
A highly efficient synthesis of the DEFG-ring system of rubriflordilactone B†
Yong
Wang
,
Yuhan
Zhang
,
Zhongle
Li
,
Zhenjie
Yang
and
Zhixiang
Xie
*
State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, 222 Tianshui South Road, Lanzhou 730000, P. R. China. E-mail: xiezx@lzu.edu.cn
First published on 18th October 2016
Abstract
A highly efficient synthesis of the DEFG-ring system of rubriflordilactone B via polar-radical-crossover cycloaddition and carbonyl allylation/lactonization is described.
Introduction
Many species of the Schisandraceae family, which includes the genera Schisandra and Kadsura, have a long history of use as traditional medicines in China. The most famous species are the dried mature fruits of Schisandra chinensis and S. sphenanthera, with the Chinese Pharmacopoeia name of “Wu-Wei-Zi” and “Nan-Wu-Wei-Zi”, respectively, utilized for the treatment of several diseases such as asthenia, insomnia, and enhancing mental and physical working capacity over two thousand years in China.1 Modern phytochemical and pharmacological studies attract great attention from the medicinal chemistry and drug discovery community owing to the remarkable medicinal functions of the Schisandraceae family. As a milestone in phytochemical investigations of the Schisandraceae species, more than 400 triterpenoids including 200 schinortriterpenoids with 16 frameworks have been isolated from fruits, vines, and stems of the plants.2 About ten years ago, the attractive architectures and biologic activities of Schisandraceae triterpenoids attracted attention from synthetic organic chemists.3 The complex oxygenated skeletons of the schinortriterpenoids also represent a formidable synthetic challenge.4 The pioneering work was first reported by Yang and co-workers in 2011 for the total synthesis of schindilactone A.5 Recently, Schilancitrilactones B and C and propindilactone G were synthesized by Tang and Yang, respectively.6Rubriflordilactones A (1) and B (2) were isolated from the leaves and stems of Schisandra rubriflora in 2006 by Sun.7 Their structure and relative configurations were established on the basis of extensive spectroscopic methods and finally confirmed by single-crystal X-ray diffraction (Fig. 1). The structural features of 1 possess a 5/5/7/6/5/6-fused hexacyclic framework containing seven chiral centers and 2 contains a 5/5/7/6/5/5-fused hexacyclic ring system with eight chiral centers. Both their structures have two oxo-quaternary carbon centers in the AB ring system. In addition, an unusual multisubstituted arene motif was embedded in both hexacyclic frameworks, relatively rare in schinortriterpenoids.2 Preliminary biological assays indicated that compound 1 showed weak anti-HIV-1 activity, and compound 2 exhibited an EC50 value of 9.75 μg mL−1 (SI = 12.39) against HIV-1 replication with low cytotoxicity. Their bioactivities and intriguing molecular architecture have drawn a great amount of attention from the synthetic community. Studies on the total synthesis of rubriflordilactones A and B have been reported by many groups.8 The enantiomer of the original proposed rubriflordilactone A was accomplished by Li's group9 in 2014 and Anderson's group10 in 2015, respectively. Very recently, Li and coworkers completed the total synthesis of rubriflordilactone B, and found the synthetic rubriflordilactone B to be identical to the reported structure of authentic rubriflordilactone B derived from an independent X-ray crystallographic analysis and clearly different from the 1H and 13C NMR spectra of the authentic samples in deuterated chloroform and pyridine. Therefore, with other proofs they speculated that the originally isolated sample of “rubriflordilactone B” was composed of two compounds, named rubriflordilactone B and pseudo-rubriflordilactone B, respectively.11
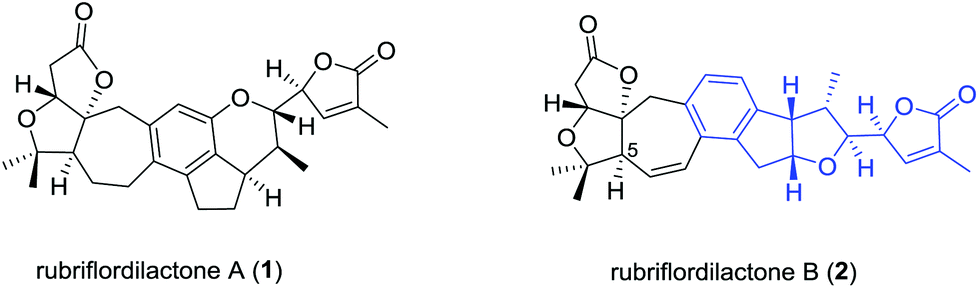 | ||
| Fig. 1 Structure of rubriflordilactones A and B. | ||
Recently we have developed a convergent synthetic route for the construction of the C-5-epi ABCDE ring system of rubriflordilactone B by using the stereoselective Mukaiyama–Michael reaction and intramolecular [2 + 2 + 2] cycloaddition of triynes.12 There are two challenges to complete the total synthesis of B: construction of the remaining ring system and revising the chirality of C-5. Here, we describe our efforts towards the construction of the remaining ring system and the synthesis of the DEFG-ring of rubriflordilactone B.
Results and discussion
Our retrosynthetic analysis is illustrated in Scheme 1. We envisioned that 2 could be made from alkene 3 and alkenol 4 through polar-radical-crossover cycloaddition (PRCC),13 which could help us to transfer chiralities form compound 4 to a tetrahydrofuran moiety. Compound 3 with the undesired configuration at the C-5 position has been synthesized in our group. As the secondary challenge, the revision of the chirality of C-5 is underway in our laboratory. Compound 5 was obtained from compound 6via the ring-closing metathesis (RCM) reaction followed by deprotection to afford compound 4. Compound 6 could be formed from the esterification of the allyl alcohol compound 7, which is a known compound.14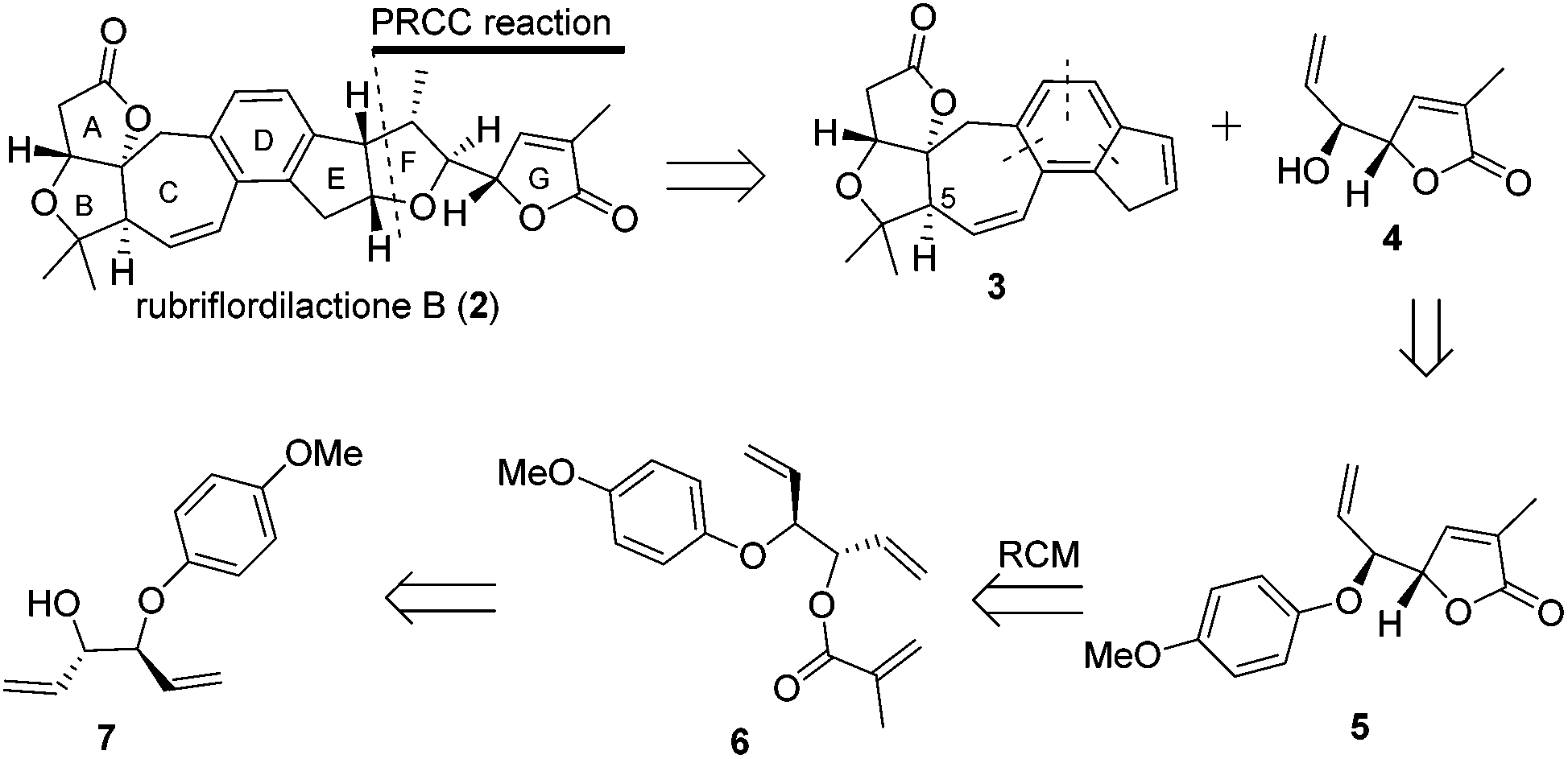 | ||
| Scheme 1 Retrosynthetic analysis of rubriflordilactone B. | ||
The synthesis of compound 4 was accomplished as illustrated in Scheme 2. The synthesis commenced from the known allyl alcohol compound 7, which was prepared from acrylaldehyde in 3 steps.14 Esterification of compound 7 with methacryloyl chloride provided compound 6 in 72% yield. The construction of the lactone was accomplished using the RCM reaction. Compound 6 was treated with 10 mol% Grubbs 2nd generation catalyst to produce compound 5, which was treated with CAN to give compound 4 smoothly.14
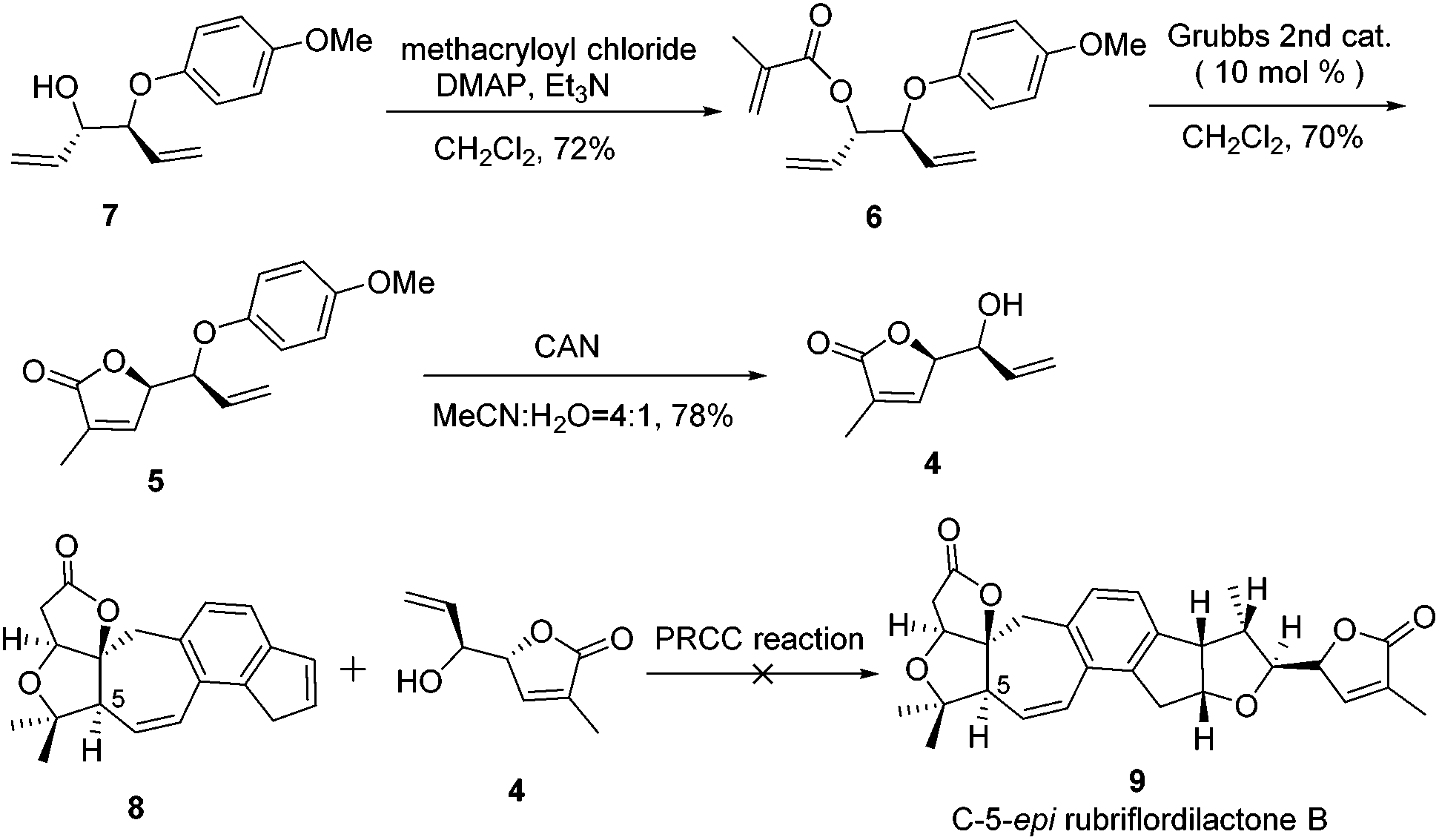 | ||
| Scheme 2 Synthesis of C-5-epi rubriflordilactone B (9). | ||
With compound 4 in hand, the PRCC reaction was investigated. Using compound 8 as an alkene partner, cycloadditions of alkene 8 and alkenol 4 were conducted following the same procedure, which was reported by Nicewicz.13 To our disappointment, the alkene partner 8 decomposed very soon under the reaction conditions. All attempts failed to furnish the desired C-5-epi rubriflordilactone B due to the instability of the alkene partner 8.
In order to shed light on the problem of this reaction, we further explored the PRCC reaction (Table 1) and selected indene as the alkene partner, which was used in the synthesis of tetrahydrofurans by catalytic polar-radical-crossover cycloaddition. Originally, we selected some chiral substrates as the alkenol partner in advantage of diastereoselective synthesis (entries 1–3). The results were disappointing. On using compound 4 as the alkenol partner, the obtained reaction product was complex (entry 1). By using compound 7 and compound 1014 as the alkenol partner, no reaction was observed (entries 2 and 3). To our delight, by using the commercial material penta-1,4-dien-3-ol as the alkenol partner, we obtained the diastereoisomer compounds 13 and 13′ in moderate yields with the dr value 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Significant NOE enhancements observed among the marked protons corroborated the above structural assignment. Fortunately, the major compound 13 with the relative configuration of the four stereogenic centers is desired.
:1. Significant NOE enhancements observed among the marked protons corroborated the above structural assignment. Fortunately, the major compound 13 with the relative configuration of the four stereogenic centers is desired.

With compound 13 in hand, our efforts were focused on the construction of the DEFG-ring system of rubriflordilactone B. As shown in Scheme 3, we first used ozone to cleave the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in compound 13. Unfortunately, we could not obtain aldehyde 14 because of the high decomposition of the substrate.15 When we carried out the same reaction under Li's conditions, we finally achieved compound 14 as expected.16 We conceived that the construction of ring G via carbonyl allylation/lactonization might be carried out through a one-pot procedure. With this in mind, ethyl 2-(bromomethyl)acrylate in THF was added to the mixture of Zn powder and compound 14 in refluxing THF affording two diastereoisomer compounds 15 and 15′, which were unstable and inseparable by column chromatography.17 Therefore, we used it without further purification. After filtration through Celite and removal of the solvents under vacuum, the residue was directly treated with the rhodium catalyst in toluene. The disubstituted exocylic double bond of 15 was successfully transferred into the trisubstituted endocyclic form.18 Fortunately, the desired compound 16 was obtained as a single stereoisomer in 73% yield over two steps. All spectroscopic data of compound 16 were consistent with the previously reported data, which was unambiguously established by X-ray analysis.8e
C in compound 13. Unfortunately, we could not obtain aldehyde 14 because of the high decomposition of the substrate.15 When we carried out the same reaction under Li's conditions, we finally achieved compound 14 as expected.16 We conceived that the construction of ring G via carbonyl allylation/lactonization might be carried out through a one-pot procedure. With this in mind, ethyl 2-(bromomethyl)acrylate in THF was added to the mixture of Zn powder and compound 14 in refluxing THF affording two diastereoisomer compounds 15 and 15′, which were unstable and inseparable by column chromatography.17 Therefore, we used it without further purification. After filtration through Celite and removal of the solvents under vacuum, the residue was directly treated with the rhodium catalyst in toluene. The disubstituted exocylic double bond of 15 was successfully transferred into the trisubstituted endocyclic form.18 Fortunately, the desired compound 16 was obtained as a single stereoisomer in 73% yield over two steps. All spectroscopic data of compound 16 were consistent with the previously reported data, which was unambiguously established by X-ray analysis.8e
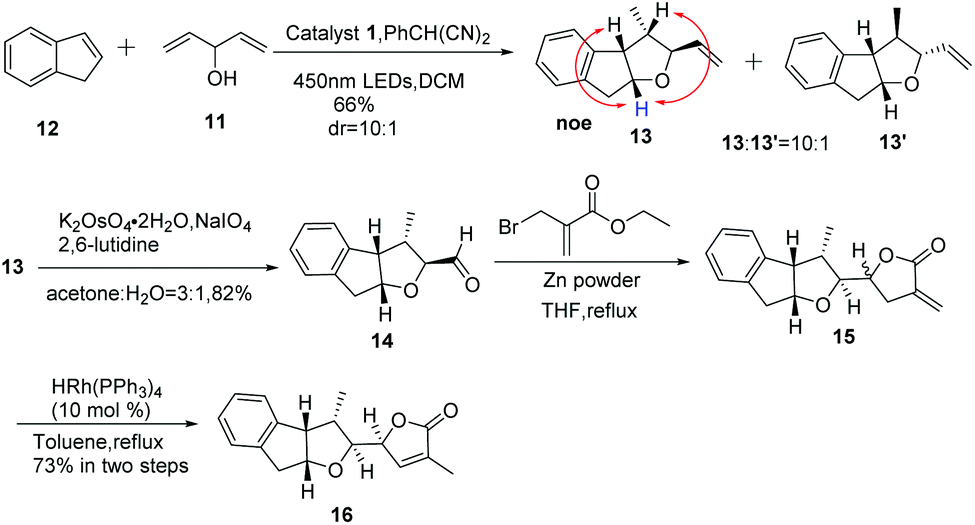 | ||
| Scheme 3 Synthesis of compound 16. | ||
Conclusions
A concise and stereoselective strategy based on polar-radical-crossover cycloaddition has been developed for the efficient assembly of the DEFG-ring system with consecutive five stereogenic centers in rubriflordilactone B from commercial and accessible materials in four steps. Presently, further studies to achieve the total synthesis of rubriflordilactone B using this strategy is still under investigation in our laboratory.Experimental
General experimental methods
Oxygen- and moisture-sensitive reactions were carried out under an argon atmosphere. Solvents were purified and dried by standard methods prior to use. All commercially available reagents were used without further purification unless otherwise noted. Column chromatography was performed on silica gel (200–300 mesh). NMR spectra were recorded on Bruker 400 MHz and Oxford 600 MHz spectrometers in CDCl3 or acetone d6. Chemical shifts are reported as δ values relative to internal chloroform (δ 7.27 for 1H NMR and 77.00 for 13C NMR) and acetone-d6 (δ 2.05 for 1H NMR and 29.92 for 13C NMR). High resolution mass spectra (HRMS) were obtained on a 4G mass spectrometer by using electrospray ionization (ESI) analyzed by quadrupole time-of-flight (Q-TOF). Optical rotations were measured on a Rudolph Autopol IV polarimeter.:1) to afford 6 (538.6 mg, 72% yield) as a colorless oil. [α]20.3D −30.0 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.92–6.86 (m, 2H), 6.83–6.80 (m, 2H), 6.16 (s, 1H), 6.00–5.84 (m, 2H), 5.59 (dd, J = 3.2, 1.6 Hz, 2H), 5.40–5.31 (m, 4H), 4.66 (t, J = 6.0 Hz, 1H), 3.77 (s, 3H), 1.96 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.3, 154.3, 152.2, 136.2, 133.6, 132.3, 125.9, 119.2, 118.7, 117.6, 114.5, 80.8, 75.2, 55.6, 18.3. IR (neat, cm−1): ν 2929, 1720, 1506, 1227, 1162; HRMS (ESIMS) calcd for C17H20O4 Na+ [M + Na]+ 311.1254, found 311.1263.
:1) to afford 5 (175.0 mg, 67%) as a colorless oil. [α]20.1D −48.0 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.13 (t, J = 1.2 Hz, 1H), 6.86–6.78 (m, 4H), 5.76–5.67 (m, 1H), 5.40–5.33 (m, 2H), 5.10–5.08 (m, 1H), 4.71 (t, J = 5.6 Hz, 1H), 3.74 (s, 3H), 1.94 (t, J = 1.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.6, 154.5, 151.3, 145.1, 132.2, 131.9, 120.6, 117.7, 117.6, 114.5, 81.1, 79.8, 55.5, 10.6. IR (neat, cm−1): ν 2953, 2928, 1761, 1506, 1224, 1061, 1037, 827; HRMS (ESIMS) calcd for C15H16O4 Na+ [M + Na]+ 283.0941, found 283.0948.
:1) to afford 4 (38.2 mg, 82%) as a yellow oil. [α]20.5D −62.0 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.02 (t, J = 2.0 Hz, 1H), 5.81–5.73 (m, 1H), 5.23 (d, J = 17.2 Hz, 1H), 5.22 (d, J = 10.4 Hz, 1H), 4.83 (dd, J = 4.0, 2.0 Hz, 1H), 4.18 (t, J = 5.6 Hz, 1H), 3.62 (br, 1H), 1.85 (d, J = 1.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.1, 145.9, 134.7, 131.1, 118.4, 83.5, 73.1, 10.4. IR (neat, cm−1): ν 3427, 1751, 1098, 1060; HRMS (ESIMS) calcd for C8H10O3 Na+ [M + Na]+ 177.0522, found 177.0527.
:1) to afford 13 and 13′ (263 mg, 66%) as a colorless oil. 131H NMR (400 MHz, CDCl3) δ 7.28–7.16 (m, 4H), 5.81–5.72 (m, 1H), 5.20–5.14 (m, 2H), 5.02 (td, J = 6.4, 2.0 Hz, 1H), 3.85 (t, J = 7.2 Hz, 1H), 3.54 (dd, J = 9.6, 7.2 Hz, 1H), 3.20 (dd, J = 17.6, 6.8 Hz, 1H), 3.10 (d, J = 17.6, 1H), 2.26–2.16 (m, 1H), 1.07 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 143.6, 140.4, 137.4, 127.0, 126.3, 125.9, 124.8, 117.4, 85.0, 82.3, 54.0, 43.8, 41.1, 12.0. IR (neat, cm−1): ν 2926, 1643, 1458, 1038, 924, 751; HRMS (ESIMS) calcd for C14H17O+ [M + H]+ 201.1274, found 201.1276.
:1) were added 2,6-lutidine (56.3 mg, 0.06 mL, 52.5 mmol, 1.0 equiv.), NaIO4 (168.5 mg, 78.8 mmol, 1.5 equiv.) and K2OsO4·2H2O (9.8 mg, 2.6 mmol, 0.05 equiv.) successively at 0 °C. The mixture was stirred for 3 h at room temperature. The reaction was quenched by the addition of saturated aqueous Na2S2O3. The mixture was extracted with EtOAc, and the organic layer was washed with brine and dried over anhydrous Na2SO4. After filtration, the solvent was removed under vacuum, and the resulting mixture was purified by silica gel chromatography (petroleum ether/ethyl acetate = 8:1) to give aldehyde 17 (87.1 mg, 82%) as a colorless liquid. 1H NMR (400 MHz, CDCl3) δ 9.64 (d, J = 2.5 Hz, 1H), 7.29–7.16 (m, 4H), 5.09 (td, J = 5.9, 1.7 Hz, 1H), 3.89–3.85 (t, J = 8.0 Hz, 1H), 3.54 (dd, J = 10.1, 2.4 Hz, 1H), 3.23–3.10 (m, 2H), 2.64–2.57 (m, 1H), 1.22 (d, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 201.8, 143.1, 139.1, 127.5, 126.4, 126.2, 125.1, 87.4, 84.5, 53.8, 40.4, 40.0, 12.4. IR (neat, cm−1): ν 3407, 2928, 1730, 1458, 1049, 1021, 750; HRMS (ESIMS) calcd for C13H14O2Na+ [M + Na]+ 225.0886, found 225.0883.
:1) to afford 16 (48.6 mg, 73% in two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 6.8 Hz, 1H), 7.23–7.18 (m, 3H), 6.98 (m, J = 1.6 Hz, 1H), 4.88 (ddd, J = 7.3, 4.9, 1.8 Hz, 2H), 3.86–3.82 (m, 1H), 3.50 (dd, J = 9.6, 2.4 Hz, 1H), 3.08 (dt, J = 31.5, 11.4 Hz, 2H), 2.83–2.74 (m, 1H), 1.95 (t, J = 2.0 Hz, 3H), 1.58 (d, J = 7.1 Hz, 3H), 1.25 (d, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.3, 146.3, 143.4, 140.1, 130.6, 127.3, 126.4, 126.1, 125.2, 83.6, 82.6, 79.8, 53.8, 40.4, 39.2, 13.3, 10.8. IR (neat, cm−1): ν 2959, 2926, 1759, 1455, 1069, 743; HRMS (ESIMS) calcd for C17H22O3N+ [M + NH4]+ 288.1594, found 288.1597.
Acknowledgements
We gratefully acknowledge financial support from the NSFC (Grant No. 21272104), the “111” Program of MOE (111-2-17), the PCSIRT (IRT_15R28), the NCET (NCET-12-0247), and the FRFCU (lzujbky-2013-ct02 and lzujbky-2012-56).Notes and references
- Pharmacopoeia of People's Republic of China, Chemical Industry Press, Beijing, 2005, vol. 1, pp. 44–45 and 169 Search PubMed.
- (a) W.-L. Xiao, R.-T. Li, S.-X. Huang, J.-X. Pu and H.-D. Sun, Nat. Prod. Rep., 2008, 25, 871 RSC; (b) Y.-M. Shi, W.-L. Xiao, J.-X. Pu and H.-D. Sun, Nat. Prod. Rep., 2015, 32, 367 RSC; (c) Y.-G. Xia, B.-Y. Yang and H.-X. Kuang, Phytochem. Rev., 2015, 14, 155 CrossRef CAS.
- Y.-F. Tang, Y.-D. Zhang, M.-J. Dai, T.-P. Luo, L.-J. Deng, J.-H. Chen and Z. Yang, Org. Lett., 2005, 7, 885 CrossRef CAS PubMed.
- (a) Y.-D. Zhang, Y.-F. Tang, T.-P. Luo, J. Shen, J.-H. Chen and Z. Yang, Org. Lett., 2006, 8, 107 CrossRef CAS PubMed; (b) D. Fischer and E. A. Theodorakis, Eur. J. Org. Chem., 2007, 4193 CrossRef CAS; (c) Y.-D. Zhang, W.-W. Ren, Y. Lan, Q. Xiao, K. Wang, J. Xu, J.-H. Chen and Z. Yang, Org. Lett., 2008, 10, 665 CrossRef CAS PubMed; (d) S. Maity, K. Matcha and S. Ghosh, J. Org. Chem., 2010, 75, 4192 CrossRef CAS PubMed.
- Q. Xiao, W.-W. Ren, Z.-X. Chen, T.-W. Sun, Y. Li, Q.-D. Ye, J.-X. Gong, F.-K. Meng, L. You, Y.-F. Liu, M.-Z. Zhao, L.-M. Xu, Z.-H. Shan, Y. Shi, Y.-F. Tang, J.-H. Chen and Z. Yang, Angew. Chem., Int. Ed., 2011, 50, 7373 CrossRef CAS PubMed.
- (a) L. Wang, H.-T. Wang, Y.-H. Li and P.-P. Tang, Angew. Chem., Int. Ed., 2015, 54, 5732 ( Angew. Chem. , 2015 , 127 , 5824 ) CrossRef CAS PubMed; (b) L. You, X.-T. Liang, L.-M. Xu, Y.-F. Wang, J.-J. Zhang, Q. Su, Y.-H. Li, B. Zhang, S.-L. Yang, J.-H. Chen and Z. Yang, J. Am. Chem. Soc., 2015, 137, 10120 CrossRef CAS PubMed.
- W.-L. Xiao, L.-M. Yang, N.-B. Gong, L. Wu, R.-R. Wang, J.-X. Pu, X.-L. Li, S.-X. Huang, Y.-T. Zheng, R.-T. Li, Y. Lu, Q.-T. Zheng and H.-D. Sun, Org. Lett., 2006, 8, 991 CrossRef CAS PubMed.
- (a) S. Maity, K. Matcha and S. Ghosh, J. Org. Chem., 2010, 75, 4192 CrossRef CAS PubMed; (b) M. F. Hossain, K. Matcha and S. Ghosh, Tetrahedron Lett., 2011, 52, 6473 CrossRef CAS; (c) S. S. Goh, H. Baars, B. Gockel and E. A. Anderson, Org. Lett., 2012, 14, 6278 CrossRef CAS PubMed; (d) B. Gockel, S. S. Goh, E. J. Puttock, H. Baars, G. Chaubet and E. A. Anderson, Org. Lett., 2014, 16, 4480 CrossRef CAS PubMed; (e) Y. Peng, S.-M. Duan and Y.-W. Wang, Tetrahedron Lett., 2015, 56, 4509 CrossRef CAS.
- (a) J. Li, P. Yang, M. Yao, J. Deng and A. Li, J. Am. Chem. Soc., 2014, 136, 16477 CrossRef CAS PubMed; (b) J. Li, P. Yang, M. Yao, J. Deng and A. Li, J. Am. Chem. Soc., 2015, 137, 12730 CrossRef CAS PubMed.
- S.-S. Goh, G. Chaubet, B. Gockel, M.-C. A. Cordonnier, H. Baars, A. W. Phillips and E. A. Anderson, Angew. Chem., Int. Ed., 2015, 54, 12618 CrossRef CAS PubMed.
- P. Yang, M. Yao, J. Li, Y. Li and A. Li, Angew. Chem., Int. Ed., 2016, 55, 6964 CrossRef CAS PubMed.
- Y. Wang, Z.-L. Li, L.-B. Lv and Z.-X. Xie, Org. Lett., 2016, 18, 792 CrossRef CAS PubMed.
- J.-M. M. Grandjean and D. A. Nicewicz, Angew. Chem., Int. Ed., 2013, 52, 3967 CrossRef CAS PubMed.
- B. M. Trost and A. Aponick, J. Am. Chem. Soc., 2006, 128, 3931 CrossRef CAS PubMed.
- (a) C. L. Chandler and A. J. Phillips, Org. Lett., 2005, 7, 3493 CrossRef CAS PubMed; (b) H.-J. Lo, Y.-K. Chang and T.-H. Yan, Org. Lett., 2012, 14, 5896 CrossRef CAS PubMed; (c) M. González, Z. Gándara, A. Martínez, G. Gómez and Y. Fall, Synthesis, 2013, 45, 1693 CrossRef; Z.-Y. Cai and H. F. DeLuca, Chin. J. Org. Chem., 2014, 34, 1582 Search PubMed.
- J. Deng, S.-Z. Zhou, W.-H. Zhang, J. Li, R.-F. Li and A. Li, J. Am. Chem. Soc., 2014, 136, 8185 CrossRef CAS PubMed.
- (a) Z.-H. Chen, Z.-M. Chen, Y.-Q. Zhang, Y.-Q. Tu and F.-M. Zhang, J. Org. Chem., 2011, 76, 10173 CrossRef CAS PubMed; (b) X.-K. Liu, J.-L. Ye, Y.-P. Ruan, Y.-X. Li and P.-Q. Huang, J. Org. Chem., 2013, 78, 35 CrossRef CAS PubMed.
- R. M. Patel, V. G. Puranik and N. P. Argade, Org. Biomol. Chem., 2011, 9, 6312 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all new compounds. See DOI: 10.1039/c6qo00241b |
| This journal is © the Partner Organisations 2017 |