
A novel spirocyclic triterpenoid and a new taraxerane triterpenoid from Teucrium viscidum†
Zheng-Yu
Li
a,
Feng-Ming
Qi
b,
De-Juan
Zhi
a,
Qiao-Ling
Hu
a,
Ying-Hong
Liu
a,
Zhan-Xin
Zhang
*a and
Dong-Qing
Fei
*a
aSchool of Pharmacy, Lanzhou University, Lanzhou 730000, People's Republic of China. E-mail: zhangzhx@lzu.edu.cn; feidq@lzu.edu.cn; Fax: +86 931 8915686; Tel: +86 931 8915686
bState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, People's Republic of China
First published on 10th October 2016
Abstract
Two new triterpenoids, teuviscins A (1) and B (2), were isolated from the whole plants of Teucrium viscidum. Compound 1 possessed a rare 7(8→9)abeo-9R-D:C-friedo-B′:A′-neo-gammacerane skeleton, and compound 2 is a taraxerane triterpene acid. Their structures were characterized by extensive spectroscopic methods (HR-ESI-MS, 1D and 2D NMR), and the absolute configurations of 1 and 2 were secured by single-crystal X-ray diffraction analyses using Cu Kα radiation. Compound 2 showed anti-AD bioactivity which delayed animals paralysis of transgenic AD Caenorhabditis elegans.
Introduction
The genus Teucrium, belonging to the family Lamiaceae, consists of nearly 300 species, which are distributed over the world, particularly in the Mediterranean basin.1,2 Many Teucrium species have long been used in folk medicine and pharmacy for their diverse biological properties, such as anti-inflammatory,3 antiacetylcholinesterase,4 antidiabetic,5–7 antifeedancy,8,9 and antimicrobial effects.10Teucrium viscidum is a perennial herb, widely distributed throughout northwest mainland China, Japan, North Korea, Myanmar, India, Indonesia, and the Philippines.11 The whole plants are used as a traditional folk medicine to treat hematemesis, traumatic injuries, pulmonary abscesses and bites from rabid dogs or venomous snakes.12 Previous phytochemical investigation on T. viscidum revealed the main presence of diterpenoids,13,14 triterpenoids, lignans, steroids,15 glucosylated coumaroyltyramine derivatives,11 and sesquiterpenoids.16 In our ongoing program to discover structurally interesting bioactive compounds from Chinese medicinal herbs,17–23 two new triterpenoids, teuviscins A (1) and B (2) (Fig. 1), were isolated from the whole plants of T. viscidum.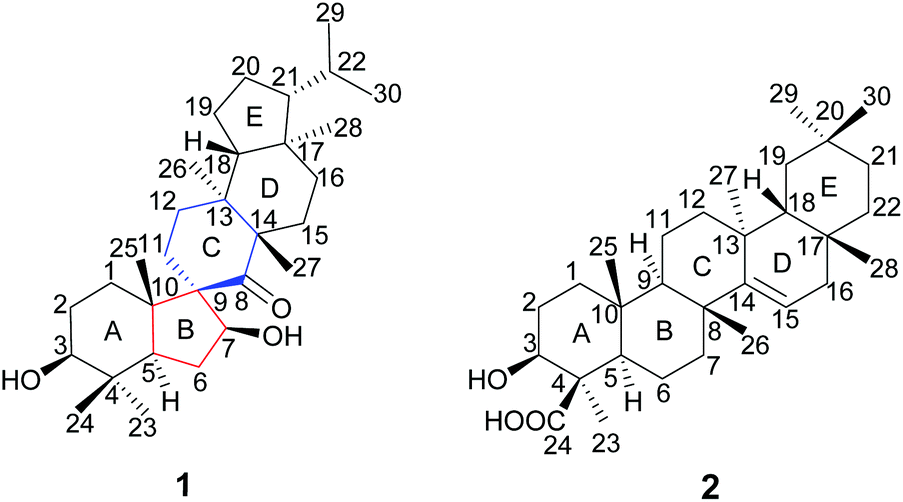 | ||
| Fig. 1 Structures of teuviscins A (1) and B (2). | ||
Compound 1 displayed a rare 7(8→9)abeo-D:C-friedo-B′:A′-neo-gammacerane skeleton, so far, to our knowledge, only one case of this skeleton was reported three decades ago.24 Compound 2 was identified as (3S,4R,5R,8R,9R,10R,13S,17R,18R)-3β-hydroxy-taraxer-14-en-24-oic acid. Their structures were elucidated on the basis of 1D and 2D nuclear magnetic resonance (NMR) spectroscopic data analyses, and single-crystal X-ray diffraction studies. Caenorhabditis elegans can display Alzheimer Disease (AD)-like symptoms of paralysis induced by Aβ toxicity after being transferred with the human gene of the Aβ1–42 downstream of the muscle promoter. Such transgenic worms have been widely used to detect the bioactivity of tested compounds for anti-AD.25–29 Therefore, we used C. elegans as an AD pathological model to evaluate the bioactivity of the new compounds in the present work. We herein describe the isolation and structure elucidation of 1 and 2, and their anti-AD bioactivities.
Results and discussion
The air-dried powder of the whole plants of T. viscidum was percolated with 95% EtOH at room temperature to give a crude extract, which was suspended in H2O and successively partitioned with EtOAc and n-BuOH. The EtOAc extract was subjected to various column chromatography techniques to afford compounds 1 and 2.Compound (1)‡§ was obtained as colorless needles (CHCl3), its molecular formula of C30H50O3 was established by HR-ESI-MS (m/z 459.3829 [M + H]+), requiring six degrees of unsaturation, m.p. 239–241 °C. The IR absorption bands at 3442, 3387, and 1657 cm−1 indicated the presence of two hydroxyl groups and one carbonyl group, respectively. Analysis of the 1H NMR data of 1 indicated the presence of two oxygenated methine protons at δH 3.27 (1H, dd, J = 10.8, 5.6 Hz) and 4.04 (1H, dd, J = 7.2, 5.1 Hz), together with six tertiary methyls at δH 0.76 (3H, s), 0.88 (3H, s), 0.90 (3H, s), 0.97 (3H, s), 1.01 (3H, s) and 1.08 (3H, s), and two secondary methyls at δH 0.83 (3H, d, J = 6.4 Hz) and 0.90 (3H, d, J = 6.4 Hz). The 13C NMR, DEPT and HSQC spectra of 1 (Table 1) suggested the presence of 30 carbons, including eight methyls, nine methylenes, six methines (including two oxygen-bearing carbons), and seven quaternary carbons (including one carbonyl group). Moreover, deducting one degree of unsaturation accounted for one ketone carbonyl, the remaining five degrees of unsaturation were indicative of the pentacyclic ring system of 1.
| 1 | 2 | |||
|---|---|---|---|---|
| Position | δ H | δ C | δ H | δ C |
| a The spectrum was recorded at 400 MHz. b The spectrum was recorded at 100 MHz. | ||||
| 1 | (α)1.42 m | 33.0 | 2.01 m | 38.3 |
| (β)1.83 m | ||||
| 2 | (α)2.25 m | 28.2 | 1.97 m | 29.4 |
| (β)1.85 m | ||||
| 3 | 3.27 dd, 10.8, 5.6 | 79.1 | 3.38 dd, 12.0, 4.4 | 78.7 |
| 4 | 38.4 | 49.8 | ||
| 5 | 1.45 m | 50.1 | 1.02 m | 57.2 |
| 6 | 2.13 m | 33.6 | 2.16 m | 42.0 |
| 7 | 4.04 dd, 7.2, 5.1 | 85.5 | (α)1.49 m | 35.7 |
| (β)1.42 m | ||||
| 8 | 221.6 | 36.4 | ||
| 9 | 51.1 | 1.02 m | 21.9 | |
| 10 | 60.0 | 39.5 | ||
| 11 | 1.30 m | 19.5 | (α)1.72 m | 18.3 |
| (β)1.50 m | ||||
| 12 | 1.26 m | 26.1 | (α)1.05 m | 37.3 |
| (β)1.35 m | ||||
| 13 | 39.2 | 29.4 | ||
| 14 | 52.5 | 158.5 | ||
| 15 | (α)1.47 m | 31.5 | 5.64 dd, 8.2, 2.8 | 117.5 |
| (β)1.56 m | ||||
| 16 | 1.42 m | 34.9 | 1.72 m | 39.2 |
| 1.69 m | ||||
| 17 | 42.8 | 38.1 | ||
| 18 | 1.58 m | 52.5 | 1.48 m | 49.5 |
| 19 | 1.77 m | 28.1 | 2.06 m | 38.3 |
| 20 | (α)0.96 m | 28.8 | 26.0 | |
| (β)1.03 m | ||||
| 21 | 0.99 m | 59.5 | 2.12 m | 21.5 |
| 22 | 0.84 m | 30.6 | 1.27 m | 34.3 |
| 23 | 0.90 s | 15.7 | 1.75 s | 25.0 |
| 24 | 0.97 s | 29.6 | 181.1 | |
| 25 | 1.01 s | 18.6 | 1.18 s | 14.1 |
| 26 | 1.08 s | 16.0 | 1.00 s | 30.4 |
| 27 | 0.88 s | 17.9 | 1.17 s | 26.0 |
| 28 | 0.76 s | 14.3 | 0.90 s | 30.5 |
| 29 | 0.83 d, 6.4 | 22.9 | 1.03 s | 21.9 |
| 30 | 0.90 d, 6.4 | 22.0 | 1.03 s | 33.8 |
The gross structure of 1 was initially deduced by comprehensive analysis of its 1D and 2D NMR spectra. The 1H–1H COSY data (Fig. 2) exhibited five structural fragments [–CH2(1)–CH2(2)–CH(3)–; –CH(5)–CH2(6)–CH(7)–; –CH2(11)–CH2(12)–; –CH2(15)–CH2(16)–; and –CH(18)–CH2(19)–CH2(20)–CH(21)–CH(22)–CH3(29)/CH3(30)], which were connected with each other and other functional groups by the HMBC experiment (Fig. 2). The 1H–1H COSY correlations of H-21/H-22/H3-29(H3-30), in combination with the HMBC correlations from H3-29 and H3-30 to C-22, gave an isopropyl moiety. The isopropyl group was shown to be connected to the C-21 by the HMBC correlations (H3-29 to C-21, and H3-30 to C-21). In the HMBC spectrum, the H2-6 showed correlations with C-9, and C-10, the H3-25 showed correlations with C-1, C-5 and C-9, besides C-9 was a quaternary carbon, these NMR data revealed a five-membered B ring. The HMBC correlations of H-7 with C-8, of H-11 with C-10, gave rise to the connectivity of the B ring and C ring joined at C-9, forming a novel spiro-[5,6] system. The C-7 position of 1 was substituted by a hydroxyl based on the HMBC correlations of H-5, H2-6 with the oxygenated carbon at δC 85.5. The long range correlation of H3-27 and C-8 led to the assignment of the carbonyl group at C-8. The remaining proton and carbon resonances, corresponding to three rings due to three degrees of unsaturation, are pretty close to those of the rings A, D, and E of spirosupinanonediol24 which was a C-9 epimer of 1, suggesting that the structures of A, D, and E rings of 1 are identical to those of spirosupinanonediol. The HMBC correlations from H3-23 to C-3 and C-5, from H3-25 to C-1, C-5 and C-10, from H3-26 to C-14 and C-18, and from H3-28 to C-16, C-18 and C-21 further confirmed the above deduction. Therefore, the planar structure of 1 was constructed as shown in Fig. 1.
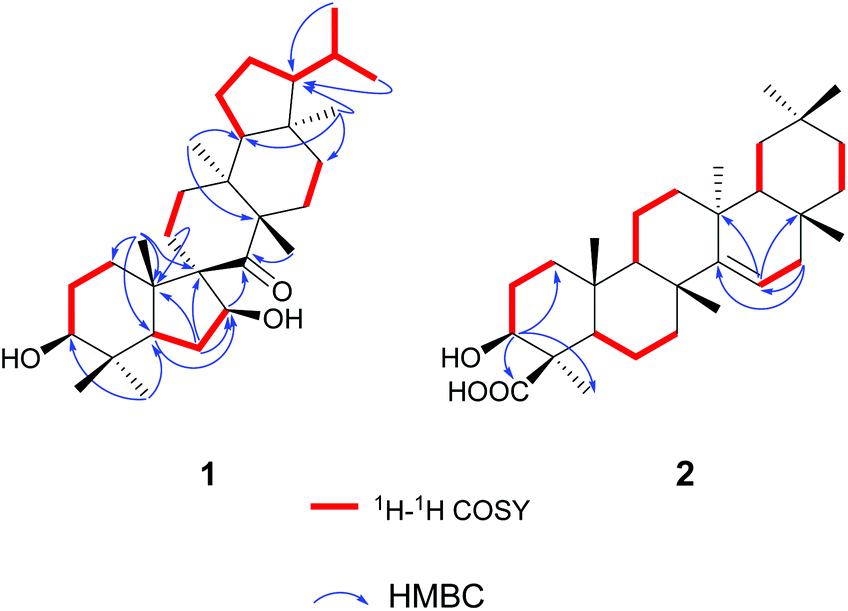 | ||
| Fig. 2 Key 2D NMR correlations of teuviscins A (1) and B (2). | ||
The relative configuration of 1 was established on the basis of the NOESY experiment (Fig. 3). For rings A and B, the NOESY correlations of H-3 with H-5 and H3-23, of H-7 with H-5, indicated that H-3, H-5, H-7 and H3-23 adopted the same orientation, arbitrarily designated as α-orientation. In addition, the correlation of H3-24 with H3-25 concluded that H3-24 and H3-25 were adopted β-orientation. The relative stereochemistry around rings C/D/E was also assigned by the NOESY correlations of H3-27 with H-18, of H-21 with H-18, of H3-28 with H3-26, indicating that H3-27 and H-18, H3-28 and H3-26 and the isopropyl group resided on the opposite side of the plane defined by the C/D/E rings. The NOESY correlation of H-7 with H2-11 led to the conclusion that the carbonyl at C-8 is opposite to H-7. This configuration necessitated a dihedral angle near 90° among rings A/B and C/D/E to maintain an anti-coplanar conformation. To our delight, compound 1 afforded high-quality crystals that allowed a successful performance of X-ray crystallography (Cu Kα radiation) (Fig. 4), which not only confirmed the structure of 1, but also determined the absolute configuration as 3S, 5R, 7S, 9R, 10S, 13S, 14S, 17R, 18R and 21R [absolute structure parameter: −0.03(10)].30 Thus, the structure of 1 was elucidated as shown, which was named teuviscin A. This rare 7(8→9)abeo-D:C-friedo-B′:A′-neo-gammacerane skeleton triterpenoid was discovered for the second time.
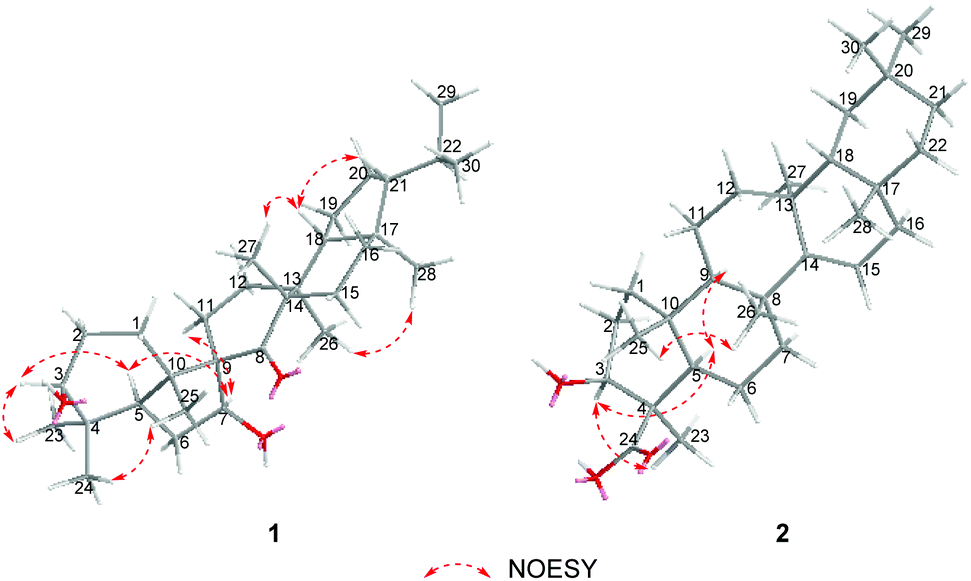 | ||
| Fig. 3 Key NOESY correlations of teuviscins A (1) and B (2). | ||
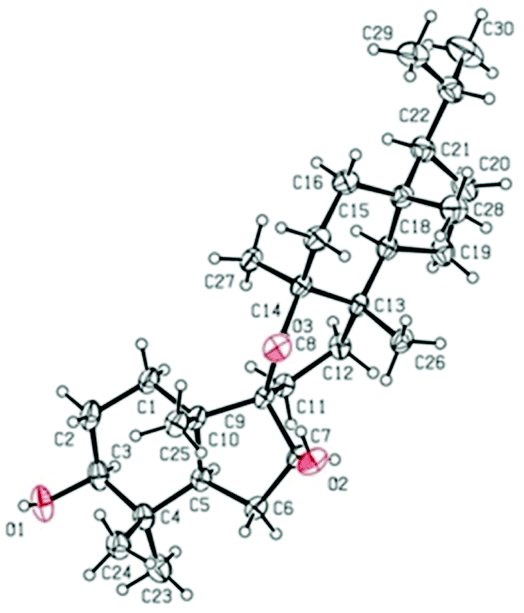 | ||
| Fig. 4 X-Ray crystal structure of compound 1. | ||
Compound (2)¶,|| was obtained as colorless lamellar crystals (C5H5N), m.p. over 300 °C. Its molecular formula was determined to be C30H48O3 from the quasi-molecular ion peak at m/z 479.3487 [M + Na]+ (calcd for C30H48O3Na, 479.3496) in the HR-ESI-MS, corresponding to seven degrees of unsaturation. The IR spectrum of 2 showed characteristic bands of hydroxyl (3404 cm−1), carboxyl (1718 cm−1), and double bond (1638 cm−1) functionalities. The 1H NMR spectrum of 2 exhibited seven tertiary methyls at δH 0.90 (3H, s), 1.00 (3H, s), 1.03 (3H, s), 1.03 (3H, s), 1.17 (3H, s), 1.18 (3H, s), and 1.75 (3H, s). The downfield region contained only two signals i.e. a double doublet at δH 3.38 (1H, J = 12.0, 4.4 Hz), and a broad double doublet at δH 5.64 (1H, J = 8.2, 2.8 Hz), which could be assigned to the hydroxyl-bearing C-3 methine and the vinylic C-15 proton by their HMBC correlations of H-3 with C-1, C-23 and C-24, of H-15 with C-13 and C-17, respectively. The 13C NMR spectrum of 2 exhibited 30 carbon resonances in agreement with the HR-ESI-MS data, which were sorted into seven methyls, ten methylenes, five methines (including one olefinic carbon, and one oxygenated carbon), as well as eight quaternary carbons (including one olefinic carbon and one carboxyl carbon). According to the NMR data of compound 2, a taraxerane triterpenoid skeleton was assumed.31
More detailed information about the structure of 2 came from the analysis of the 2D NMR data. In the 1H–1H COSY spectrum, the cross-peaks of H2-1/H2-2/H-3, H-5/H2-6/H2-7, H-9/H2-11/H2-12, H-15/H2-16, H-18/H2-19, and H2-21/H2-22 suggested the presence of six key fragments. In the HMBC spectrum, the H-3 methine proton resonating at δH 3.38 showed cross-peaks with the methyl C-23 and the carboxyl carbon C-24, revealing that the carboxyl carbon was attached to C-4. The correlations of H2-16 with two vinylic carbons C-14 and C-15, of H-15 with C-13 and C-17, led to the assignment of a Δ14,15 double bond. Thus, the planar structure of compound 2 was determined.
The relative configuration of 2 was elucidated on the basis of the NOESY spectrum and MM2 minimize energy modeling by Chem3D. The NOESY correlations of H-3 with H3-23, H-5, of H-9 with H-5, of H3-25 with H3-26 and the Chem3D modeling are depicted in Fig. 3, where H-3, H-5, H-9, and H3-23 were in α-orientation, and H3-25 and H3-26 were in β-orientation. In addition, the various chiral centers were consistent with other taraxerane triterpenoids.31 Ultimately, to confirm the above assignments and the configuration, a single X-ray crystallographic analysis using an anomalous scattering of Cu Kα radiation was carried out and confirmed the structure of compound 2 as depicted (Fig. 5). The absolute configuration of 2 was determined as 3S, 4R, 5R, 8R, 9R, 10R, 13S, 17R, and 18R through the refinement of Flack's parameter [0.12(14)].30 Thus, compound 2 was elucidated as (3S,4R,5R,8R,9R,10R,13S,17R,18R)-3β-hydroxy-taraxer-14-en-24-oic acid, which was named teuviscin B.
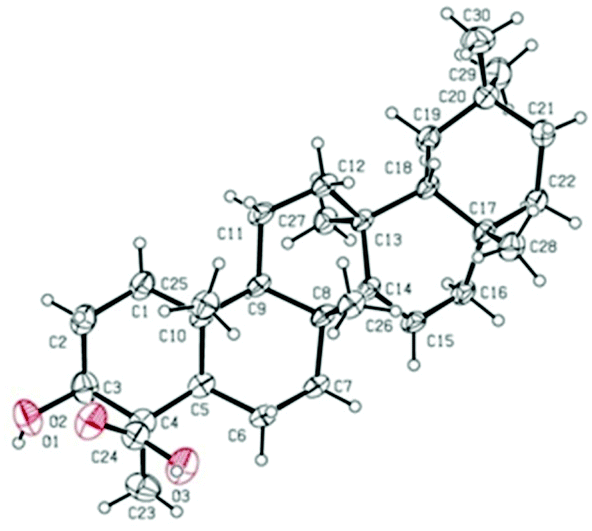 | ||
| Fig. 5 X-Ray crystal structure of compound 2. | ||
The anti-AD bioactivities of compounds 1 and 2 were evaluated using the AD pathological model. The results (Fig. 6) showed that compound 1 did not delay worm paralysis, but 2 significantly delayed animals paralysis of transgenic AD C. elegans, suggesting that 2 possessed anti-AD bioactivity.
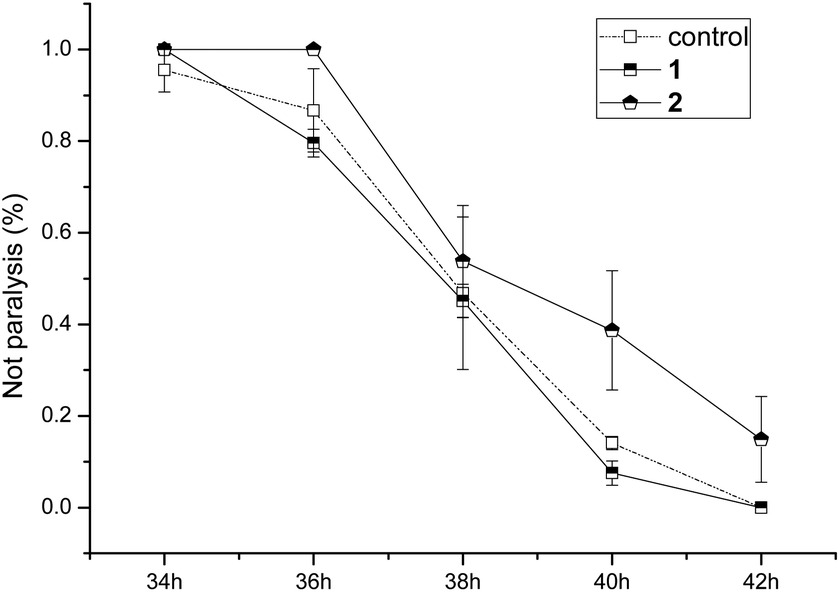 | ||
| Fig. 6 Bioactivity assay of compounds 1 and 2 for delaying paralysis in transgenic Alzheimer disease C. elegans. | ||
Conclusions
In conclusion, we isolated and identified two new triterpenoids teuviscins A (1) and B (2) from the whole plants of T. viscidum. Their structures were elucidated by extensive spectroscopic techniques. The absolute configurations of the new compounds were determined by means of X-ray diffraction analysis. Compound 1 displayed a rare 7(8→9)abeo-D:C-friedo-B′:A′-neo-gammacerane skeleton; to our knowledge, only one case of this skeleton was reported three decades ago. To date, there is no suitable synthesis routine for spirocyclic triterpenoids. The novel structure could attract interest of synthetic organic chemists. Compound 2 was a taraxerane triterpenoid. The anti-AD bioactivities of two new compounds were tested using the AD pathological model. Compound 2 exhibited potent anti-AD bioactivity.Acknowledgements
The work was supported by the National Natural Science Foundation of China (no. 31670350, 21402075 and 21102065), and the Fundamental Research Funds for the Central Universities of China (no. lzujbky-2015-56 and lzujbky-2015-58).Notes and references
- N. Djabou, M. J. Battesti, H. Allali, J. M. Desjobert, L. Varesi, J. Costa and A. Muselli, Phytochemistry, 2011, 72, 1390 CrossRef CAS PubMed.
- H. Henchiri, B. Bodo, A. Deville, L. Dubost, L. Zourgui, A. Raies, P. Grellier and L. Mambu, Phytochemistry, 2009, 70, 1435 CrossRef CAS PubMed.
- M. Tariq, A. M. Ageel, M. A. Al-Yahya, J. S. Mossa and M. S. Al-Said, Int. J. Tissue React., 1989, 11, 185 CAS.
- I. Orhan and M. Aslan, J. Ethnopharmacol., 2009, 122, 327 CrossRef PubMed.
- A. J. Alonso-Castro, R. Zapata-Bustos, J. Romo-Yañez, P. Camarillo-Ledesma, M. Gómez-Sánchez and L. A. Salazar-Olivo, J. Ethnopharmacol., 2010, 127, 1 CrossRef PubMed.
- F. U. Afifi, B. Al-Khalidi and E. Khalil, J. Ethnopharmacol., 2005, 100, 314 CrossRef CAS PubMed.
- M. A. Esmaeili and R. Yazdanparast, J. Ethnopharmacol., 2004, 95, 27 CrossRef PubMed.
- J. Coll and Y. Tandrón, Phytochemistry, 2004, 65, 387 CrossRef CAS PubMed.
- J. López-Olguín, M. C. de la Torre, F. Ortego, P. Castañera and B. Rodríguez, Phytochemistry, 1999, 50, 749 CrossRef.
- E. Darabpour, H. Motamedi and S. M. S. Nejad, Asian Pac. J. Trop. Med., 2010, 3, 124 CrossRef.
- H. W. Lv, M. D. Zhu, J. G. Luo and L. Y. Kong, J. Nat. Prod., 2014, 77, 200 CrossRef CAS PubMed.
- State Administration of Traditional Chinese Medicine, Zhong Hua Ben Cao, Shanghai Science and Technology Publishing Company, Shanghai, China, 1999, p. 6239.
- E. Fujita, I. Uchida and T. J. Fujita, J. Chem. Soc., Chem. Commun., 1973, 793 RSC.
- M. Node, M. Sai and E. Gujita, Phytochemistry, 1981, 20, 757 CrossRef CAS.
- X. C. Hao, J. W. Zhang, G. X. Xia, Y. B. Xue, Z. W. Luo, Y. Y. Si, G. M. Yao and Y. H. Zhang, Molecules, 2013, 18, 1262 CrossRef CAS PubMed.
- X. C. Hao, J. W. Zhang, G. Q. Zhan, Y. B. Xue, Z. W. Luo, G. M. Yao and Y. H. Zhang, Biochem. Syst. Ecol., 2013, 51, 78 CrossRef CAS.
- D. Q. Fei, L. L. Dong, F. M. Qi, G. X. Fan, H. H. Li, Z. Y. Li and Z. X. Zhang, Org. Lett., 2016, 18, 2844 CrossRef CAS PubMed.
- Z. X. Zhang, H. H. Li, F. M. Qi, L. L. Dong, Y. Hai, G. X. Fan and D. Q. Fei, RSC Adv., 2014, 4, 30059 RSC.
- H. H. Li, F. M. Qi, L. L. Dong, G. X. Fan, J. M. Che, D. D. Guo, Z. X. Zhang and D. Q. Fei, Phytochem. Lett., 2014, 10, 304 CrossRef CAS.
- L. L. Dong, H. Y. Xiong, F. M. Qi, H. H. Li, G. X. Fan, J. M. Che, D. D. Guo, Z. X. Zhang and D. Q. Fei, J. Asian Nat. Prod. Res., 2015, 17, 415 CrossRef CAS PubMed.
- G. X. Fan, D. J. Zhi, H. Ren, Z. Y. Li, Q. L. Hu, Y. H. Liu, Z. X. Zhang and D. Q. Fei, Nat. Prod. Commun., 2016, 11, 497 Search PubMed.
- G. X. Fan, L. L. Dong, H. H. Li, Z. Y. Li, Z. X. Zhang and D. Q. Fei, Chem. Nat. Compd., 2016, 52, 754 CrossRef CAS.
- Z. X. Zhang, F. M. Qi, H. H. Li, L. L. Dong, Y. Hai and D. Q. Fei, Bull. Korean Chem. Soc., 2014, 35, 641 CrossRef CAS.
- S. Matsunaga, R. Morita, T. Ishida, M. Inoue, M. Shigi and A. Miyamae, J. Chem. Soc., Chem. Commun., 1984, 1128 RSC.
- S. Abbas and M. Wink, Phytomedicine, 2010, 17, 902 CrossRef CAS PubMed.
- C. D. Link, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9368 CrossRef CAS.
- Y. Wu, Z. Wu, P. Butko, Y. Christen, M. P. Lambert, W. L. Klein, C. D. Link and Y. Luo, J. Neurosci., 2006, 26, 13102 CrossRef CAS PubMed.
- L. Diomede, G. Cassata, F. Fiordaliso, M. Salio, D. Ami, A. Natalello, S. M. Doglia, A. De Luigi and M. Salmona, Neurobiol. Dis., 2010, 40, 424 CrossRef CAS PubMed.
- L. Xin, R. Yamujala, Y. Wang, H. Wang, W. H. Wu, M. A. Lawton, C. Long and R. Di, PLoS One, 2013, 8, e63874 CAS.
- H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 876 CrossRef.
- V. S. Chaturvedula, J. K. Schilling, S. Malone, J. H. Wisse, M. C. Werkhoven and D. G. Kingston, Planta Med., 2003, 69, 271 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR, HR-ESI-MS, and IR spectra of compounds 1 and 2. CCDC 1498102 and 1498101. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00460a |
| ‡ Teuviscin A (1): colorless needles (CHCl3); m.p. 239–241 °C; [α]20D +30 (c 0.10, CHCl3); IR (KBr) νmax: 3442, 3387, 2941, 2871, 1736, 1657, 1459, 1396, 1261, 1120 and 1098 cm−1; HR-ESI-MS: m/z 459.3829 [M + H]+ (calcd for C30H51O3, 459.3833); 1H and 13C NMR data, see Table 1. |
§ Crystal data for 1: C30H50O3, Mr = 458.70, orthorhombic, space group P212121, a = 6.52391(10) Å, b = 12.9427(2) Å, c = 31.7963 (6) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 2684.79 (8) Å3, T = 155 K, Z = 4, d = 1.135 g cm−3, μ(CuKα) = 0.54 mm−1, F(000) = 1016, crystal dimensions 0.14 × 0.12 × 0.11 mm were used for measurement on an Agilent Technologies SuperNova, Dual source, EOS CCD with mirror optics, using graphite monochromated Cu Kα radiation (λ = 1.54184 Å). The total of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 673 reflections measured, 5019 independent reflections (Rint = 0.024). The final R1 value was 0.0381 (I > 2σ(I)). The final wR(F2) value was 0.1006 ((I > 2σ(I)). The final R1 value was 0.0436 (all data). The final wR(F2) value was 0.0965 (all data). The goodness of fit on F2 was 1.023. Crystallographic data for the structure of compound 1 have been deposited with the Cambridge Crystallographic Data Centre (deposition no. 1498102). 673 reflections measured, 5019 independent reflections (Rint = 0.024). The final R1 value was 0.0381 (I > 2σ(I)). The final wR(F2) value was 0.1006 ((I > 2σ(I)). The final R1 value was 0.0436 (all data). The final wR(F2) value was 0.0965 (all data). The goodness of fit on F2 was 1.023. Crystallographic data for the structure of compound 1 have been deposited with the Cambridge Crystallographic Data Centre (deposition no. 1498102). |
| ¶ Teuviscin B (2): colorless lamellar crystals; m.p. over 300 °C; [α]20D +90 (c 0.10, CHCl3); IR (KBr) νmax: 3404, 2927, 1718, 1687, 1638, 1473, 1375, 1248, 1135 and 1041 cm−1; HR-ESI-MS: m/z 479.3487 [M + Na]+ (calcd for C30H48O3Na, 479.3496); 1H and 13C NMR data, see Table 1. |
| || Crystal data for 2: C30H48O3·C5H5N, Mr = 535.78, monoclinic, space group P21, a = 7.4550 (5) Å, b = 11.3624 (7) Å, c = 18.4877 (13) Å, α = 90.00°, β = 92.585(6)°, γ = 90.00°, V = 1564.44 (18) Å3, T = 274 K, Z = 2, d = 1.137 g cm−3, μ(CuKα) = 0.545 mm−1, F(000) = 588, crystal dimensions 0.21 × 0.15 × 0.14 mm were used for measurement on an Agilent Technologies SuperNova, Dual source, EOS CCD with mirror optics, using graphite monochromated Cu Kα radiation (λ = 1.54184 Å). The total of 11623 reflections measured, 5591 independent reflections (Rint = 0.026). The final R1 value was 0.0567 (I > 2σ (I)). The final wR(F2) value was 0.1616 ((I > 2σ(I)). The final R1 value was 0.0628 (all data). The final wR(F2) value was 0.1539 (all data). The goodness of fit on F2 was 1.040. Crystallographic data for the structure of compound 2 have been deposited with the Cambridge Crystallographic Data Centre (deposition no. 1498101). |
| This journal is © the Partner Organisations 2017 |