DOI:
10.1039/C6QI00120C
(Research Article)
Inorg. Chem. Front., 2016,
3, 1236-1244
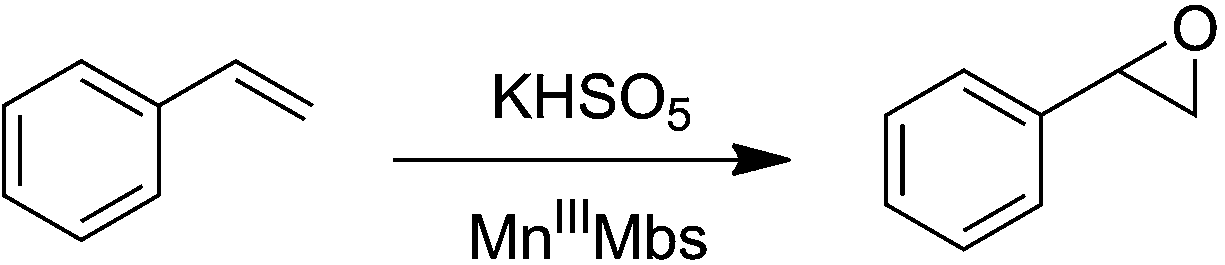
Manganese protoporphyrin IX reconstituted myoglobin capable of epoxidation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C bond with Oxone®†
C bond with Oxone®†
Received
9th May 2016
, Accepted 11th August 2016
First published on 12th August 2016
Abstract
Replacement of the native heme cofactor by manganese protoporphyrin IX (MnPPIX) to reconstitute manganese myoglobin (MnIIIMb) is an important approach to investigate the reactivity of the Mn center inside protein scaffolds. However, unlike the Mn porphyrin synthetic model compounds, MnPPIX reconstituted myoglobins (MnIIIMb) have no reactivity in the epoxidation of styrene using H2O2, which was attributed to the low reactivity of the MnIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediate after homocleavage of the O–O bond in manganese peroxide. To address this issue, we herein chose Oxone® (2KHSO5·KHSO4·K2SO4), a well-known oxidant undergoing O–O bond heterocleavage. After screening 7 mutants and wild-type MnIIIMb, we found that the L29H/F43H mutant could generate a new species ([MnIVO]+˙), tentatively assigned by using UV-vis and EPR spectra, through heterocleavage of the O–O bond. Computational docking showed hydrogen bonds between three distal histidines (H64, L29H and F43H) and anions, which increase the binding affinity to persulfate. With Oxone® as the oxidant, MnIIIMb (L29H/F43H) showed the highest reactivity toward the epoxidation of styrene, different from that with the H2O2 oxidant. This work demonstrates the first example of MnPPIX reconstituted Mb which could catalyze styrene epoxidation and provides new insights to further explore the reactivity of the Mn center in protein scaffolds.
O intermediate after homocleavage of the O–O bond in manganese peroxide. To address this issue, we herein chose Oxone® (2KHSO5·KHSO4·K2SO4), a well-known oxidant undergoing O–O bond heterocleavage. After screening 7 mutants and wild-type MnIIIMb, we found that the L29H/F43H mutant could generate a new species ([MnIVO]+˙), tentatively assigned by using UV-vis and EPR spectra, through heterocleavage of the O–O bond. Computational docking showed hydrogen bonds between three distal histidines (H64, L29H and F43H) and anions, which increase the binding affinity to persulfate. With Oxone® as the oxidant, MnIIIMb (L29H/F43H) showed the highest reactivity toward the epoxidation of styrene, different from that with the H2O2 oxidant. This work demonstrates the first example of MnPPIX reconstituted Mb which could catalyze styrene epoxidation and provides new insights to further explore the reactivity of the Mn center in protein scaffolds.
Introduction
Manganese (Mn) is an important redox cofactor widely found in many metalloproteins or metalloenzymes such as the oxygen evolution cluster (OEC) in photosynthetic system II,1,2 superoxide dismutases (SOD),3 and catalases.4 In sharp contrast to iron (Fe), few Mn protoporphyrin complexes have been used as cofactors in naturally redox metalloproteins, although their synthetic analogues such as Mn(III) and Fe(II) porphyrins displayed comparable catalytic reactivity in oxygen atom transfer reactions.5–10 To decipher their different reactivities inside protein scaffolds, replacing the heme cofactor with Mn analogues in heme containing metalloproteins thus became an emerging approach to deepen the understanding of the structural relationship of redox reactivity, expand the repertoire of metalloenzyme catalyzed reactions and even enrich our knowledge of nature's choice for metal cofactors.
Toward this goal, Mn protoporphyrin IX (MnPPIX) reconstituted heme containing metalloproteins, including horseradish peroxidase,11–13 cytochrome c peroxidase,14,15 microperoxidase-8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16–19 and myoglobin,20–27 have been extensively studied. Especially inside myoglobin, the comparative studies of the structure,20 electrochemical properties,21,28 Raman spectra,23 electron paramagnetic resonance (EPR),24,29 and redox reactivity22,25–27,30 of MnPPIX with heme provided valuable insights to understand the relationship between the intrinsic electronic structures and the redox reactivities. Prominently, Watanabe,31 Hayashi32 and Lu33–35 reported that replacement of heme with Mn porphycene or salen complexes afforded new Mn myoglobins capable of catalyzing oxygen atom transfer reactions even in good enantioselectivity, which revealed the potential applications of Mn reconstituted myoglobins in organic catalysis. Despite the tremendous progress made in the past few years, most MnPPIX reconstituted metalloproteins exhibited peroxidase like reactivity,36–38 other than the reactivity in oxygen atom transfer reactions such as epoxidation of the CC bond, sulfoxidation and hydroxylation of C–H bonds.39 To address this issue, we herein reported the first example of MnPPIX reconstituted myoglobin (L29H/F43H), capable of catalyzing epoxidation of the CC bond (styrene) with Oxone®.
16–19 and myoglobin,20–27 have been extensively studied. Especially inside myoglobin, the comparative studies of the structure,20 electrochemical properties,21,28 Raman spectra,23 electron paramagnetic resonance (EPR),24,29 and redox reactivity22,25–27,30 of MnPPIX with heme provided valuable insights to understand the relationship between the intrinsic electronic structures and the redox reactivities. Prominently, Watanabe,31 Hayashi32 and Lu33–35 reported that replacement of heme with Mn porphycene or salen complexes afforded new Mn myoglobins capable of catalyzing oxygen atom transfer reactions even in good enantioselectivity, which revealed the potential applications of Mn reconstituted myoglobins in organic catalysis. Despite the tremendous progress made in the past few years, most MnPPIX reconstituted metalloproteins exhibited peroxidase like reactivity,36–38 other than the reactivity in oxygen atom transfer reactions such as epoxidation of the CC bond, sulfoxidation and hydroxylation of C–H bonds.39 To address this issue, we herein reported the first example of MnPPIX reconstituted myoglobin (L29H/F43H), capable of catalyzing epoxidation of the CC bond (styrene) with Oxone®.
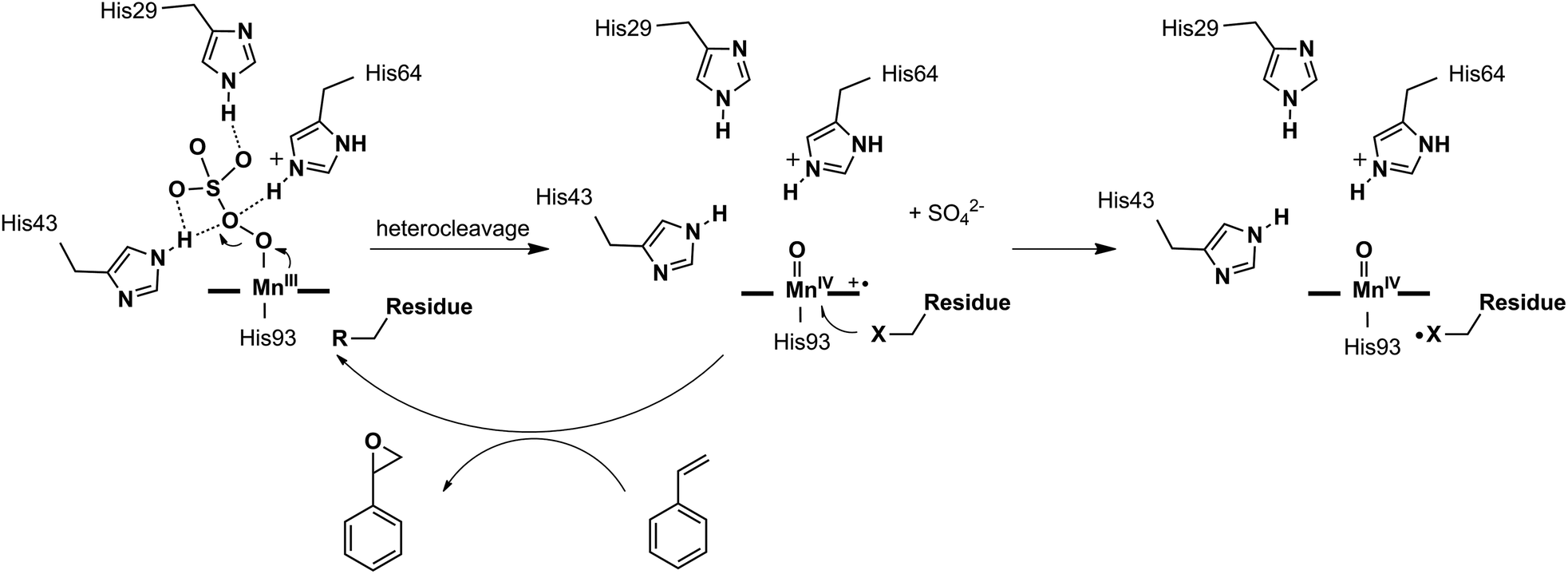
Different reactivities between the active intermediates [FeIVO] (less active, Comp. II) and [FeIVO]+˙ (more active, Comp. I) have been found in myoglobins, which are generated through homo- and heterocleavage of the O–O bond in iron(III) peroxide, respectively. Especially for the latter, the [FeIVO]+˙ species was believed to be the real active intermediate in many catalytic oxygen atom transfer reactions.40–43 However, MnPPIX had been reported to generate a low active [MnIVO] intermediate inside the myoglobin scaffold, which could not oxidize styrene but ABTS, with typical reactivity like peroxidases.38 Thus, we hypothesized that the intermediate [Mn-OOH] inside myoglobin tends to undergo homocleavage of the O–O bond. Thus, looking for an appropriate oxidant to produce active [MnVO] or [MnIVO]+˙ intermediate(s) through heterocleavage of the O–O bond is of importance. To test this hypothesis, we herein chose Oxone® as the oxidant, because it is well-known to react with the Mn center through heterocleavage of the O–O bond.5,6,44–47 Moreover, Oxone® has low pKa and exists in the anionic form under physiological conditions, which is favorable to bind the Mn cation center. To optimize the secondary coordination sphere, we designed several mutants (L29H, F43H, H64F, L29H/H64F, F43H/H64F, L29H/F43H and L29H/F43H/H64F). As shown in UV-vis spectroscopic titration, only the L29H/F43H mutant (His3 MnIIIMb) showed the formation of the Mn-oxo type intermediate with a broad absorption centered at ca. 623 nm, which might be arising from the MnIV-oxo cation radical. EPR studies for the reaction between His3 MnIIIMb and Oxone® showed both the radical signal at g = 2.00 and the MnIV signal at g = 4.30. These results clearly suggested the generation of different Mn-oxo intermediates in Mb (L29H/F43H) between Oxone® and H2O2. Computational docking of the HSO5− ion to the cavity of His3 Mb (PDB code: 4FWZ)48 demonstrates that three distal histidines bind the persulfate anion through hydrogen bonds, which might increase the binding affinity as well as promote the O–O bond cleavage. Interestingly, the reactivity assay for epoxidation of styrene demonstrated that MnIIIMb (L29H/F43H) with Oxone® has the highest reactivity (48 ± 2 × 10−3 turnover per min) and the main product is styrene epoxide. Therefore, this work provides an access to understand the oxidation mechanism for MnPPIX inside myoglobin scaffolds and explore the potential application in oxygen atom transfer reactions.
Results and discussion
Reactions of MnIIIMbs and Oxone®
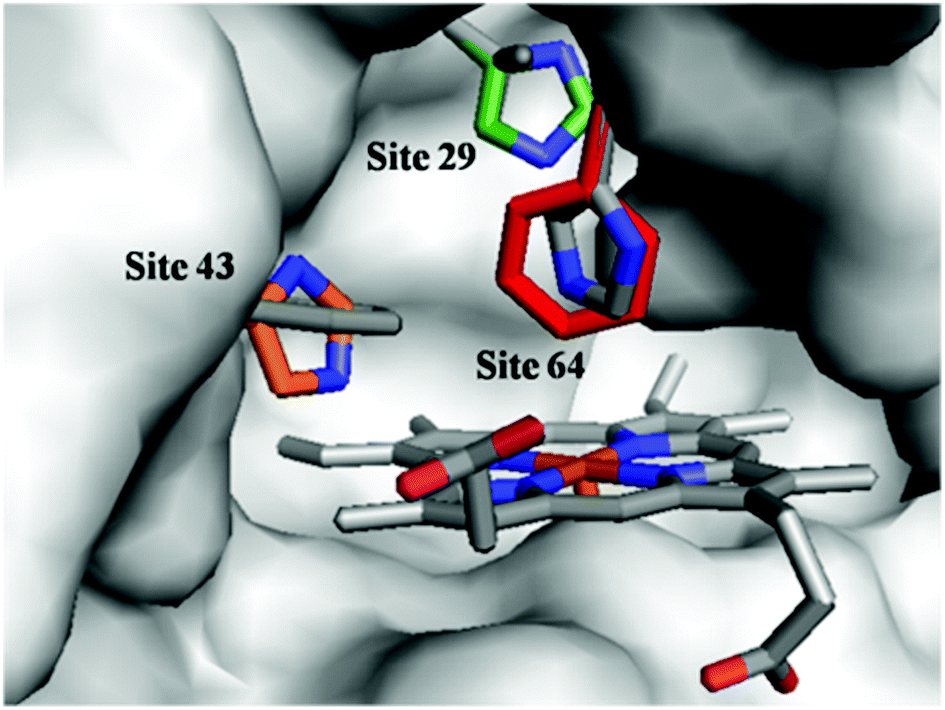
To optimize the secondary coordination sphere for the reaction between the Mn center and Oxone®, we herein focused on the different combinations of distal histidines (Fig. 1 and Table 1) such as Leu-29, Phe-43, and His-64, according to our and other's previous reports.33,38,41,49 Construction of these Mn Mb mutants was carried out following our previous procedures, and UV-vis absorption and mass spectra were identical.38
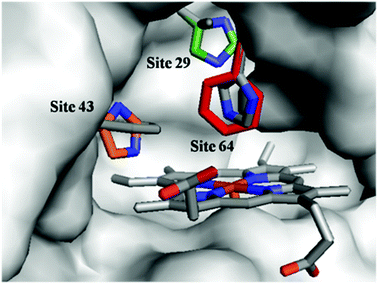 |
| | Fig. 1 Structures of the heme pocket of sperm whale myoglobin (gray). And the residues mutated in our studies are also shown: His-29 (green), His-43 (orange), and Phe-64 (red). All N atoms are marked in blue. The PDB code for wild-type Mb is 1JP6. Structures of mutants were reproduced by PyMol. | |
Table 1 Residues at sites 29, 43, and 64 of wild-type Mb and different mutants
| Myoglobin mutants |
Residues |
| Site 29 |
Site 43 |
Site 64 |
| Wild-type |
Leu |
Phe |
His |
| F43H |
Leu |
His |
His |
| L29H |
His |
Phe |
His |
| H64F |
Leu |
Phe |
Phe |
| L29H/F43H (His3) |
His |
His |
His |
| F43H/H64F |
Leu |
His |
Phe |
| L29H/H64F |
His |
Phe |
Phe |
| L29H/F43H/H64F |
His |
His |
Phe |
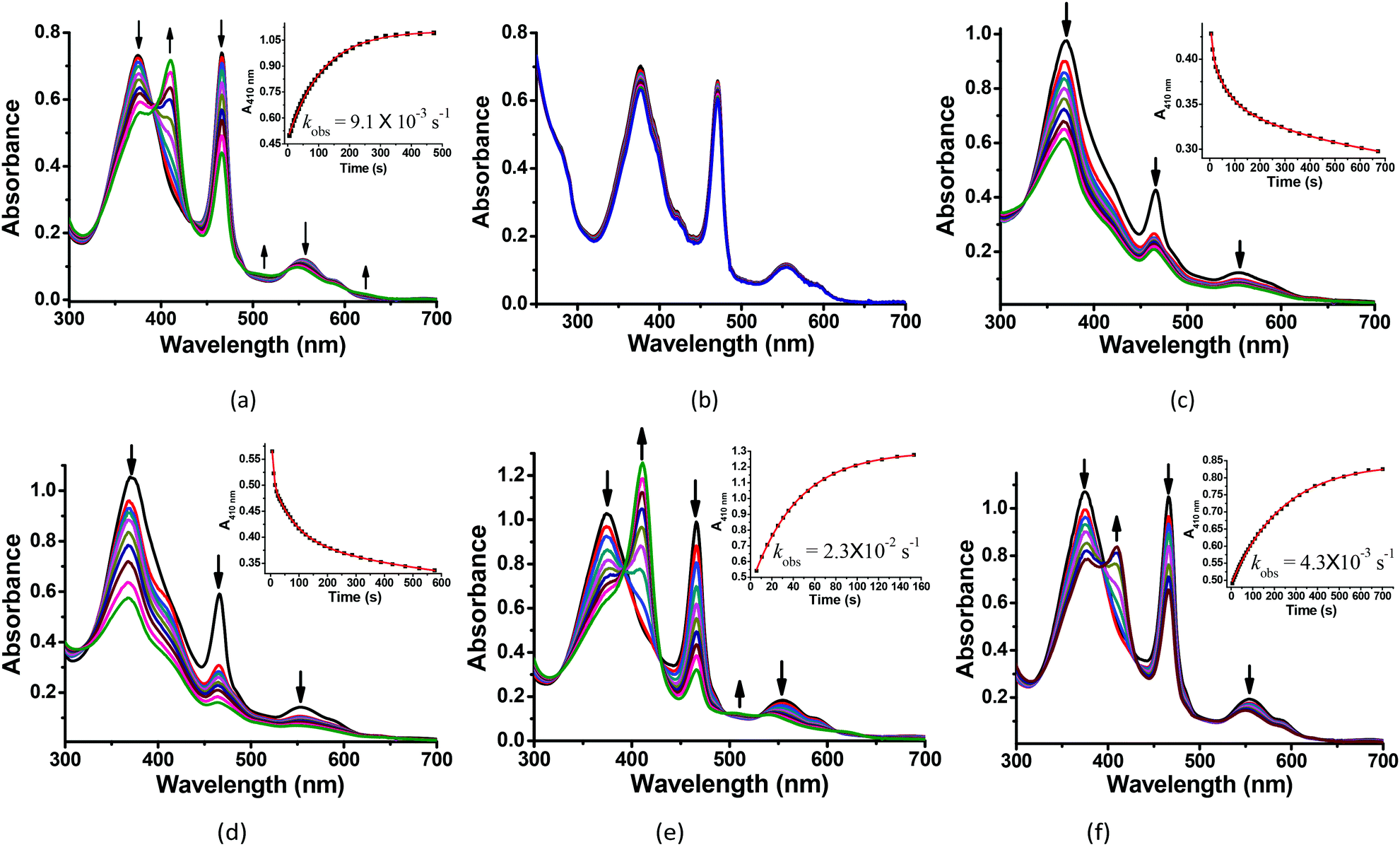
The reactions of MnIIIMbs with 100 eq. Oxone® (containing 200 eq. KHSO5) were carried out and monitored by UV-vis absorption spectroscopy in 50 mM potassium phosphate buffer (pH = 7.4) at 20 °C. As shown in Fig. 2a, b and S2–7,† only the L29H/F43H mutant (His3 MnIIIMb) displayed a decrease in the absorption at 376, 471 and 550 nm, and an appearance of a single Soret band at 411 nm. In Fig. 2a, the reaction between His3 MnIIIMb and Oxone® resulted in nearly isosbestic conversion in the initial stage (0–500 s) and the kinetic trace obeyed pseudo first-order kinetics. This clearly indicates that the combination of three distal histidines (L29H, F43H and H64) is more effective to activate the reaction with Oxone® compared to the other mutants and wild-type MnIIIMb. Notably, a broad shoulder centered at 623 nm appeared, indicating the formation of the MnIV porphyrin π cation radical according to those found in some synthetic porphyrinoid ligands.50–52 In our previous work using H2O2 to oxidize His3 MnIIIMb,38 no such broad shoulder at ca. 623 nm was observed. It is worth noting that Stone and co-workers reported similar spectral changes when they used m-CPBA as the two-electron oxidant.30 To confirm that the new species is Oxone® dependent, we reduced the amount of Oxone® to 20 equiv. (Fig. S1†) and found that a smaller amount of new species formed with a slower reaction rate (kobs) of 2.8 × 10−3 s−1 (vs. 9.1 × 10−3 s−1 with 200 equiv. KHSO5, reaction time: 0–500 s). This suggested the generation of an intermediate like the MnIV-oxo cation radical or the protein radical using the Oxone® oxidant.
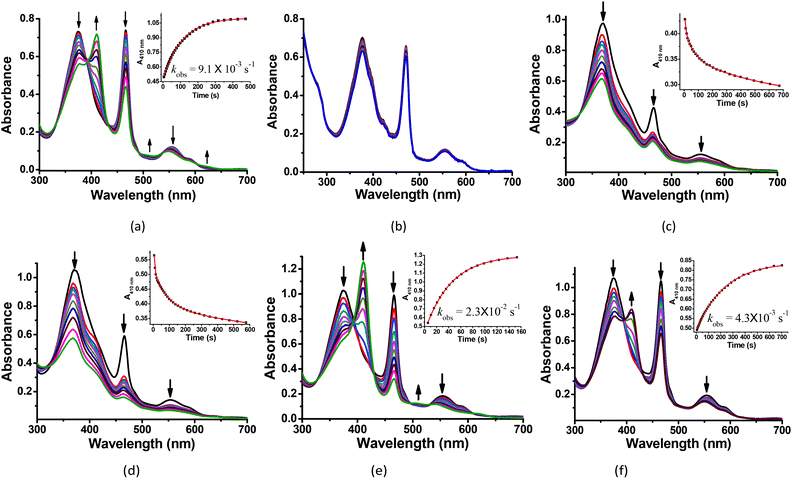 |
| | Fig. 2 Kinetic studies of 30 μM (a) His3 MnIIIMb and (b) wild-type MnIIIMb reacted with 200 equiv. KHSO5 in PBS buffer (pH = 7.4). And 30 μM His3 MnIIIMb reacted with 200 equiv. KHSO5 at (c) pH = 5.1, (d) pH = 6.0, (e) pH = 7.0, and (f) pH = 8.0. Inset: Time course plots of absorbance at ca. 410 nm, attributing to the formation of the MnO intermediate. | |
We then investigated the pH effect (pH 5.1, 6.0, 7.0 and 8.0) on the reaction between Oxone® and His3 MnIIIMb, and UV-vis spectra are shown in Fig. 2c–f. At low pH values (5.1 and 6.0), UV-vis spectra showed fast disappearance of Soret and Q bands, with a slight increase of absorption at 411 nm. This might be due to MnPPIX bleaching or slipping out of the protein scaffold. We ascribed the low stability to the protonation of distal histidines (pKaca. 6.0), which likely alters the H-bonding interactions in the distal pocket or even the configuration of the protein scaffold. In contrast to that, higher pH values (7.0, 7.4 and 8.0) showed the typical UV-vis spectra of the MnIV-oxo cation radical with a negligible absorption decrease, indicating that high valent [MnVO] or [MnIVO]+˙ species may be stabilized under basic conditions.53 When pH ≥ 7, the reaction rates (first order kinetic rate) also decreased along with the increase of pH. Under the incubation with 6 mM Oxone, His3 MnIIIMb displays much faster (2.3 × 10−2 s−1 at 20.0 °C) at pH 7 than those at higher or lower pH.
Docking HSO5− to His3 Mb
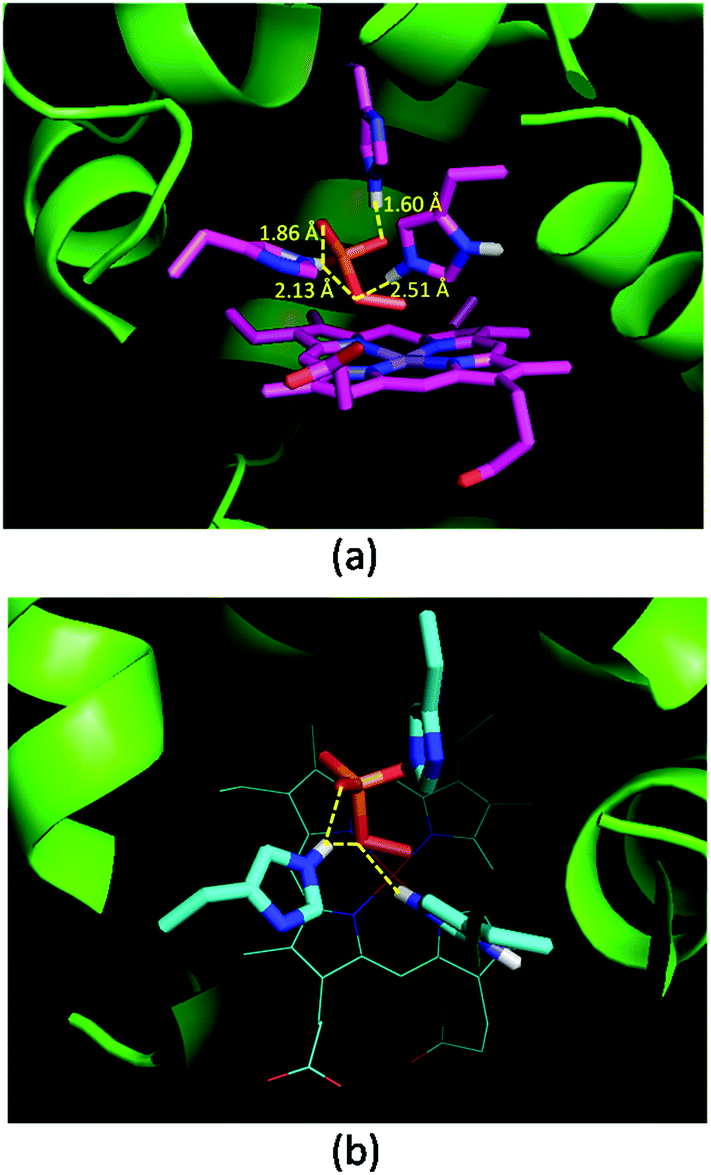
Oxone® is a mixture of KHSO5·0.5KHSO4·0.5K2SO4 and the active species is potassium hydrogen persulfate (KHSO5). To understand the role of distal histidines (L29H, F43H and H64) in activating Oxone®, we carried out a computational study for docking HSO5− to the His3 cavity above the MnPPIX in myoglobin. The model myoglobin we chose is His3 Mb (PDB code: 4FWZ) reported by Lu and co-workers.48 We replaced the heme cofactor by the MnPPIX complex and removed water molecules in the crystal data. For the low pKa of hydrogen persulfate, the proton was supposed to transfer to His-64. As shown in Fig. 3, hydrogen bonding interactions are observed between terminal oxygens and His-29 and His-43, with the distance of 1.60 and 1.86 Å, respectively. The proximal oxygen in hydrogen persulfate could form hydrogen bonds with His-43 and protonated His-64, with the distance of 2.13 and 2.51 Å, respectively. The distance between the H atom of His64 and the O atom of hydrogen persulfate proximal to the Mn center is 3.15 Å, which is a little longer than the normal hydrogen bond. And the N–H in His64 was directly towards the uncoordinated O atom. Thus, we considered that the H-bond was only between His-64 and the distal O atom of peroxide. Thus, this docking experiment tentatively interprets that three distal histidines facilitate HSO5− binding to the cavity above MnPPIX through cooperative hydrogen bonding, which might assist the consequent O–O bond heterocleavage.
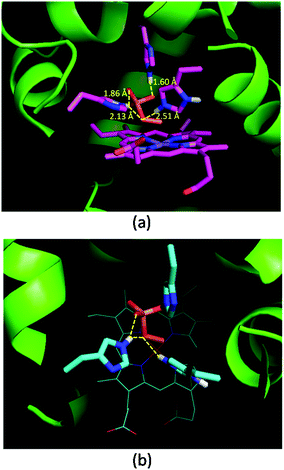 |
| | Fig. 3 Docking results of His3 MnIIIMb and HSO5−. (a) Side view, (b) top view. Hydrogen bonds and distances of three histidines and oxygens in HSO5− are labelled in (a). | |
EPR studies for the reaction of His3 MnIIIMb and Oxone®
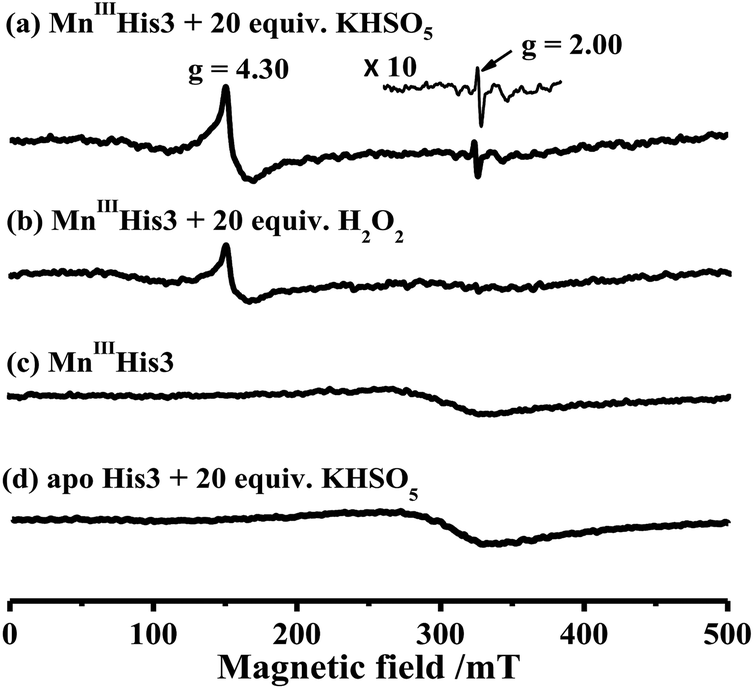
To characterize intermediates in the reaction between Oxone® and His3 MnIIIMb, we carried out EPR experiments to monitor the reaction process according to the work by Yonetani54 and Hori.55 In this study, we used H2O2 as a control. 10 eq. Oxone® (containing 20 eq. KHSO5) was added to the solution of His3 MnIIIMb (1.0 mM) in PBS buffer (pH 7.4). The reaction lasted for 3 min at 4 °C, and then quickly frozen with liquid nitrogen. As shown in Fig. 4a, His3 MnIIIMb reacted with KHSO5 and showed a MnIV signal at g = 4.30 and a characteristic sharp signal at g = 2.00 which was assigned to the protein radical or porphyrin π cation radical. Similarly, 20 eq. of H2O2 was used with the same procedure, and only the MnIV signal at g = 4.30 was observed (Fig. 4b), consistent with the previously reported homocleavage of the O–O bond of Mn peroxide.27,38,56 As shown in Fig. 4c, in the absence of oxidant, no signal was observed at both g = 2.00 and 4.30.
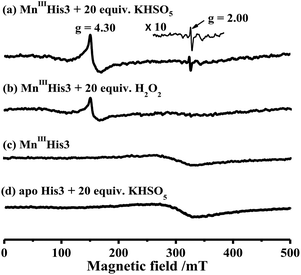 |
| | Fig. 4 X-band EPR spectra of (a) 1.0 mM His3 MnIIIMb in 50 mM PBS buffer (pH 7.4), and (b) in the presence of 20 equiv. H2O2 or (c) 20 equiv. KHSO5, and (d) apo-His3 Mb in the presence of 20 equiv. KHSO5. The EPR spectra are recorded at −170 °C. | |
In addition, the reaction between Oxone® and apo-myoglobin did not afford such a radical signal at g = 2.00 (Fig. 4d). Thus, the signal at g = 2.00 could be assigned to either protein radicals such as Tyr and Trp through one-electron oxidation11 or porphyrin π cation radicals. Nevertheless, these EPR results clearly indicate the different Mn-oxo species generated by Oxone® from that by H2O2.
Post-oxidation of protein scaffold by Oxone®
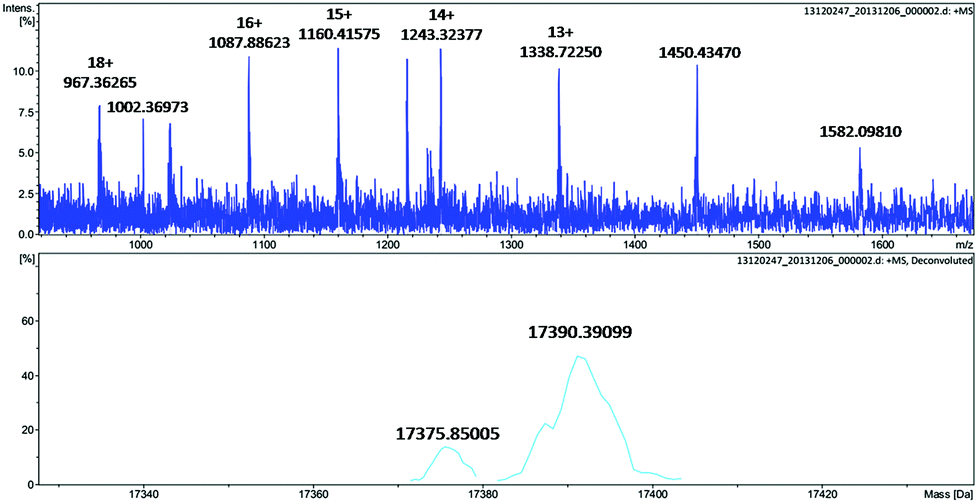
To further identify the oxidation of the protein scaffold by Oxone®, we performed ESI-MS spectrometry to characterize the molecular weight immediately after the reaction of Oxone® and His3 MnIIIMb for 3 min. The fitting results show two peaks at 17376 and 17390 Da (Fig. 5), which correspond to the addition of 2 and 3 oxygen atoms to the His3 Mb (M = 17345 Da), respectively. This indicates that the protein scaffold was oxidized with the addition of 2 or 3 oxygen atoms using 20 eq. Oxone®. The reaction of Oxone® with apo-myoglobin was used as control (Fig. S8†) and no oxidized protein was found in ESI-MS, indicating that the oxidation of the protein scaffold by KHSO5 only occurs in the presence of the MnPPIX cofactor.
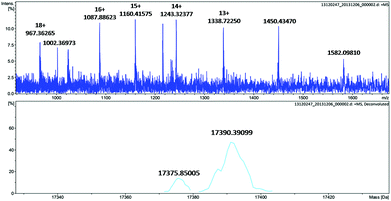 |
| | Fig. 5 ESI-MS of His3 MnIIIMb after reacting with 20 equiv. KHSO5 for 3 min. | |
We then used LC-MS/MS to identify the oxidized amino acid residues. His3 MnIIIMb was reacted with Oxone® for 3 min. H2O2 was used as a control oxidant, and apoprotein was taken as a blank to exclude the possible oxidation occurring in LC-MS/MS treatments. The normalized peak areas were calculated by the ratio of oxidized/unoxidized peptides, which was used to estimate the degree of oxidization. As shown in Tables 2 and 3 peptides were found to be oxidized by KHSO5. We found that the fragmentation patterns of the peptide were HPGDFGADAQGAM(Ox)NK, as shown in Fig. S9 and 10.† According to the peptide sequences, oxidation mainly occurred at Met-55, Met-131 and Tyr-151. It's noteworthy that reacting with H2O2 or apo-His3 Mb in the presence of KHSO5 does not generate remarkable oxidations. They only show slightly increasing contents on Met-55. This indicates that a quick post-oxidation by a high reactive Mn-oxo intermediate with Oxone® could oxidize the residues inside the protein scaffold, which is in accordance with the EPR experimental results. These three residues were distant from the Mn center and localized near the surface, which was probably because of the radical transfer through some residues from the cavity to the surface.
Table 2 Comparison of oxidized peptides found in trypsin autolysis products
| Peptides |
Observed m/z |
Theoretical m/z |
Delta |
Normalized Area |
| Apo-His3 Mb + Oxone® |
His3 MnIIIMb |
His3 MnIIIMb + Oxone® |
His3 MnIIIMb + H2O2 |
HPGDFGADAQGA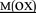 NK NK |
766.52 |
766.81 |
−0.29 |
3.2 × 106 |
4.6 × 106 |
1.1 × 107 |
3.9 × 106 |
HLKTEAE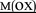 K K |
368.38 |
368.43 |
−0.05 |
3.8 × 104 |
3.6 × 104 |
1.2 × 106 |
4.7 × 104 |
YKELG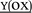 QG QG |
487.37 |
487.53 |
−0.16 |
1.5 × 106 |
1.5 × 106 |
4.3 × 106 |
1.6 × 106 |
Table 3 Reaction rate of different proteins catalyzed by styrene epoxidationa
| Entry |
Catalyst |
Rateb |
|
Reaction conditions: 0.2 mM proteins, 4 mM styrene and KHSO5 in 50 mM PBS buffer (pH = 7.4) at 4 °C, reacting for 1 h.
The unit of rate is 10−3 turnover per min.
The epoxidation product was not detected.
Reaction with H2O2 instead of KHSO5 as oxidants.
|
| 1 |
No catalyst |
N.D.c |
| 2 |
MnPPIX |
N.D.c |
| 3 |
Apo-His3 Mb |
N.D.c |
| 4 |
His3 MnIIIMb |
48 ± 2 |
| 5 |
His3 FeIIIMb |
15 ± 2 |
| 6 |
F43H MnIIIMb |
11 ± 1 |
| 7 |
Wild-type MnIIIMb |
5.4 ± 0.4 |
| 8 |
His3 MnIIIMb (with H2O2)d |
N.D.c |
Epoxidation activity
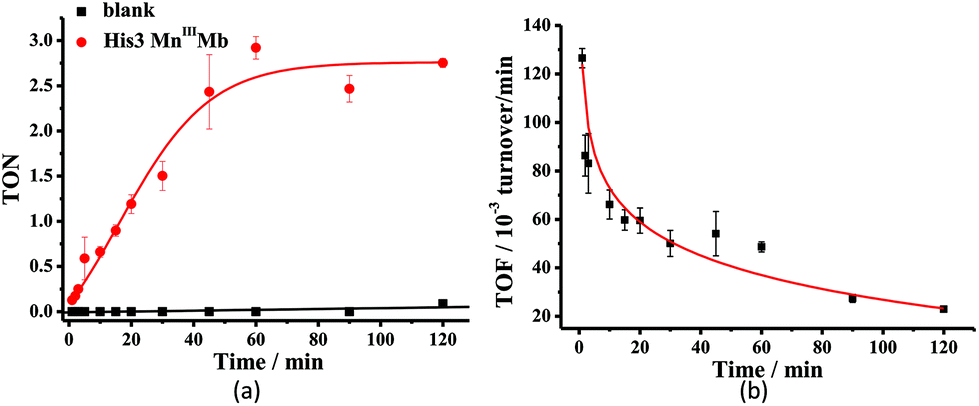
Mn porphyrins are well known catalysts in oxygen-atom transfer (OAT),6–10 hydrogen-atom transfer (HAT),57,58 and electron transfer (ET) reactions,59,60 and MnV-oxo or/and [MnIVO]+˙ species were proposed as active intermediates. Since the reaction of Oxone® and Mn His3 Mb could generate more active Mn-oxo species, it is very important to investigate the reactivity toward oxygen atom transfer reactions. In this work, we chose styrene as the substrate, and Oxone® as the oxidant to examine the catalytic reactivity of these MnIIIMbs (Scheme 1, Table 3). As shown in Fig. S11,† styrene epoxide is the main product. And the time course experiment of styrene epoxidation catalyzed by His3 MnIIIMb (Fig. 6a) shows that the reaction ends in about 1 h. The initial TOF (turnover frequency) is up to 1.30 × 10−1 under the same conditions. Changing Oxone® to H2O2, His3 MnIIIMb showed no reactivity toward epoxidation of styrene (entry 8).
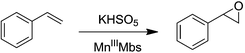 |
| | Scheme 1 Styrene epoxidation by KHSO5. | |
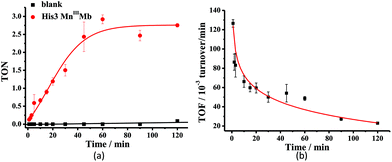 |
| | Fig. 6 Time course plots of (a) turnover number (TON) with or without His3 MnIIIMb, and (b) turnover frequency (TOF) of styrene epoxidation catalyzed by His3 MnIIIMb. TON and TOF were calculated based on the amount of styrene. The initial TOF was calculated at 1 min. | |
Among the MnIIIMb mutants and wild-type MnIIIMb, His3 MnIIIMb shows the highest reaction rate (48 ± 2 × 10−3 turnover per min, entry 4,) with Oxone® as the oxidant, which is 8.9 times higher than wild-type MnIIIMb (5.4 ± 0.4 × 10−3 turnover per min, entry 7), 4.4 times higher than F43H MnIIIMb (11 ± 1 × 10−3 turnover per min, entry 6), and even 3.2 times higher than His3 FeIIIMb (15 ± 2 × 10−3 turnover per min, entry 5). Other mutants are not reactive toward epoxidation of styrene under these reaction conditions. The trend observed in the epoxidation of styrene is similar to that in the UV-vis spectra for the reactions between MnIIIMbs and Oxone®, suggesting the correlation between the formation of Mn-oxo intermediate(s).
We also investigated the pH effect on the epoxidation of styrene, since the formation of MnIV/V-oxo was pH dependent. As shown in Fig. S12,† at pH 7.0 and pH 7.4, the reactivity toward the epoxidation of styrene is much higher than those obtained at lower pH (<7) or higher pH (>7.4), which is quite in line with the UV-vis studies for pH effects. In addition, we examined the enantioselectivity of styrene epoxide catalyzed by His3 MnIIIMb and low enantioselectivity (<10%) was detected. Finally, we used ethylbenzene as the substrate to test the reactivity of His3 MnIIIMb and Oxone® and no oxidation of the C–H bond was obtained. Nevertheless, the reactivity of His3 MnIIIMb and Oxone® toward styrene epoxidation reveals the potential of this protocol for further application in oxygen atom transfer reactions.
The possible reaction mechanism
In this work, we demonstrated the ability of Mn reconstituted Mb to oxidize the CC bond using Oxone® as the oxidant. We proposed the possible mechanism of the generation and subsequent reactions of the Mn-oxo intermediate as in Scheme 2. The coordination of HSO5− ions to Mn was stabilized by hydrogen bonds with His-29, His-43, and His-64. Then the MnV-oxo intermediate was generated by O–O bond heterocleavage. This highly active intermediate could oxidize substrates such as styrene, or oxidized residues like tyrosine to form protein radicals. On the other hand, distal histidine plays a critical role in activating Oxone® as we mentioned above. Previously, Watanabe and co-workers revealed that the location of single distal histidine affects the activation process of H2O2 in myoglobin.41 Recently, we reported the cooperative effect of dual distal histidines at positions 64 and 43 on the activation of H2O2 with the Mn center within the myoglobin scaffold, which might be due to the higher pKa of H2O2 binding to Mn than that of the Fe center.38 For the effects of three distal histidines, Lin and co-workers reported that Cu binding to His3 Mb inhibits the peroxidase activity, which thus suggests their essential role in the activation of the oxidant.61 In this work, we demonstrated the essential role of three distal histidines in activating Oxone, a two-electron oxidant, via heterocleavage of the O–O bond. This is in line with the previous studies and highlights the importance of distal histidines in designing artificial metal oxidases. Thus, the next step to improve this catalytic system would include how to avoid the decomposition of MnPPIX and self-oxidation of protein scaffolds, which are further important to enhance the reactivity and reaction turnover numbers.
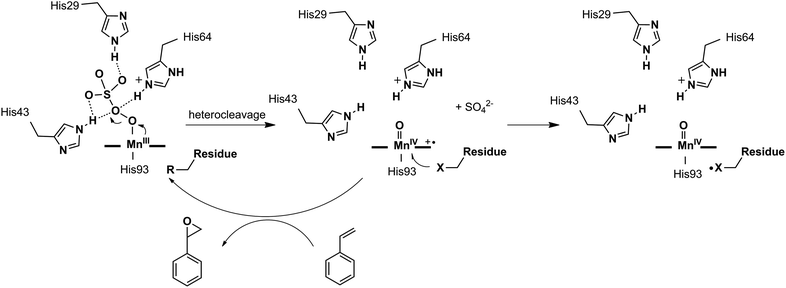 |
| | Scheme 2 Proposed mechanism of the formation of the Mn-oxo intermediate, styrene epoxidation, and oxidation of protein amino acid residues. | |
Conclusions
Altogether, we described the first example of design of manganese reconstituted myoglobin, which is reactive toward oxygen atom transfer reactions with Oxone® as the oxidant. There are two critical factors to determine this kind of reactivity. One is to find an appropriate oxidant, which undergoes heterocleavage of the O–O bond of Mn peroxide to generate more active [MnIVO]+˙ intermediate(s) other than the low reactive MnIVO intermediate generated by H2O2. The next is the secondary coordination sphere, which is constructed by three distal histidines (L29H, F43H and H64) in this work. Compared to our previous work in which two histidines (H64 and F43H) could bind H2O2 and deprotonate proximal hydrogen and generate MnIVO,38 the activation of Oxone® required three distal histidines to form a cooperative hydrogen bond with the terminal oxygen atom. Obviously, more charge separation between the O–O bond is favourable to such bond hetero-cleavage. More importantly, His3 MnIIIMb has the ability to oxidize styrene, which reveals the perspective on engineering metalloproteins by changing metal cofactors and tuning the secondary coordination sphere, and brings new insights into design of more effective metalloproteins with functions not found in nature.
Materials and methods
General
All chemicals including protoporphyrin IX, Oxone® and styrene were purchased from Alfa Aesar and used without further purification. Manganese(III) protoporphyrin IX and different Mbs were prepared according to the previous reports.38 ESI-MS spectra were recorded with a Bruker Apex IV FTMS spectrometer. LC-MS/MS was performed on a LTQ Velos Pro dual-pressure linear ion trap liquid Chromatography Mass Spectrometer. UV-vis spectra were recorded with an Agilent 8453 UV-Vis spectrometer. Continuous wave X-band EPR spectra were recorded on a Bruker ER200D-SRC X-band spectrometer with irradiation at 9.042 GHz and 7.99 mW. Gas chromatography was performed on an Agilent 7890A gas chromatograph with an FID detector. The yield was detected by using a 30 m × 0.32 mm2 HP-5 column, and the enantioselectivity was determined by using a 30 m × 0.25 mm2 Cyclodex-B column.
Preparation of proteins
Mutations of the desired residues were performed by the polymerase chain reaction-based technique. Heme proteins were expressed and purified according to previous reports.33,49,62 The extraction of heme and incorporation of manganese(III) protoporphyrin IX to apomyoglobin were performed according to the literature.26,35,63–65 All the mutants were confirmed by ESI-MS.
Kinetic studies with KHSO5
The reactions were initiated by using 10 equiv. Oxone® (20 equiv. KHSO5) in 50 mM potassium phosphate buffer, monitored by using a UV-vis spectrometer. The final concentration of protein was 30 μM reacting at 5 °C. The kinetic traces at 410 nm for MnO Por were used for calculating the pseudo first-order rates kobs by fitting to a single-exponential eqn (1):| | | y = y0 + a (1 − e−x/t) | (1) |
where x is the reaction time, and y is the absorbance at λ = 410 nm. kobs = −1/t.
Docking of HSO5− and His3 Mb
Preparation of ligand and receptor.
The His3 Mb protein scaffold was obtained from the Protein Data Bank (PDB code: 4FWZ). Before docking, water molecules were removed, and the HSO5− ion was optimized by using the Gaussian 09 package.66 The optimization was performed by the DFT/B3LYP method, with the 6-31G (d) basis set for all atoms.
Docking.
We performed docking of HSO5− into His3 Mb using AutoDock (version 4.2.3) and AutoDockTools (version 1.5.4). The grids (8.25 Å × 12.75 Å × 12 Å) were set around the heme-binding site containing the distal ligands and axial ligand. Three distal residues, His-29, His-43, and His-64, were set as flexible parts, other residues were considered to be rigid. In the docking program, the Lamarckian genetic algorithm method was used. The number of individuals in population was 150, the maximum number of energy evaluations was set as 2500000, and the maximum of generations was 27000, and other parameters were used with default values.
EPR experiments
The EPR spectra were recorded at −170 °C in PBS buffer (pH 7.4). 1.0 mM protein was mixed with 20 equiv. H2O2 or 10 equiv. Oxone® (containing 20 equiv. KHSO5) in buffer. The mixed solution was kept for 3 min and then frozen with liquid nitrogen for EPR measurement.
Styrene epoxidation
In the time course experiment, 0.2 mM proteins were mixed with 20 equiv. styrene in 50 mM potassium phosphate buffer (pH 7.4). The reactions were initiated with 10 equiv. Oxone®, reacting at 4 °C for 0–2 h. The reaction was quenched with 20 eq. Na2S2O3 at a specific time. Then CH2Cl2 was added to extract the organic components. The CH2Cl2 solution was dried with anhydrous Na2SO4, concentrated to about 300 μL, and detected by GC. 20 equiv. of chlorobenzene were used as an internal reference. The epoxide product was also confirmed by 1H NMR, and its retention time was confirmed by using styrene oxide purchased.
In the screening of different proteins, we use the same conditions and processes but quench the reaction after 1 h. The mixture was also extracted with CH2Cl2, dried with anhydrous Na2SO4, and detected by GC.
Acknowledgements
This project was supported by the National Scientific Foundation of China (grant no. 20971007, 21101169, 21271013, and 21571007) and the National Key Basic Research Support Foundation of China (NKBRSFC) (2010CB912302, 2015CB856300, and 2015CB856301).
Notes and references
- J. Barber and J. W. Murray, Coord. Chem. Rev., 2008, 252, 233 CrossRef CAS.
- H. Dau and M. Haumann, Coord. Chem. Rev., 2008, 252, 273 CrossRef CAS.
- V. Daier, D. Moreno, C. Duhayon, J.-P. Tuchagues and S. Signorella, Eur. J. Inorg. Chem., 2010, 965 CrossRef CAS.
- S. Signorella and C. Hureau, Coord. Chem. Rev., 2012, 256, 1229 CrossRef CAS.
- F. Campaci and S. Campestrini, J. Mol. Catal. A: Chem., 1999, 140, 121 CrossRef CAS.
- W. Nam, I. Kim, M. H. Lim, H. J. Choi, J. S. Lee and H. G. Jang, Chem. – Eur. J., 2002, 8, 2067 CrossRef CAS.
- H. M. Neu, T. Yang, R. A. Baglia, T. H. Yosca, M. T. Green, M. G. Quesne, S. P. De Visser and D. P. Goldberg, J. Am. Chem. Soc., 2014, 136, 13845 CrossRef CAS PubMed.
- P. Leeladee and D. P. Goldberg, Inorg. Chem., 2010, 49, 3083 CrossRef CAS PubMed.
- H. Srour, P. L. Maux and G. Simonneaux, Inorg. Chem., 2012, 51, 5850 CrossRef CAS PubMed.
- K. A. Prokop, H. M. Neu, S. P. De Visser and D. P. Goldberg, J. Am. Chem. Soc., 2011, 133, 15874 CrossRef CAS PubMed.
- R. J. Nick, G. B. Ray, K. M. Fish, T. G. Spiro and J. T. Groves, J. Am. Chem. Soc., 1991, 113, 1838 CrossRef CAS.
- K. K. Khan, M. S. Mondal and S. Mitra, J. Chem. Soc., Dalton Trans., 1996, 1059 RSC.
- K. K. Khan, M. S. Mondal and S. Mitra, J. Chem. Soc., Dalton Trans., 1998, 4, 533 RSC.
- X. Wang and Y. Lu, Biochemistry, 1999, 38, 9146 CrossRef CAS PubMed.
- A. Gengenbach, S. Syn, X. Wang and Y. Lu, Biochemistry, 1999, 38, 11425 CrossRef CAS PubMed.
- D. W. Low, S. Abedin, G. Yang, J. R. Winkler and H. B. Gray, Inorg. Chem., 1998, 37, 1841 CrossRef CAS.
- C. Veeger, J. Inorg. Biochem., 2002, 91, 35 CrossRef CAS PubMed.
- J.-L. Primus, M. G. Boersma, D. Mandon, S. Boeren, C. Veeger, R. Weiss and I. M. C. M. Rietjens, J. Biol. Inorg. Chem., 1999, 4, 274 CrossRef CAS PubMed.
- J.-L. Primus, S. Grunenwald, P.-L. Hagedoorn, A.-M. Albrecht-Gary, D. Mandon and C. Veeger, J. Am. Chem. Soc., 2002, 124, 1214 CrossRef CAS PubMed.
- Z. N. Zahran, L. Chooback, D. M. Copeland, A. H. West and G. B. Richter-Addo, J. Inorg. Biochem., 2008, 102, 216 CrossRef CAS PubMed.
- I. Taniguchi, C.-Z. Li, M. Ishida and Q. Yao, J. Electroanal. Chem., 1999, 460, 245 CrossRef CAS.
- Y.-W. Lin, N. Yeung, Y.-G. Gao, K. D. Miner, S. Tian, H. Robinson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8581 CrossRef CAS PubMed.
- N.-T. Yu and M. Tsubaki, Biochemistry, 1980, 19, 4647 CrossRef CAS PubMed.
- F. Masuya and H. Hori, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1993, 1203, 99 CrossRef CAS.
- J. L. Heinecke, J. Yi, J. C. M. Pereira, G. B. Richter-Addo and P. C. Ford, J. Inorg. Biochem., 2012, 107, 47 CrossRef CAS PubMed.
- M. S. Mondal, S. Mazumdar and S. Mitra, Inorg. Chem., 1993, 32, 5362 CrossRef CAS.
- M. S. Mondal and S. Mitra, Biochim. Biophys. Acta, 1996, 1296, 174 CrossRef.
- R. Lin, C. E. Immoos and P. J. Farmer, J. Biol. Inorg. Chem., 2000, 5, 738 CrossRef CAS PubMed.
- M. Horitani, H. Yashiro, M. Hagiwara and H. Hori, J. Inorg. Biochem., 2008, 102, 781 CrossRef CAS PubMed.
- K. L. Stone, J. Hua and H. Choudhry, Inorganics, 2015, 3, 219 CrossRef CAS.
- M. Ohashi, T. Koshiyama, T. Ueno, M. Yanase, H. Fujii and Y. Watanabe, Angew. Chem., Int. Ed., 2003, 42, 1005 CrossRef CAS PubMed.
- K. Oohora, Y. Kihira, E. Mizohata, T. Inoue and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 17282 CrossRef CAS PubMed.
- J.-L. Zhang, D. K. Garner, L. Liang, D. A. Barrios and Y. Lu, Chem. – Eur. J., 2009, 15, 7481 CrossRef CAS PubMed.
- D. K. Garner, L. Liang, D. A. Barrios, J.-L. Zhang and Y. Lu, ACS Catal., 2011, 1, 1083 CrossRef CAS PubMed.
- J. R. Carey, S. K. Ma, T. D. Pfister, D. K. Garner, H. K. Kim, J. A. Abramite, Z. Wang, Z. Guo and Y. Lu, J. Am. Chem. Soc., 2004, 126, 10812 CrossRef CAS PubMed.
- A. Tsai, C. Wei, H. K. Baek, R. J. Kulmacz and H. E. Van Wart, J. Biol. Chem., 1997, 272, 8885 CrossRef CAS PubMed.
- E. S. Ryabova and E. Nordlander, Dalton Trans., 2005, 1228 RSC.
- Y.-B. Cai, X.-H. Li, J. Jing and J.-L. Zhang, Metallomics, 2013, 5, 828 RSC.
- M. H. Gelb, W. A. Toscano Jr. and S. G. Sligar, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 5758 CrossRef CAS.
- S. Ozaki, T. Matsui and Y. Watanabe, J. Am. Chem. Soc., 1996, 118, 9784 CrossRef CAS.
- T. Matsui, S. Ozaki, E. Liong, G. N. Phillips Jr. and Y. Watanabe, J. Biol. Chem., 1999, 274, 2838 CrossRef CAS PubMed.
- T. Matsui, S. Ozaki and Y. Watanabe, J. Am. Chem. Soc., 1999, 121, 9952 CrossRef CAS.
- S. Ozaki, M. P. Roach, T. Matsui and Y. Watanabe, Acc. Chem. Res., 2001, 34, 818 CrossRef CAS.
- B. Meunier, Chem. Rev., 1992, 92, 1411 CrossRef CAS.
- D. Chatterjee, A. Sikdar, V. R. Patnam, A. Theodoridis and R. van Eldik, Dalton Trans., 2008, 3851 RSC.
- J. T. Groves, J. Lee and S. S. Marla, J. Am. Chem. Soc., 1997, 119, 6269 CrossRef CAS.
- N. Jin and J. T. Groves, J. Am. Chem. Soc., 1999, 121, 2923 CrossRef CAS.
- K. D. Miner, A. Mukherjee, Y.-G. Gao, E. L. Null, I. D. Petrik, X. Zhao, N. Yeung, H. Robinson and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 5589 CrossRef CAS PubMed.
- J. L. Anderson, J. Ding, R. D. McCulla, W. S. Jenks and D. W. Armstrong, J. Chromatogr. A, 2002, 946, 197 CrossRef CAS PubMed.
- H. M. Neu, R. A. Baglia and D. P. Goldberg, Acc. Chem. Res., 2015, 48, 2754 CrossRef CAS PubMed.
- P. Leeladee, R. A. Baglia, K. A. Prokop, R. Latifi, S. P. De Visser and D. P. Goldberg, J. Am. Chem. Soc., 2012, 134, 10397 CrossRef CAS PubMed.
- C. J. Bougher, S. Liu, S. D. Hicks and M. M. Abu-Omar, J. Am. Chem. Soc., 2015, 137, 14481 CrossRef CAS PubMed.
- N. Jin, J. L. Bourassa, S. C. Tizio and J. T. Groves, Angew. Chem., Int. Ed., 2000, 39, 3849 CrossRef CAS.
- T. Yonetani and T. Asakura, J. Biol. Chem., 1969, 244, 4580 CAS.
- H. Hori, M. Ikeda-Saito and T. Yonetani, Biochim. Biophys. Acta, 1987, 912, 74 CrossRef CAS.
- A. J. Allentoff, J. L. Bolton, A. Wilks, J. A. Thompson and P. R. Ortiz de Montellano, J. Am. Chem. Soc., 1992, 114, 9744 CrossRef CAS.
- D. E. Lansky and D. P. Goldberg, Inorg. Chem., 2006, 45, 5119 CrossRef CAS PubMed.
- D. Janardanan, D. Usharani and S. Shaik, Angew. Chem., Int. Ed., 2012, 51, 4421 CrossRef CAS PubMed.
- C. Crestini, A. Pastorini and P. Tagliatesta, Eur. J. Inorg. Chem., 2004, 4477 CrossRef CAS.
- A. Rezaeifard, M. Jafarpour, S. Rayati and R. Shariati, Dyes Pigm., 2009, 80, 80 CrossRef CAS.
- Y.-W. Lin, S.-S. Dong, J.-H. Liu, C.-M. Nie and G.-B. Wen, J. Mol. Catal. B: Enzym., 2013, 91, 25 CrossRef CAS.
- J. A. Sigman, B. C. Kwok and Y. Lu, J. Am. Chem. Soc., 2000, 122, 8192 CrossRef CAS.
- B. A. Springer and S. G. Sligar, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8961 CrossRef CAS.
- F. W. J. Teale, Biochim. Biophys. Acta, 1959, 35, 543 CrossRef CAS.
- W. R. Fisher, H. Taniuchi and C. B. Anfinsen, J. Biol. Chem., 1973, 248, 3188 CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, G. Z. J. Bloino, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.2), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qi00120c |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 NK
NK K
K QG
QG