
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic valorisation of glycerol via a dual NHC-catalysed telescoped reaction†
A.
Axelsson
,
A.
Antoine-Michard
and
H.
Sundén
 *
*
Chemistry and Chemical Engineering, Chalmers University of Technology, Kemivägen 10, 412 96 Göteborg, Sweden. E-mail: sundenh@chalmers.se
First published on 23rd March 2017
Abstract
A general telescoped reaction for the NHC-catalysed carbonation and aerobic esterification of glycerol and 2-amino-2-methylpropane-1,3-diol has been developed. The reaction provides highly functionalised glycerol derivatives in good to excellent yields (up to 95%) using low catalyst loadings and ambient conditions.
Our current dependence on fossil resources represents a major obstacle in the transition towards sustainable development.1 It is estimated that up to 95% of the carbon-containing molecules needed to sustain daily life are derived from petrochemical sources.2 Hence, the valorisation of biomass into fuels, bulk and fine chemicals as well as pharmaceuticals, is of utmost importance.2–4 Moreover, in efforts to circumvent direct competition with food production the use of waste-streams instead of first generation biomass is preferred.5 For this reason, glycerol is an excellent feedstock since it is obtained as a by-product from the production of biodiesel. Consequently, there is currently a large surplus of glycerol readily available.6 Conversion of glycerol into commodity and fine chemicals is typically performed using metal catalysts.7–10 In contrast, organocatalytic functionalisation of glycerol is not commonly utilized. A possible explanation for this is that transition metal catalysts are typically considered to be more reactive than organocatalysts. Nevertheless, organocatalysts have many virtues such as low-cost, low-toxicity and high stability and can thus be considered as competitive alternatives to their metal counterpart. Moreover, the increasingly tough restrictions regarding metal contaminants in consumer-products further favours organocatalysis.11
Previous organocatalytic transformations of glycerol have shown that glycerol carbonate can be synthesised from glycerol and dialkyl carbonates or urea using N-heterocyclic carbenes (NHCs), quaternary ammonium salts and Brønsted-Lowry bases (Scheme 1A).12–16 Additionally, acylation of glycerol is possible by transesterification of fatty methyl esters with either phosphazene- or guanidine-based catalysts, via Fisher esterification catalysed by sulfonic acids, and by acetate-catalysed reactions with anhydrides (Scheme 1B).17–21 Both homogenous and heterogeneous sulfonic acids have also found use in the conversion of glycerol into a variety of ethers and acetals (Scheme 1C).22–24
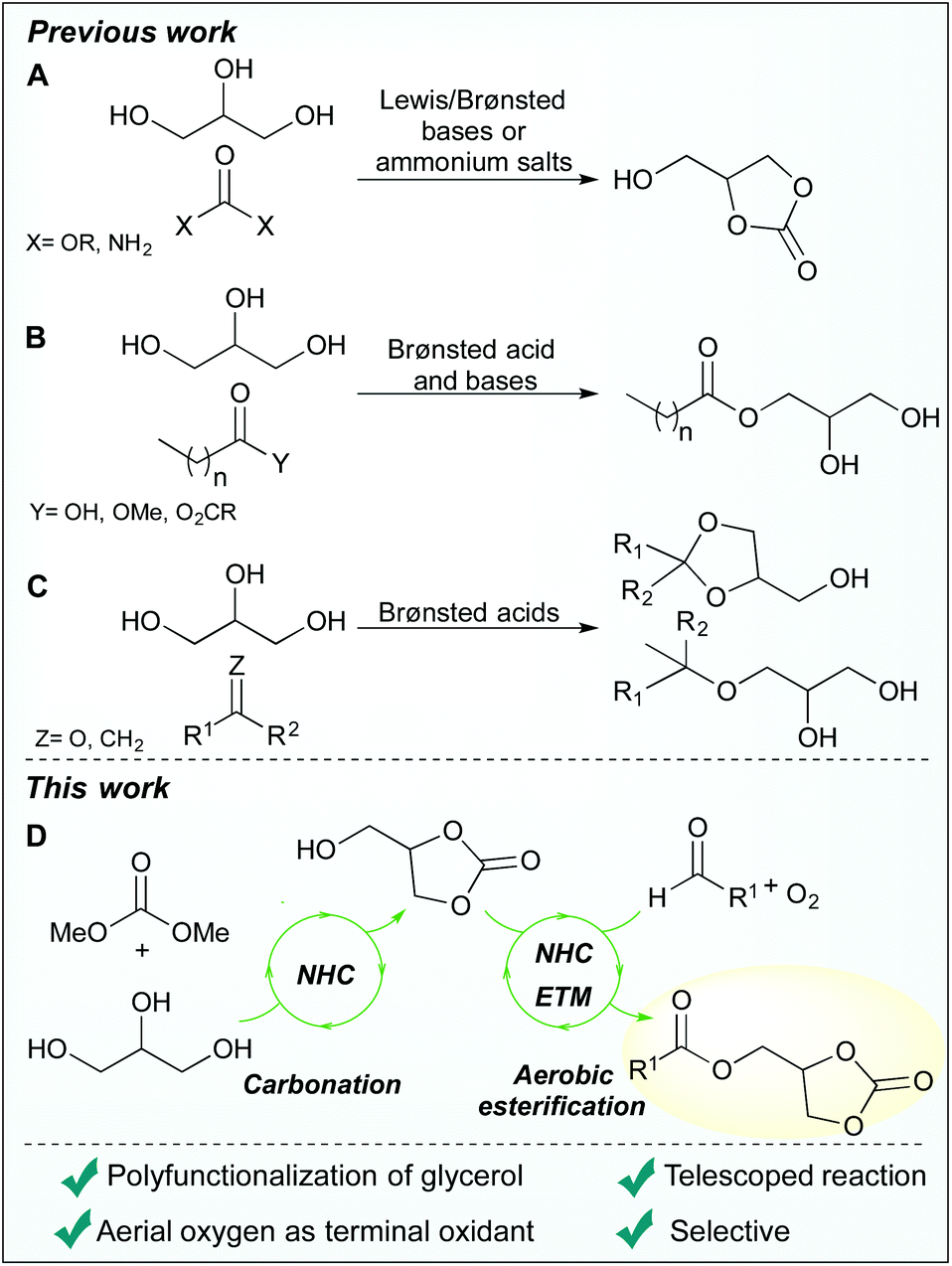 | ||
| Scheme 1 Previous organocatalytic strategies for glycerol valorisation: transcarbonation (A), acylation (B) and acetal or ether formation (C) and the current approach combining carbonation and aerobic esterification in a telescoped reaction (D). | ||
While NHC-catalysis offers a multitude of reaction paths25–28 and has proven compatible with glycerol, to date, the use of NHC-catalysis in glycerol valorisation is limited to carbonation reactions. As our research interest include sustainable NHC-catalysis, we set out to merge oxidative NHC-catalysis29,30 with glycerol upgrading. Instead of employing conventional methods that require stoichiometric amounts of high molecular weight oxidants, such as the Kharasch oxidant 7,31,32 an aerobic approach was envisioned, as previously reported by us and others.33–40 More precisely, it was hypothesised that aerobic esterification of glycerol would be possible by using a biomimetic strategy comprised of electron transport mediators (ETMs), as pioneered by the Bäckvall group.41–45 The ETMs work in concert by enabling a low energy path for electrons to flow from the substrate to oxygen (O2), circumventing the unfavourable reaction kinetics associated with direct O2-oxidations.45 Moreover, it was predicted that a dual functionalisation of glycerol, comprising a sequential NHC-catalysed carbonation followed by an aerobic esterification, would be possible by using a telescoped reaction strategy (Scheme 1D). This approach offers several advantages including: direct valorisation of glycerol, high atom economy, improved pot-economy,46 and the incorporation of the carbonate group as a valuable synthetic handle.12
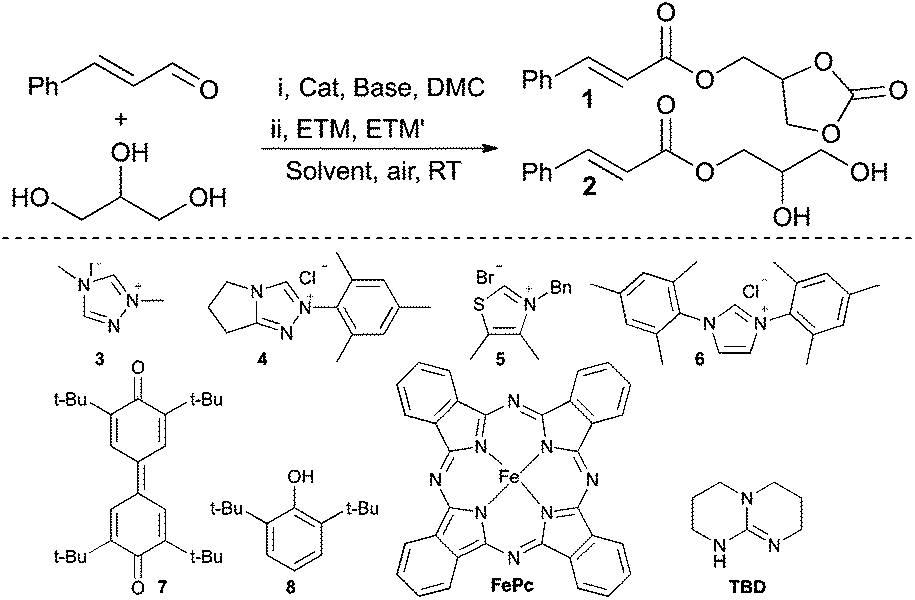
An initial experiment with a telescoped approach that included a one-pot sequential carbonation, using dimethyl carbonate (DMC), and aerobic esterification was successful providing the unsaturated carbonate (1) in 89% yield (Table 1, entry 1). It should be noted that dimethyl carbonate is considered a benign reagent as it can be obtained from CO2, is nontoxic and biodegradable.47 A survey of different NHC-catalysts showed that the thiazolium based catalyst 5 was inactive (entry 3), while 4 and 6 provided 1 in slightly lower yields compared to when 3 was employed (entries 2 and 4). This is due to increased carboxylic acid formation and incomplete consumption of cinnamaldehyde, respectively. The choice of base also proved important and the use of K2CO3 or Et3N in place of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) resulted in no conversion of starting material (entries 5 and 6). This effect can be rationalised by the fact that the essential in situ FePc-catalysed aerobic oxidation of 8 to 7 is also base-catalysed.31 Thus, by replacing 8 with 7 the loading of TBD can be decreased from 0.5 to 0.2 equivalents with maintained yield (entry 7), albeit with prolonged reaction time (24 h). Moreover, when the reaction is performed under a nitrogen atmosphere 1 is obtained in only 5%, showcasing that aerial oxygen truly is the terminal oxidant (entry 8). Omission of the NHC-precatalyst in the carbonation step results in the incomplete conversion of glycerol with subsequent side product formation, such as, 2, the secondary- and the diester (14% determined by 1H NMR) suggesting that this step is also NHC-catalyzed. Synthesis of 2via a direct aerobic esterification of glycerol, omitting the carbonation step, was possible and 2 was obtained in 87% yield (entry 9). However, due to the formation of side products (the secondary ester and the diester, ca. 10%), it was not possible to obtain the pure product in a satisfactory manner. Regardless of concentration and solvent selection the amount of side products remained around 10%. For example, when acetone was used as the reaction solvent a competing aldol reaction was observed and 2 was obtained in 81% yield (entry 10).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat | ETM | ETM′ | Base | Solvent | Yielda (%) | |
| 1 | 2 | ||||||
a Determined by 1H NMR with durene as internal standard.
b i, Glycerol (1.1 eq.), TBD (0.5 eq.), NHC (2 mol%), solvent/dimethyl carbonate 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. ii, Cinnamaldehyde (0.5 mmol), FePc (0.5 mol%), 8 (2 mol%).
c As in footnote b, but with 0.2 eq. TBD and 0.01 eq. of 7.
d Last step performed under a nitrogen atmosphere.
e Glycerol (4 eq.), TBD (0.5 eq.), 3 (2 mol%), cinnamaldehyde (0.5 mmol), FePc (0.5 mol%), 8 (2 mol%). :2. ii, Cinnamaldehyde (0.5 mmol), FePc (0.5 mol%), 8 (2 mol%).
c As in footnote b, but with 0.2 eq. TBD and 0.01 eq. of 7.
d Last step performed under a nitrogen atmosphere.
e Glycerol (4 eq.), TBD (0.5 eq.), 3 (2 mol%), cinnamaldehyde (0.5 mmol), FePc (0.5 mol%), 8 (2 mol%).
|
|||||||
| 1b | 3 | 8 | FePc | TBD | MeCN | 89 | |
| 2b | 4 | 8 | FePc | TBD | MeCN | 86 | |
| 3b | 5 | 8 | FePc | TBD | MeCN | 0 | |
| 4b | 6 | 8 | FePc | TBD | MeCN | 81 | |
| 5b | 3 | 8 | FePc | K2CO3 | MeCN | 0 | |
| 6b | 3 | 8 | FePc | Et3N | MeCN | 0 | |
| 7c | 3 | 7 | FePc | TBD | MeCN | 88 | |
| 8b,d | 3 | 8 | FePc | TBD | MeCN | 5 | |
| 9e | 3 | 8 | FePc | TBD | MeCN | 87 | |
| 10e | 3 | 8 | FePc | TBD | Acetone | 81 | |
Having successfully identified mild conditions (Table 1 entry 1) that enabled selective dual functionalisation of glycerol with aerial oxygen as the terminal oxidant, the scope of this transformation was investigated (Scheme 2). The reaction proved general, and both electron-donating (9–11, 16) and electron-withdrawing (12–15) substituents were tolerated on the cinnamaldehyde scaffold. Compound 9, isolated in 95% yield, is worth highlighting since 4-methoxycinnamates are commonly used in sunscreens and the glycerol ester has been investigated as a more benign sunscreen agent.48 Moreover, polyaromatics were well tolerated and anthracene ester 17 could be obtained in 76% yield. Initial reactions with aliphatic enals were sluggish. However, by using 4 mol% of the more active catalyst 4 2-hexenal yielded the corresponding ester 18 in 64% yield. Benzaldehydes proved less reactive than cinnamaldehydes and required 4 mol% (instead of 2 mol%) of catalyst 3 to react efficiently. With the increase in catalyst loading both electron rich and electron poor aromatic as well as heteroaromatic aldehydes worked well, affording the corresponding products (19–25) in moderate to good yields (51–79%).
 | ||
| Scheme 2 The scope of the aerobic glycerol functionalisation. a(i) Glycerol (1.1 eq.), TBD, 3, MeCN/DMC 5:2. (ii) Aldehyde (0.5 mmol), FePc (0.5 mol%), 8 (2 mol%). b1.6 grams isolated. c4 mol% of catalyst 4 used. d4 mol% of catalyst 3 used. | ||
For instance, piperonal derived ester 23 could be obtained in 79% yield. Moreover, this reaction is scalable and α,β-unsaturated ester 1 could be obtained in 87% yield and 1.6 grams.
Encouraged by the successful functionalisation of glycerol, the compatibility of the developed system with the aminoalcohol 2-amino-2-methyl-1,3-propanediol (26) was investigated (Scheme 3). The approach was successful and both electron rich and electron poor enals could be transformed into the corresponding 2-oxooxazolidine esters in excellent yields. For instance, cinnamaldehyde and 4-chlorocinnamaldehyde both reacted well and provided the corresponding α,β-unsaturated esters in 92% and 91% yield respectively (27 and 30).
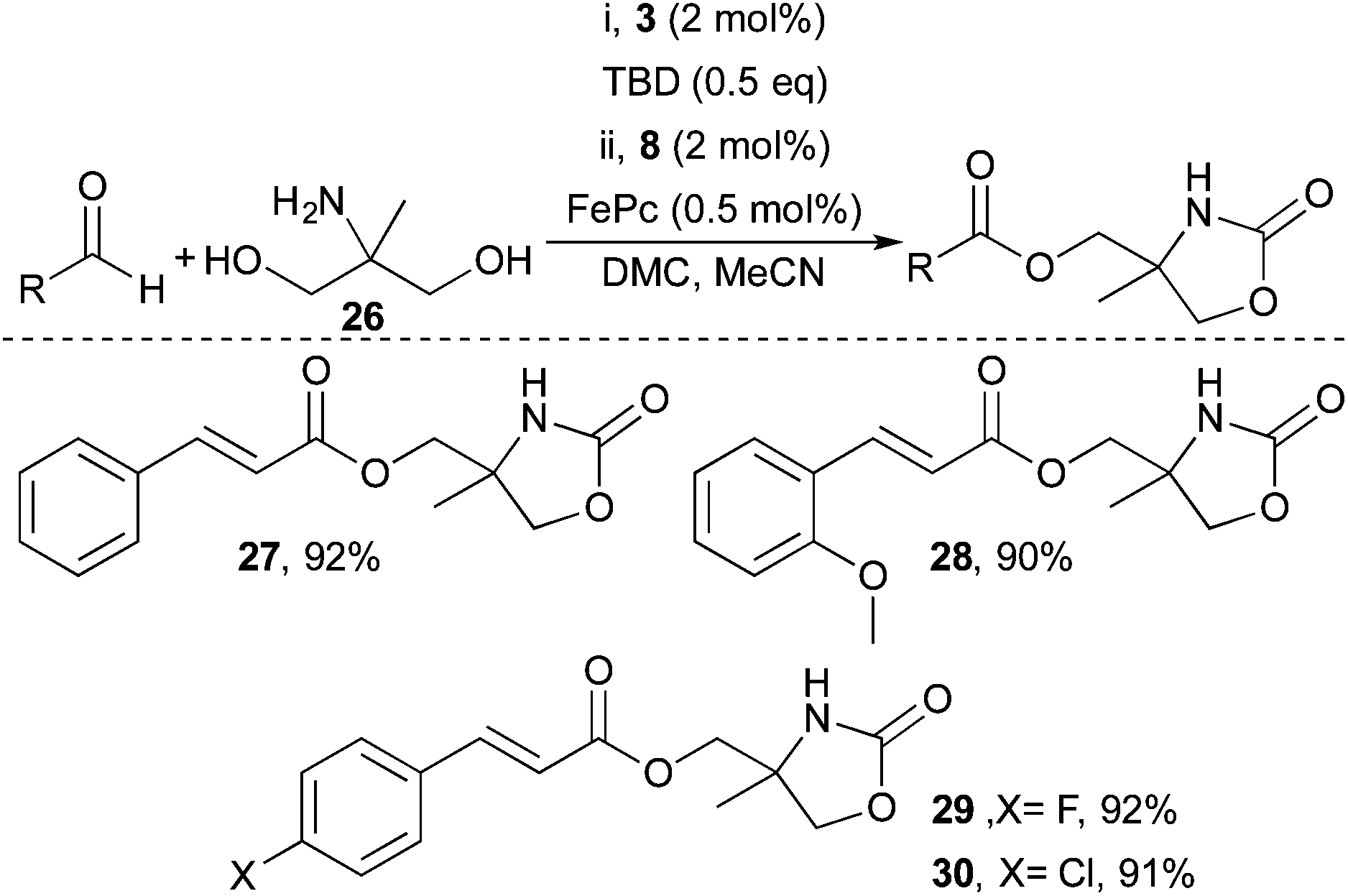 | ||
| Scheme 3 Selective modification of aminopolyol 26 by telescoped carbamate formation and aerobic esterification. (i) 26 (1.0 eq.), 3, TBD, MeCN/DMC 5:2. (ii) Aldehyde (0.5 mmol), FePc, 8, isolated yields. | ||
Ring opening of glycerol carbonate and its derivatives have obtained a lot of attention, for example, in sustainable synthesis of pharmaceuticals and polymers.9,49,50 Consequently, the herein developed procedure could generate several new glycerol carbonate building blocks for use as green monomers or for further manipulation. With this in mind, the nucleophilic ring opening of the 2-oxo-1,3-dioxolan group with piperidine was investigated (Scheme 4). Inspired by reports from Kleij et al.,51,52 it was possible to selectively activate the less electrophilic carbonate in the presence of the unsaturated ester using 5 mol% of TBD, yielding acyclic carbamate 31 in 83% yield and good regioselectivity (Scheme 4). Usage of higher loadings of TBD or other solvents (MeCN, THF) leads to formation of the amide as a side product.
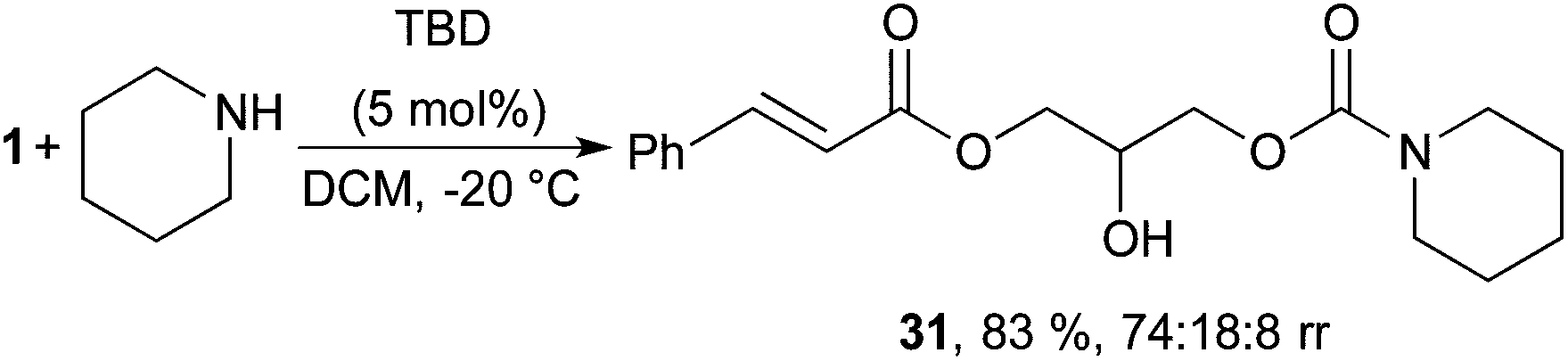 | ||
| Scheme 4 Organocatalytic ring opening of 1 to form acyclic carbamate 31. 1 (0.2 mmol), piperidine (1.2 eq.), isolated yield, regioisomeric ratio determined by 1H NMR of crude material, major isomer separable by flash chromatography. | ||
The proposed catalytic cycle starts with addition of the NHC (I) to aldehyde II forming the Breslow intermediate (III) (Scheme 5). Intermediate III is oxidized by the coupled system of ETMs to the acyl azolium IV. Glycerol carbonate (V), formed by NHC/TBD catalysis in the first step of the telescoped reaction, adds as a nucleophile to IV forming the product (VI) and regenerating the NHC.
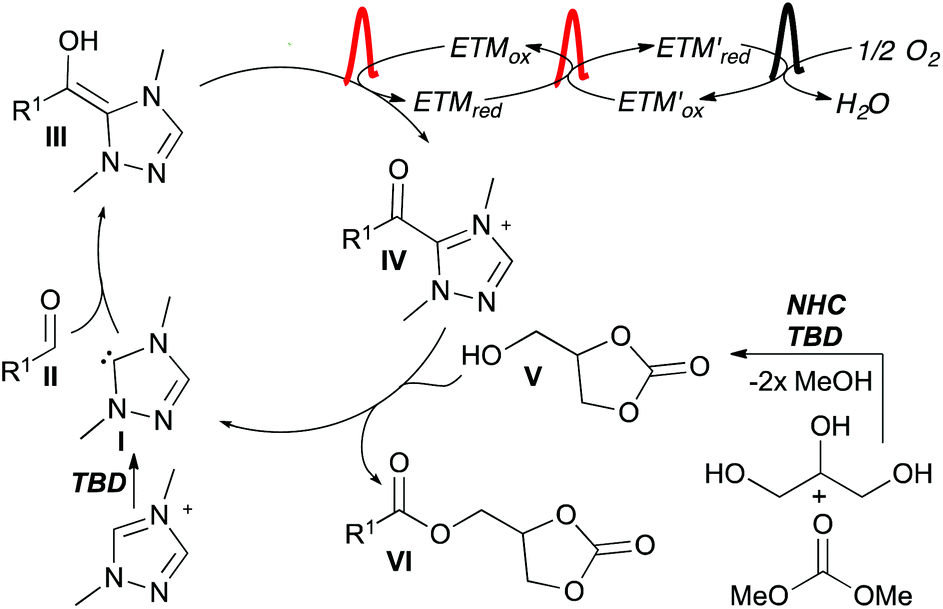 | ||
| Scheme 5 Proposed catalytic cycle. | ||
In summary, a telescoped protocol for the NHC-catalysed carbonation and aerobic esterification of glycerol has been developed. The presented method gives access to highly functionalised glycerol derivatives from sustainable resources such as glycerol, dimethyl carbonate and aerial oxygen. The reaction occurs under ambient conditions, is high yielding, scalable and has a broad scope that tolerates (hetero)aromatic aldehydes, aromatic and aliphatic α,β-unsaturated aldehydes. The method could also be further extended to amino-polyols, affording 2-oxooxazolidine esters in excellent yields. Lastly, chemoselective activation of the less electrophilic carbonate was demonstrated, showcasing the use of the obtained products. These results demonstrate the possibility to extend the role of NHC-catalysis in glycerol valorisation beyond carbonation reactions, enabling access to new sustainable building blocks for chemical synthesis.
Funding from the Swedish Research Council (VR and Formas) are gratefully acknowledged. Furthermore, we thank the C. F. Lundström foundation for supporting the running costs of this project.
References
-
G. G. Brundtland, Our common future, The World Commision on Environmental Development, Oxford, 1987 Search PubMed
.
- C. H. Christensen, J. Rass-Hansen, C. C. Marsden, E. Taarning and K. Egeblad, ChemSusChem, 2008, 1, 283–289 CrossRef CAS PubMed
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed
- J. H. Clark, J. Chem. Technol. Biotechnol., 2007, 82, 603–609 CrossRef CAS
-
M. Pagliaro and M. Rossi, The Future of Glycerol, Royal Society of Chemistry, Cambridge. U.K, 2nd edn, 2010 Search PubMed
- C.-H. Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC
- A. Said Stålsmeden, J. L. Belmonte Vázquez, K. van Weerdenburg, R. Rae, P.-O. Norrby and N. Kann, ACS Sustainable Chem. Eng., 2016, 4, 5730–5736 CrossRef
- A. M. Truscello, C. Gambarotti, M. Lauria, S. Auricchio, G. Leonardi, S. U. Shisodia and A. Citterio, Green Chem., 2013, 15, 625–628 RSC
- J. Bensemhoun and S. Condon, Green Chem., 2012, 14, 2595–2599 RSC
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC
- M. O. Sonnati, S. Amigoni, E. P. Taffin de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC
- J. A. Stewart, R. Drexel, B. Arstad, E. Reubsaet, B. M. Weckhuysen and P. C. A. Bruijnincx, Green Chem., 2016, 18, 1605–1618 RSC
- P. U. Naik, L. Petitjean, K. Refes, M. Picquet and L. Plasseraud, Adv. Synth. Catal., 2009, 351, 1753–1756 CrossRef CAS
- H. Mutlu, J. Ruiz, S. C. Solleder and M. A. R. Meier, Green Chem., 2012, 14, 1728–1735 RSC
- J. R. Ochoa-Gomez, O. Gomez-Jimenez-Aberasturi, C. Ramirez-Lopez and B. Maestro-Madurga, Green Chem., 2012, 14, 3368–3376 RSC
- G. Kharchafi, F. Jerome, J.-P. Douliez and J. Barrault, Green Chem., 2006, 8, 710–716 RSC
- F. Jerome, G. Kharchafi, I. Adam and J. Barrault, Green Chem., 2004, 6, 72–74 RSC
- J. n. Pérez-Pariente, I. Díaz, F. Mohino and E. Sastre, Appl. Catal., A, 2003, 254, 173–188 CrossRef
- X. Liu, H. Ma, Y. Wu, C. Wang, M. Yang, P. Yan and U. Welz-Biermann, Green Chem., 2011, 13, 697–701 RSC
- B. Ren, M. Rahm, X. Zhang, Y. Zhou and H. Dong, J. Org. Chem., 2014, 79, 8134–8142 CrossRef CAS PubMed
- K. Klepáčová, D. Mravec and M. Bajus, Appl. Catal., A, 2005, 294, 141–147 CrossRef
- J. Deutsch, A. Martin and H. Lieske, J. Catal., 2007, 245, 428–435 CrossRef CAS
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed
- N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed
- X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed
- C. E. I. Knappke, A. Imami and A. J. von Wangelin, ChemCatChem, 2012, 4, 937–941 CrossRef CAS
- S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem. – Eur. J., 2013, 19, 4664–4678 CrossRef CAS PubMed
- M. S. Kharasch and B. S. Joshi, J. Org. Chem., 1957, 22, 1439–1443 CrossRef CAS
- S. D. Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef PubMed
- R. S. Reddy, J. N. Rosa, L. F. Veiros, S. Caddick and P. M. P. Gois, Org. Biomol. Chem., 2011, 9, 3126–3129 CAS
- E. G. Delany, C.-L. Fagan, S. Gundala, A. Mari, T. Broja, K. Zeitler and S. J. Connon, Chem. Commun., 2013, 49, 6510–6512 RSC
- E. G. Delany, C.-L. Fagan, S. Gundala, K. Zeitler and S. J. Connon, Chem. Commun., 2013, 49, 6513–6515 RSC
- J. Zhao, C. Mück-Lichtenfeld and A. Studer, Adv. Synth. Catal., 2013, 355, 1098–1106 CrossRef CAS
- L. Ta, A. Axelsson and H. Sunden, Green Chem., 2016, 18, 686–690 RSC
- A. Axelsson, E. Hammarvid, L. Ta and H. Sunden, Chem. Commun., 2016, 52, 11571–11574 RSC
- D. Xie, D. Shen, Q. Chen, J. Zhou, X. Zeng and G. Zhong, J. Org. Chem., 2016, 81, 6136–6141 CrossRef CAS PubMed
- A. Axelsson, L. Ta and H. Sunden, Synlett, 2017 DOI:10.1055/s-0036-1588395
- J. E. Bäckvall, A. K. Awasthi and Z. D. Renko, J. Am. Chem. Soc., 1987, 109, 4750–4752 CrossRef
- J.-E. Bäckvall, R. B. Hopkins, H. Grennberg, M. Mader and A. K. Awasthi, J. Am. Chem. Soc., 1990, 112, 5160–5166 CrossRef
- J. Piera, K. Närhi and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2006, 45, 6914–6917 CrossRef CAS PubMed
- J. Wöltinger, J.-E. Bäckvall and Á. Zsigmond, Chem. – Eur. J., 1999, 5, 1460–1467 CrossRef
- J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed
- R. A. Holser, T. R. Mitchell, R. E. Harry-O'kuru, S. F. Vaughn, E. Walter and D. Himmelsbach, J. Am. Oil Chem. Soc., 2008, 85, 347–351 CrossRef CAS
- C. Duval, N. Kébir, R. Jauseau and F. Burel, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 758–764 CrossRef CAS
- G. Rokicki, P. Rakoczy, P. Parzuchowski and M. Sobiecki, Green Chem., 2005, 7, 529–539 RSC
- W. Guo, J. Gónzalez-Fabra, N. A. G. Bandeira, C. Bo and A. W. Kleij, Angew. Chem., Int. Ed., 2015, 54, 11686–11690 CrossRef CAS PubMed
- S. Sopeña, V. Laserna, W. Guo, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Adv. Synth. Catal., 2016, 358, 2172–2178 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and 1H NMR, 13C NMR and 19F NMR, COSY NMR, HSQC NMR, HMBC NMR, IR and HRMS data. See DOI: 10.1039/c7gc00471k |
| This journal is © The Royal Society of Chemistry 2017 |