
Liquid/liquid extraction of biomass-derived lignin from lignocellulosic pretreatments†
Serafin
Stiefel‡
ab,
Davide
Di Marino‡
ab,
Armin
Eggert
c,
Ivo Robert
Kühnrich
ab,
Markus
Schmidt
c,
Philipp M.
Grande
d,
Walter
Leitner
d,
Andreas
Jupke
c and
Matthias
Wessling
*ab
aAVT.CVT, Turmstr. 46, 52064 Aachen, Germany. E-mail: manuscripts.cvt@avt.rwth-aachen.de
bDWI - Leibnizinstitute for Interactive Materials, Forckenbeckstr. 50, 52074 Aachen, Germany
cAVT.FVT, Wüllnerstr. 5, 52062 Aachen, Germany
dInstitut für Technische und Makromolekulare Chemie (ITMC), RWTH Aachen University, Worringer Weg 1, 52074 Aachen, Germany
First published on 12th October 2016
Abstract
Despite the rapid progress in the field of biomass fractionation and lignin valorization, no industrial process for chemical utilization of lignin has yet been established. One major step in that direction has been made with the advent of biorefineries and new biomass fractionation methods that deliver a relatively clean lignin stream, allowing a more efficient recovery and utilization of this fraction. However, the transfer of lignin from the fractionation solvent to a different medium for subsequent valorization has been largely disregarded so far. In this work, we demonstrate the use of a green liquid/liquid-extraction to transfer lignin from the organic phase of the OrganoCat process into differently concentrated alkaline solutions for further utilization. We show that alkaline solutions of pH 13 and 14 are able to almost completely extract the OrganoCat lignin from the organic phase but that this extraction might be accompanied by changes in the molecular structure of lignin, here shown by a change in the apparent molecular weight distribution.
Even though lignin valorization to value-added chemicals has been intensively studied in the past, the technology has not yet arrived in today's chemical industry. With the advent of biorefineries, however, a crucial step towards industrial lignin utilization has been achieved.1 Novel technologies for biomass pretreatment deliver lignin streams that are relatively pure and more easily recoverable than classical fractionation methods from the pulp & paper industry.2–4 In addition, the fractionation process is one of the major driving forces for overall production costs, so that new pretreatment technologies have a major impact on the biorefinery profitability.5 Still, there is a largely disregarded issue concerning the majority of the utilized fractionation methods: most organic solvents that generally dissolve the lignin fraction of biomass are unsuited for subsequent conversion processes.6 As such, an exchange of the fractionation medium with the valorization medium is necessary. Currently, there are two main methods to realize a solvent exchange: (I) precipitation of lignin with an anti-solvent followed by drying2,7 or (II) evaporation of the organic solvent.8 In both cases, lignin is subsequently redissolved in the valorization medium. Evaporation of the organic solvent has the drawback of a relatively high-energy demand. In addition, the effect of evaporation on the lignin properties has not yet been investigated, but might be detrimental for further conversion processes. Precipitation on the other hand requires a large amount of anti-solvent, which makes it impossible to recycle the organic solvent back to the fractionation process without subsequent purification. Solvent recovery is one of the main driving forces for increased energy consumption.8 In order to avoid these drawbacks, the direct transfer of lignin by liquid/liquid-extraction from the organic solvent from the fractionation process into the solvent which is utilized for the valorization seems prudent, but has not yet been systematically investigated. To demonstrate the principle, we show the application of an L/L-extraction to transfer lignin from 2-methyltetrahydrofuran (2-MTHF), the organic phase of the OrganoCat pretreatment (termed raffinate), into an alkaline solution (termed extract).
The OrganoCat process is a green fractionation method that uses oxalic acid as a catalyst for the depolymerization of hemicellulose and 2-MTHF as a solvent for the lignin fraction.3,9 This fractionation process is especially promising for the inclusion in biorefinery concepts for several reasons: oxalic acid is non-toxic and less corrosive than other catalysts, the mild process conditions result in relatively low energy requirements and the use of 2-MTHF meets environmental criteria due to its origin from biomass and its recyclability.3,10 The extraction into an alkaline solution is promising as several valorization methods utilize lignin in an alkaline solution for further conversion, such as electrochemical processes or organic catalysis.11–26
In the present work, the possibility of a liquid–liquid-extraction to transfer lignin from the organic phase of the OrganoCat process into an aqueous sodium hydroxide solution to make it accessible for subsequent valorization has been investigated. Fig. 1 shows the proposed link between the fractionation and the lignin valorization.
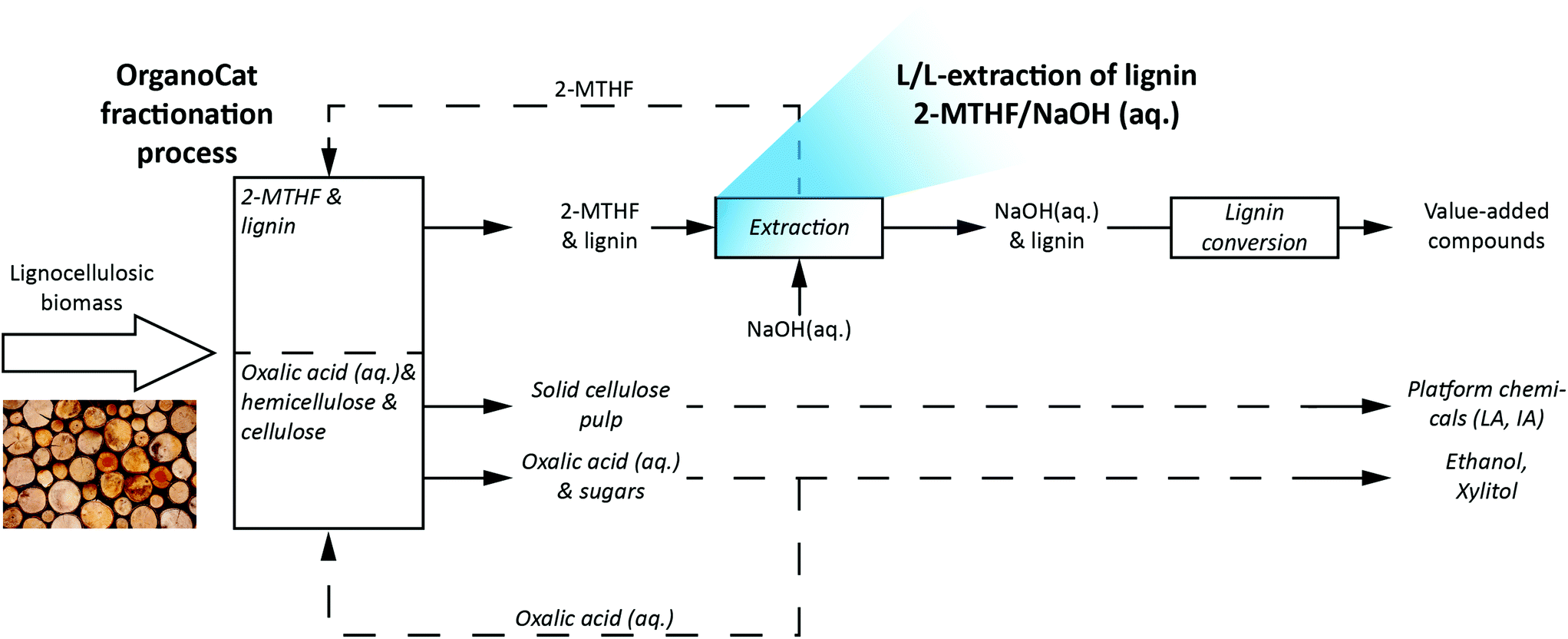 | ||
| Fig. 1 The OrganoCat process for the fractionation of lignocellulosic biomass with the subsequent liquid/liquid-extraction of lignin from 2-MTHF into an alkaline solution. Lignin can be subsequently treated by different processes for the production of value-added compounds, while the solid cellulose pulp can be transformed into platform chemicals such as levulinic acid (LA) and itaconic acid (IA). | ||
For an efficient as well as green liquid–liquid extraction, the two utilized solvents should fulfill several requirements: low mutual solubility, fast demixing and low toxicity, just to name a few. The mutual solubility of 2-MTHF and 0.1 M sodium hydroxide solution at T = 30 °C was determined by quantitative 1H-NMR spectroscopy. The areas of characteristic peaks (methyl group of 2-MTHF and the hydroxide ion) were integrated and the ratio of these areas was evaluated via calibration curves. 1H-NMR spectra and the evaluated data can be found in the ESI.†
The solubility of 2-MTHF in 0.1 M sodium hydroxide solution has been determined as follows:
| corg→aq. = 118.9 g Laq. NaOH−1 or worg→aq. = 0.119. |
The measured solubility of 11.9 wt% of 2-MTHF in 0.1 M aqueous NaOH solution at 30 °C is slightly higher than the value reported by Stephenson (11.4 wt% at 29.5 °C). The calculation of the solubility of aqueous sodium hydroxide in 2-MTHF results in
| caq.→org = 42.2 g L2-MTHF−1 or waq.→org = 0.05. |
The calculated solubility of 0.1 M sodium hydroxide solution in 2-MTHF of 42.2 g L−1 and 5.0 wt% is again slightly higher than the solubility of water in 2-MTHF with 4.3 wt% at 29.5 °C.27 As such, the addition of NaOH to the aqueous phase shows only a minor influence on the miscibility of water and 2-MTHF.
For an industrial process, low volumes of sodium hydroxide should be applied in the liquid–liquid extraction in order to minimize the loss of 2-MTHF and achieve higher solute concentrations in the extract phase, favouring valorization. The solubility of aqueous NaOH in 2-MTHF is relatively low, so that a direct recycle of 2-MTHF into the OrganoCat process might be feasible. However, the possible interaction of the alkaline fraction in 2-MTHF with oxalic acid in the aqueous fractionation phase needs to be investigated.
To investigate the influence of the alkalinity of the extract on the extraction effectivity, extracts with NaOH concentrations corresponding to pH values between 11 and 14 have been prepared. In order to show the versatility of the extraction process, two different lignin types have been used in the experiments: OrganoCat lignin, the actual intended substrate for this process, and Kraft lignin (Sigma Aldrich, 370959), to investigate the suitability for other substrates. Prior to the experiments, 2-MTHF and the aqueous sodium hydroxide solutions have been brought into contact and were stirred for 72 hours in order to allow the two phases to equilibrate. Dry OrganoCat or Kraft lignin was then dissolved in the saturated organic phase with a concentration of 15 g L−1, which corresponds to the loading of 2-MTHF with lignin in the OrganoCat process. 3 ml of lignin dissolved in 2-MTHF and 3 mL of 2-MTHF-saturated aqueous sodium hydroxide solution were filled in 15 ml centrifuge tubes and rotated in a water bath for two hours at 30 °C in order to allow the phases to reach a thermodynamic equilibrium. Afterwards the samples were centrifuged at 1400g for 10 minutes to accelerate the separation process (Rotanta 460r, Hettich). In order to ensure that the step of drying and redissolving the lignin in 2-MTHF does not influence the extraction effectivity, lignin samples have also been taken directly from the OrganoCat process and were subjected to the L/L-extraction without intermediate drying. The lignin concentration in the aq. sodium hydroxide solution after extraction was determined via UV/vis-absorbance measurements at a wavelength of λ = 280 nm (Thermo Scientific, Genesys 10S) and evaluated via calibration curves. One has to keep in mind that the UV absorbance of the different lignin fractions will deviate from their bulk properties. As such, the distribution of mass over the two phases might not completely correlate with the distribution of the UV absorbance.
The alkalinity of the aqueous solution shows a major influence on the partition of OrganoCat and Kraft lignin between the organic and the aqueous phase. For the OrganoCat lignin, pH values of 12 and lower cause a majority of lignin to remain in the organic phase. An increase of the pH to 13 and above shifts the equilibrium strongly in favor of the alkaline extract. For Kraft lignin, the equilibrium already started to shift in favor of the aqueous phase at a pH of 12. In the case of Kraft lignin and an extract of pH 12, even after centrifugation a stable emulsion persists at the two-phase boundary. Fig. 2 shows the samples for pH 11 to 14 after two hours of equilibration. The kinetics of the extraction has not yet been determined.
 | ||
| Fig. 2 Two-phase systems of 2-MTHF/aq. NaOH with 15 g L−1 OrganoCat/Kraft lignin after 2 hours of equilibration at 25 °C. At pH 11, both lignin types primarily dissolve in the organic phase, as indicated by the darkened solution. At pH 12, the Kraft lignin starts to dissolve in the aqueous phase whereas the OrganoCat lignin largely remains in the 2-MTHF. At pH 13 and above, both lignin types primarily dissolve in the aqueous phase. | ||
The amount of extracted lignin, as determined by UV spectroscopy, can be found in Table 1. At pH 11, only 4.05% can be found in the aqueous phase, corresponding to a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P-value of −1.37, whereas at pH 13 already 94.64% are extracted from the 2-MTHF (logP = 1.25). logP represents the logarithm of the distribution coefficient, which is defined as the solute concentration of the extract divided by the solute concentration of the raffinate in equilibrium. An increase to pH 14 shows no further increase of extraction efficiency. There is no discernible difference between the OrganoCat lignin that was dried after the fractionation and then redissolved in saturated 2-MTHF and the OrganoCat samples that were used as received from the OrganoCat fractionation. The control experiment with commercially available Kraft lignin showed an even better extraction compared to the OrganoCat lignin, with already 67.24% extracted at pH 12 (logP = 0.31). Table 1 also shows the logarithm of the respective partition coefficients of all samples. To exclude ionic strength influencing the partition of lignin within the two phases, the extraction was performed again at pH 12 and with addition of NaCl to have the ionic strength of a pH 14 NaOH solution. The experiment yielded a low amount of extracted lignin, similarly to the standard pH 12 extraction, indicating that ionic strength has no major influence on the extraction.
P-value of −1.37, whereas at pH 13 already 94.64% are extracted from the 2-MTHF (logP = 1.25). logP represents the logarithm of the distribution coefficient, which is defined as the solute concentration of the extract divided by the solute concentration of the raffinate in equilibrium. An increase to pH 14 shows no further increase of extraction efficiency. There is no discernible difference between the OrganoCat lignin that was dried after the fractionation and then redissolved in saturated 2-MTHF and the OrganoCat samples that were used as received from the OrganoCat fractionation. The control experiment with commercially available Kraft lignin showed an even better extraction compared to the OrganoCat lignin, with already 67.24% extracted at pH 12 (logP = 0.31). Table 1 also shows the logarithm of the respective partition coefficients of all samples. To exclude ionic strength influencing the partition of lignin within the two phases, the extraction was performed again at pH 12 and with addition of NaCl to have the ionic strength of a pH 14 NaOH solution. The experiment yielded a low amount of extracted lignin, similarly to the standard pH 12 extraction, indicating that ionic strength has no major influence on the extraction.
| Extract pH [−] | Lignin type | Extracted lignin [%] | logP [−] |
|---|---|---|---|
| 11 | OrganoCat, dried | 4.05 | −1.37 |
| 12 | OrganoCat, dried | 6.48 | −1.16 |
| 13 | OrganoCat, dried | 94.64 | 1.25 |
| 14 | OrganoCat, dried | 93.23 | 1.14 |
| 14 | OrganoCat, untreated | 95.02 | 1.28 |
| 11 | Kraft, dried | 12.71 | −0.64 |
| 12 | Kraft, dried | 67.24 | 0.31 |
| 13 | Kraft, dried | 95.13 | 1.29 |
| 14 | Kraft, dried | 96.61 | 1.46 |
In order to investigate a potential change in molecular weight during extraction and to determine the extraction efficiency as a function of the molecular weight, size exclusion chromatography (SEC) measurements have been utilized. The extract was used as obtained from the experiment. Analyses were performed on an Agilent 1200 system, equipped with a refractive index detector (DawnEOS, Wyatt Technology) and a UV detector (VWD-UV, Agilent) at a wavelength of λ = 280 nm. The stationary phase comprised a pre-column (8 × 50 mm) and three MCX gel columns (8 × 300 mm, particle diameter: 5 μm, nominal pore width: 1000 Å). The mobile phase was 0.1 mol L−1 sodium hydroxide and 0.01 wt% sodium azide at a flow rate of 1 mL min−1 at 40 °C. Calibration was achieved using narrowly distributed poly(styrene sulfonate) standards.
The SEC measurements show that low-molecular weight compounds are already partially extracted at pH 11 and 12, whereas the high-molecular weight compounds are only extracted at pH 13 and above. These results correspond to the results of the UV absorbance measurements. However, the SEC diagrams show a change in the molecular weight distribution between pH 13 and pH 14. In the pH 14 sample, a significant amount of high-molecular-weight fractions seems to be absent, whereas an increase in low-molecular-weight compounds can be observed (Fig. 3). The same behaviour was observed with Kraft lignin, except that a significant amount of high-molecular-weight compounds was already dissolved at pH 12 (see ESI Fig. SI 1†). One possible explanation for this observation is the occurrence of a chemical reaction of the OrganoCat lignin in highly alkaline solutions, leading to partial cleavage of the intramolecular bonds. The fact that a strong base catalyzes lignin fragmentation is not new; however, normally these processes only occur at temperatures above 270 °C.28–31 Another possibility is that the organic solvent present in the aqueous solution induces a change in lignin conformation, leading to a change in hydrodynamic radius32 or aggregation behaviour33 and thus to smaller apparent molecular weights. Further investigations are necessary in order to elucidate the phenomena acting on lignin in such two-phase systems. As such, one can completely remove the OrganoCat lignin from the fractionation solvent, possibly accompanied by changes in the molecular structure. However, even if such changes are proven in future investigations, their consequences for the subsequent lignin valorization steps might even be positive. Furthermore, partition coefficients of carbohydrates (xylose) in the OrganoCat process were measured by dissolving xylose in water and by using 2-MTHF as an extractant. The resulting partition coefficient of xylose in the 2-MTHF–water mixture is logP = 0.0046, indicating that almost no sugar is extracted into the organic phase. Hence, lignin only has traces of sugar. Formation of sodium oxalate may also represent a challenge during the extraction. However, the solubility of sodium oxalate in aqueous solution is rather high (37 g L−1, 20 °C). The maximum possible oxalate concentration in the 2-MTHF phase would be ca. 4.5 g L−1 and no precipitation is observed under the applied conditions.
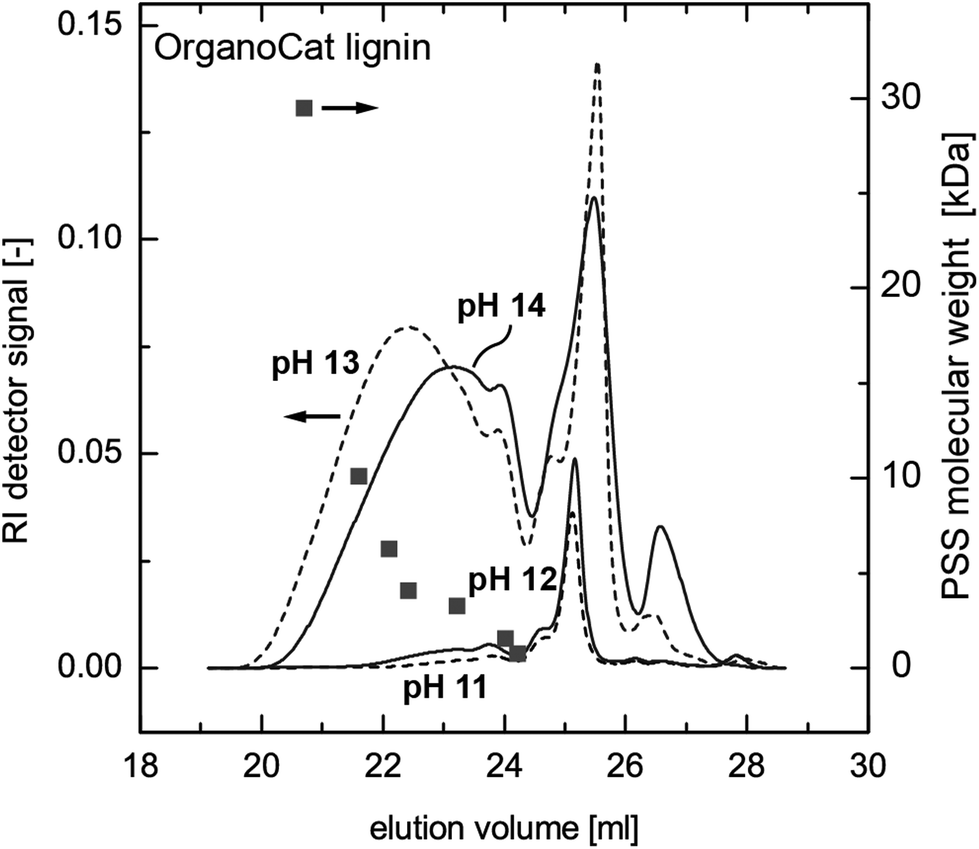 | ||
| Fig. 3 Size exclusion chromatograms for the extracted OrganoCat samples for an extract pH between 11 and 14. With an alkaline extract of pH 11 and 12, low-molecular weight compounds around elution times of 25 minutes are primarily extracted from the 2-MTHF. An increase to pH 13 also extracts the high-molecular weight fractions between elution times of 19 to 24 minutes. A further increase of the extract to pH 14 shows a decrease in molecular weight, which might signify a chemical reaction of the lignin molecule during the extraction. | ||
Conclusions
Lignin was successfully extracted from 2-MTHF, the organic phase of the OrganoCat fractionation process, into an aqueous sodium hydroxide solution. The effect of salt addition on the miscibility of 2-MTHF and water has been determined and was shown to only have a minor influence. At a pH value of 13 and above the extraction was almost complete, so that the lignin-depleted 2-MTHF can theoretically be recycled back to the fractionation process. However, at a high extract alkalinity of pH 14 the SEC elugram shows a change in the apparent molecular weight distribution of the OrganoCat lignin, a sign that the extraction might be accompanied by a chemical reaction. This work focuses on showing the difference in the size distribution of lignin as an effect of extraction/dissolution in NaOH solution, which is visible via size exclusion chromatography (SEC). Further characterization of the lignin would be beyond the scope of this communication and should be part of a follow-up study. Nonetheless, L/L-extraction showed to be a promising green approach for lignin transfer between solvents with low miscibility.Acknowledgements
This work was supported by the Alexander-von-Humboldt Foundation and the Cluster of Excellence “Tailor-made Fuels from Biomass” funded by the Excellence Initiative of the German federal and state governments. Part of the work was performed at the Center for Chemical Polymer Technology, which is supported by the EU and the federal state of North Rhine-Westphalia (grant no. EFRE 30 00 883 02). Davide Di Marino appreciates the financial support of the Marie Curie Action in the framework of SuBiCat (FP7). We thank Ines Bachmann-Remy and Prof. Jürgen Klankermayer for their assistance in NMR measurements. We thank Rainer Haas for the SEC measurements.References
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna and M. Keller, Science, 2014, 344, 1246843 CrossRef PubMed.
- X. Zhao, K. Cheng and D. Liu, Appl. Microbiol. Biotechnol., 2009, 82, 815–827 CrossRef CAS PubMed.
- T. vom Stein, P. M. Grande, H. Kayser, F. Sibilla, W. Leitner and P. Dominguez de Maria, Green Chem., 2011, 13, 1772–1777 RSC.
- J. Wildschut, A. T. Smit, J. H. Reith and W. J. J. Huijgen, Bioresour. Technol., 2013, 135, 58–66 CrossRef CAS PubMed.
- B. Yang and C. E. Wyman, Biofuels, Bioprod. Biorefin., 2008, 2, 26–40 CrossRef CAS.
- J. Zakzeski, P. Bruijnincx, A. Jongerius and B. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed.
- P. J. de Wild, W. J. J. Huijgen and H. J. Heeres, J. Anal. Appl. Pyrolysis, 2012, 93, 95–103 CrossRef CAS.
- J. Viell, A. Harwardt, J. Seiler and W. Marquardt, Bioresour. Technol., 2013, 150, 89–97 CrossRef CAS PubMed.
- P. M. Grande, J. Viell, N. Theyssen, W. Marquardt, P. Dominguez de Maria and W. Leitner, Green Chem., 2015, 17, 3533–3539 RSC.
- F. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., 2010, 122, 5642–5646 CrossRef.
- S. Nanayakkara, A. F. Patti and K. Saito, Green Chem., 2014, 16, 1897–1903 RSC.
- H. Deng, L. Lin, Y. Sun, C. Pang, J. Zhuang, P. Ouyang, Z. Li and S. Liu, Catal. Lett., 2008, 126, 106–111 CrossRef CAS.
- H. Deng, L. Lin, Y. Sun, C. Pang, J. Zhuang, P. Ouyang, J. Li and S. Liu, Energy Fuels, 2009, 23, 19–24 CrossRef CAS.
- M. J. Robinson, L. J. Wright and I. D. Suckling, J. Wood Chem. Technol., 2000, 20, 357–373 CrossRef CAS.
- H. Korpi, P. J. Figiel, E. Lankinen, P. Ryan, M. Leskelä and T. Repo, Eur. J. Inorg. Chem., 2007, 2007, 2465–2471 CrossRef.
- V. Tarabanko, N. Fomova, B. Kuznetsov, N. Ivanchenko and A. Kudryashev, React. Kinet. Catal. Lett., 1995, 55, 161–170 CrossRef CAS.
- K. Kervinen, M. Allmendinger, M. Leskelä, T. Repo and B. Rieger, Phys. Chem. Chem. Phys., 2003, 5, 4450–4454 RSC.
- K. Kervinen, H. Korpi, J. Gerbrand Mesu, F. Soulimani, T. Repo, B. Rieger, M. Leskelä and B. M. Weckhuysen, Eur. J. Inorg. Chem., 2005, 2005, 2591–2599 CrossRef.
- S. Stiefel, C. Marks, T. Schmidt, S. Hanisch, G. Spalding and M. Wessling, Green Chem., 2016, 18, 531–540 RSC.
- S. Stiefel, J. Lölsberg, L. Kipshagen, R. Möller-Gulland and M. Wessling, Electrochem. Commun., 2015, 61, 49–52 CrossRef CAS.
- F. G. Sales, L. C. A. Maranhão, N. M. L. Filho and C. A. M. Abreu, Chem. Eng. Sci., 2007, 62, 5386–5391 CrossRef CAS.
- H. Zhu, Y. Chen, T. Qin, L. Wang, Y. Tang, Y. Sun and P. Wan, RSC Adv., 2014, 4, 6232–6238 RSC.
- H. Zhu, L. Wang, Y. Chen, G. Li, H. Li, Y. Tang and P. Wan, RSC Adv., 2014, 4, 29917–29924 RSC.
- J. Zhang, H. Deng and L. Lin, Molecules, 2009, 14, 2747–2757 CrossRef CAS PubMed.
- Y.-m. Zhang, Y. Peng, X.-l. Yin, Z.-h. Liu and G. Li, J. Chem. Technol. Biotechnol., 2014, 89(12), 1954–1960 CrossRef CAS.
- R. Katahira, A. Mittal, K. McKinney, X. W. Chen, M. P. Tucker, D. K. Johnson and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 1474–1486 CrossRef CAS.
- R. M. Stephenson, J. Chem. Eng. Data, 1992, 37, 80–95 CrossRef CAS.
- J.-M. Lavoie, W. Baré and M. Bilodeau, Bioresour. Technol., 2011, 102, 4917–4920 CrossRef CAS PubMed.
- V. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. Li and J. A. Lercher, Chem. – Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed.
- R. Beauchet, F. Monteil-Rivera and J. Lavoie, Bioresour. Technol., 2012, 121, 328–334 CrossRef CAS PubMed.
- A. Toledano, L. Serrano and J. Labidi, J. Chem. Technol. Biotechnol., 2012, 87, 1593–1599 CrossRef CAS.
- T. Garver and P. Callaghan, Macromolecules, 1991, 24, 420–430 CrossRef CAS.
- M. Norgren and H. Edlund, Curr. Opin. Colloid Interface Sci., 2014, 19, 409–416 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SEC elugrams for the extracted Kraft lignin, the molecular weight of both lignins as well as details concerning the methods and results of NMR measurements. See DOI: 10.1039/C6GC02270G |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |