
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
6-exo-trig Michael addition-lactonizations for catalytic enantioselective chromenone synthesis†
Rifahath M.
Neyyappadath
,
David B.
Cordes
 ,
Alexandra M. Z.
Slawin
and
Andrew D.
Smith
*
,
Alexandra M. Z.
Slawin
and
Andrew D.
Smith
*
EaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
First published on 19th January 2017
Abstract
The catalytic enantioselective 6-exo-trig Michael addition-lactonization of enone-acid substrates to form cis-chromenones with high diastereo- and enantiocontrol was developed using the commercially available isothiourea tetramisole. An acidic workup proved necessary to minimize product epimerization and maximize product er, providing cis-chromenones in excellent yield, and with excellent diastereo- and enantioselectivity.
The development of catalytic processes that allow the preparation of valuable heterocyclic frameworks from readily prepared starting materials under mild conditions is of widespread importance.1 A range of enantioselective methods that fulfil these goals has been developed.2 In recent years the catalytic use of C(1)-ammonium enolates,3 particularly those using carboxylic acids as starting materials,4 has been popularized following the intramolecular enantioselective nucleophile-catalyzed aldol lactonization (NCAL) methodology developed by Romo for the synthesis of stereodefined β-lactones.5 In this area, 5-exo-ring closure to prepare the corresponding carbo- and heterocyclic ring systems is commonplace (Fig. 1a), and this strategy has been applied successfully for the construction of complex molecular targets.6 To date, only limited isolated examples of this approach for the formation of 6-membered ring systems have been developed,7 all of which use cinchona alkaloids as catalysts. In previous work we developed an isothiourea-catalyzed8,9 5-exo-Michael addition-lactonization approach to 5-membered carbo- and heterocycle synthesis from enone acids (Fig. 1b).10 In this manuscript the application of this methodology for the preparation of 6-membered heterocycles is reported for the first time, allowing the synthesis of cis-chromenones11 in up to 99
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 98:2 er (Fig. 1c).
:1 dr and 98:2 er (Fig. 1c).
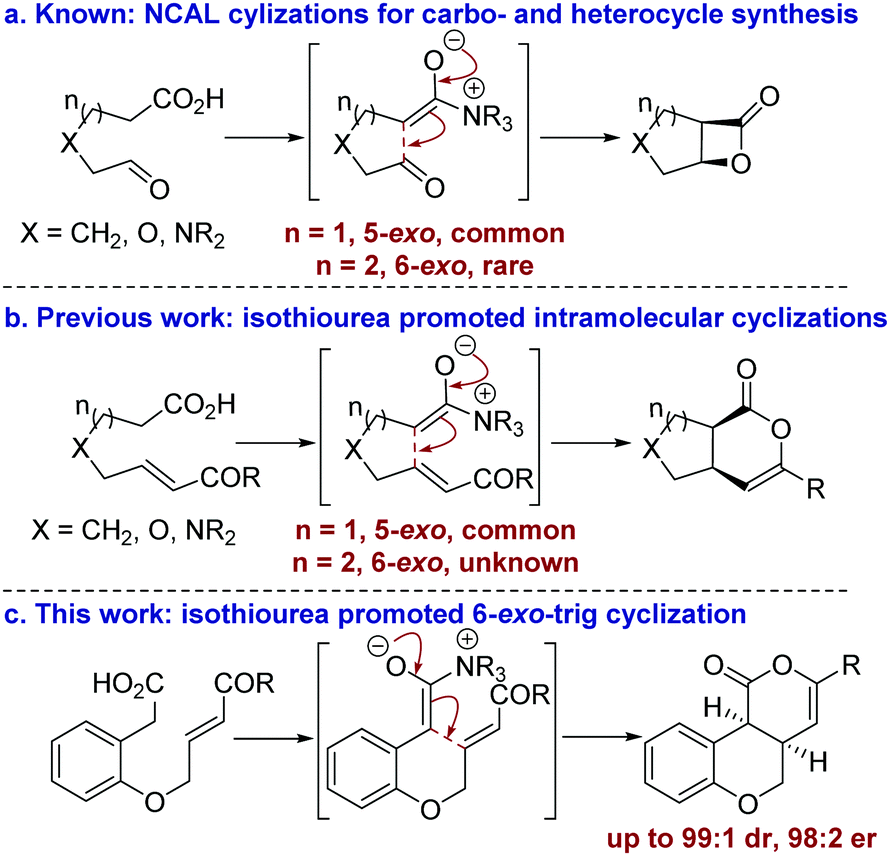 | ||
| Fig. 1 Summary of ammonium enolate promoted intramolecular catalytic enantioselective carbo- and heterocycle formation. | ||
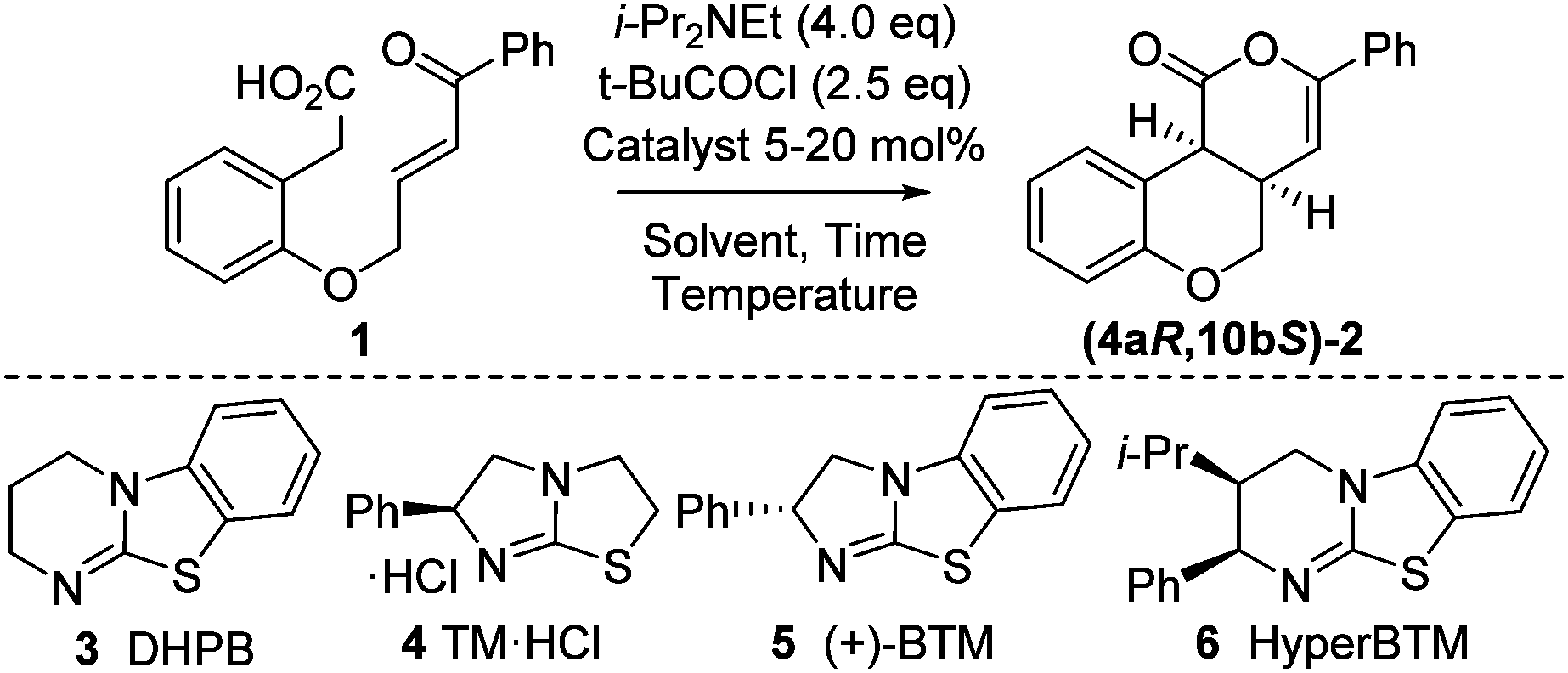
Initial model studies probed the cyclization of enone acid 1 to chromenone 2, with 1 readily prepared in three steps from 2-hydroxyphenylacetic acid.12 Treatment of 1 with pivaloyl chloride and i-Pr2NEt gave the corresponding mixed anhydride in situ, which was subsequently treated with isothiourea catalysts 3 to 6 and evaluated for the proposed cyclization (Table 1, entries 1–4). Achiral DHPB13 gave the desired cis-chromenone 2 in 85% yield and >99:1 dr. Screening of a small range of chiral isothioureas 4–6 indicated the use of tetramisole 4 and its benzannulated counterpart, BTM 5, showed promising enantioselectivity (∼87:13 er, entries 2 and 3). Subsequent optimization through variation of solvent, temperature and base12 showed that performing the reaction at 0 °C in CHCl3 with excess i-Pr2NEt (1.5 equiv. for mixed anhydride formation, followed by an additional 2.5 equiv.) gave highest observed dr and er (entries 6–9). Lowering the catalyst loading to 5 mol% using tetramisole 4 gave 2 in 85% yield, >99:1 dr and 93:7 er, with BTM 5 giving lower conversion and isolated product yield even after extended reaction times (entries 10 and 11).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | T (°C) | Time (h) | Yielda (%) | drb (cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :trans) :trans) |
erc (4aR,10bS:4aS,10bR) |
| a Isolated yield. b Measured by 1H NMR spectroscopy of crude reaction product. c Measured by chiral HPLC (major cis-diastereoisomer). d CH2Cl2 (0.1 M). e CHCl3 (0.1 M). f CHCl3 (0.05 M). | ||||||
| 1d | 3 (20) | rt | 16 | 85 | >99:1 |
Racemic |
| 2d | 4 (20) | rt | 16 | 84 | >99:1 |
87:13 |
| 3d | 5 (20) | rt | 16 | 84 | >99:1 |
13:87 |
| 4d | 6 (20) | rt | 16 | 69 | >99:1 |
57:43 |
| 5d | — | rt | 16 | nil | — | — |
| 6e | 5 (20) | rt | 16 | 85 | >99:1 |
7:93 |
| 7f | 5 (20) | rt | 16 | 84 | >99:1 |
7:93 |
| 8e | 5 (20) | 0 | 4 | 87 | >99:1 |
7:93 |
| 9e | 5 (20) | −10 | 16 | 83 | >99:1 |
6:94 |
| 10e | 4 (5) | 0 | 4 | 85 | >99:1 |
93:7 |
| 11e | 5 (5) | 0 | 16 | 65 | >99:1 |
7:93 |
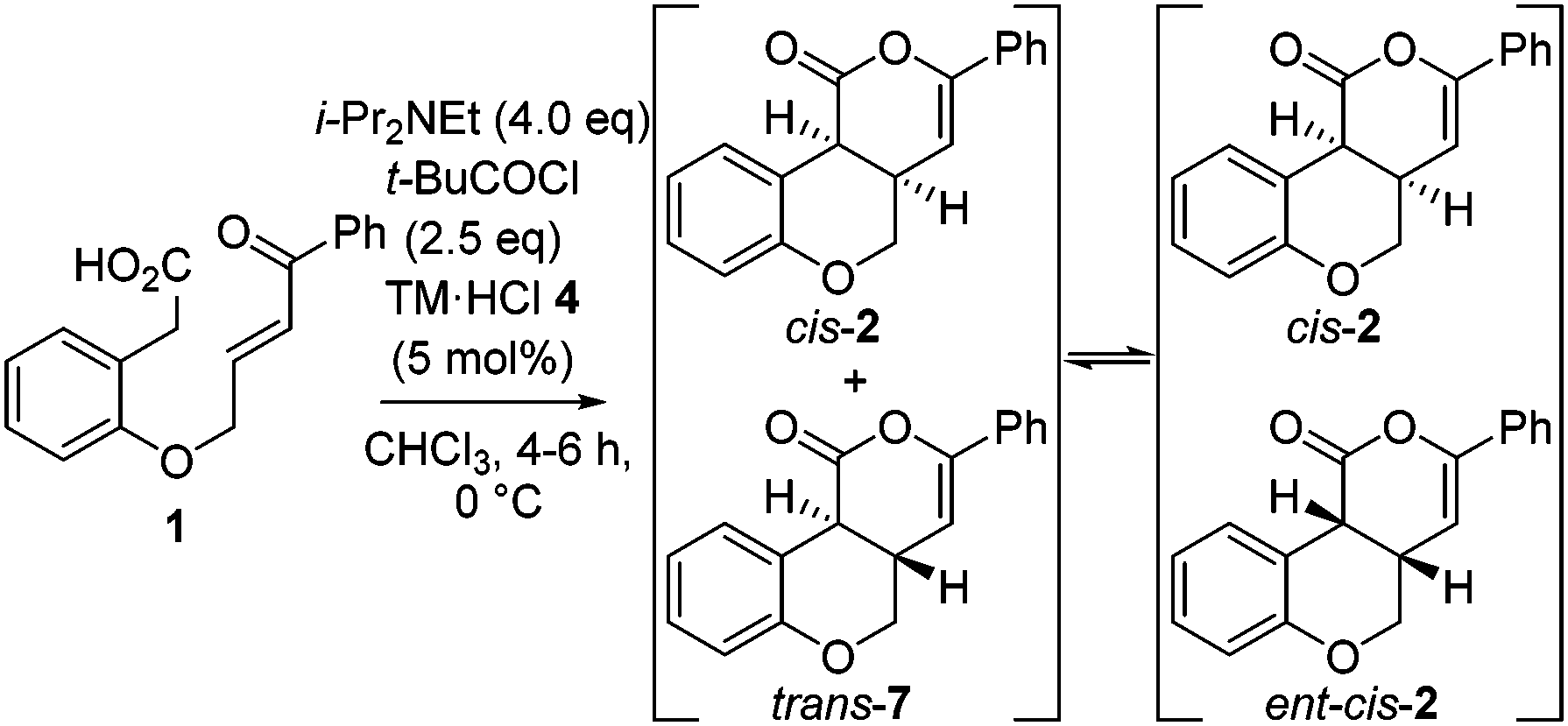
Further investigation monitored product dr and er with reaction conversion and time (Table 2). These studies indicated the dr of the product remained constant (92:8 dr cis:trans) up to full conversion, but increased to 99:1 upon extended reaction times. Furthermore, the er of the major cis-product decreased from 99:1 er (up to full conversion) to 93:7 er with time.12 These observations are consistent with base catalyzed-epimerization of the minor trans-diastereoisomer (4aS,10bS)-7 to ent-cis-(4aS,10bR) 2, resulting in increased product dr but lower product er. Consistent with this epimerization process, treatment of an 80:20 mixture of trans-7:cis-2 with i-Pr2NEt gave cis-2 in >99:1 dr.14
To circumvent product epimerization and maximize product er incorporation of an acidic aqueous work-up protocol was essential. For example, carrying the reaction out at 0 °C, followed by work up with H2O at rt gave 2 in 85% yield, >99:1 dr and 93:7 er. However, work up with 0.1 M HCl at 0 °C gave 2 in 93:7 dr, with purification giving 2 as a single diastereoisomer in 70% yield and 98:2 er (Scheme 1).
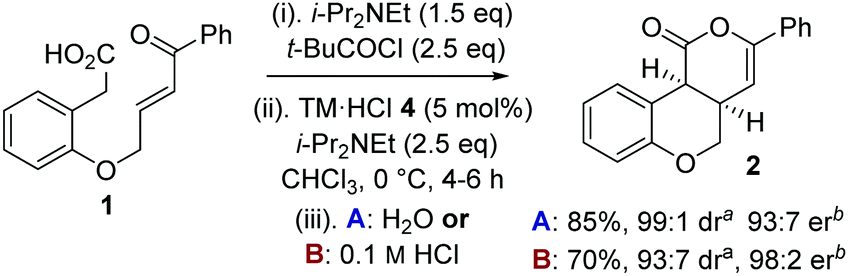 | ||
| Scheme 1 Optimized procedure. aMeasured by 1H NMR spectroscopy of crude reaction product. bMeasured by chiral HPLC (major cis-diastereoisomer). | ||
With an optimized protocol established, the generality of this process was investigated (Table 3). The tolerance of this methodology to variation within the enone portion was initially probed, with all starting materials prepared from the corresponding 2-hydroxy arylacetic acid through O-allylation, ozonolysis and Wittig olefination.12 Using the 0.1 M HCl work up protocol generally high product er and dr was observed.15 Notable trends within this series showed that incorporation of halogen (4-FC6H48 and 4-ClC6H49) substituents, as well as electron-donating (4-MeOC6H410 and 4-MeC6H411) and 2-naphthyl substituents 15 gave the desired cis-chromenones in excellent enantioselectivity (97:3 to 98:2 er). Incorporation of electron-withdrawing 4-CF3C6H4or 3,5-(CF3)2C6H3 substituents was also tolerated, giving 12 with marginally reduced enantioselectivity and 13 in moderate 37% yield. Incorporation of an aliphatic enone led to decreased reactivity, requiring high catalyst loadings (20 mol%) to promote this transformation (29% isolated yield at 46% conversion), giving 14 as a single diastereoisomer in moderate 71:29 er.16 The relative and absolute configuration within 9 was unambiguously confirmed by X-ray crystal structure analysis,17 with the absolute configuration of all other products assigned by analogy.

The generality of this methodology was further investigated using different substituents within the aromatic tether (Table 4). Variation of the aromatic tether, incorporating substitution with electron-donating (5-Me, 4-OMe), halogen (4-F) and naphthyl groups gave cis-chromenones 16–22 with excellent enantioselectivity (95:5 to 98:2 er). Notably, incorporation of 4-OMe substituents on the aromatic tether (to give 17 and 21) showed decreased reactivity, with the reaction taking extended reaction times (12–14 h) to reach >98% conversion, but still proceeded with excellent enantioselectivity.
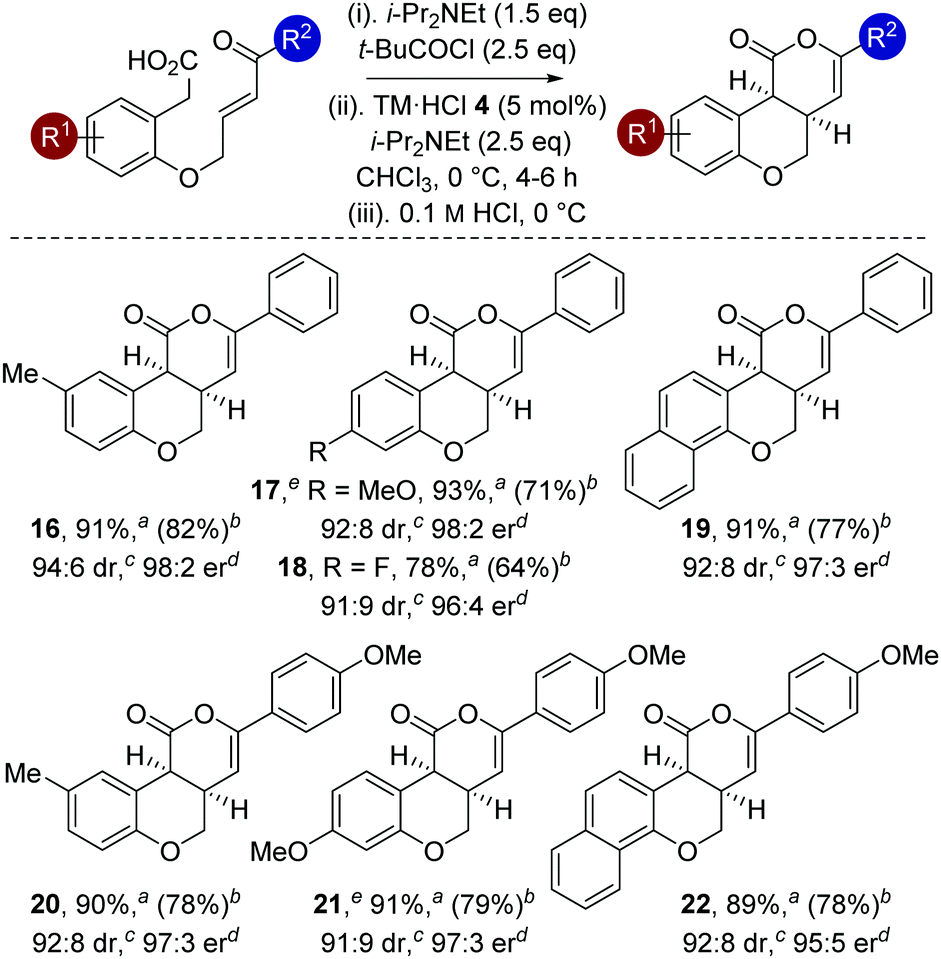
Reaction scale-up and subsequent product derivatization was investigated. On a one-gram scale, complete conversion of 1 to 2 was observed using only 2.5 mol% catalyst within 6 h to give 2 in 86% isolated yield as a single diastereoisomer and 98:2 er.
The synthetic utility of the products was then explored through a range of derivatizations (Scheme 2). Ring-opening of 2 with either methanol, morpholine or benzylamine gave the corresponding cis-dihydrobenzopyrans 23–25 in excellent yield, dr and er. Treatment of cis-chromenone 2 with Pd/C and H2 (1 atm) led to hydrogenation and hydrogenolysis, giving acid 26 in excellent yield. Alternatively, treatment of a recrystallized sample of 2 (>99:1 er) with m-CPBA, followed by p-TSA, gave the 5-membered lactone1827 in excellent yield and stereocontrol [96:4 dr, >99:1 er]. Recrystallization from 10% EtOAc in hexane gave 27 in >99:1 dr, >99:1 er and 82% yield. The relative and absolute configuration of 27 was confirmed by single crystal X-ray structure analysis.17
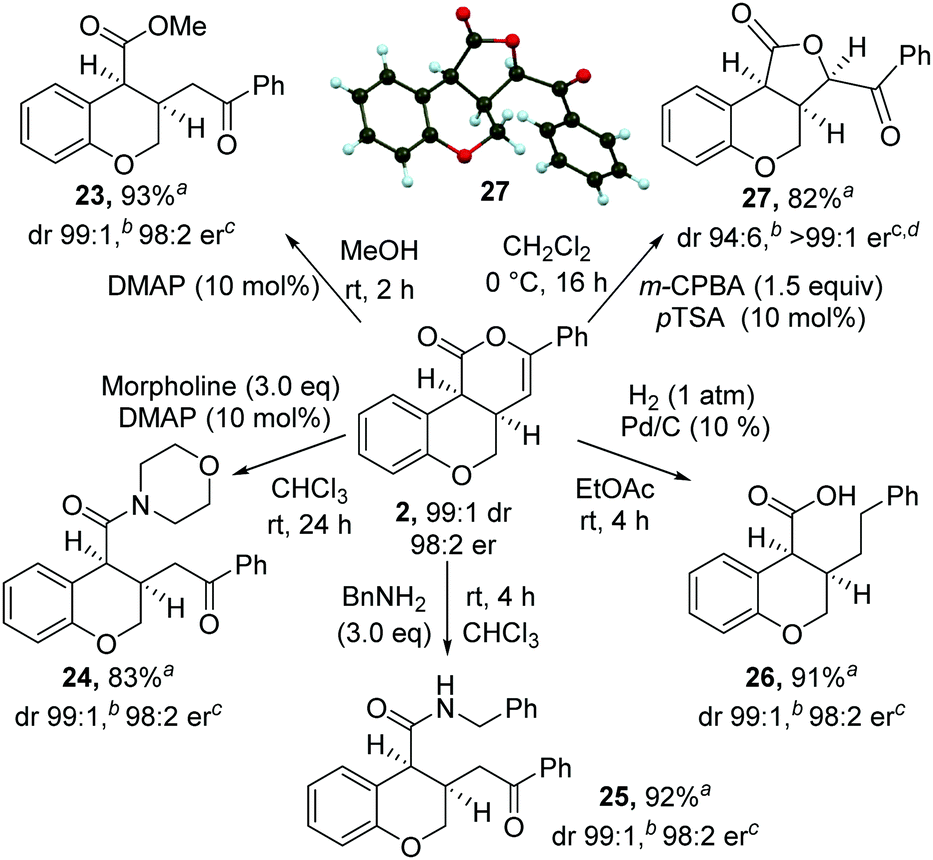 | ||
| Scheme 2 Product derivatization. aIsolated yield of major diastereoisomer (>99:1 dr). bMeasured by 1H NMR spectroscopy of crude reaction product. cMeasured by chiral HPLC (major cis-diastereoisomer). dStarting material 2 was >99:1 dr and 99:1 er. | ||
The mechanism of the isothiourea-catalyzed reaction, shown for the cyclization of enone-acid 1 to 2, is postulated to proceed via in situ formation of mixed anhydride 28 (Scheme 3). Nucleophilic addition of isothiourea 4 to 28 gives acyl isothiouronium ion intermediate 29, with deprotonation generating (Z)-ammonium enolate 30. Subsequent intramolecular 6-exo-trig Michael addition to the tethered enone generates intermediate 31, with lactonization giving cis-chromenone 2 and regenerating the catalyst 4. A simplistic model to rationalize the observed diastereo- and enantiocontrol utilizes a stabilising n0 to σC–S* interaction19 between the enolate oxygen and the sulfur of the isothiouronium ion to restrict the conformation of the (Z)-enolate,20 forcing the stereodirecting phenyl substituent to adopt a pseudoaxial orientation to minimize 1,2-strain. Subsequent 6-exo-trig Michael addition occurs anti- to this stereodirecting group as represented by pre-transition state assembly 32, with the two-prostereogenic centres along the developing C–C bond adopting a staggered array to minimize non-bonding interactions.
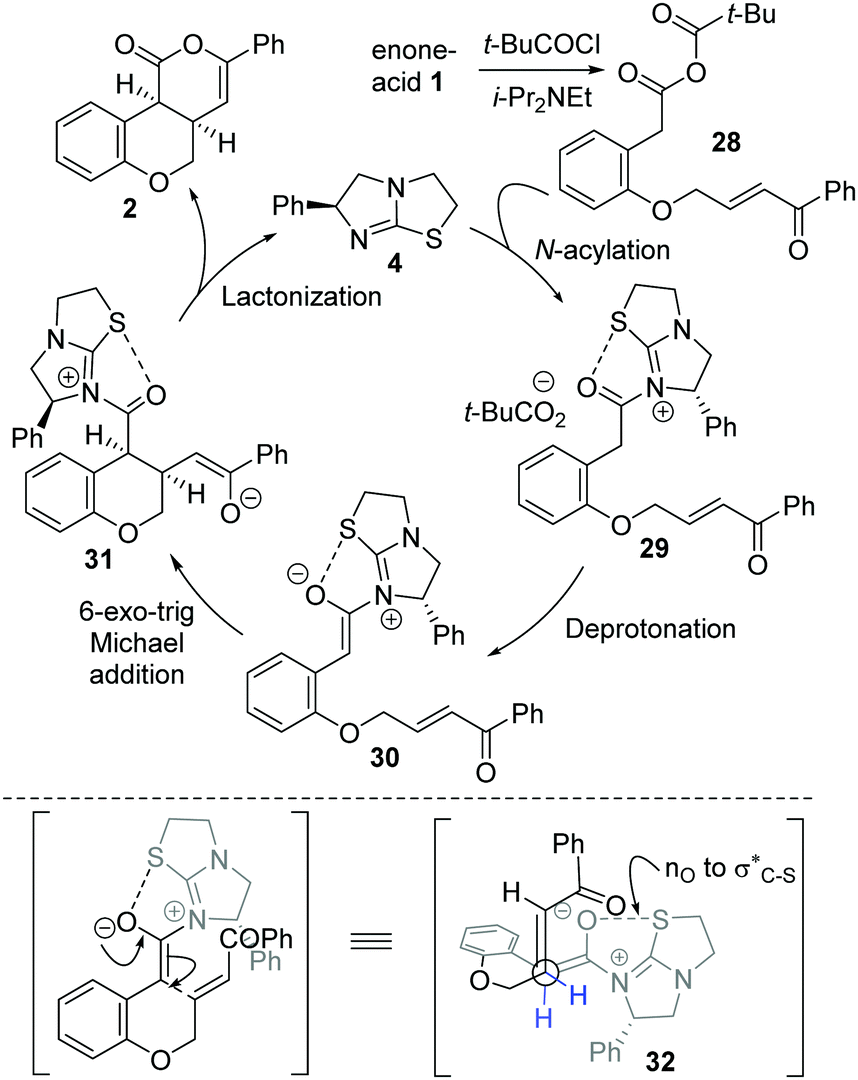 | ||
| Scheme 3 Proposed mechanism and stereochemical rationale. | ||
In conclusion, the catalytic enantioselective synthesis of cis-chromenones has been achieved using commercially available tetramisole as a catalyst. This method provides a range of cis-chromenone derivatives in high yield with excellent diastereo- and enantiocontrol (up to 99:1 dr and 98:2 er). On-going studies in this laboratory are focused on further applications of Lewis base organocatalysts in enantioselective catalysis.
We thank the EPSRC Centre for Doctoral Training in Critical Resource Catalysis (CRITICAT, grant code EP/L016419/1, RMNP) for funding. The European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013) ERC Grant Agreement No. 279850 is also acknowledged. ADS thanks the Royal Society for a Wolfson Research Merit Award. We also thank the EPSRC UK National Mass Spectrometry Facility at Swansea University.
Notes and references
- For select reviews see: (a) M. Álvarez-Corral, M. Muñoz-Dorado and I. Rodriguez-Garcia, Chem. Rev., 2008, 108, 3174–3198 CrossRef PubMed; (b) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed; (c) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed; (d) B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937–2980 CrossRef CAS PubMed.
- For select reviews see: (a) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156–1171 CrossRef CAS PubMed; (b) S. Ponra and K. C. Majumdar, RSC Adv., 2016, 6, 37784–37922 RSC; (c) L. M. Stanley and M. P. Sibi, Chem. Rev., 2008, 108, 2887–2902 CrossRef CAS PubMed; (d) J. Royer, M. Bonin and L. Micouin, Chem. Rev., 2004, 104, 2311–2352 CrossRef CAS PubMed.
- M. J. Gaunt and C. C. C. Johansson, Chem. Rev., 2007, 107, 5596–5605 CrossRef CAS PubMed.
- L. C. Morrill and A. D. Smith, Chem. Soc. Rev., 2014, 43, 6214–6226 RSC.
- G. S. Cortez, R. L. Tennyson and D. Romo, J. Am. Chem. Soc., 2001, 123, 7945–7946 CrossRef CAS PubMed.
- (a) Y. Yokota, G. S. Cortez and D. Romo, Tetrahedron, 2002, 58, 7075–7080 CrossRef CAS; (b) H. Henry-Riyad, C. Lee, V. C. Purohit and D. Romo, Org. Lett., 2006, 8, 4363–4366 CrossRef CAS PubMed; (c) G. Ma, H. Nguyen and D. Romo, Org. Lett., 2007, 9, 2143–2146 CrossRef CAS PubMed; (d) W. Zhang, A. S. Matla and D. Romo, Org. Lett., 2007, 9, 2111–2114 CrossRef CAS PubMed; (e) H. Nguyen, G. Ma, T. Gladysheva, T. Fremgen and D. Romo, J. Org. Chem., 2011, 76, 2–12 CrossRef CAS PubMed; (f) G. Liu and D. Romo, Angew. Chem., Int. Ed., 2011, 50, 7537–7540 CrossRef CAS PubMed; (g) C. A. Leverett, V. C. Purohit, A. G. Johnson, R. L. Davis, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2012, 134, 13348–13356 CrossRef CAS PubMed.
- (a) G. S. Cortez, S. H. Oh and D. Romo, Synthesis, 2001, 1731–1736 CrossRef CAS; (b) S. H. Oh, G. S. Cortez and D. Romo, J. Org. Chem., 2005, 70, 2835–2838 CrossRef CAS PubMed; (c) K. A. Morris, K. M. ArendtS., H. Oh and D. Romo, Org. Lett., 2010, 12, 3764–3767 CrossRef CAS PubMed; (d) D. Sikriwal and D. K. Dikshit, Tetrahedron, 2011, 67, 210–215 CrossRef CAS.
- For reviews on isothiourea catalysis (a) J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 RSC; (b) J. Merad, J.-M. Pons, O. Chuzel and C. Bressy, Eur. J. Org. Chem., 2016, 5589–5610 CrossRef CAS.
- For seminal work on isothiourea catalysis (a) X. Li and V. B. Birman, Org. Lett., 2006, 8, 1351–1354 CrossRef PubMed; (b) H. Jiang, X. Li, L. Guo, E. W. Uffman and V. B. Birman, J. Am. Chem. Soc., 2006, 128, 6536–6537 CrossRef PubMed; (c) M. Kobayashi and S. Okamoto, S, Tetrahedron Lett., 2006, 47, 4347–4350 CrossRef CAS; (d) X. Li and V. B. Birman, Org. Lett., 2008, 10, 1115–1118 CrossRef PubMed; (e) Y. Zhang and V. B. Birman, Adv. Synth. Catal., 2009, 351, 2525–2529 CrossRef CAS PubMed; (f) C. Joannesse, C. P. Johnston, C. Concellón, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914–8918 CrossRef CAS PubMed.
- (a) D. Belmessieri, L. C. Morrill, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2714–2720 CrossRef CAS PubMed; (b) D. Belmessieri, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2013, 15, 3472–3475 CrossRef CAS PubMed; (c) D. Belmessieri, A. de la Houpliere, E. D. D. Calder, J. E. Taylor and A. D. Smith, Chem. – Eur. J., 2014, 20, 9762–9769 CrossRef CAS PubMed; (d) D. G. Stark, P. Williamson, E. R. Gayner, S. F. Musolino, R. W. F. Kerr, J. E. Taylor, A. M. Z. Slawin, T. J. C. O’Riordan, S. A. Macgregor and A. D. Smith, Org. Biomol. Chem., 2016, 14, 8957–8965 RSC.
- For an overview see: (a) R. Pratab and V. J. Ram, Chem. Rev., 2014, 114, 10476–10526 CrossRef PubMed . For catalytic routes see: ; (b) J. Chen, J. Chen, F. Lang, X. Zhang, L. Cun, J. Zhu, J. Deng and L. Liao, J. Am. Chem. Soc., 2010, 132, 4552–4553 CrossRef CAS PubMed; (c) A. M. Hardman-Baldwin, M. D. Visco, J. M. Wieting, C. Stern, S. Kondo and A. E. Mattson, Org. Lett., 2016, 18, 3766–3769 CrossRef CAS PubMed.
- See ESI† for substrate synthesis, optimization and epimerization.
- V. B. Birman, X. Li and Z. Han, Org. Lett., 2007, 9, 37–40 CrossRef CAS PubMed and 9(c).
- Epimerization of the 80:20 trans:cis mixture gave exclusively cis-2 (>99:1 dr and 38:62 er (4aR,10bS:4aS,10bR)). See ESI† for details.
- To test the generality of the acidic work up to minimize the epimerization process, a number of reactions were quenched using either H2O at rt or 0.1 M HCl at 0 °C. See ESI† for full information.
- After work-up a 54:46 mixture of starting material acid enone and 14 was observed in the crude reaction mixture. We hypothesise that competitive deprotonation may retard reactivity in this case.
- Crystallographic data obtained for 9 and 27 has been deposited with the Cambridge Crystallographic Data Centre and the supplementary data can be found via CCDC 1510311 and 1510312 respectively.
- Z. Fu, X. Wu and Y. R. Chi, Org. Chem. Front., 2016, 3, 145–149 RSC.
- For 1,5-S⋯O interactions as a control element in isothiourea catalysis see (a) M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492–4495 CrossRef CAS PubMed; (b) P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288–3291 CrossRef CAS PubMed; (c) E. R. T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6916–6927 Search PubMed; (d) Y. Nagao, S. Miyamote, M. Miyamoto, H. Takeshige, K. Hayashi, S. Sano, M. Shiro, K. Yamaguchi and Y. Sei, J. Am. Chem. Soc., 2006, 128, 9722–9729 CrossRef CAS PubMed.
- For discussions on the origin of S⋯O interactions, see: (a) B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed; (b) X. Zhang, Z. Gong, J. Li and T. Lu, J. Chem. Inf. Model., 2015, 55, 2138–2153 CrossRef CAS PubMed; (c) R. C. Reid, M.-K. Yau, R. Singh, J. Lim and D. P. Fairlie, J. Am. Chem. Soc., 2014, 136, 11914–11917 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; characterization data for novel compounds; 1H and 13C NMR spectra, HPLC traces,12 and X-ray crystallographic data files compounds 9 and 27 (CIF). CCDC 1510311 and 1510312. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc10178j |
| This journal is © The Royal Society of Chemistry 2017 |