in a different way: CW (the same carbon employed as the support in the preparation of the here studied 5 wt% Pt/C catalyst) is activated in steam, while CChemi is activated in H3PO4. The INS spectra of the two carbons are shown in Fig. 5a. The spectrum of CChemi is almost double the intensity with respect to that of CW, but the shape of the spectra is very similar. This means that the nature of the hydrogen species located at the edges of the sp2 graphitic domains and their relative abundance are the same for the two differently treated carbons, but hydrogen species are more abundant in CChemi. This was attributed to the lower sizes of sp2 graphitic domains in CChemi with respect to CW, induced by the acid treatment.1 Both carbons were employed as supports for Pd-based catalysts (Pd/CW and Pd/CChemi). Fig. 5b compares the INS spectrum of CW with that of the corresponding catalyst Pd/CW. Also in this case, as for the Pt/C catalyst investigated in this work, the peaks associated to the C–H in-plane and out-of-plane bending modes are slightly less intense in the spectrum of the catalyst, indicating that at least a fraction of the C–H terminations are involved in the Pd deposition.1 In particular, the band at 880 cm−1, indicative of regular borders, is the most affected one, indicating that the Pd NPs are mainly located at the regular edges of the sp2 graphitic domains.2,3 A similar trend was observed also for the Pd/CChemi catalyst. Finally, considering that most of the supports are almost transparent to neutrons, INS can be used to characterize many metal-supported catalysts. For example, we have investigated also a 5 wt% Pt/Al2O3 catalyst, that will be the subject of a successive paper. Regarding the second question, in principle INS can be employed to study every adsorbed molecule as all nuclei contribute, with no selection rules, to the overall INS spectrum. However, we have to remember that the main parameter determining the intensity of the INS spectrum is the scattering cross section of the nuclei present in the chosen probe molecule: that is for 1H one order of magnitude greater with respect to all other nuclei (the scattering cross section of 1H is 82.0 barn).4 This implies that the technique is still very suitable for molecules having a large fraction of hydrogen atoms, such as CH4, NH3, C2H4, etc. Conversely, if the selected molecule does not contain hydrogen atoms (CO, NO, CO2, etc.) and if the investigated sample contains a non-negligible amount of hydrogen atoms, then the INS spectrum will be dominated by hydrogen features, that most likely will obscure the vibrational features of the probe molecule.
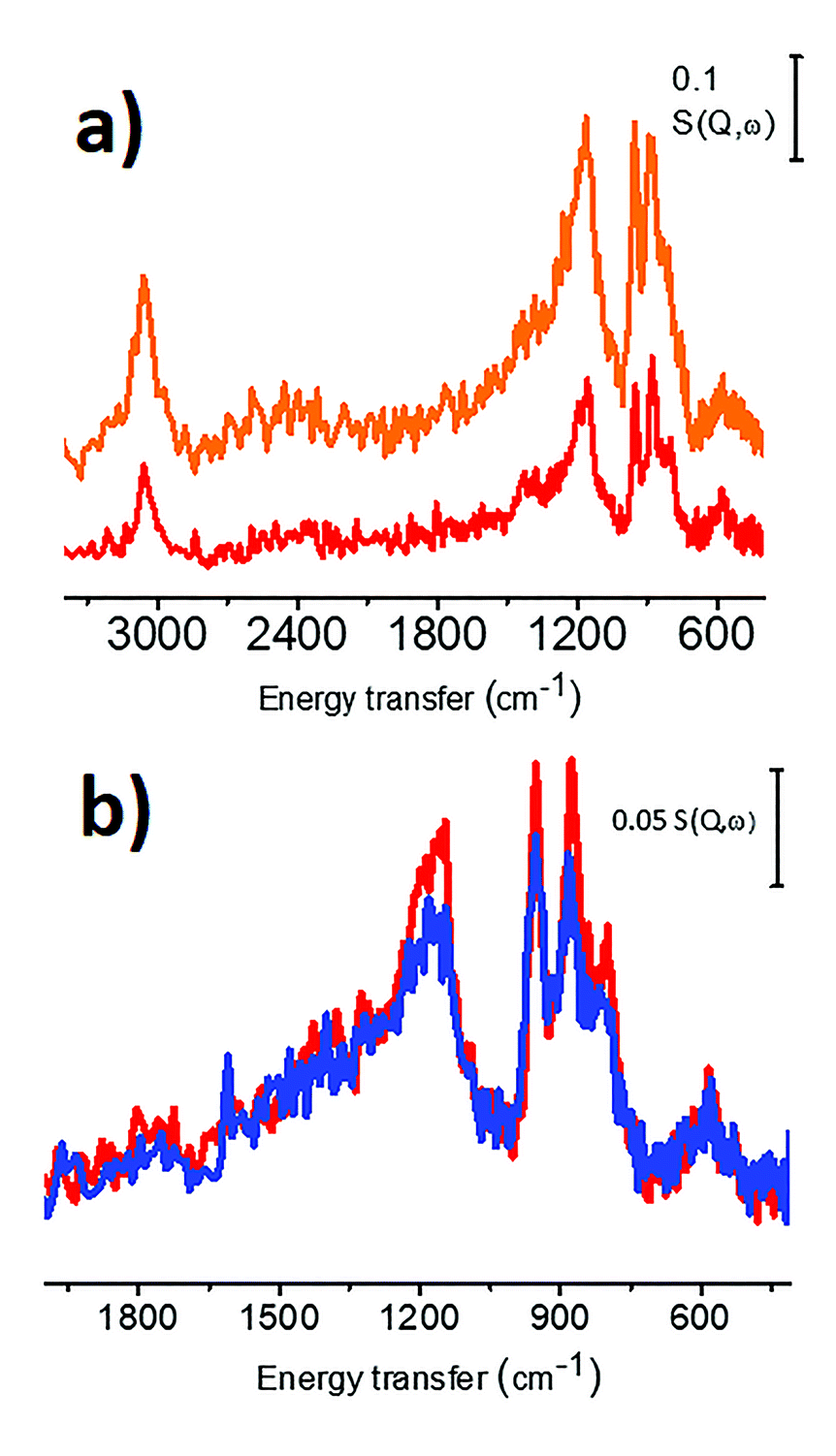 | ||
| Fig. 5 Part (a) INS spectra of Cw (red spectrum) and CChemi (orange spectrum). Part (b) INS spectra of Cw (red) and of the corresponding Pd/Cw catalyst (blue). Previously unpublished figure, reporting data published in ref. 1. | ||
1 A. Lazzarini, A. Piovano, R. Pellegrini, G. Leofanti, G. Agostini, S. Rudic, M. R. Chierotti, R. Gobetto, A. Battiato, G. Spoto, A. Zecchina, C. Lamberti and E. Groppo, Catal. Sci. Technol., 2016, 6, 4910–4922.
2 P. W. Albers, J. Pietsch, J. Krauter and S. F. Parker, Phys. Chem. Chem. Phys., 2003, 5, 1941.
3 A. Piovano, A. Lazzarini, R. Pellegrini, G. Leofanti, G. Agostini, S. Rudic, A. L. Bugaev, C. Lamberti and E. Groppo, Adv. Condens. Matter Phys., 2015, 2015, 803267.
4 V. F. Sears, Methods Exp. Phys., 1986, 23, 521–550.
David Willock commented: The images of the graphitic edge sites you show are always zig-zag in nature. There is also the possibility of armchair termination of a graphitic sheet1 as shown in Fig. 6. We have previously considered armchair terminations as more easily functionalised for the improved adhesion of metal particles.2
 | ||
| Fig. 6 Armchair termination of a graphitic sheet. | ||
1 A. P. Seitsonen, A. M. Saitta, T. Wassmann, M. Lazzeri, F. Mauri, Phys. Rev. B, 2010, 82, 115425.
2 P. R. Davies, R. Burgess, C. Buono, R. J. Davies, T. Legge, A. Lai, R. Lewis, D. J. Morgan, N. Robinson, D. J. Willock, J. Catal., 2015, 323, 10.
Michele Carosso responded: You are perfectly right. The TOC image shows a zig-zag terminated graphitic sheet. This was done for simplicity, and perhaps does not fully represent the real case, where also arm-chair terminated borders are present. The INS spectra reported in our work (DOI: 10.1039/c7fd00214a) indicate that all the borders (regular and irregular, zig-zag and arm-chair) are affected by Pt deposition. However, the C–H band most affected is that located at 880 cm−1, which is attributed to regular zig-zag borders. This is why we conclude that the Pt NPs are mainly located at the zig-zag edges.
Carlo Lamberti commented: The “solo”, “duo” and “trio” nomenclature has been given in the specialized literature1–4 to out-of-plane C–H bending modes of specific structures located at the borders of the sp2 domains, depending on the number of adjacent aromatic C–H groups which vibrate out-of-plane in the fused rings.
1 M. Zander, Polycycliche aromaten, Teubner, Stuttgart, 1995.
2 A. Centrone, L. Brambilla, T. Renouard, L. Gherghel, C. Mathis, K. Mullen and G. Zerbi, Carbon, 2005, 43, 1593–1609.
3 A. Piovano, A. Lazzarini, R. Pellegrini, G. Leofanti, G. Agostini, S. Rudit, A. L. Bugaev, C. Lamberti and E. Groppo, Adv. Condens. Matter Phys., 2015, 2015, 803267.
4 A. Lazzarini, R. Pellegrini, A. Piovano, S. Rudic, C. Castan-Guerrero, P. Torelli, M. R. Chierotti, R. Gobetto, C. Lamberti and E. Groppo, Catal. Sci. Technol., 2017, 7, 4162–4172.
Katerina Soulantica asked: Would an exchange of H2 by D2 help to distinguish between free, chemisorbed and physisorbed species?
Michele Carosso answered: Analogously to the optical spectroscopies (IR and Raman), also in INS the energy of a vibrational motion depends on the mass of the oscillator (the same vibrational mode involving 1H will be located at a higher energy than that involving 2H); this means that sending D2 over an H2-loaded sample can in principle help in discriminating weakly-bonded to strongly-bonded hydrogen species. However, attention must be paid to the problem related to the band intensities. The intensity of an INS band is directly proportional to the total bound scattering cross sections (stotal) of the nuclei involved in the vibration: stotal of 1H is one order of magnitude greater than that of 2H (82.0 barn and 7.64 barn, respectively).1 Hence, the vibrational mode of a species involving hydrogen that has been replaced by deuterium, will appear in the INS spectrum, at a different frequency, but with a much lower intensity, which makes its detection questionable. Replacing in a selective way some hydrogen species with deuterium could be useful in order to weaken their INS features and hence to highlight some other hydrogen species that we want to focus on.
1 V. F. Sears, Methods Exp. Phys., 1986, 23, 521–550.
Stanley Lai enquired: Can you speculate or hypothesise on the effect of the different H-species on the activity of the catalysts? For example, do you expect all species to play a role under catalytic conditions, where there will be competitive adsorption from the reactant, product and, for solution-phase reactions, solvent molecules? This competitive adsorption may be a particularly important consideration for the weaker, physisorbed H-species. Similarly, would you expect to see different regimes in reactivity, similarly to different regimes in H-species (Fig. 3)?
Michele Carosso responded: Based on INS data only, we cannot provide any speculation on the reactivity of the different hydrogenous species formed upon H2 adsorption on our 5 wt% Pt/C catalyst, at least for two reasons: (1) the INS measurements are performed at 20 K, and at this temperature no reaction would occur; (2) a single spectrum takes several hours to be collected, preventing the study of Pt-hydride dynamics. Hence, INS spectroscopy is a powerful tool for the quantitative investigation of the Pt-hydride species, but it provides a “static” picture of the studied system. However, we have experimental evidences on that: (1) the relative abundance of the Pt hydrides changes with the hydrogen coverage, i.e. the Pt-hydrides have a dynamic behavior as a function of the hydrogen pressure and (2) only a fraction of the Pt-hydrides are involved in catalysis. These evidences are derived from IR experiments performed on a similar 5 wt% Pt/Al2O3 catalyst, and have not been published yet.
Revana Chanerika said: You have hydrogen chemisorbed over steps and corners as the defect sites. What evidence do you have (or have you imaged various areas using miscoscopy techniques) to safely conclude that there are no kinks present where there could be chemisorption of hydrogen occurring as well?
Michele Carosso replied: According to TEM analysis, the 5 wt% Pt/C catalyst studied in this work is characterized by Pt NPs with an average particle size of ca. 2 nm. So small NPs surely contain many defects on the surfaces, including steps and corners, as well as kinks. The presence of such defects can be highlighted by means of FT-IR spectroscopy of adsorbed CO, which is a method that has been largely employed in the past to characterize both single crystal Pt surfaces and supported Pt NPs.1–8 The interaction of CO with the Pt surface is dominated by the back-donation of electron density from the d-orbitals of the metal to the 2π* orbital of CO. The extension of this phenomenon depends on the coordination of the adsorption site, being greater for low-coordinated (i.e. more defective) adsorption sites. Hence, the analysis of the ν(CO) absorption bands provides a qualitative indication of the adsorption sites present on Pt surfaces. Unfortunately, the application of this method to carbon-supported catalysts is hampered by the highly absorbing nature of the carbon support. However, we employed this approach to investigate the surface properties of a 5 wt% Pt/Al2O3 catalyst, that, according to TEM and CO chemisorption experiments, is very similar to the 5 wt% Pt/C investigated in the present work. Fig. 7 shows the FT-IR spectrum of CO adsorbed at room temperature on that catalyst, in the 2125–1950 cm−1 range, which includes the interval typical for linearly adsorbed CO on Pt surfaces. The sharp absorption band centred at 2087 cm−1 is assigned to CO linearly adsorbed on Pt terrace sites,1–8 while the series of bands in the 2070–1950 cm−1 range is related to CO linearly adsorbed on a series of low-coordinated Pt defect sites, among which are steps, corners and kinks. All of these defect sites are potential adsorbing sites for hydrogen. However, it is extremely difficult to discriminate by using INS between atomic hydrogen chemisorbed on the different Pt defect sites. In conclusion, we cannot exclude the presence of chemisorbed hydrogen also on kinks.
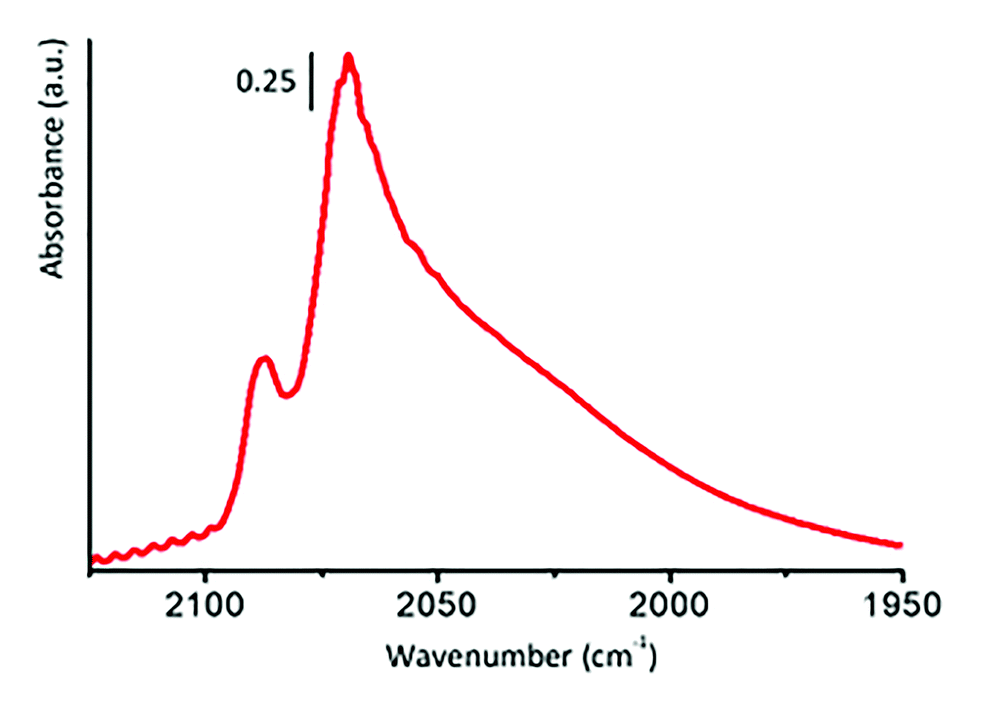 | ||
| Fig. 7 FT-IR spectrum of CO adsorbed at room temperature on a 5 wt% Pt/Al2O3 catalyst similar to the Pt/C investigated in this work. | ||
1 A. Crossley and D. A. King, Surf. Sci., 1977, 68, 528–538.
2 B. E. Hayden and A. M. Bradshaw, Surf. Sci., 1983, 125, 787–802.
3 B. E. Hayden, K. Kretzschmar, A. M. Bradshaw and R. G. Greenler, Surf. Sci., 1985, 149, 394–406.
4 F. M. Hoffmann and A. M. Bradshaw, J. Catal., 1976, 44, 328–331.
5 K. Horn and J. Pritchard, J. Phys., 1977, 4, 164–171.
6 H. J. Krebs and H. Lueth, Appl. Phys., 1977, 14, 337–342.
7 R. A. Shigeishi and D. A. King, Surf. Sci., 1976, 58, 379–396.
8 D. M. Haaland, Surf. Sci., 1987, 185, 1–14.
Bruce Gates asked: Would you please share your perspectives on what’s new in XANES for catalyst characterization?
Carlo Lamberti responded: From a historical point of view, already at the end of the fifties, XANES spectroscopy had been used to benchmark many transition metal compounds, classifying their spectra according to the atomic structure and valence of the metal element in the compound. In that context the chemical shift of the edge with valence of the metal was observed. This fingerprint classification was used by Van Nordsthand to identify the structural/valence form of elements in catalysts,1 which are usually so highly dispersed that their X-ray diffraction patterns cannot be measured. This means that a decade before the correct theory of EXAFS was established,2 XANES had already been able to provide relevant information on materials of relevance in catalysis. Then, for several decades, the level of understanding of the XANES spectra didn’t improve significantly. Starting from the early nineties, the first reliable theoretical simulations of the XANES spectra appeared.3,4 Nowadays, very accurate codes are available based on either the full multiple scattering5–9 or the finite difference method,10,11 the latter allowing us to go beyond the muffin tin approximation. With these advanced codes it is possible to perform structural refinement starting from an initial estimated cluster (even of large size) and modifying the type of ligands, number of ligands, bond distances and bond angles until the best agreement is obtained between experiment and theory.11–16 Moreover, machine learning approaches are being actively developed17 and may result in a real revolution in XANES data analysis in some years or decades.
Finally, advanced multivariate analysis (e.g., Multivariate Curve Resolution, MCR) of in situ/operando XANES datasets, in combination with DFT-assisted simulation of XANES spectra, can be used to understand the number and the nature of different species present in a catalyst and to follow their mutual transformation along an activation process or a reaction path.18 These approaches have great potential in going beyond the inherent limitations dictated by the average character of the XANES technique, facilitating the discrimination among active and spectator absorber-containing species in a working catalyst.
The quantitative structural determination by simulation of XANES spectra from the DFT optimized structure is the only X-ray based available approach in cases where the catalytic active site is highly diluted (preventing EXAFS spectra to be collected with sufficiently high signal to noise ratio), a scenario that is quite common in catalysis. The same holds for more concentrated catalysts monitored under operando conditions in cases where XAS spectra must be collected with a very short acquisition time to follow the kinetics of the reaction. Indeed, modern beamlines equipped with oscillating channel-cut monochromators and fast ionization chambers offer time-resolution down to 10 ms.19,20 20 ms time-resolution has been recently achieved using new fluorescence-detected XAS setup.21 For energy-dispersive beamlines, even 100 ps time-resolution was reported.22
I conclude this brief perspective by underlining two directions where I foresee remarkable improvements in the understanding of a working catalyst. The first is the development of the operando setups equipped with thin SiC or SiN membrane windows able to operate in the soft X-ray range up to ambient pressure,23,24 making the L- and M-edge of metals accessible, as well as the K-edges of relevant light elements (C, O, N, etc.). The second concerns the development of X-ray emission spectroscopy (XES), which probes the occupied density of states (DOS), nicely complementing the unoccupied-DOS usually measured in XANES experiments. The combination of XANES/XES experiments provides the complete DOS characterization of the system.25–29 The relevance of such combined XANES/XES experiments is so important that more and more beamlines at synchrotrons are starting to be equipped with XES spectrometers.
1 R. A. Van Nordsthand, Adv. Catal., 1960, 12, 149–187.
2 D. E. Sayers, E. A. Stern and F. W. Lytle, Phys. Rev. Lett., 1971, 27, 1204–1207.
3 A. Bianconi, J. Garcia, M. Benfatto, A. Marcelli, C. R. Natoli and M. F. Ruizlopez, Phys. Rev. B, 1991, 43, 6885–6892.
4 Z. Y. Wu, M. Benfatto and C. R. Natoli, Phys. Rev. B, 1992, 45, 531–534.
5 M. Benfatto, A. Congiu-Castellano, A. Daniele and S. D. Longa, J. Synchrot. Radiat., 2001, 8, 267–269.
6 M. Benfatto, S. Della Longa and C. R. Natoli, J. Synchrot. Radiat., 2003, 10, 51–57.
7 J. J. Rehr and A. L. Ankudinov, Coord. Chem. Rev., 2005, 249, 131–140.
8 J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Phys. Chem. Chem. Phys., 2010, 12, 5503–5513.
9 J. J. Kas, K. Jorissen and J. J. Rehr, in X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, ed. J. A. van Bokhoven and C. Lamberti, John Wiley and Sons, Chichester, UK, 2016, pp. 51–72.
10 Y. Joly and S. Grenier, in X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, ed. J. A. van Bokhoven and C. Lamberti, John Wiley and Sons, Chichester, UK, 2016, pp. 73–97.
11 S. A. Guda, A. A. Guda, M. A. Soldatov, K. A. Lomachenko, A. L. Bugaev, C. Lamberti, W. Gawelda, C. Bressler, G. Smolentsev, A. V. Soldatov and Y. Joly, J. Chem. Theory Comput., 2015, 11, 4512–4521.
12 D. Gianolio, E. Groppo, J. G. Vitillo, A. Damin, S. Bordiga, A. Zecchina and C. Lamberti, Chem. Commun., 2010, 46, 976–978.
13 E. Borfecchia, S. Maurelli, D. Gianolio, E. Groppo, M. Chiesa, F. Bonino and C. Lamberti, J. Phys. Chem. C, 2012, 116, 19839–19850.
14 Y. Tulchinsky, C. H. Hendon, K. A. Lomachenko, E. Borfecchia, B. C. Melot, M. R. Hudson, J. D. Tarver, M. D. Korzynski, A. W. Stubbs, J. J. Kagan, C. Lamberti, C. M. Brown and M. Dinca, J. Am. Chem. Soc., 2017, 139, 5992–5997.
15 E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Chem. Sci., 2015, 6, 548–563.
16 C. Barzan, A. Piovano, L. Braglia, G. A. Martino, C. Lamberti, S. Bordiga and E. Groppo, J. Am. Chem. Soc., 2017, 139, 17064–17073.
17 J. Timoshenko, D. Y. Lu, Y. W. Lin and A. I. Frenkel, J. Phys. Chem. Lett., 2017, 8, 5091–5098.
18 A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Chem. Sci., 2017, 8, 6836–6851.
19 O. Müller, M. Nachtegaal, J. Just, D. Lützenkirchen-Hecht and R. Frahm, J. Synchrotron Rad., 2016, 23, 260–266.
20 V. Briois, C. La Fontaine, S. Belin, L. Barthe, T. Moreno, V. Pinty, A. Carcy, R. Girardot and E. Fonda, J. Phys.: Conf. Ser., 2016, 712, 012149.
21 A. A. Guda, A. L. Bugaev, R. Kopelent, L. Braglia, A. V. Soldatov, M. Nachtegaal, O. V. Safonova and G. Smolentsev, J. Synchrotron Rad., 2018, 25, 989–997.
22 S. Pascarelli, O. Mathon, T. Mairs, I. Kantor, G. Agostini, C. Strohm, S. Pasternak, F. Perrin, G. Berruyer, P. Chappelet, C. Clavel and M. C. Dominguez, J. Synchrotron Rad., 2016, 23, 353–368.
23 C. Castan-Guerrero, D. Krizmancic, V. Bonanni, R. Edla, A. Deluisa, F. Salvador, G. Rossi, G. Panaccione, and P. Torelli, Rev. Sci. Instrum., 2018, 89, 054101
24 F. Borgatti, P. Torelli, M. Brucale, D. Gentili, G. Panaccione, C. Castan Guerrero, B. Schäfer, M. Ruben and M. Cavallini, Langmuir, 2018, 34, 3604–3609.
25 P. Glatzel and U. Bergmann, Coord. Chem. Rev., 2005, 249, 65–95.
26 J. Singh, C. Lamberti and J. A. van Bokhoven, Chem. Soc. Rev., 2010, 39, 4754–4766.
27 F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559.
28 K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028.
29 J. A. van Bokhoven and C. Lamberti, X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, John Wiley and Sons, Chichester, UK, 2016.
Cynthia Friend opened general discussion of Carlo Lamberti’s, Annette Trunschke’s and Michele Carosso’s papers: A variety of tools for in situ/operando characterization has been described today. What are the most versatile tools and which specific combinations provide the most insight in total?
Annette Trunschke responded: Adequate techniques depend also on the catalyst system and the reaction to be studied, but definitely a combination of complementary techniques is required. The high spatial resolution of electron microscopy provides new insights into the nanostructure of metal particles, but should always be combined with techniques performed in operando that yield integral information concerning the entire sample. The challenging aspect is that not seldom only a minor number of surface species might be relevant for high activity and/or selectivity and it is difficult to distinguish these species. The most sensitive tool is perhaps the application of probe reactions in combination with spectroscopy.
Michele Carosso replied: For our purpose (a quantitative characterization of chemisorbed and physisorbed hydrogen species at the Pt and C surfaces) INS surely represents the election technique for a series of reasons: (1) it is highly selective toward hydrogen (1H presents the highest scattering cross section among all nuclei)1, (2) most of the supports are almost transparent to neutrons, and (3) in principle, all of the vibrational modes involving hydrogen-containing species can be detected by INS. However, the intensity of a band in an INS spectrum depends also on the displacement of the H-containing species with respect to the equilibrium position. This is the reason why we clearly observe the bending modes of linear, bridged and hollow hydrides, but we fail in observing the stretching modes of linear hydrides. For this reason, INS has to be coupled with other characterization techniques to obtain an overall picture of the nature and abundance of chemisorbed hydrogen species. For example, by FT-IR spectroscopy we can clearly observe stretching modes of linear hydrides on Pt/Al2O3 samples.2–7
1 V. F. Sears, Methods Exp. Phys., 1986, 23, 521–550.
2 W. A. Pliskin and R. P. Eischens, Z. Phys. Chem., 1960, 24, 11–23.
3 D. D. Eley, D. M. Moran and C. H. Rochester, Trans. Faraday Soc. , 1968, 64, 2168–2180.
4 M. Primet, J. M. Basset, M. V. Mathieu and M. Prettre, J. Catal., 1973, 28, 368–375.
5 T. Szilagyi, J. Catal., 1990, 121, 223–227.
6 L. T. Dixon, R. Barth and J. W. Gryder, J. Catal., 1975, 37, 368–375.
7 J. P. Candy, P. Fouilloux and M. Primet, Surf. Sci., 1978, 72, 167–176.
Carlo Lamberti answered: I recently carried out a bibliometric study, using the ISI WoS database, covering the time period from 1985–September 2016 and combining the keywords catalysis OR catalyst with keywords allowing me to determine 18 different specific spectroscopies (the study does not cover diffraction techniques or microscopies). More than 150,000 papers were found, testifying to the need that the catalysis community has for supporting their catalytic studies with spectroscopic characterizations. The most used techniques are: IR 32.0%; NMR 21.0%, XPS 15.1%; Raman, 8.9%; UV-Vis 7.8%; EPR 4.3%; photoluminescence 2.7%, EXAFS 2.5%; XANES 1.3%; Mössbauer 1.5%, INS 0.2%; NEXAFS 0.15%; XES 0.05%. It is evident that such numbers are defined by several interconnected aspects that are: (i) the cost of the instrumentation (low for e.g. IR, UV-Vis; moderate for Raman and EPR; higher for NMR, XPS, EELS, HREELS); (ii) the applicability to a large variety of different systems (high for e.g. IR, UV-Vis, EXAFS/XANES/NEXAFS; moderate for NMR; low for EPR; very low for Mössbauer); (iii) the accessibility to the instrumentation (high for e.g. IR, UV-Vis, NMR, EPR; moderate for Raman, XPS; low for EXAFS/XANES/NEXAFS; very low for XES and INS); (iv) the possibility to easily work under in situ and operando conditions (high for e.g. IR, UV-Vis, EXAFS/XANES, XES; moderate for NMR, EPR, Mössbauer; very low for NEXAFS and electron-based spectroscopies); (v) the complexity in the interpretation of the experimental results; (vi) the richness of the obtainable information.
Besides all of these numbers, it is evident that the characterization of an active center inside a catalyst requires a multitactical approach able to shed light on its structural, energetic, electronic and vibrational properties. Only once these four aspects have been clarified and unified in a harmonious picture the understanding process may be considered accomplished. Several techniques are available to investigate each of the four mentioned aspects and the most (or the more) appropriate one(s) depend on the investigated sample. Indeed, one should always try to find the most relevant technique to a particular system and a particular phenomenon. For the study that I presented here (DOI: 10.1039/c7fd00211d), it was important to combine short- and long-range order sensitive techniques, as EXAFS and XRD, respectively, to highlight the differences in the crystalline core and in the amorphous shell regions of the nanoparticles and to use a technique sensitive to the unoccupied density of states (XANES) to discriminate between Pd carbide and Pd hydrate phases.
A final point that should not be overlooked is the support that DFT theory provides to the experiments. DFT is no longer required just for validating a reaction mechanism defining the energetic barriers of each intermediate step, but has become a fundamental help in the in-depth understanding of the different spectroscopic techniques.
1. E. Borfecchia, L. Mino, E. Groppo, S. Bordiga, A. L. Bugaev, A. Budnyk, K. A. Lomachenko, A. A. Guda, M. A. Soldatov, A. V. Soldatov, C. Lamberti, Stud. Surf. Sci. Catal., 2017, 177, 221–284.
Alexander Genest asked: Given the impurities, hydrogen and carbon, discussed here to be inside Pd/Pt particles and their known large effect on catalysis, could you comment on how likely it is that there can be experiments on pure Pd or Pt surfaces? What is to be expected, when experiments do not report on impurities?
Annette Trunschke answered: Contamination of catalysts by very small amounts of impurities cannot be completely avoided when high-performance catalysts are studied. Impurities are not only introduced during synthesis. Trace compounds in the feed gases might be accumulated on or in the catalyst during catalytic testing as well. Therefore an impact of unexpected components that might act as promoters should always be taken into account.
Michele Carosso responded: Considering that the 5 wt% Pt/C catalyst studied in our paper is a catalyst for hydrogenation reactions of industrial relevance, I believe that we cannot speak about hydrogen as an impurity. H2 is present in the reaction feed mixture, and platinum-hydride species formed upon H2 chemisorption are the effective species involved in the reduction of the substrate of interest, they are real reactants. Impurities could have both a negative or a positive effect on catalytic performances: of course, a contaminant could decrease the available surface area for the reaction to take place on, decreasing the catalyst activity and leading to catalyst deactivation. On the other hand, a contaminant could improve the selectivity of a catalyst towards a specific product: the most famous example in this sense is the Lindlar’s catalyst, for the selective hydrogenation of alkynes to alkenes. Regarding the second part of the question, impurities-free experiments can be certainly due in low-scale, laboratory conditions, where the amounts of studied sample are generally very low and contaminants could prevent the observation of a certain phenomenon. On the other hand, impurities are much more difficult to be avoided in reactions performed in industrial plants, where a compromise between costs and yields has to be reached.
Carlo Lamberti replied: I would not use the word “impurities” to describe the hydrogen and carbon atoms that interact with the Pd or Pt nanoparticles in the studies reported in these two papers (DOI: 10.1039/c7fd00211d and 10.1039/c7fd00214a), because they come from the reagent feed and they must be present if we want to run a hydrogenation reaction. Besides this semantic comment, in agreement with what’s just been stated by Annette Trunschke, I believe that it is possible to realize “clean” experiments on the laboratory scale, meaning experiments where the level of impurities is below the detection limit of the most accurate techniques. This of course depends on the accuracy of the authors of the study, but it is clearly the way it should be. Experiments concerning samples prepared on the industrial scale is a different story. The impurity control in samples prepared on the several ton-scale cannot be the same as for samples prepared in the g- or sub-g-scale. This is particularly true when the support used for the catalyst preparation is not a “clean” material such as silica, alumina, titania, etc. purchased from Sigma-Aldrich or other competitors, but is a natural carbon. Notwithstanding this intrinsic limitation, even working on the industrial scale with a “dirty” support such as active carbon of natural origin, it is possible to avoid the contamination of the active metal phase by working properly. It is even possible to verify in a spent catalyst if has been contaminated by a non-pure reagent feed during several months of work inside an industrial plant. As an example, analyzing spent Pd/C catalysts coming from different industrial plants it has been possible to determine S or Pb contamination in the active phase and Mo, Cr, Fe, Ti and Al contamination in the support.1 The detection limit of that study was down to a few tens of ppb.
1 R. Pellegrini, G. Agostini, E. Groppo, A. Piovano, G. Leofanti and C. Lamberti, J. Catal., 2011, 280, 150–160.
Michael Bowker remarked: In consideration of the point that many catalyst materials are well characterised and ‘clean’ we must note that in the papers this morning we have heard that the lattice expansion due to the presence of hydrides and carbides is very small. Hence it may be that a lot of experiments, especially with Pd, are not on pure metal nanoparticles at all, but on dilute carbides or hydrides. This is especially the case in the hydrogenation reactions for which Pd is widely used in academia and industry.
Hans-Joachim Freund responded: I would like to add, we have investigated in the past, whether and how hydrogen in Pd influences reactions. The important point is to be able to clearly assign the present hydrogen to be inside the particle. This can be done by using a technique called resonant-Nuclear-Reaction-Analysis (r-NRA).1
1 M. Wilde, K. Fukutani, W. Ludwig, B. Brandt, J. H. Fischer, S. Schauermann and H. J. Freund, Angew. Chem. Int. Ed., 2008, 47, 9289.
Valerii Bukhtiyarov added: I have a small comment on the discussion about the role of impurities introduced in the stages of catalyst preparation or as result of catalytic reaction. It is an important topic, but hopefully in many cases a catalyst continues to work in spite of the accumulation of impurities. As for the development of new methods for catalyst investigations, it is a really important matter, since it helps us to get new insight into the reasons for catalytic performance. So I appreciate very much the use of INS to study active hydrogen species on Pt/carbon catalysts. However, discussing the data available with new methods, we have to get knowledge about the chemical state and structure of the samples produced with other routine techniques. Maybe it is better, at least in the first steps of the method development, to use well-characterized model catalysts. Another point which should be clarified is the sensitivity of INS to the content of metal. In real catalysts the loading of noble method is usually less than 1 wt%.
Michele Carosso responded: Thank you for the comment. I agree with you when you say that well-characterized model catalysts should be the starting point for the development of any methods applied to the study of “real” catalysts, and also that new methods should be coupled with other routine characterization techniques. The 5 wt% Pt/C catalyst investigated in this work by INS has been indeed characterized with a multitude of other (well-established) techniques. Concerning the sensitivity of INS to the content of the metal, it must be noticed that the first INS studies on the interaction between H2 and noble metal surfaces were performed on samples containing a very high metal loading. For example, in the pioneering works by Albers and Parker1–3, and also by other authors4–6 Pt/C samples with a Pt loading ranging from 20 wt% to 60 wt% were considered. Although far from the metal loadings usually adopted in catalysis, these works demonstrated the potential of the INS technique in catalysis. To the best of our knowledge our sample is the lowest loaded Pt/C system investigated by INS so far. 5 wt% is a typical metal loading for catalysts used in hydrogenation reactions for producing fine chemicals.
1 P. Albers, E. Auer, K. Ruth and S. F. Parker, J. Catal., 2000, 196, 174–179.
2 P. W. Albers, M. Lopez, G. Sextl, G. Jeske and S. F. Parker, J. Catal., 2004, 223, 44–53.
3 S. F. Parker, C. D. Frost, M. Telling, P. Albers, M. Lopez and K. Seitz, Catal. Today, 2006, 114, 418–421.
4 A. J. Renouprez and H. Jobic, J. Catal., 1988, 113, 509–516.
5 H. Asada, T. Toya, H. Motohashi, M. Sakamoto and Y. Hamaguchi, J. Chem. Phys., 1975, 63, 4078–4079.
6 P. C. H. Mitchell, A. J. Ramirez-Cuesta, S. F. Parker and J. Tomkinson, J. Mol. Struct., 2003, 651–653, 781–785.
Carlo Lamberti responded: I agree with your statement that a catalyst contaminated with impurities may still continue working, and I would even add that in few cases these impurities may be responsible for its high activity or selectivity. In such cases impurities are named promoters. So, when we speak about impurities, we do not assume that they always have a negative effect on the catalytic properties.
I will now answer the first part of your comment. Yes, I fully agree with your statement that the development of a new technique requires first the use of model systems to validate the experimental, methodological and interpretation aspects of the experiment. Notwithstanding this point, I will not define an INS study of hydrogen adsorption on Pt/C nanoparticles (NPs) a novel technique as it was pioneered by Albers and Parker almost twenty years ago.1–5 The novelty of the study of Carosso (DOI: 10.1039/c7fd00214a), is related to the fact that INS spectra have been collected on Pt/C samples containing an order of magnitude less metal than in the past1–5 and that the quality of the spectra, in terms of both energy resolution and signal/noise ratio, allowed the authors to obtain relevant information not achievable with the spectra present in the published literature. Moreover, the Pt/C catalyst investigated in the paper of Carosso et al. (DOI: 10.1039/c7fd00214a), has been deeply investigated by CO chemisorption, TEM, Pt L3 XANES and EXAFS techniques, that combine and complement the H2-adsorption measurements and INS spectra reported here, so they cannot be considered as “unknown” materials.
Concerning the second part of your comment, it is true that in real catalysts the loading of noble metal is often less than 1 wt%, however, a 5 wt% Pt/C system represents a catalyst of industrial relevance, prepared by the Chimet company. I believe that a 5 wt% Pt/C represents the loading limit to obtain high quality INS data with the neutron fluxes and the detector efficiency available today. A 1 wt% Pt/C sample will require different investigation techniques.
1 P. Albers, E. Auer, K. Ruth and S. F. Parker, J. Catal., 2000, 196, 174–179.
2 P. W. Albers, H. Klein, E. S. Lox, K. Seibold, G. Prescher and S. F. Parker, Phys. Chem. Chem. Phys., 2000, 2, 1051–1058.
3 P. W. Albers, M. Lopez, G. Sextl, G. Jeske and S. F. Parker, J. Catal., 2004, 223, 44–53.
4 S. F. Parker, J. W. Taylor, P. Albers, M. Lopez, G. Sextl, D. Lennon, A. R. McInroy and I. W. Sutherland, Vib. Spectrosc., 2004, 35, 179–182.
5 S. F. Parker, C. D. Frost, M. Telling, P. Albers, M. Lopez and K. Seitz, Catal. Today, 2006, 114, 418–421.
Rosa Arrigo asked: Considering that the time resolution achieved with synchrotron radiation during operando and in situ studies is not comparable with the time scale of a catalytic reaction, how can we make sure that we are observing relevant dynamics and not just spectators? Would you encourage the application of free electron laser for catalysis?
Annette Trunschke answered: Spectator species might be distinguished by recording the response of spectroscopic signatures upon perturbation of the system by changing external parameters such as concentration, pressure or temperature. Operando experiments in which spectroscopic features and information concerning functional properties of a catalyst (activity, selectivity) are recorded at the same time in the same experiment need to be performed under fluctuating conditions. Modulation excitation spectroscopy has been used to improve time resolution in FTIR spectroscopy. Vibrational action spectroscopy employing infrared radiation from a free-electron laser might be applied in the future to sensitively probe surface species in the presence of similar bulk modes, but considerable method development is necessary in this field.
Carlo Lamberti responded: The time resolution of an XANES spectrum of a real sample (not a metal foil) can move from few minutes (using a standard step-scan acquisition mode) through a fraction of a second (using a quick-EXAFS acquisition mode)1 down to few milliseconds (using a dispersive acquisition mode)2. I believe that these time resolutions are perfectly suitable to follow the time-dependent perturbations that can be induced on working catalysts changing either the catalyst temperature or the feed flux.3,4 The problem of spectators is another story, that is not specifically related to synchrotron-based spectroscopies or to time resolved experiments. It is however a real problem, that can influence all kinds of catalyst characterization and may bias our interpretations or even lead to wrong conclusions. The presence of spectators may be difficult to spot. Often several standard or some advanced experiments are required to detect and quantify spectators. This can be done by changing the metal loading on the catalyst, and so changing the ratio between spectators and actors.5,6 Alternatively, advanced approaches such as modulated excitation experiments7–12 or steady-state isotope transient kinetic analysis13,14 can be adopted. Finally, as far as free electron lasers (FELs) are concerned, I foresee that they will allow breakthrough understanding of photocatalysts in terms of investigation of the exited states reached by the system upon photon absorption following the laser pump/X-ray probe approach.15–18
1 M. Nachtegaal, O. Müller, C. König and R. Frahm, in X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, ed. J. A. van Bokhoven and C. Lamberti, John Wiley and Sons, Chichester, UK, 2016, pp. 155–183.
2 O. Mathon, I. Kantor and S. Pascarelli, in X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, ed. J. A. van Bokhoven and C. Lamberti, John Wiley and Sons, Chichester, UK, 2016, pp. 185–212.
3 S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736–1850.
4 C. Lamberti and J. A. van Bokhoven, in X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, ed. J. A. van Bokhoven and C. Lamberti, John Wiley and Sons, Chichester, UK, 2016, pp. 353–383.
5 G. Leofanti, M. Padovan, M. Garilli, D. Carmello, A. Zecchina, G. Spoto, S. Bordiga, G. T. Palomino and C. Lamberti, J. Catal., 2000, 189, 91–104.
6 G. Leofanti, M. Padovan, M. Garilli, D. Carmello, G. L. Marra, A. Zecchina, G. Spoto, S. Bordiga and C. Lamberti, J. Catal., 2000, 189, 105–116.
7 C. F. J. Konig, J. A. van Bokhoven, T. J. Schildhauer and M. Nachtegaal, J. Phys. Chem. C, 2012, 116, 19857–19866.
8 D. Ferri, M. A. Newton, M. Di Michiel, S. Yoon, G. L. Chiarello, V. Marchionni, S. K. Matam, M. H. Aguirre, A. Weidenkaff, F. Wen and J. Gieshoff, Phys. Chem. Chem. Phys., 2013, 15, 8629–8639.
9 D. Ferri, M. A. Newton, M. Di Michiel, G. L. Chiarello, S. Yoon, Y. Lu and J. Andrieux, Angew. Chem. Int. Edit., 2014, 53, 8890–8894.
10 G. L. Chiarello and D. Ferri, Phys. Chem. Chem. Phys., 2015, 17, 10579–10591.
11 O. Martin, C. Mondelli, A. Cervellino, D. Ferri, D. Curulla-Ferre and J. Perez-Ramirez, Angew. Chem. Int. Edit., 2016, 55, 11031–11036.
12 V. Marchionni, D. Ferri, O. Krocher and A. Wokaun, Anal. Chem., 2017, 89, 5802–5810.
13 M. El-Roz, P. Bazin, M. Daturi and F. Thibault-Starzyk, ACS Catal., 2013, 3, 2790–2798.
14 C. Ledesma, J. Yang, D. Chen and A. Holmen, ACS Catal., 2014, 4, 4527–4547.
15 L. X. Chen, Faraday Discuss., 2003, 122, 315–329.
16 C. Bressler and M. Chergui, Annu. Rev. Phys. Chem., 2010, 61, 263–282.
17 L. X. Chen, X. Zhang and M. L. Shelby, Chem. Sci., 2014, 5, 4136–4152.
18 E. Borfecchia, C. Garino, L. Salassa and C. Lamberti, Philos. Trans. R. Soc. A-Math. Phys. Eng. Sci., 2013, 371, 40.
Michael Bowker added: Spectators are a problem, especially in catalysis with a low loading of metal on a support. There may be spectators on the support, which might dominate spectroscopies such as IR, and which may not respond to dynamic changes in the gas phase. However, there may also be a similar, but reactive species on the metal which is masked by the dominant support species. An example of this is ethanol synthesis on Rh catalysts where the acetate intermediate is on both the metal and the support, but IR doesn't respond fast to dynamic changes in reaction conditions, even though the Rh species is a crucial intermediate in the reaction.
Simone Gallarati opened general discussion of the paper by Yaroslav Odarchenko: GISAXS/GIXD were used to characterise Au NPs with regular periodicity deposited by dip coating on a flat single crystal SiO2/Si(111) substrate. While GISAXS/GIXD are here very powerful techniques to probe the surface chemistry of the catalyst, they seem limited to model systems of planar, ordered nanoparticles. Could this approach be extended in any way to more “realistic” and less ordered systems such as powders/supported NPs? In particular, if one wishes to investigate the structure of core–shell particles and probe the catalyst at different sampling depths, could these techniques be employed?
Yaroslav Odarchenko replied: The main limitation of the GISAXS/GIXD methods is that they require special sample preparation, i.e. a flat substrate. Thus the bulk (powder) catalyst cannot be used even in the form of a pellet, since the surface roughness will be too large. As soon as the surface is flat (typically roughness <1 nm), there is no requirement for how the nanoparticles are arranged on the surface. We’ve fabricated ordered nanoparticle arrays in order to avoid ‘cross-talk’ between individual particles and possible sintering/coalescence. For an example of GISAXS analysis of a ‘less’ ordered system please previous work,1 where Au supported on TiO2 was prepared by physical vapour deposition, resulting in random particle position on the crystal surface and eventually sintering during the catalytic testing. The GISAXS/GIXD is very useful for probing core–shell particles,2 subsurface morphology3 or multilayered films.4, 5 For example, shell thickness and core size for diblock-copolymer micelles filled with silica were calculated from GISAXS.2 The depth sensitivity is mainly achieved by varying the incident angle.4
1 I. Laoufi, M. C. Saint-Lager, R. Lazzari, J. Jupille, O. Robach, S. Garaudée, G. Cabailh, P. Dolle, H. Cruguel and A. Bailly, J. Phys. Chem. C, 2011, 115, 4673–4679.
2 A. Frömsdorf, A. Kornowski, S. Pütter, H. Stillrich and L. T. Lee, Small, 2007, 3, 880–889.
3 D. James Martin, D. Decarolis, Y. I. Odarchenko, J. J. J. Herbert, T. Arnold, J. Rawle, C. Nicklin, H.-G. Boyen and A. M. Beale, Chem. Commun., 2017, 53, 5159–5162.
4 J. Wang, Y. I. Odarchenko, M. Defaux, J. Lejnieks, D. V. Ahokhin, H. Keul, D. A. Ivanov, M. Möller and A. Mourran, Macromolecules, 2013, 46, 6159–6168.
5 I. Saito, T. Miyazaki and K. Yamamoto, Macromolecules, 2015, 48, 8190–8196.
Maurits Boeije commented: For this method, a flat substrate with a homogeneously distributed arrangement of nanoparticles is needed. The authors show a very elegant way of obtaining such a sample, while retaining a narrow size distribution. A drawback of this technique is that the preparation can be costly and the time for synthesizing such samples is rather long. Another approach of obtaining samples suitable for both GISAXS and TEM measurements, is by depositing (size selected) nanoparticles onto a substrate from the gas phase, using particle sizes from 1 to 10 nm. We show that spark ablation technology is a fast and clean technique that can simplify sample preparation in these cases. Because the deposition takes place at room temperate and at ambient pressure, the technique is easy to use. Limited post-processing is needed because no surfactants or solvents are introduced (Fig. 8 and 9).
 | ||
| Fig. 8 Au nanoparticles deposited on Si, deposited for 5 min. | ||
 | ||
| Fig. 9 Au nanoparticles deposited on Si, deposited for 100 min. | ||
Yaroslav Odarchenko responded: Thank you for your suggestion. It would be very interesting to compare the particle uniformity and particle size distribution for the 2D model catalyst prepared by reverse polymer encapsulation and spark ablation. My main concern is the control of the interparticle distance by the spark ablation deposition, since in our case it is conveniently controlled by the dip coating velocity and block copolymer length. Although it was mentioned in the reply to the previous question that the regular nanoparticle arrangement is not a prerequisite for the operando GISAXS/GIXD analysis, the possibility to control the distance between the particles in the nanoarray could be critical in order to minimise the interactions between particles during heat treatment or catalytic reaction.
Graham Hutchings asked: Using SiO2 for the Au catalysed CO oxidation is not the optimum support as it is not reducible and indeed you only observe activity at 300 °C. The model in Fig. 7 is very similar to models previously proposed except that with SiO2 you cannot have defects that are necessary for the low temperature oxidation of CO. Is it possible to use a single crystal of TiO2 as Au/TiO2 is a very active catalyst for CO oxidation?
Yaroslav Odarchenko replied: I strongly agree with your comment. The main reason for using Si with a native SiO2 layer was the fact that the fabrication of the 2D model catalyst using reverse micelle encapsulation was optimised for this type of support. Also it is a relatively inexpensive single crystal substrate (in comparison with TiO2 or Al2O3 single crystals) that meets the requirements (roughness and size) for the GISAXS/GIXD analysis. I have also to add that this project continues and we have successfully prepared flat single- and bi-metal nanoparticulate catalysts using polished rutile TiO2 (110) single crystals.
Carlo Lamberti enquired: In the cartoon model reporting the structural transformations of a gold nanoparticle (NP) supported on SiO2/Si(111) during CO oxidation (Fig. 7 (DOI: 10.1039/c8fd00007g)) you propose a model of a bimodal sphere with metal gold on top and gold oxide at the gold-SiO2 interface. Where comes the evidence of the Au2O3 phase? From SAXS I guess, as I do not believe that the Au2O3 phase is sufficiently ordered to provide Bragg peaks in the WAXS pattern. Do you plan to perform also an XANES study to confirm the presence of an oxidized fraction of gold?
Yaroslav Odarchenko responded: The evidence for the Au2O3 phase comes from the X-ray photoelectron spectroscopy (XPS) analysis and grazing incidence X-ray diffraction (GIXD) data collected during butadiene hydrogenation using the same system.1 It has to be mentioned that the butadiene hydrogenation was performed at 473 K, which is 100 K lower than the temperature used for CO oxidation in this work.
1 D. J. Martin, D. Decarolis, Y. I. Odarchenko, J. J. J. Herbert, T. Arnold, J. Rawle, C. Nicklin, H.-G. Boyen and A. M. Beale, Chem. Commun., 2017, 53, 5159–5162.
Toru Murayama said: I would like to ask about the size of the gold nanoparticle and the reaction temperature. In the manuscript, the average size of the gold nanoparticle was 9 nm. Based on the previous report, the catalytic activity for CO oxidation increased drastically with less than 5 nm of particle size. If the authors can prepare the smaller size of gold nanoparticle, that would be better. The reaction temperature tested was 573 K as in Fig. 6. This temperature is very high for nanoparticulate gold catalysts. In this temperature region, surface reactions on gold have been reported. Therefore, I would like to ask about the evidence of the dissociation of oxygen at the gold-support interface shown in Fig. 7 and conclusions. If the authors could calculate the apparent activation energy in their experimental conditions, that value would help to understand the reaction mechanism.
Yaroslav Odarchenko answered: I agree that according to the literature the catalytic activity of gold during CO oxidation increases for the smaller particle sizes, i.e. below 5 nm. In this proof of principle work the choice of 9 nm nanoparticle size was a compromise between sensitivity of the experimental methods (surface X-ray scattering) and catalyst reactivity. The other factor that determined the particle size was simply availability of the highly monodispersed commercial polymer precursors required for the reverse micelle encapsulation method used in this study. We are currently preparing new samples with the smaller nanoparticles by using shorter PS-P2VP block copolymers. Calculation of the apparent activation energy would help to confirm the suggested reaction mechanism, however CO oxidation was performed only at one temperature, which is not sufficient to determine the energy.
Scott Rogers asked: Following on from previous questions regarding the limitations of the technique, would it be possible to acquire good quality data on nanoparticles that averaged ∼ 2 nm, or would you need a particle larger than the penetration depth of the X-rays (∼ 3 nm) in order to minimise signal loss?
Yaroslav Odarchenko replied: One should be able to acquire reasonably good Grazing Incidence Small Angle X-ray Scattering (GISAXS) and Grazing-Incidence X-ray Diffraction (GIXD) data using gold nanoparticles of ∼ 2 nm in size. Moreover, the GISAXS data have been reported before for 2.1 nm gold nanoparticles on TiO2.1 The X-ray diffraction signal from the crystalline FCC phase for 1.8 nm gold nanoparticles on a mesoporous silica/graphene oxide powdered catalyst suggests that GIXD analysis is also feasible (Fig. 22). However, in our previous work we have been able to detect the signal from gold oxide in addition to the metallic phase for 9 nm particles using GIXD, which could be problematic for particles as small as 2–3 nm.3 I would like to add a remark regarding the signal loss. In the grazing incidence X-ray analysis, penetration depth will not be limited by the particle size, since the angle between the incoming X-rays and the sample surface is very small (below the Brewster angle) the surface sensitivity is achieved. For our case it means that the reflected/refracted X-ray beam becomes confined in the monolayer of gold nanoparticles at the substrate surface and the effective sample thickness is increasing from nm to cm (depending on the sample and incidence angle). Also one has to mention that the X-ray beam size was 0.3 × 0.3 mm2, which is larger than the gold monolayer thickness.
1 I. Laoufi, M. C. Saint-Lager, R. Lazzari, J. Jupille, O. Robach, S. Garaudée, G. Cabailh, P. Dolle, H. Cruguel and A. Bailly, J. Phys. Chem. C, 2011, 115, 4673–4679.
2 L. Peng, J. Zhang, S. Yang, B. Han, X. Sang, C. Liu, X. Ma and G. Yang, Chem. Commun., 2015, 51, 4398–4401.
3 D. James Martin, D. Decarolis, Y. I. Odarchenko, J. J. J. Herbert, T. Arnold, J. Rawle, C. Nicklin, H.-G. Boyen and A. M. Beale, Chem. Commun., 2017, 53, 5159–5162.
Michael Bowker enquired: Concerning the preparation of your materials, we must remember that the behaviour of silicon isn’t simple, since it normally forms a native oxide layer of 1 or 2 layers under ambient conditions and oxidises more deeply at higher temperatures. You say little in the paper about the state of the Si/SiO2 surface; by your plasma oxidation do you sputter the surface or oxidise it? It is quite possible for the Si during oxidation to grow around the Au nanoparticles, as shown by us,1 albeit in thermal oxidation at higher temperatures.
1 M. Bowker, J. J. Crouch, A. F. Carley, P. R. Davies, D. J. Morgan, G. Lalev, S. Dimov and D.-T. Pham, J. Phys. Chem. C, 2013, 117, 21577–21582.
Yaroslav Odarchenko responded: Thank you for your question. We have performed XPS analysis to estimate the SiO2 layer thickness after the plasma oxidation and consequent annealing at 300 °C, which can be found in our recent paper.1 The XPS data indicate that the volume surrounding Au nanoparticles is mostly composed of a native oxide layer (thickness around 3–4 nm).
1 D. James Martin, D. Decarolis, Y. I. Odarchenko, J. J. J. Herbert, T. Arnold, J. Rawle, C. Nicklin, H.-G. Boyen and A. M. Beale, Chem. Commun., 2017, 53, 5159–5162.
Parag Shah opened discussion of the paper by Valerii Bukhtiyarov: Have you investigated how the CO binds to the Au/Pd nanoparticles? Does it change from bridging/terminal binding as the temperature increases?
Valerii Bukhtiyarov replied: In our previous work1 we showed that XPS spectra could be used for determining bridging/terminal binding CO molecules with Pd(111). The difference between the C1s position was 0.7 eV originating from C1s binding energy values of 285.5 eV for the bridge bonding molecule and 286.2 eV for the on-top one. The O1s spectra were not analyzed in that work due to their overlapping with Pd4d spectrum, which is again the case for the Pd-Au system used here. Application of the carbon containing support in this study (HOPG) results in a great masking effect also in the C1s spectra. So for this specific system, Pd-Au/HOPG, XPS cannot be used for investigation of the change from bridging/terminal binding of CO as the temperature increases.
1 V. V. Kaichev, I. P. Prosvirin, V. I. Bukhtiyarov, H. Unterhalt, G. Rupprechter and H.-J. Freund, J. Phys. Chem. B, 2003, 107, 3522–3527.
Haoliang Huang asked: My question mainly refers to the XPS deconvolution in Fig. 5. The Pd 3d spectra were deconvoluted into Pd alloy, Pd0, Pd-CO and Pd cluster, but they are more or less in the zero oxidation state. Can XPS provide such subtle detail of the chemical environment? I also notice you haven't included Pd surface oxide and asymmetric features of metallic Pd, which would affect your fitting results. Can you comment on this?
Valerii Bukhtiyarov answered: I agree with you that deconvolution of Pd3d spectra is not so straightforward due to closeness of the binding energies of different components. Nevertheless, analysis of the numerous literature data indicates that in the case of synchrotron radiation-based XPS, when we have the best resolution of the spectra, the species discussed in the paper, Pdalloy, Pdmetal, and Pd–CO, can be discriminated. Furthermore, the species have different temperature-dependent behavior, which is in full agreement with the literature data. For metallic Pd species, we indeed used an asymmetric feature again in full agreement with the literature data for a bulk palladium sample. As for Pd oxide, we did not discuss its location, because we have no reliable basis to discriminate this.
Philip R. Davies said: In Fig. 5 you identify a number of palladium states which are carefully assigned based on your previous work. It is interesting that the high binding energy state (at 337.2 eV) you assign to localised small Pd clusters do not sinter on heating but I notice that these species are not present after heating and then cooling back to room temperature. Can you comment on what has happened to these states?
Valerii Bukhtiyarov replied: Honestly speaking, this high BE species is observed in all experiments for many metals deposited on a defective surface of HOPG, Ag, Au, Pd, etc., but we could not find any correlation of its intensity with parameters of the sample preparation or with treatment conditions. For example, the Pd 3d spectrum from the Pd-Au high sample taken after heating up to 250 °C following cooling down to RT (the spectra are not presented in the paper) contains this feature. So we continue to insist on assignment of this feature to metallic clusters on some defects. Moreover, Granozzi and his colleagues in their recent work1 revealed the additional spectral component with a binding energy of 338.1 eV when palladium was deposited on N-HOPG and attributed this state to Pd nanoclusters decorating surface defect sites.
1 M. Favaro, G. A. Rizzi, S. Nappini, E. Magnano, F. Bondino, S. Agnoli and G. Granozzi, Surf. Sci., 2016, 646, 132–139.
Carlo Lamberti asked: With the experimental set-up that you have used for this experiment, is it possible to collect the O(1s) XPS peak from the CO molecule adsorbed on the gold nanoparticles and to discriminate that signal from that coming from the CO molecules in the reaction atmosphere?
Valerii Bukhtiyarov answered: You are absolutely right. The original idea of application of HOPG with a low content of oxygen-containing impurities as a model support was indeed an attempt to use the O1s XPS peak to study the nature of such adsorbates of oxygen and CO and we did it for silver particles on HOPG in the course of the ethylene epoxidation study. We could identify two different oxygen species (nucleophilic and electrophilic) and observe the influence of Ag particle size on their relative population. We can also see that for the O1s binding energy range > 531 eV the species from oxygen groups located on the graphite surface appear. The signals at BE = 531.6–532.2 eV were assigned to the groups having a C=O bond (carbonyl, carboxyl and ester groups), and the signals at BE > 533 eV, to the groups having a C–O bond (hydroxyl, carboxyl and ether groups). It is evident that the O1s signals from CO adsorbed both on Pd and on Au sites would be overlapped with the HOPG-originated signals. Furthermore, specifically for the palladium-gold system the overlapping of the Pd4d spectrum with the O1s signal makes the analysis of adsorbed CO species in our experiments impossible. You are absolutely right. The original idea of the application of HOPG with a low content of oxygen-containing impurities as a model support was indeed an attempt to use the O1s XPS peak to study the nature of such adsorbates of oxygen and CO and we did it for silver particles on HOPG in the course of the ethylene epoxidation study. We could identify two different oxygen species (nucleophilic and electrophilic) and observe the influence of Ag particle size on their relative population. We can also see that for the O1s binding energy range > 531 eV the species from oxygen groups located on the graphite surface appear. The signals at BE = 531.6–532.2 eV were assigned to the groups having a C=O bond (carbonyl, carboxyl and ester groups), and the signals at BE > 533 eV, to the groups having a C–O bond (hydroxyl, carboxyl and ether groups). It is evident that the O1s signals from CO adsorbed both on Pd and on Au sites would be overlapped with the HOPG-originated signals. Furthermore, specifically for the palladium-gold system the overlapping of the Pd4d spectrum with the O1s signal makes the analysis of adsorbed CO species in our experiments impossible.
Emma K. Gibson enquired: I was really interested to see that you saw that the AuPd alloy is on the surface above 150 °C. We did a similar study on CO oxidation over a AuPd bimetallic catalyst using combined XAFS/DRIFTS but found (from the DRIFTS) that the gold disappears from the surface at roughly this temperature.1 Is it possible that the differences we observe are due to the different catalyst synthesis that we employed?
1 E. K. Gibson, A. M. Beale, C. R. A. Catlow, A. Chutia, D. Gianolio, A. Gould, A. Kroner, K. M. H. Mohammed, M. Perdjon, S. M. Rogers, and P. P. Wells, Chem. Mater., 2015, 27, 3714–3720.
Valerii Bukhtiyarov responded: Thank you very much for your question. I think you are correct that the differences arise from the synthetic procedure. If I am not mistaken, calcination at 300 °C was used as the final treatment of your Pd-Au bimetallic catalyst in your work. However we have shown previously1 that annealing under vacuum at 400 °C is necessary to form the alloy structure. This conclusion has been based on the dependence of the Au:Pd ratio on temperature (Fig. 7). It has been shown that the ratio increases when the temperature rises from 200 to 400 °C and remains constant at higher temperatures. This, along with the decrease in the Au4f and Pd3d binding energy values, has led us to the conclusion that formation of the alloy occurs at T ≥ 400 °C. At 300 °C the way to alloy only begins, but does not finish. I would also like to note that calcination in air compared with annealing under vacuum was used in our work.
1 A. V. Bukhtiyarov, I. P. Prosvirinab and V. I. Bukhtiyarov, Appl. Surf. Sci., 2016, 367, 214–221.
Cynthia Friend asked: Your Au:Pd is quite high in the materials you studied, perhaps accounting for the segregation of Pd. The distribution of Pd under reaction conditions will depend on the synthesis and the composition. Furthermore, your reaction mixture appears to be very oxygen rich based on the formation of PdO during the reaction. Can you comment on whether you studied the effect of the reactant gas composition?
Valerii Bukhtiyarov replied: I absolutely agree with your comment that the effect of Pd segregation has to be dependent on Au:Pd ratio in the catalyst and the ratio between CO and O2 in reaction mixture. Indeed, we should sufficiently increase the number of Pd atoms to block the alloy surface and thus to deactivate low-temperature gold activity in CO oxidation. So, in the case of a sample with small Pd content we can expect some activity in the room temperature range. Again, variation of the CO to O2 ratio in the reaction mixture can decrease (or even prevent) the PdO formation. You are right that these responses should be studied in more detail and we are planning to do so in the future.
Michael Bowker remarked: In relation to the AuPd nano-alloy system what we really need is a phase diagram, since a small amount of Pd on a lot of gold tends to alloy into the bulk, whereas high concentrations lead to significant segregation, and vice versa for Au in Pd.
Valerii Bukhtiyarov responded: I would like to add also that we should take into account the size of the alloy particles. If the particle size is so small that we can count the number of atoms (less than few hundreds), then we have not enough Pd atoms to block the surface completely. Increasing the amount of Pd atoms will segregate them on the surface and cover it. However, if the size of the alloy particles moves to several nanometers, the ratio of surface to bulk atoms goes to 10%. Then, such an amount of palladium in Pd-Au alloy particles (∼ 10%) will be high enough to block completely the metal particle surface with Pd.
Graham Hutchings asked: Can you use your methodology to make core shell structures with a Au shell and Pd core, or a Pd shell and Au core and then anneal them to get the random alloys. Could these then be used for a reaction other than CO oxidation as the addition of Pd to Au typically decreases the activity for this reaction. Perhaps the approach suggested by Andrew Logsdail in his poster would be a possibility.1
1 R. Su, R. Tiruvalam, A. J. Logsdail, Q. He, C. A. Downing, M. T. Jensen, N. Dimitratos, L. Kesavan, P. P. Wells, R. Bechstein, H. H. Jensen, S. Wendt, C. R. A. Catlow, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2014, 8, 3490–3497.
Valerii Bukhtiyarov replied: Definitely, core–shell structures with an Au shell and Pd core, or a Pd shell and Au core can be prepared with our methodology. Subsequent heating of the core–shell structures will transform them to the random alloys. All of these structures can be applied then to study a reaction other than CO oxidation. For example, just now, our paper, which is devoted to the propyne hydrogenation reaction on HOPG-supported Pd-Au bimetallic catalysts, is being considered for publication in ACS Catalysis. Parahydrogen-enhanced nuclear magnetic resonance (NMR) spectroscopy has been used as a tool for the investigation. In this study we could show that bimetallic PdAualloy and Aushell-Pdcore samples demonstrate 2–5 orders of magnitude higher activity in pairwise hydrogen addition in comparison with the Pd sample. Moreover, it is shown that the Aushell-Pdcore structure of bimetallic PdAu particles supported on HOPG is much more active and selective in the propyne hydrogenation reaction than PdAualloy one.
Roy Johnston opened general discussion of the paper by Rene Nome: I am interested in the composition dependence of the segregation that you have reported. At low gold concentration you state that the hollow AgAu nanoparticles are gold rich at the surface. Do you mean the outer surface only or both the outer and inner surfaces, or can the inner surface composition not be determined? I note that in your synthetic method the hollow nanoparticles are heated to 100 °C, but they are not annealed at higher temperatures (unlike in the reported simulations). Have you tried annealing the hollow particles? Finally, I have a cautionary comment concerning the molecular dynamics simulations. The simulations you report use Embedded Atom Model potentials for gold and silver. Our experience is that, for low Au concentrations, DFT calculations predict that Au occupies surface sites (in line with your experimental observations), whereas empirical potentials over-stabilise surface Ag (as in your simulated annealing results).
Rene Nome replied: In answer to the first part of your question, based on 3D compositional mapping using energy dispersive X-ray tomography within the scanning transmission electron microscope, surface segregation occurs on both the inner and outer surfaces.1 Indeed, surface segregation on both the inner and outer surfaces is of crucial importance, and it also affects catalytic activity1 and the electron-phonon coupling times reported here.
In answer to the next part of your question, Pedro Camargo, with whom I discussed this matter, indicated that with respect to annealing, they have performed a study heating bimetallic AgAu nanoparticles and observing compositional variations and surface segregation in situ using electron microscopy.2
Regarding the last part of your question, Rodrigo Albuquerque, with whom I discussed this matter said that this is an important point and there may be many reasons for this discrepancy. One of them is clearly the EAM parameters, as you said. However, the way the structures were produced is also very important. In the MD simulations, the annealed structures with Ag-rich surfaces were generated after heating up to about 900 K, while in the laboratory the AgAu nanoshells were produced at 373 K, where Au was inserted via galvanic replacement using a Ag seed. This means that experimentally Au was first placed directly onto the surface of the Ag seed. After this happens, further Au migration, which influences segregation, will strongly depend on the temperature, as well as on the cohesive energy of Au (3.8 eV) and Ag (2.95 eV), and on their surface energies (78 meV A−2 for Ag and 97 meV A−2 for Au). It is difficult to know at this stage if the experimental temperature (373 K) allowed for atomic rearrangement of the surface via Au or Ag migration, i.e., if the experiment produces a kinetically or thermodynamically stable nanoshell. Probably DFT-MD would help answer this question.
1 T. J. A. Slater, A. Macedo, S. L. M. Schroeder, M. G. Burke, P. O’Brien, P. H. C. Camargo and S. J. Haigh, Nano Lett., 2014, 14, 1921.
2 E. A. Lewis, T. J. A. Slater, E. Prestat, A. Macedo, P. O’Brien, P. H. C. Camargo and S. J. Haigh, Nanoscale, 2014, 6, 13598.
Sabrina Simoncelli said: Surface defects allow electrons to scatter more efficiently in the lattice, therefore, did you observe a correlation between the number of surface defects and the electron-phonon coupling times in the investigated hollow AgAu nanoparticles?
Rene Nome answered: For reference, in the case of perfectly disordered solid alloy AgAu nanoparticles, a linear correlation between electron-phonon coupling times and composition (inner and surface composition) has been reported.1 In our present work, in going from solid to hollow AgAu nanoparticles, we report a nonlinear dependence of electron-phonon coupling times on composition. Based on the known surface segregation behavior as a function of AgAu composition reported previously for the same nanoparticles,2 our results indicate longer electron-phonon coupling times with increasing surface Ag defects in the composition-range investigated here.
1 M. Broyer, E. Cottancin, J. Lermé , M. Pellarin, N. Del Fatti, F. Vallée, J. Burgin, C. Guillon and P. Langot, Faraday Discuss., 2008, 138, 137–145.
2 T. J. A. Slater, A. Macedo, S. L. M. Schroeder, M. G. Burke, P. O’Brien, P. H. C. Camargo and S. J. Haigh, Nano Lett., 2014, 14, 1921.
Sabrina Simoncelli enquired: Besides the catalytic field, your results are also very interesting for the hot-electron community, where having longer electron-phonon coupling times would be beneficial to allow electrons to react before thermalization. Have you considered performing experiments to correlate hot-electron reactivity with electron-phonon coupling times in your system?
Rene Nome answered: The linear relationship between electron-phonon coupling times and pump-pulse energy reported in the present work holds in the weak perturbation limit. By extrapolating the linear fit to zero pump energy, we have determined intrinsic electron-phonon coupling times for each composition, which is the main point of this paper. We have performed additional pump-probe spectroscopy experiments at higher pump energies than those reported here. However, at higher pump energies the relationship between electron-phonon coupling times and pump energy is not linear (not shown). At higher energies, depending on the pump energy, the measured electron-phonon coupling times are longer, similar or shorter than the values reported here. We are currently pursuing additional experiments, simulations and analysis to better understand the response at higher pump energies
Francesca Baletto enquired: In your simulations, as reported in Fig. 6 of the paper, the initial hollow clusters have an empty region about 3 nm which is going to be preserved up to the melting point. A few considerations:
Could you observe any radial breathing of the cluster during the melting? From Fig. 6(c) there is a little variation of the cluster diameter. Nonetheless it seems that the internal cavity has a lower radius due to the presence of Ag atoms mainly.
Rene Nome responded: In the simulations, we have not characterized radial breathing mode vibrations during melting, although that is a very interesting study that may be performed by calculating the power spectrum from the Fourier transform of the thermal fluctuations (valid for small displacements). Alternatively, these vibrations can be modelled explicitly with density functional theory or with coupled two-temperature model/molecular dynamics (TTM-MD) simulations (see previous work1,2 for examples of such simulations applied to Ag clusters and Au nanorods, respectively). In the ultrafast spectroscopy experiments, the oscillations observed in the pump-probe transients were assigned to acoustic breathing modes, and we have observed these oscillations for all of the pump energies reported, which are well below the pump energies needed to promote nanoparticle melting.
1 C. M. Lethiec, L. R. Madison and G. C. Schatz, J. Phys. Chem. C, 2016, 120, 20572.
2 Y. Gan, Z. Sun and Z. Chen, Phys. Chem. Chem. Phys., 2016, 18, 22590.
Francesca Baletto commented: The number of counts (of the order of 10−5) looks strange for a such big clusters.
Rene Nome responded: Rodrigo Albuquerque, with whom I discussed this matter, indicated that this was generated by placing one single ghost atom at (0, 0, 0) and by calculating the pair distribution function (RDF) between this ghost atom and Au or Ag. Doing the RDF in the conventional way for e.g. the pair Au–Ag produces indeed much higher peaks in the RDF curve.
Francesca Baletto asked: How many independent simulations have been performed to get the caloric curve in Fig. 6(d)?
Rene Nome replied: Rodrigo Albuquerque, with whom I discussed this matter, commented that only one simulation was done for the caloric curve, since we didn’t want to make a quantitative determination of the melting point, for which monitoring e.g., mean square displacements would be more appropriate than monitoring V × T.
Francesca Baletto enquired: Could you plot the difference in the potential energy surface? Could you comment on a change of 103 eV between the alloyed and the core shell ordering? The value is the one the reader can extrapolate at 300 K from Fig. 6(d).
Rene Nome responded: Lower energies mean more stable structures; at higher energies, which are thermally accessible at higher temperatures, degeneracy increases and more atomic arrangements are equally likely (thus increasing disorder). About the change in energy, this quantity is much larger than the thermal fluctuations at 300 K, and so is the energetic barrier that separates these two states. The MD results thus indicate that the alloyed ordering is more stable than the core shell one; however the change in energy required to cross the barrier indicates the high stability of the core shell structure as well. Experimentally, pump intensity-dependent ultrafast spectroscopy studies performed at higher pump energies could be used to investigate the core-shell/alloyed nanoparticle transformations but we do not believe such transformations occur with the low pump energies employed in the present work.
Francesca Baletto asked: What happens if you cool down the melted clusters? Could you recover the formation of any cavity?
Rene Nome answered: In the simulations, it depends on the highest temperature reached. If the upper temperature limit is below the melting temperature, then upon cooling the cavity structure is maintained and cavity size is recovered. On the other hand, if the nanoparticle is heated above the melting temperature, then the energetically favorable downhill pathway is that of the alloyed structure, and no cavity formation is recovered. In the ultrafast spectroscopy experiments reported here, we have measured intensity-dependent pump–probe transients in the 23 °C to 90 °C temperature range without any change in structure from core shell to alloyed, which would have significantly modified the decay constant and oscillation periods recorded.
Francesca Baletto questioned: Could you estimate the smaller size of the cavity to be stable at 0.75 their melting temperature?
Rene Nome replied: Rodrigo Albuquerque, with whom I discussed this matter, indicated that for the alloyed nanoshell with 20% Au the inner diameter of the cavity was about 7.5 nm (at 300 K it is about 7.7 nm).
Aram Bugaev asked: From the SEM images that you have reported it seems that you have different degrees of agglomeration for the particles of different compositions. Can you account for this effect in your theoretical calculations of the optical spectra?
Rene Nome answered: In our studies in solution, we have optimized sample concentration to avoid aggregation. Thus, we have used dilute samples to perform steady-state and time-resolved spectroscopic studies in the linear absorbance regime, so we believe there are no nanoparticle aggregation effects in the optical studies reported here. For future work, it would be interesting to perform spectroscopic studies at a higher nanoparticle number to study aggregation effects in solution. About simulating optical spectra of nanoparticle aggregates, one may use generalized Mie theory1,2 or explicit finite-element modeling as performed here, extended to include two or more particles.
1 J. E. L. Villa, D. P. dos Santos and R. J. Poppi, Microchim. Acta, 2016, 183, 2745–2752.
2 Y. Xu, Appl. Opt., 1995, 34, 4573–4588.
Laura Torrente-Murciano enquired: It is my understanding that your data indicates that above the melting temperature, the AgAu hollow particles melt forming solid particles, however, are the particles fully stable up to that point? Do you observe any agglomeration or changes in the surface?
Rene Nome replied: Rodrigo Albuquerque, with whom I discussed this matter, indicated that the particles are stable ca. 150–200 K below the melting point. For instance, the AgAu nanoshell with a random distribution of atoms (20% Au) at 800 K is roughly the same for the period of time that we investigated (10 ns) without imploding the cavity or decreasing its size. Some slow deformations (breathing motion) of the cavity occur at this temperature. At 700 K there are even less geometrical modifications within 10 ns. At 900 K, on the other side, the inner diameter decreases from 7.7 to 6.6 nm in 10 ns, but in this case the potential energy decreases in a stair-like shape, every new horizontal region becoming stable for a longer time. This means that even at 900 K the cavity diameter might become stabilised at a lower value, but a considerably longer MD simulation must be made to check it. The hollow nanoparticles clearly show less organised surfaces at higher temperatures (and below the melting point). For instance, one can easily identify 100 or 111 planes at room temperature, while just below the melting point such surfaces become much more difficult to identify; as a consequence the Radial Distribution Function (RDF) becomes broader and has fewer peaks at higher temperatures (this is also related to your question about liquid-like properties at high temperatures). The surface becomes more Ag rich at higher temperatures and below the melting point (e.g., at 900 K).
Laura Torrente-Murciano asked: Solid particles, especially at high temperature, present liquid-like properties (one of the reasons behind their high mobility). Do you observe similar behaviour in the hollow particles?
Rene Nome answered: Rodrigo Albuquereque, with whom I discussed this matter, indicated that at high temperatures the hollow (nano)particles show some plasticity of the cavity, where slight deformations can be observed. Also, the radial distribution functions (RDFs) at high temperatures show broader and fewer peaks. The molten particles (T > 1000 K) show only 3 peaks as compared with about 10 peaks (range 0–10 Å) for particles at room temperature. Based on the RDFs one could say that there is indeed a slight liquid character at higher temperatures for these particles.
Andrea Russell commented: I am interested in the fact that you’ve used the Galvanic displacement method to prepare your AgAu nanoparticles and used a solution of AuCl4− as the source of the Au. Do you see any evidence for the precipitation of AgCl? Later in this meeting Laura Torrente-Murciano (DOI: 10.1039/c8fd00001h) is going to show for the AgPd system that using K2PdCl4 as the Pd source resulted in exactly this problem. I would like to suggest that some of the stability you observe for your system is due to the presence of AgCl. It would be interesting to repeat your study using a different Au salt as the precursor.
Rene Nome responded: Pedro Camargo, with whom I discussed this matter, said that after the synthesis, the nanoparticles are washed through successive centrifugation cycles with supernatant decantation employing NaCl saturated solution. This procedure removes any AgCl that could have been formed during synthesis. In this case, with excess Cl−, there is formation of AgCl2− complex, which is soluble. Performing the synthesis with another precursor that does not contain chloride could be an interesting study but we do not believe there is chloride on the nanoparticle products reported here.
Revana Chanerika asked: Silver is very mobile. What procedure do you use (apart from calcination) to stabilise the particle sizes and prevent agglomeration before introducing Au?
Rene Nome answered: Pedro Camargo, with whom I discussed this matter, indicated that the nanoparticles are made by wet-chemistry methods. The control over size stabilization and aggregation prevention is performed employing PVP, which is added in the reaction medium during the Ag NP synthesis. In this case, PVP acts as a stabilizing ligand. We have polymeric or steric NP stabilization, thus preventing their aggregation.
Maurits Boeije opened discussion of the paper by Aram Bugaev: The Rietveld refinement of the powder diffraction pattern shows good correspondence with the model. However, from the article it is not clear what the crystal structure is, because the Wyckoff positions do not correspond to the space group and the provided input does not completely correspond to Fig. 2. Are you sure the values that are provided are correct? In addition, the peaks at 9.4 and 13.2 degrees gradually split, beginning around 200 K. They clearly stand out from the background and indicate a second-order phase transition, corresponding to a lowering of symmetry. What implications does this have for the MOF and its behavior?
Aram Bugaev replied: We confirm that all values obtained from the Rietveld refinement are correct. The Wyckoff positions do exactly correspond to the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and the atomic coordinates of each atom remain close to the initial ones. Thus, the crystal data provided in Table 1 are complete and consistent. In the paper (DOI: 10.1039/c7fd00224f), the atomic structure presented in Fig. 2 is also correct and is fully based on the data provided in Table 1. However, this structure is aimed to give the reader an idea of what the linker connecting the two secondary building units looks like. For example, atomic occupancy for C3 and C4 atoms is about 0.5. This reduction comes from the fact that rotation of the linker results in the change of its Wyckoff position from 96j to 192l. This happens because in a real sample, different linkers can be rotated in different directions. However, the aim of Fig. 2 is to show only one linker and, therefore, we have put C3 and C4 atoms in one of the two possible equivalent positions and colored them as if they had an occupancy factor ∼1. For any further details on the refinement, please refer to the CIF-file attached in the Supplementary Files.
m space group and the atomic coordinates of each atom remain close to the initial ones. Thus, the crystal data provided in Table 1 are complete and consistent. In the paper (DOI: 10.1039/c7fd00224f), the atomic structure presented in Fig. 2 is also correct and is fully based on the data provided in Table 1. However, this structure is aimed to give the reader an idea of what the linker connecting the two secondary building units looks like. For example, atomic occupancy for C3 and C4 atoms is about 0.5. This reduction comes from the fact that rotation of the linker results in the change of its Wyckoff position from 96j to 192l. This happens because in a real sample, different linkers can be rotated in different directions. However, the aim of Fig. 2 is to show only one linker and, therefore, we have put C3 and C4 atoms in one of the two possible equivalent positions and colored them as if they had an occupancy factor ∼1. For any further details on the refinement, please refer to the CIF-file attached in the Supplementary Files.
As we have described in the paper (DOI: 10.1039/c7fd00224f), all XRD peaks are shifted towards higher 2θ due to a small decrease of the lattice parameter. The two X-ray diffraction patterns are shown here in Fig. 10 in a standard I (2θ) representation for clarity: they represent the UiO-67-Pd MOF at the beginning (black) and at the end (red) of the H2-temperature programmed reduction (TPR) treatment. One should also consider that these data were collected at the synchrotron with the wavelength λ = 0.51353(1) and that the cell parameter of UiO-67 is about 27 Å. Due to this fact, the observed peaks in the 2θ region from 8 to 16° correspond to high hkl indexes. This region contains about 40 independent reflections, and the changes of their intensity can be mistaken for the peak splitting, especially looking at the 2D map shown in Fig. 4 of the main text (DOI: 10.1039/c7fd00224f). For example, the ostensible splitting at 13.4° is the result of an increase in relative intensity of the (11 3 3) reflection. In addition, at 9.2° there is a (6 4 4) reflection, which does not split. Although the appearance of a new peak in this region is observed, it is difficult to assign this peak to any phase, because all other reflections are fitted by the standard UiO-67 structure. It is worth noticing that this additional peak interestingly disappears after cooling down to room temperature.
 | ||
| Fig. 10 X-ray diffraction patterns of the UiO-67-Pd sample taken during reduction in hydrogen at 33 °C (black) and 300 °C (red). | ||
Jiaguang Zhang asked: It was mentioned that the NPs are prepared by 1/10 functionalising ligands, less than 1 Pd atom in the cage, is there any model for how these Pd atoms travel in the framework? The size of the NP is bigger than the size of the cage - any explanation?
Aram Bugaev answered: We did not introduce any special model to describe the travel of Pd atoms through the pores of UiO-67, however the mobility of small Pd ions in the big pores (1.6 nm for the octahedral cavities) (DOI: 10.1039/c7fd00224f) resulting in the formation of metal clusters seems to be quite a natural effect, and the process should be analogous to the formation of metal nanoparticles on the standard supports. TEM images show that only a small part of the cages contains Pd clusters, which correlates with the fact that only 10% of linkers have been functionalized by Pd.
The average size of the NPs determined by using EXAFS is 2.1 nm, which implies that the left side of the particle size distribution is compatible with the octahedral cavity diameter of a defect-free UiO-67 framework. Moreover, it should be considered that EXAFS data are weighted by the weight of the particles and not on their number. This means that the small fraction of particles which is formed outside the pores on the outer surface of UiO-67 crystals considerably shifts the average size to bigger values. The co-existence of particles with different sizes leads to a relatively big error in the determination of the average coordination number, which corresponds to a particle size in the range from 1.6 to 3.2 nm as shown in Fig. 6c of the main text (DOI: 10.1039/c7fd00224f).
Yaroslav Odarchenko remarked: There are 2 XRPD peaks (around 9.4° and 13.2°) that behave differently upon heating as in Fig. 4b. They seem to split and one of the components moves to the lower 2θ that could correspond to the expansion of the MOF lattice in certain crystallographic planes.
Aram Bugaev responded: The explanation of such behavior was already given in the reply to the question before last. All of the peaks of the UiO-67 structure behave in a similar way and shift slightly to the higher 2θ angles due to the decrease of the lattice parameter. At 2θ = 13.2° we have observed an increase in intensity of the (11 3 3) reflection. To show that all peaks behave in a similar way, two XRPD patterns at the beginning and in the end of H2-TPR treatment have been shown in the previously mentioned response.
Carlo Lamberti opened general discussion of the papers by Valerii Bukhtiyarov, Rene Nome and Aram Bugaev: I would like to come back to the possible location of the Pt nanoparticles (NPs) after the H2-reduction of the Pt-functionalized UiO-67-Pt metal-organic framework (MOF) (DOI: 10.1039/c7fd00224f). The NP belonging to the left side of size distribution reported in the inset of Fig. 7b (DOI: 10.1039/c7fd00224f), can be hosted in the octahedral cavities of UiO-67 (16 Å in diameter).1 I believe that also a fraction of those having a diameter larger than 16 Å may still be located inside the MOF framework. Indeed, as underlined by Bruce Gates in his Introductory Lecture (DOI: 10.1039/c8fd00076j), MOFs of the UiO-66 and -67 category may contain a quite large fraction of defects (missing linkers and/or missing inorganic cornerstones) that lead to larger internal defective cavities.2–7 Actually, defect sites may act as preferential anchoring sites for the metal NP. Obviously, the fraction of the NP with a larger diameter must be hosted outside the MOFs crystals.
1 S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14, 1614–1626.
2 L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mat., 2011, 23, 1700–1718.
3 G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mat., 2014, 26, 4068–4071.
4 C. Atzori, G. C. Shearer, L. Maschio, B. Civalleri, F. Bonino, C. Lamberti, S. Svelle, K. P. Lillerud and S. Bordiga, J. Phys. Chem. C, 2017, 121, 9312–9324.
5 V. V. Butova, A. P. Budnyk, A. A. Guda, K. A. Lomachenko, A. L. Bugaev, A. V. Soldatov, S. M. Chavan, S. Oien-Odegaard, U. Olsbye, K. P. Lillerud, C. Atzori, S. Bordiga and C. Lamberti, Cryst. Growth Des., 2017, 17, 5422–5431.
6 D. Yang, V. Bernales, T. Islamoglu, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2016, 138, 15189–15196.
7 D. Yang, M. A. Ortuno, V. Bernales, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2018, 140, 3751–3759.
Graham Hutchings addressed Carlo Lamberti: I am trying to understand the catalysis data presented in Fig. 9b of your paper. The activity of the catalyst appears to increase with time when reacted at 25 °C and 40 °C. If so why is this happening and is the catalyst structure unchanged after the reaction, i.e. is the MOF structure intact?
Carlo Lamberti answered: XRD and N2-adsorpton isotherms confirm that both the crystallinity ant the internal surface area of the Pd-functionalized MOF are not deteriorated after the ethylene hydrogenation reaction. The stepwise changes in the catalyst activity reported in Fig. 9b (DOI: 10.1039/c8fd00224f) are due to a temperature increase of the experiment, while the non-perfectly constant behavior of the m/z = 30 signal is due to a small drift of the MS signals along the experiment. The MS data reported in this operando IR experiment should not be treated in a quantitative way. These data are not normalized by any reference signal (e.g. He) and the increase of the signal assigned to ethylene may originate from the drift in the response of the mass spectrometer.
Laura Torrente-Murciano addressed Aram Bugaev: What is the degree of accessibility to the Pd nanoparticle system? Your data indicates that the Pd nanoparticles are entrapped within the cages of the MOF crystal, however, not all of them might be accessible to reactants. It is possible that only the particles entrapped close to the MOF surface are accessible. This might be tested by modifying the Pd loading in the system and testing whether the same turn-over values (e.g. moles converted per mol of metal per time) are achieved (which it should if everything else, especially the metal particle size, is constant).
Aram Bugaev answered: This a very critical question for any catalytic system, to determine if all kinds of Pd-nanoparticles (NPs) are active and participate in the reaction. The accessibility of Pd NPs which are inside the cages is supported by many experimental evidences: (i) we observe the formation of palladium carbide which is a consequence of the interaction of palladium NPs with the hydrocarbon molecules (DOI: 10.1039/c7fd00211d);1–2 (ii) we observed full conversion at 80 °C with the following flux conditions: 3 ml min−1 H2, 3 ml min−1 C2H2 and 44 ml min−1 of He, using 10 mg of the sample inside the 1.5 capillary during operando XAS and XRPD measurements. This activity is comparable with supported Pd NPs with much higher Pd loadings, and is unlikely to be achieved if only the particles located outside the MOF are participating in the reaction. Finally, the width of the windows of the MOF cages is much bigger than the size of the substrate used for our test reaction, leaving almost free access to the NPs faces.
1 A. L. Bugaev, A. A. Guda, A. Lazzarini, K. A. Lomachenko, E. Groppo, R. Pellegrini, A. Piovano, H. Emerich, A. V. Soldatov, L. A. Bugaev, V. P. Dmitriev, J. A. van Bokhoven and C. Lamberti, Catal. Today, 2017, 283, 119–126.
2 A. L. Bugaev, O. A. Usoltsev, A. A. Guda, K. A. Lomachenko, I. A. Pankin, Yu. V. Rusalev, H. Emerich, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, J. Phys. Chem. C, 2018, 122, 12029–12037.
Katharina Brinkert asked: Could you comment on the stability of the MOF-Pd system? Have you studied degradation mechanisms?
Aram Bugaev replied: We have chosen the UiO-67 material for this study because of the exceptional thermal and chemical resistance of this class of MOFs,1–4 already mentioned by Bruce Gates in his Introductory Lecture (DOI: 10.1039/c8fd00076j). The stability of the UiO-67-Pd system was confirmed via two independent methods. First, temperature-dependent XRPD data collected in situ together with XAS spectra (Fig. 4b of the main text (DOI: 10.1039/c7fd00224f)) indicate that the material does not lose its crystallinity even at 300 °C under H2 flow and after the formation of Pd nanoparticles (NPs). In addition, we have performed N2 adsorption measurements (shown in Fig. 11) for the UiO-67 MOF with and without Pd functionalization, as well as after reduction in H2 at 300 °C, obtaining BET surface areas of 2618, 2398 and 2156 m2 g−1, respectively. As the theoretical surface area of non-functionalized UiO-67 is 2850 m2 g−1 (calculated using the grand canonical Monte Carlo method with a probe of radius = 1.82 Å3), the obtained values are compatible with an almost perfect UiO-67 starting material (2618, m2 g−1, black curve in Fig. 11), that loses a first fraction of its BET area because of the insertion of 1/10 of bulky PdCl2bpydc linker (2398 m2 g−1, blue curve in Fig. 11) and a second fraction upon formation in the pores of Pd NPs (2156 m2 g−1, red curve in Fig. 11). This behavior mirrors what was already observed for the Pt-functionalized UiO-67 system.5,6 Summarizing, the combined XRPD and volumetric studies demonstrate that the Pd-functionalized UiO-67 framework, subjected to the H2-TPR treatment, keeps its crystallinity and that it loses a small fraction of its surface area (about 10%), which is compatible with the formation of Pd NPs insides its pores.
 | ||
| Fig. 11 N2 adsorption isotherms of UiO-67 with 10% of modified ligands non-functionalized (black squares) and functionalized by Pd before (blue triangles) and after (red circles) activation in H2 at 300 °C. | ||
1 J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851.
2 L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718.
3 S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14, 1614–1626.
4 G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mat., 2014, 26, 4068–4071.
5 L. Braglia, E. Borfecchia, K. A. Lomachenko, A. L. Bugaev, A. A. Guda, A. V. Soldatov, B. T. L. Bleken, S. Oien-Odegaard, U. Olsbye, K. P. Lillerud, S. Bordiga, G. Agostini, M. Manzoli and C. Lamberti, Faraday Discuss., 2017, 201, 277–298.
6 L. Braglia, E. Borfecchia, A. Martini, A. L. Bugaev, A. V. Soldatov, S. Oien-Odegaard, B. T. Lonstad-Bleken, U. Olsbye, K. P. Lillerud, K. A. Lomachenko, G. Agostini, M. Manzoli and C. Lamberti, Phys. Chem. Chem. Phys., 2017, 19, 27489–27507.
Michael Bowker remarked: A point for general discussion. With the new and rapid developments in nano-imaging techniques such as HRTEM, acTEM which now have in situ, operando capabilities, do you think synchrotron techniques may become less necessary in the field of catalysis? It seems that now with TEM we can analyse even the surface layers both chemically and structurally while carrying out reactions on real, powdered catalysts.
Cynthia Friend responded: With regard to Michael Bowker’s question about TEM vs. X-ray techniques: in my opinion, they are distinct and complementary. TEM probes single particles and usually only a few are imaged in detail. On the other hand, X-ray tools average over the ensemble of catalyst particles and have the possibility of being atom specific (i.e. enabling compositional analysis) and being more sensitive to light elements and surface sensitive.
Graham Hutchings added: Adding to the debate, TEM as a technique is making huge advances at present especially with the design of detectors but there are reactions that are of interest, e.g acetylene hydrochlorination, that are not at present suitable for in situ TEM. So other in situ techniques have to be used at present.
Richard Catlow remarked: Despite the very rapid progress in microscopy, there will be a continuing need for X-ray based techniques, both diffraction and spectroscopy, as the latter is giving ensemble average information while the former gives images of specific species. The two classes of technique are complementary.
Justin Hargreaves asked: In the application of in situ TEM, are there any concerns about the formation and effects of reactive plasmas?
Michael Bowker answered: Yes, certainly, high power inputs can generate excited and dissociated species. Care needs to be taken with this and lower power TEMs are being developed.
Rosa Arrigo added to the discussion: With respect to the question asked by Michael Bowker on whether in situ transmission electron microscopy (TEM) will overtake synchrotron-based techniques in the future, I agree with Richard Catlow on the importance of applying complementary in situ characterization techniques in catalysis. As an imaging technique, in situ TEM will be very powerful to observe structural dynamics with millisecond time resolution. However, my feeling is that the analytical part (electron energy loss spectroscopy EELS) will be very challenging to perform in situ. I would like to bring to attention our work,1 in which we compare NK edge absorption spectra of carbon materials measured by EELS in a TEM and by synchrotron-based soft X-ray NEXAFS. Therein, it is very clear that the energy resolution achieved by EELS was not comparable to the resolution achieved by NEXAFS. Although by using a monochromated electron beam it is possible to achieve the same energy resolution as in NEXAFS, the occurrence of beam damage and the detection limit of the instruments will be limiting factors for the success of such an in situ experiment. Additionally, EELS is not a surface sensitive technique and therefore synchrotron-based surface sensitive methodologies will play an important role in catalysis.
1 R. Arrigo, M. E. Schuster, Z. Xie, Y. Yi, G. Wowsnick, L. L. Sun, K. E. Hermann, M. Friedrich, P. Kast, M. Hävecker, A. Knop-Gericke and R. Schlögl, ACS Catal., 2015, 5, 2740–2753.
Carlo Lamberti said: TEM instruments equipped with XRF detectors and electron energy loss instrumentation and microfocus beamlines at synchrotron facilities are important instruments that allow space resolved speciation of compositional, structural and electronic properties of catalysts. TEM provides a much better spatial resolution, but the focusing performances of synchrotron sources have increased tremendously in the last decade, and will further improve with the diffraction-limited fourth generation of storage rings.1 On the other hand synchrotrons provide a much higher energy resolution of the collected XANES spectra and allow work at high pressures in the gas phase and in the liquid phase. I foresee that, in the near future, the improvements of both instruments will progressively reduce the relative spatial resolution, energy resolution and pressure gaps between the two facilities.
1 L. Mino, E. Borfecchia, J. Segura-Ruiz, C. Giannini, G. Martínez-Criado and C. Lamberti, Rev. Mod. Phys., 2018, 90, 025007.
Wilke Dononelli addressed Rene Nome: One very interesting result from your paper is, that depending on the used concentration of Au in your core–shell nanoparticles, Au is either found at the core, or the shell. I was wondering why it will not be the same metal independent of the concentration that is found at the core or in the shell. E.g. there is a study by Hoppe et al. that indicates that silver was most stable at a subsurface layer of a gold single crystal surface, if no adsorbates are found at the surface.1 They also found that no real segregation was found in these single crystals. Could you please explain why you find your nanoparticles as core–shell systems and not as perfectly dispersed nanoparticles? Could you suggest an explanation, what is the driving force for the change of the metal in the core and in the shell?
1 S. Hoppe and S. Müller, J. Appl. Phys., 2017, 122, 235303.
Rene Nome replied: Indeed, depending on the initial Au concentration used, surface segregation in the hollow bimetallic core–shell AgAu nanoparticles investigated here changes from Au rich to Ag rich.1 In the present work, we report that the influence of initial Au concentration used is also manifested in the resulting ultrafast pump-probe transients. These findings allowed us to correlate the AgAu composition and catalytic activity with the intrinsic electron-phonon coupling times reported herein. We believe the core–shell structures correspond to a metastable state in the underlying energy landscape, and that there is a high energetic barrier separating the core–shell and alloyed states such that the core shell systems are stable. About the driving force underlying nanostructure formation, single-nanoparticle mechanistic studies of the galvanic exchange reaction between Ag and Au3+ have characterized the stochastic nature of this reaction, in addition to identifying a critical structural event associated with void size formation.2 After void formation, hollow nanostructures are formed spontaneously and rapidly.
1 E. A. Lewis, T. J. A. Slater, E. Prestat, A. Macedo, P. O’Brien, P. H. C. Camargo and S. J. Haigh, Nanoscale, 2014, 6, 13598.
2 J. G. Smith, Q. Yang and P. K. Jain, Angew. Chem. Int. Ed., 2014, 126, 2911.
Andrea Russell commented: This really just returns to my previous question, in that as you increase the amount of Au, you are also increasing the amount of chloride ions and will end up with more AgCl in the sample. So, as I've already stated, I think that it would be very interesting to compare to samples prepared in the absence of chloride ions.
Michael Bowker asked Valerii Bukhtiyarov: Is gas phase activation by X-rays or by secondary electron emission playing a role in affecting the chemistry observed in these experiments?
Valerii Bukhtiyarov answered: The question about the influence of X-rays or secondary electrons is quite general to the researchers who carry out in situ or operando XPS experiments. There is a routine method which has been often used by researchers who work in the field of NAP XPS. They perform two experiments with and without X-rays and try then to compare the results of such experiments and to scrutinize the differences. Honestly speaking, I am not aware of any indications of such effect even for the case when the nature of adsorbed or reactive species has been characterized. To support this statement I can cite such investigations as attempts to fix CO dissociation over transition metals or to identify weakly bound oxygen adspecies over gold caused by ambient pressures. All of them were unsuccessful. So I am quite sure that in our investigation we have chemistry behavior rather than X-ray effects.
Carlo Lamberti opened discussion of Hans-Joachim Freund’s paper: My question is related to the part of your paper1 dealing with the Phillips catalyst. Although this industrial catalyst has been known since the fifties,2 the nature of the reduced Cr sites (in terms of molecular structure, local geometry and oxidation state) is still the subject of a long, ongoing, debate.3–12 In this regard your approach may be revolutionary in understanding the structure of the active site. Do you believe you may also be able to identify the oxidation state of chromium during ethylene polymerization?
1 Q. Pan, L. Li, S. Shaikhutdinov, Y. Fujimori, M. Hollerer, M. Sterrer and H.-J. Freund, Faraday Discuss., 2017, Doi: 10.1039/c7fd00209b.
2 J. P. Hogan and R. L. Banks, US Pat., 2.825.721, 1958.
3 B. M. Weckhuysen, I. E. Wachs and R. A. Schoonheydt, Chem. Rev., 1996, 96, 3327–3349.
4 E. Groppo, C. Lamberti, S. Bordiga, G. Spoto and A. Zecchina, Chem. Rev., 2005, 105, 115–183.
5 E. Groppo, C. Prestipino, F. Cesano, F. Bonino, S. Bordiga, C. Lamberti, P. C. Thune, J. W. Niemantsverdriet and A. Zecchina, J. Catal., 2005, 230, 98–108.
6 G. Agostini, E. Groppo, S. Bordiga, A. Zecchina, C. Prestipino, F. D’Acapito, E. van Kimmenade, P. C. Thune, J. W. Niemantsverdriet and C. Lamberti, J. Phys. Chem. C, 2007, 111, 16437–16444.
7 M. P. McDaniel, Adv. Catal., 2010, 53, 123–606.
8 E. Groppo, K. Seenivasan and C. Barzan, Catal. Sci. Technol., 2013, 3, 858–878.
9 M. P. Conley, M. F. Delley, G. Siddiqi, G. Lapadula, S. Norsic, V. Monteil, O. V. Safonova and C. Coperet, Angew. Chem. Int. Edit., 2014, 53, 1872–1876.
10 M. F. Delley, F. Nunez-Zarur, M. P. Conley, A. Comas-Vives, G. Siddiqi, S. Norsic, V. Monteil, O. V. Safonova and C. Coperet, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11624–11629.
11 C. Brown, J. Krzystek, R. Achey, A. Lita, R. Fu, R. W. Meulenberg, M. Polinski, N. Peek, Y. Wang, L. J. V. de Burgt, S. Profeta, A. E. Stiegman and S. L. Scott, ACS Catal., 2015, 5, 5574–5583.
12 C. Barzan, A. Piovano, L. Braglia, G. A. Martino, C. Lamberti, S. Bordiga and E. Groppo, J. Am. Chem. Soc., 2017, 139, 17064–17073.
Hans-Joachim Freund answered: As you correctly point out, there is an ongoing debate about the structure and electronic properties of the active Phillips catalyst. It is the aim of our studies to contribute to this debate, in the sense that we hope to be able to image the active site and through a combination of experiments elucidate the electronic structure.
Andrea Russell asked: I note that your results show that the edges of the Au particles are charged and this made me think back to the paper presented earlier by Yaroslav Odarchenko (DOI: 10.1039/c8fd00007g) where they saw that water was activated on the support at the edges of the nanoparticles. Would you be able to put these two observations together? In other words, does the charge on the edge of the metal nanoparticles modify the local electronic structure of the support and does this play a role in the activation of the water? I’m including Yaroslav Odarchenko in this question, as he may also wish to comment.
Yaroslav Odarchenko responded: It is a very good point. I can add that one has to be careful with the charging of the nanoparticles with the X-ray beam during the measurement. In our GISAXS/GIXD experiments we have not observed any direct correlation between presence/absence of the X-rays and catalyst activity (i.e. water activation).
Hans-Joachim Freund answered: We have not studied water adsorption on our systems. We do plan to do so in connection with CO2 reaction at the rim as alluded to in our present paper. Due to the localization of electrons at the rim, I could imagine that due to the long range of electrostatic interactions the adsorption of water may be influenced.
Bruce Gates enquired: You presented evidence of a new class of samples, hydroxylated silica layers, for the investigation of supported catalytic species. You incorporated chromium sites on the silica layers; when you made these samples, did you determine how the chromium precursor reacts with OH groups and demonstrate the removal of OH groups? Can you regulate the density of the OH groups and thereby regulate the coverage of the surface with Cr?
Hans-Joachim Freund responded: The hydroxylation is accomplished by electron excitation of condensed water and depends strongly on exposure. We will be ultimately able to control the amount by understanding the details of the electron excitation mechanism and the parameters the excitation depends on. Studies in this direction are in progress.
Bruce Gates asked: Do you see products of the reaction of your precursor with the surface OH groups, splitting off some organic species as the metal was anchored to the surface?
Hans-Joachim Freund replied: We are performing studies in this direction, and there are some very preliminary results on the structure of the anchored precursor but it is too early for detailed conclusions.
Bob Tooze enquired: Chromium based complexes with specific phosphine ligands have been shown to give selective oligomerisation of ethene to yield 1-hexene or 1-octene. There is ongoing debate about the mechanism, namely metallacycle versus chain growth, this has also been postulated to involve two chromium centres by some authors, do you think it would be possible to observe intermediates to help this debate?
Hans-Joachim Freund replied: In principle we should be able to contribute to answering this question, since the approach is independent of the complex used. The requirement is, of course, that the complex may be supplied through evaporation.
Graham Hutchings asked: Following up on the point raised by Bruce Gates, you introduce OH groups onto silica by reacting water. Have you investigated using D2O and does this change the reactivity and change the dispersion of the Cr?
Hans-Joachim Freund answered: We have done this. There is no problem with replacing the water used with heavy water. For mechanistic studies such labelling could be very useful. I would like to point out that we cannot only label the adsorbed molecules we could also label the silica film itself by preparing it using 18O2 .
Bruce Gates enquired: Your sample seems to open the way to comparisons with catalysts on high-area hydroxylated supports, such as silica gel and mesoporous silicas. What opportunities do you see for such comparisons?
Hans-Joachim Freund responded: We are limited at present to the double layer silica films. However, the film comes in a crystalline and a vitreous form and we think that the vitreous one contains structural motifs that typically occur in gels. This could perhaps open up possibilities in the direction of the posed question. Also, since we have demonstrated that the film may be removed from the substrate after preparation it might be possible in the future to stack these films on a substrate in order to mimic closer resembling bulk supports.
Graham Hutchings asked: Considering the flat gold structures where CO is activated at the peripheral sites, what happens if you introduce CO along with the CO2? Would the CO compete for the peripheral sites and switch off the reaction and maybe CO could be a useful chemical probe?
Hans-Joachim Freund responded: According to previous studies of CO on those sites Lin et al.1 have studied the adsorption of CO on those systems. CO2 is bound much more strongly, so I would not expect that CO could really compete for sites. If the particles were not charged the situation would certainly be different.
1 X. Lin, B. Yang, H.-M. Benia, P. Myrach, M. Yulikov, A. Aumer, M. A. Brown, M. Sterrer, O. Bondarchuk, E. Kieseritzky, J. Rocker, T. Risse, H.-J. Gao, N. Nilius, and H.-J. Freund, J. Am. Chem. Soc., 2010, 132, 7745–7749.
Bruce Gates questioned: Could you say more about the dimerization of ethylene on your sample? Does the reaction turn over (can you remove the dimer and repeat the process)? Do you have any evidence of higher oligomers than dimers?
Hans-Joachim Freund replied: We do not have any detailed information of the mechanism of the dimerization. The evidence stems from temperature programmed desorption studies monitoring the relevant masses and IRAS measurements. We do not have any evidence for higher oligomers than dimers so far.
Valerii Bukhtiyarov opened discussion of the paper by Stig Helveg: In your TEM study, specially prepared supported vanadia catalysts were used. Did you characterise these catalysts with other physical methods, for example with XPS? What was the oxidation state of vanadium?
Stig Helveg answered: We have previously studied vanadium loaded anatase TiO2 as SCR catalysts.1 In this work electron energy loss spectroscopy was used to address the spatial distribution of vanadium as well as its oxidation state. The measurements showed a very homogeneous vanadium distribution and an oxidation state with a strong dependency of the oxygen partial pressure, implying that in situ conditions are mandatory to obtain relevant insight about the vanadium.
1 M. Ek, Q. M. Ramasse, L. Arnarson, P. Georg Moses and S. Helveg, Nature Comm., 2017, 8, 305.
Carlo Lamberti asked: Non-stoichiometry is quite an issue for TiO2, do you think that the advanced techniques shown in your paper (DOI: 10.1039/c7fd00222j) (combined TEM/STM at atomic resolution) offer some chance to get insights on the structural environment around an oxygen vacancy?
Stig Helveg replied: In our present work the STM definitely resolved point defects in the (001) surface. Other STM studies of rutile TiO2 also offered a characterization of O vacancies, OH groups etc. With TEM , the characterization is more challenging due to the projection geometry. Our paper discusses that the reduced contrast of atomic columns at the step edge could reflect a lower content of either O and Ti atoms compared to the neighboring atomic columns.
Carlo Lamberti addressed Graham Hutchings: I fully agree with you that the cost of a synchrotron is some order of magnitude higher than the costs of an environmental TEM, however the costs of synchrotrons are paid by the whole community. As a consequence, for a group that has no environmental TEM facility in its department, it may be easier to get access to synchrotron light rather than to environmental TEM.
Graham Hutchings replied: At present I agree this is the situation but soon I envisage that high end TEM may have similar national facilities for access to environmental TEM. Of course access to skilled microscopists will be essential to ensure the most is obtained from any session.
Said Said asked Stig Helveg: In relation to discrepancies, can you comment on the choice/importance of the substrate for imaging?
Stig Helveg answered: We studied the anatase-TiO2 (001) surface on nanoparticles and single-crystals and the results show reasonably good agreement on the structural features on the two types of samples. As we discuss the article, slight differences between the two samples could be due to the difference in preparation methods or the sample size.
David Willock enquired: You showed very nice atomically resolved images of step edges on the surface of titania. We are more used to seeing "fuzzy" regions on the edge of such materials in TEM images and assume that these show amorphous regions on the surface of the oxide. Could you comment on the conditions required to obtain clear images? What is the requirement for ordering in the Z-direction to see step edges clearly and how was beam damage of the oxide avoided?
Stig Helveg replied: To obtain the atomically resolved images, we employed low-dose-rate imaging under oxidizing conditions and describe in the paper how this approach was optimized to suppress beam-induced sample alternations. We have described detection schemes like this in several recent papers.1,2 Abandoning the optimized detection scheme can definitely result in more fuzzy regions of the sample surface as also shown in the paper.
1 S. Helveg, C. F. Kisielowski. J. R. Jinschek, P. Specht, G. Yuan and H. Freib, Micron, 2014, 68, 176–185.
2 S. Helveg, J. Catal., 2015, 328, 102–110.
Hans-Joachim Freund asked: Did you do STM and TEM on the same sample?
Stig Helveg answered: The present STM and TEM measurements were done using an extended single crystal and a commercially available powder, respectively. However, it would in principle be possible to conduct both types of measurements on the same sample as long as it obeys the geometric constraints; that is, the sample should be sufficient plane and wide for top-view imaging with the STM and the sample should be sufficiently thin for side-view imaging in TEM. So it should be possible to do STM and TEM on the same sample in orthogonal viewing directions but it will require more elaborate sample preparation than is employed in the present work.
Annette Trunschke enquired: High-performing catalysts are often very complex in terms of chemical composition and/or structure. Does the spatial resolution of electron microscopy represent an advantage in describing any possible local inhomogeneity on the surface of such a catalyst or predominate technical or fundamental limits in analyzing such samples?
Stig Helveg responded: The electron microscopy stands out by offering atomic-resolution information about the individual nanoparticles including their surface structure and dynamic behavior as illustrated in our paper. This insight complements ensemble-averaged information obtained from various photon-based techniques as well as information obtained from simpler surface science model systems. Relating the insight from electron microscopy to the global catalyst performance is definitely challenging in cases where the catalyst is characterized by broad distributions in size, shape, composition and locations. In cases where the catalyst heterogeneity is low, such as for the catalyst discussed by Bruce Gates, the link between structure and performance is obviously more straightforward to make. In general, a close interplay with the photon-techniques and other methods should be beneficial to continue to address the issue of catalyst heterogeneity.
Bruce Gates asked: Would you please share your perspective on the effectiveness of national user facilities for the characterization of complex catalytic materials with TEM? What do you think it takes in practice for such a facility to be effective in helping non-experienced users to do meaningful characterization of such materials to determine the structural features that are important for catalysis?
Stig Helveg answered: To obtain chemically relevant information with the more advanced electron microscopy techniques can be a delicate matter at present. First, a set of observations is needed that address the heterogeneity of the catalyst structure and composition in sufficient detail. Second, the catalyst may have to be examined under different reaction conditions or with a palette of complementary electron microscopy techniques that may be technically complex to execute. Third, it has become clear that the electron illumination needed for generating high-resolution information in general induces alterations in the catalysts. Hence it becomes mandatory to evaluate the role of the electron beam on the measurements. We have recently discussed strategies that may help suppressing such beam-induced sample alterations by using low-dose-rate imaging and dose-fractionating techniques.1,2 As a result, the advanced electron microscopy examinations, in particular in situ studies, require extended measurements sessions, which may extend beyond the few days that are often provided by the user facilities, and require support from specialized scientists as well.
1 S. Helveg, C. F. Kisielowski. J. R. Jinschek, P. Specht, G. Yuan and H. Freib, Micron, 2014, 68, 176–185.
2 S. Helveg, J. Catal., 2015, 328, 102–110.
Cynthia Friend commented: User facilities for in situ electron microscopy can be effective. The critical factors are to have sufficient time for experiments, including repeat visits, careful thought and preparation for experiments (including ex situ TEM and catalyst pretreatment) and collaboration with expert scientific staff at the facilities. Experiments and objectives also need to be carefully thought out. The requirements are similar to synchrotron experiments.
Yaroslav Odarchenko addressed Stig Helveg: In your in situ TEM experiment the annealing temperature was up to 870 K, however it’s know that the anatase phase is metastable and starts to transform to rutile at temperatures as low as 673 K.1 Could you please tell us whether you have observed any presence of the rutile phase? For example, we have observed a significant increase in conductivity of the single crystal rutile TiO2 (110) upon annealing at 773 K and our X-PEEM data suggests co-existance of rutile and anatase. Have you considered of using rutile or other cheaper alternative to anatase (e.g. rutile doped with Ti for the catalytic applications)?
1 D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874.
Stig Helveg replied: We focussed on anatase TiO2 which is commonly used in industrial-style SCR catalysts. The images show that the anatase structure remains, so I expect that the time-scale of the electron microscopy is not sufficient to monitor the phase transition.
Francesca Baletto addressed Hans-Joachim Freund: A comment/question on Fig. 9 of the paper, which shows STM images of gold nanoparticles. The average mean island is measured as a function of the annealing temperature. Is the reduction of the mean area with T after the 2D-3D transition a sign of a different wettability of the cluster? Could you estimate/measure how the nearest and second nearest neighbour change with the annealing temperature?
Hans-Joachim Freund answered: The reduced area as seen in the STM images indicates that Au atoms from the island rim may migrate on top of the particle at increasing temperature, thus leading to three-dimensional particles. We have not attempted to estimate the nearest or next nearest neighbour distances between particles.
Roy Johnston added: My question is for Hans-Joachim Freund and follows on from the question of Francesca Baletto. Assuming that the electron transfer from the underlying silver substrate to the MgO-supported gold nanoparticles stabilises the 2D structure relative to 3D, if the thickness of the MgO film is gradually increased, would you expect the 2D-3D transition temperature to decrease?
Hans-Joachim Freund responded: We have investigated the dependence of particle formation on the MgO layer thickness. We have not investigated the temperature dependence for different thicknesses. In previous work1 it is demonstrated that for three layers initially a flat Au particle morphology is observed while for 8 layers only three dimensional particles are observed from the lowest temperature on.
1 M. Sterrer et al., Phys. Rev. Lett., 2007, 98, 206103.
Kassim Badmus asked: What is the best method/procedure that can be used for the determination of the surface charge?
Hans-Joachim Freund answered: I had mentioned that in special cases you may be able to count electrons (in the case of small clusters of Au on MgO by knowing the occupied and unoccupied states, see also Au on alumina1). In general, XPS, if analysed properly, may give you an indication. Also, certain variants of scanning probe techniques give you information on surface potentials connected to surface charges. So, the answer is, there is no best method, but a combination of methods will provide an indication.
1 N. Nilius et al., Phys. Rev. Lett., 2008, 100, 096802.
Conflicts of interest
Maurits Boeije declared that the presented data was obtained using a commercial product, produced by VSPARTICLE and the presenter is an employee of this company. There are no other conflicts to declare.| This journal is © The Royal Society of Chemistry 2018 |