
Reverse Watson–Crick G–G base pair in G-quadruplex formation†
Soma
Mondal
,
Jyotsna
Bhat
,
Jagannath
Jana
,
Meghomukta
Mukherjee
and
Subhrangsu
Chatterjee
*
Bose Institute, Centenary Campus, Department of Biophysics, P-1/12 CIT Scheme VIIM, Kankurgachi, Kolkata-54, India. E-mail: subhro_c@jcbose.ac.in; Web: http://bic.boseinst.ernet.in/subhro_c/ Tel: +91-33-2569-3340
First published on 9th November 2015
Abstract
A stable intermediate dimeric G-rich form as a precursor of tetrameric G-quadruplex structures has been detected via MALDI-TOF spectrometry. Molecular dynamics simulation offered detailed insights at the atomic level, assigning reverse Watson–Crick G–G base pairing (not Hoogsteen) in the G-rich dimer. In support of this, cisplatin formed a stable adduct by binding to the dimeric G-rich structure, eliminating the possibility of G–G Hoogsteen hydrogen bond formation.
Besides the canonical B-DNA that has a right handed double helical structure stabilized by Watson–Crick base pairing,1 repetitive G-rich sequences can fold into non-canonical structures known as G-quadruplexes. A G-quadruplex structure consists of π–π stacking of planar G-tetrads stabilized by Hoogsteen type hydrogen bonds.2,3 G-rich sequences are abundantly distributed in the telomeric region of human DNA and in the promoters of many oncogenes,4–7 thus playing a significant role in many biological processes.8,9 G-quadruplexes can be formed from single strands (intramolecular), two strands (bimolecular) and four strands (tetramolecular).10 Furthermore, intermolecular G-quadruplexes are gaining importance nowadays for engineering nano-molecular devices owing to their stable and rigid three dimensional scaffolds and inherent electronic properties.11–14 In addition, intermolecular G-quadruplex structures bring in frontier concepts in developing anti-cancer15 and anti-HIV therapeutics16 for the near future. Among all of the intermolecular G-quadruplex structures, tetramolecular G-quadruplex structures emerged as a new vision due to their symmetry and thermodynamic stability. The mechanism of tetramolecular G-quadruplex formation has been already investigated17,18 through kinetics19,20 and thermodynamic studies.21,22 Structures adopted by short G-rich sequences containing one flanking base at the 3′ or 5′ position or at both ends have been shown to form parallel quadruplex structures.23 From the recent literature on intermolecular G-quadruplex formation, it was observed that there has been no scientific clue and no evidence at the atomic level on how an intermolecular G-quadruplex forms. The possible G–G base pairing conformation and pathway of forming a G–G Hoogsteen hydrogen bonded base pairing state in the three dimensional scaffolds of quadruplexes, have not been extensively mined out. Now, here in this report we obtained results that showed that monomeric to dimeric G-rich DNA formation can happen through G–G reverse Watson–Crick type hydrogen bonding. The closed, stabilized G–G reverse Watson–Crick base pairs in the dimeric form open up to form active functional G–G Hoogsteen base pairs which act as a building block of a G-quartet structure [Scheme 1]. Sharma et al. have previously reported the participation of G–G reverse Watson–Crick hydrogen bonds in multi-modality for edge interactions24 in RNA. We studied four DNA sequences listed in [Table S1, ESI†] using NMR, CD spectroscopy, gel electrophoresis and MALDI-TOF spectrometry. To support these experimental findings, we further performed molecular dynamics simulations.
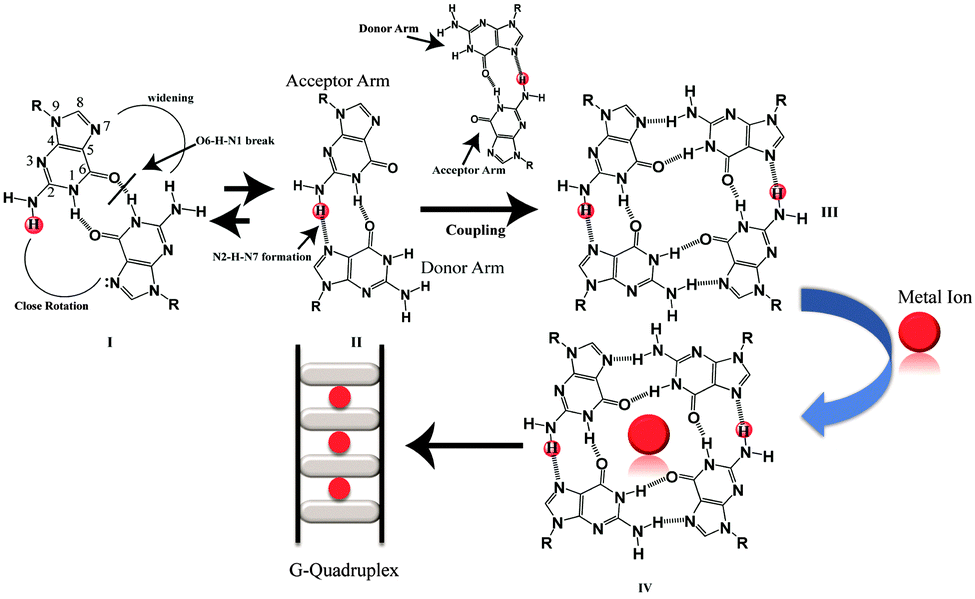 | ||
| Scheme 1 Proposed mechanism for G-quartet formation from G–G reverse Watson–Crick G–G base-pairs. | ||
CD is mainly used to reveal information regarding the conformation of G-quadruplex structures. The CD spectra of all of the GG4 sequences were recorded after overnight incubation at 4 °C [Fig. S1(A) and (C), ESI†]. The CD spectrum of GG4(A) shows a positive peak around 260 nm characteristic of a parallel quadruplex, whereas that of GG4(T) shows a positive peak around 285 nm with a minimum around 261 nm which indicates an antiparallel type structure.25 The CD spectrum of GG4(C) displays two positive peaks: one at 285 nm, and the other at 256 nm which is the characteristic peak of C rich duplex DNA.26 Interestingly, for G rich sequences, Laure et al.27 have shown that for the sequences where 7 ≤ n ≤ 13 (n = no. of guaninyl residues) the G-tract remains unfolded, therefore although GG4(G) shows a positive peak around 260 nm, it remains single-stranded. GG4(AT) exhibits a positive peak around 262 nm, quite different from the summation of the GG4(A) and GG4(T) CD spectra, which indicates the existence of other conformations including those of GG4(A) and GG4(T). In contrast, GG4(GC) displays a positive peak around 260 nm which is similar to the summation of the ellipticities of both GG4(C) and GG4(G), indicating the presence of duplex conformations of GG4(C) and a single-stranded conformation of GG4(G). Next, the CD spectra of the GG4 sequences were taken after two months of incubation at 4 °C [Fig. S1(B) and (D), ESI†]. GG4(A) and GG4(T) both show a positive peak near 260 nm accompanied by an enhancement in the CD signal, thus GG4(T) undergoes structural transition from an antiparallel type structure to a parallel stranded G-quadruplex structure. In contrast, the spectral patterns for GG4(C), GG4(G) and GG4(GC) remain unaltered after two months, indicating the presence of a stable duplex and a single strand. In the case of GG4(AT), after two months of incubation, the CD spectrum shows a positive peak near 262 nm; strikingly, this spectrum resembles the spectrum generated from the summation of the individual ellipticities of GG4(A) and GG4(T). This observation directly confirms the existence of primary conformations of GG4(A) and GG4(T) in their mixtures. MALDI-TOF mass spectrometry28,29 was employed for these sequences to unravel the intermediates that build the stable quartet conformation of the GG4 sequences with time. The MALDI-TOF spectra were recorded after the sequences were annealed in dibasic ammonium citrate and incubated at 4 °C overnight with successive time intervals of 1 month and 2 months, respectively. The G-quadruplex conformation remains the same in both the K+ and NH4+ solutions [Fig. S2, ESI†]. When the MALDI-TOF spectra were recorded after overnight incubation, we find an equilibrium between the monomer and dimer for all of the sequences except GG4(G) which exists as a monomer [Fig. S3 and Table S3, ESI†]. Interestingly, for GG4(C), a stable trimer (triplex) is also detected. A mixed strand stoichiometry is detected for the GG4(AT) dimer [Fig. S3(E), ESI† inset]. In GG4(GC) we observed an equilibrium between the GG4(C) monomeric and duplex forms but no mixed duplex is detected involving monomeric GG4(G) and GG4(C). The spectrum recorded after 1 month of incubation reveals the formation of trimers (less amount) for all sequences along with monomers and dimers except for GG4(G) which still exists as a monomer [Fig. S4 and Table S4, ESI†]. A mixed strand stoichiometry is again detected for the GG4(AT) dimer [Fig. S4(E), ESI† inset]. GG4(GC) reveals no mixed conformation. The spectrum recorded after two months of incubation reveals the formation of tetramers with very low intensity for GG4(A), GG4(T) and GG4(AT) [Fig. 1(A) inset, (B) inset and (E) inset] in MALDI-TOF spectrometry. When GG4(A) tetramer formation is just initiated, GG4(T) comprised a proper tetramolecular G-quadruplex bound to three ammonium ions. In addition, we detect some slipped G-quadruplex structures,17,18 consisting of none, one and two ammonium ions for the GG4(A), GG4(T) and GG4(AT) sequences. In the course of time, there is an enhancement in the signal in the MALDI footprints for the dimers and trimers of all of the sequences [Fig. 1 and Table S5, ESI†]. Now, in MALDI-TOF spectrometry, GG4(A) and GG4(T) both showed that the presence of the dimer was much higher than that of the trimer as well as the tetramer.
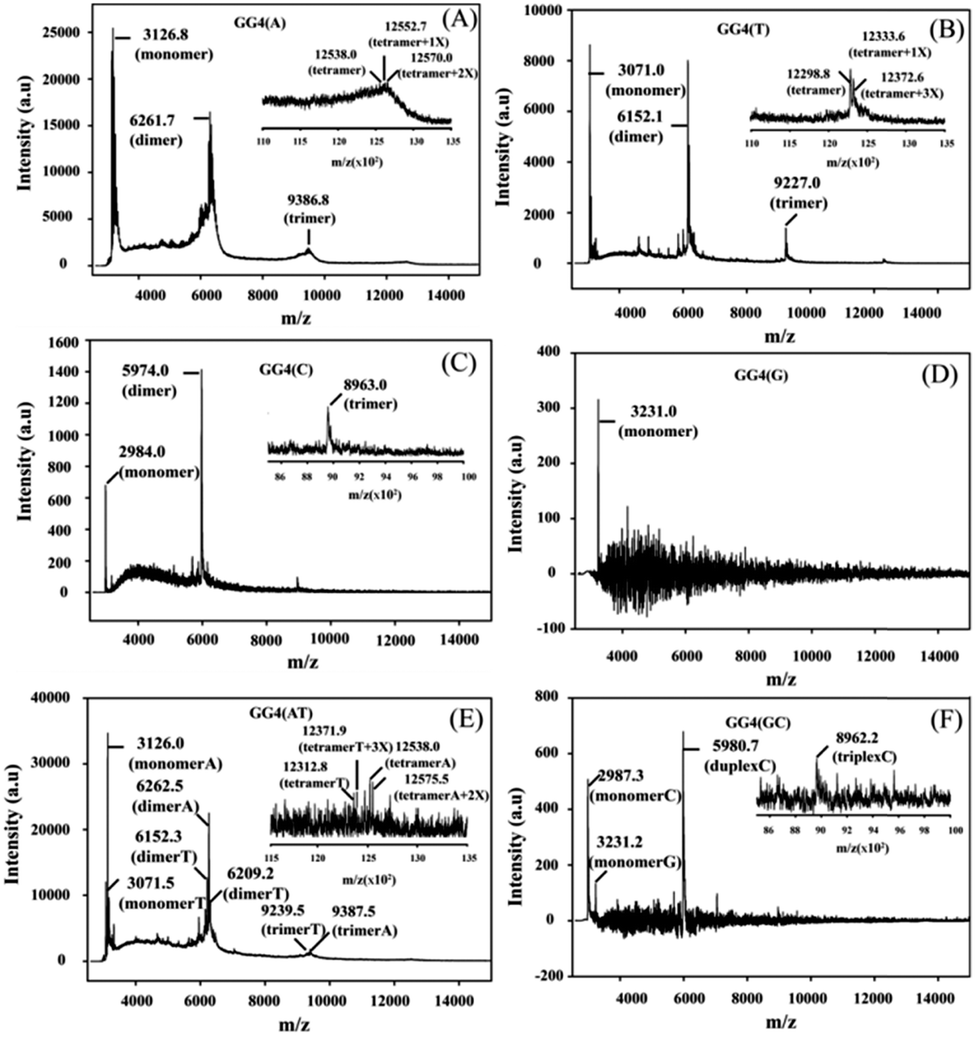 | ||
| Fig. 1 MALDI-TOF spectra of the GG4 sequences after two months of incubation at 4 °C using 3-HPA as the matrix: (A) GG4(A), inset [zoomed region of the GG4(A) tetramer]; (B) GG4(T), inset [zoomed region of the GG4(T) tetramer]; (C) GG4(G); (D) GG4(C), inset [zoomed region of the GG4(C) triplex]; (E) GG4(AT), inset [zoomed region of the GG4(AT) tetramer]; and (F) GG4(GC), inset [zoomed region of the GG4(C) triplex]. X = ammonium ion. | ||
Although the intermolecular structures are highly concentration dependent, one dimensional proton spectra were recorded at 10 °C to find out the nature of the secondary structures adopted by the GG4 series. The imino protons appear around 11.0 ppm [Fig. S5(A), ESI†] for GG4(A), GG4(T) and GG4(AT), indicating the formation of G-quadruplex structures.17 For GG4(C) and GG4(GC), imino protons are detected around 13.0 ppm [Fig. S5(B), ESI†], suggesting the formation of duplex structures.30 The single-stranded conformation of GG4(G) is evidenced with no appearance of imino signals in the down field region [Fig. S5(B), ESI†]. Gel electrophoresis data revealed the slow migration rate of GG4(A), GG4(T) and GG4(AT) followed by GG4(C) and GG4(GC), indicating the presence of G-quadruplex and duplex forms, respectively. For GG4(G), there is no band seen indicating the single-stranded nature of GG4(G) [Fig. S6, ESI†] (ethidium bromide does not bind with single-stranded DNA).
We further continued our studies with GG4(A), GG4(T) and GG4(AT) as these sequences have the potential to form G-quadruplex structures. The two most probable mechanisms for tetramolecular G-quadruplex formation reported to date are: (i) monomer–dimer–tetramer in which the coupling of the dimer is the rate determining step,19 and (ii) monomer–dimer–trimer–tetramer where the tetramer formation is the rate determining step.18 We mainly focused on the dimeric structure obtained in high yield in mass spectrometry, and whose structural elucidation is not very clear. We simulated the dimeric G-rich structure (duplex) consisting of G–G Hoogsteen base pairs, keeping the strand orientation in parallel directions to provide detailed insights at the atomic level. In the simulation, we observed the rearrangement of the hydrogen bonding pattern of the G–G base pairs. In the first phase, the G–G bases get unpaired and the guanine bases are opened up. During a further production run, G–G pairing is re-established by means of reverse Watson–Crick hydrogen bonding31 and remained stable throughout the period [Fig. 2(A)], (the structural transition can be clearly seen in the AVI movie provided in the ESI†). It has already been reported that to form parallel duplex structures, a reverse Watson–Crick base pair is more favoured than normal Watson–Crick bonding.32 As seen in Fig. 2(C), for the first 10 ns, the total number of hydrogen bonds is lower [GG4(A):3, GG4(T):2 and GG4(AT):3 (average values)]; however, hydrogen bonding is increased with the progression of the simulation run. During a 40–50 ns simulation period, the total number of hydrogen bonds increased to GG4(A):4, GG4(T):7 and GG4(AT):8 (average values). Also these hydrogen bonds are stable as illustrated by the hydrogen bond occupancy values [Table S6, ESI†]. B-factor analyses of all of the atoms showed that the G-region of all of the three systems are most stable compared to the terminal bases [Fig. 2(B)]. MMPBSA energy calculation suggested that the total energy content of the system is decreasing during the simulation thus resulting into more stable conformations [Table S7, ESI†]. The change in total energy in GG4(AT) with respect to time is shown in [Fig. 2(D), Fig. S9, ESI†]; also the corresponding change in the (donor G)N2H–N7(acceptor G) distance is represented in the three dimensional energy landscape of the conversion of Hoogsteen pairs to Watson–Crick pairs [Fig. 2(D) inset]. Similar results are observed for GG4(A) and GG4(T).
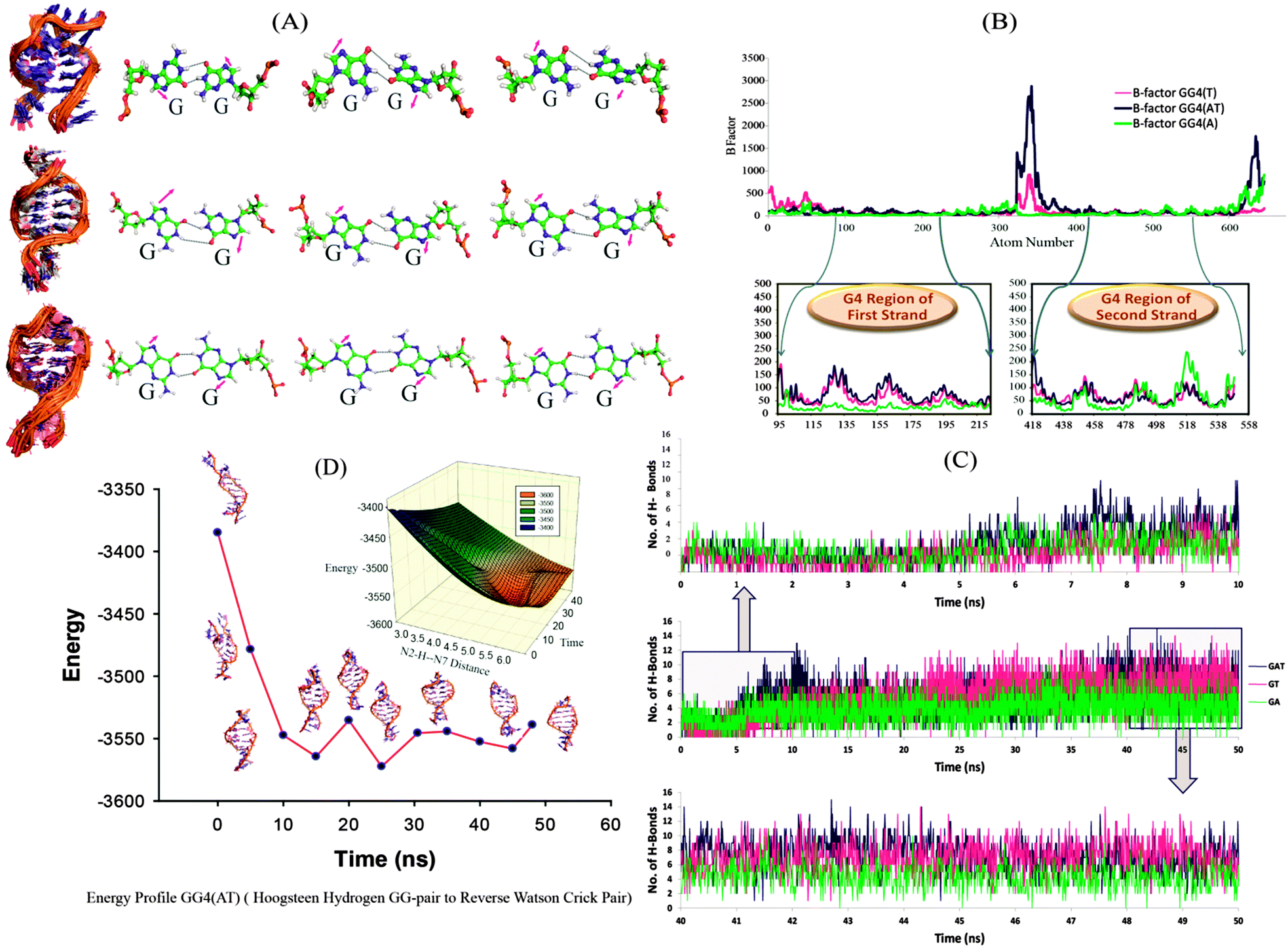 | ||
| Fig. 2 (A) Ensemble structure of GG4A (top), GG4T (middle) and GG4AT (bottom) along with reverse Watson–Crick hydrogen bonding. (B) The B-factor estimation of each atom is illustrated in the top region, and the G rich regions of the first and second strands of GG4T, GG4AT and GG4A are enlarged for detailed understanding. (C) Total number of hydrogen bonds at each time step over the entire simulation run (50 ns), the up arrow points at the enlarged graph of the number of hydrogen bonds during the first 10 ns simulation run and the down arrow points at the enlarged graph of the number of hydrogen bonds during the last 10 ns simulation run. (D) Change in the total energy in GG4AT with respect to time and the corresponding change in the N2H–N7 distance (inset). | ||
From the molecular dynamics simulation in the explicit solvent for all dimeric G rich structures (GG4(A), GG4(T), GG4(AT)) it is very well understood that reverse Watson–Crick hydrogen bonding involved in G–G base pairs stabilizes the dimeric form of DNA. The simulation, though, was augmented with G–G Hoogsteen base pairing; reverse Watson–Crick G–G paired dimers become the end result. This observation was validated by the MALDI-TOF spectrum of the G–G dimer in the presence of cisplatin (molar mass 300 g mol−1). As cisplatin binds to the N7 atoms of two consecutive guanines,33 the dimeric G–G pair has to be in a reverse Watson–Crick hydrogen bonded conformation to facilitate the cisplatin DNA adduct formation. Here we could find a signal of a G–G dimer bound to one and two cisplatin moieties as shown in [Fig. 3(A–C)]. This experimental observation is aligned with that of the theoretical molecular dynamics simulation. Interestingly, in G–G pairing, the imidazole rings of both Gs are not involved in the base-pairing. If the N1–H–O6 hydrogen bond in the pairing is cleaved, the formation of the N(2)–H–N7 bond simultaneously occurs to save the energy loss. This Watson–Crick pairing switched to Hoogsteen pairing process opens one acceptor and a donor arm which ultimately couple with another G–G Hoogsteen base-pair to form a G quadruplex structure [Scheme 1, S2, ESI†]. The central N–H–O hydrogen bond being the backbone of the switch of O6–H–N1 to N2–H–N7 causes the coupling of the pairs of dimers (GG) to produce quadruplexes. In a quadruplex structure there are two types of hydrogen bonds:34 (i) N1(donor G)–H–O6(acceptor G) and (ii) N7(acceptor G)–H–N2(donor G). The central N–H–O signal is sharp in NMR spectroscopy, confirming the slow exchange of the central proton. On the contrary, the peripheral N2–H–N7 does not appear in NMR, confirming a fast exchange.
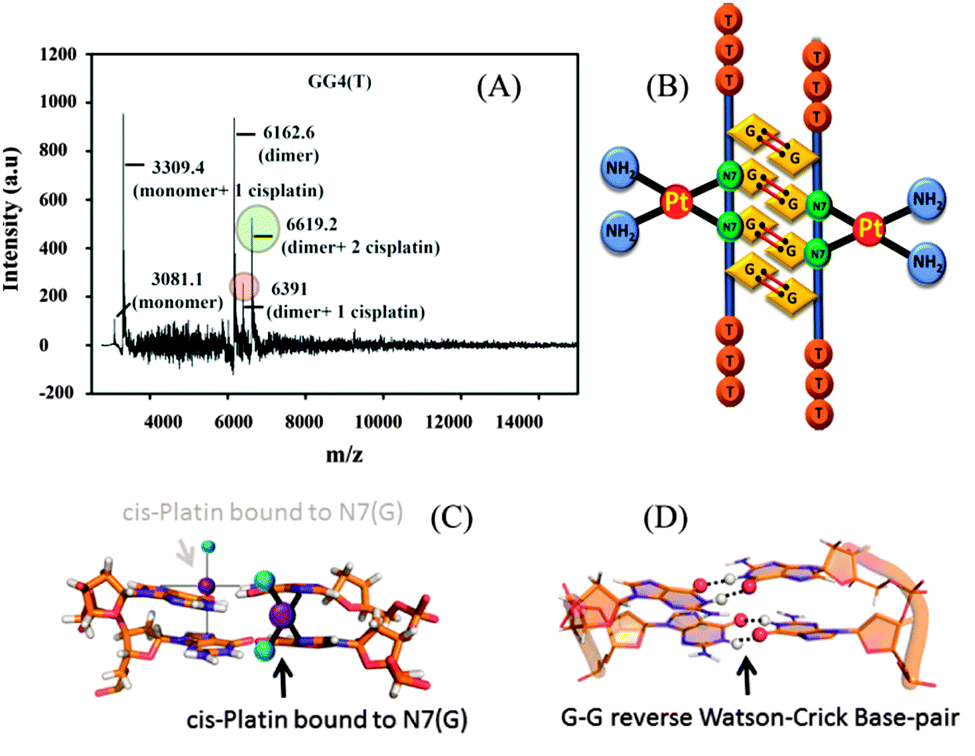 | ||
| Fig. 3 (A) MALDI-TOF spectrum of GG4(T) with cisplatin, (B) model structure of cisplatin bound to the dimeric structure of GG4(T), (C) cisplatin bound to N7 of guanine, and (D) the G–G reverse Watson–Crick base pair. | ||
In short it is proven that the stable dimeric G rich form is dictated by G–G reverse Watson–Crick bonded base pairs. However, a reverse Watson–Crick G–G pair cannot lead to the formation of G quartet structures without switching to Hoogsteen pairing. This emphasizes that GGreverse Watson–Crick converts to GGHoogsteen functional pairing, which in turn couples with another GGHoogsteen pair to form a tetramolecular G-quadruplex structure. Thus GGreverse Watson–Crick is conceptualized as the dormant, locked conformation of the G–G base pair whereas GGHoogsteen is the open, active, functional form.
SC would like to thank the DST Ramanujan Fellowship. SM, JB, JJ and MM would like to thank UGC, DBT, CSIR and DST, Govt of India, for their fellowships. SC thanks the DBT research grant BT/PR6627/GBD/27/440/2012. SC thanks Mr Amarendra Nath Biswas for carrying out the MALDI-TOF experiments.
Notes and references
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- J. Choi and T. Majima, Chem. Soc. Rev., 2011, 40, 5893–5909 RSC.
- A. T. Phan, V. Kuryavyi and D. J. Patel, Curr. Opin. Struct. Biol., 2006, 16, 288–298 CrossRef CAS PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- E. Y. Lam, D. Beraldi, D. Tannahill and S. Balasubramanian, Nat. Commun., 2013, 4, 1796 CrossRef PubMed.
- A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593–11598 CrossRef CAS PubMed.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- K. Hirashima and H. Seimiya, Nucleic Acids Res., 2015, 43, 2022–2032 CrossRef CAS PubMed.
- M. C. Chen, P. Murat, K. Abecassis, A. R. Ferré-D'Amaré and S. Balasubramanian, Nucleic Acids Res., 2015, 43, 2223–2231 CrossRef CAS PubMed.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS PubMed.
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed.
- P. Alberti, A. Bourdoncle, B. Saccà, L. Lacroix and J. L. Mergny, Org. Biomol. Chem., 2006, 4, 3383–3391 CAS.
- A. M. Chiorcea-Paquim, P. V. Santos, R. Eritja and A. M. Oliveira-Brett, Phys. Chem. Chem. Phys., 2013, 15, 9117–9124 RSC.
- O. Doluca, J. M. Withers, T. S. Loo, P. J. Edwards, C. González and V. V. Filichev, Org. Biomol. Chem., 2015, 13, 3742–3748 CAS.
- P. J. Bates, D. A. Laber, D. M. Miller, S. D. Thomas and J. O. Trent, Exp. Mol. Pathol., 2009, 86, 151–164 CrossRef CAS PubMed.
- A. Rajendran, M. Endo, K. Hidaka, P. L. Tran, J. L. Mergny, R. J. Gorelick and H. Sugiyama, J. Am. Chem. Soc., 2013, 135, 18575–18585 CrossRef CAS PubMed.
- C. Bardin and J. L. Leroy, Nucleic Acids Res., 2008, 36, 477–488 CrossRef CAS PubMed.
- F. Rosu, V. Gabelica, H. Poncelet and E. De Pauw, Nucleic Acids Res., 2010, 38, 5217–5225 CrossRef CAS PubMed.
- J. R. Wyatt, P. W. Davis and S. M. Freier, Biochemistry, 1996, 35, 8002–8008 CrossRef CAS PubMed.
- J. L. Mergny, A. De Cian, A. Ghelab, B. Saccà and L. Lacroix, Nucleic Acids Res., 2005, 33, 81–94 CrossRef CAS PubMed.
- J. Gros, A. Aviñó, J. Lopez de la Osa, C. González, L. Lacroix, A. Pérez, M. Orozco, R. Eritja and J. L. Mergny, Chem. Commun., 2008, 2926–2928 RSC.
- P. L. Tran, A. De Cian, J. Gros, R. Moriyama and J. L. Mergny, Top. Curr. Chem., 2013, 330, 243–273 CrossRef CAS PubMed.
- J. Zhou, G. Yuan, J. Liu and C. G. Zhan, Chemistry, 2007, 13, 945–949 CrossRef CAS PubMed.
- P. Sharma, A. Mitra, S. Sharma, H. Singh and D. Bhattacharyya, J. Biomol. Struct. Dyn., 2008, 25, 709–732 CAS.
- S. Paramasivan, I. Rujan and P. H. Bolton, Methods, 2007, 43, 324–331 CrossRef CAS PubMed.
- M. Morikawa, K. Kino, T. Oyoshi, M. Suzuki, T. Kobayashi and H. Miyazawa, Biomolecules, 2014, 4, 140–159 CrossRef PubMed.
- L. Joly, F. Rosu and V. Gabelica, Chem. Commun., 2012, 48, 8386–8388 RSC.
- N. Nagesh, A. Krishnaiah, V. M. Dhople, C. S. Sundaram and M. V. Jagannadham, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 303–315 CAS.
- L. Yuan, T. Tian, Y. Chen, S. Yan, X. Xing, Z. Zhang, Q. Zhai, L. Xu, S. Wang, X. Weng, B. Yuan, Y. Feng and X. Zhou, Sci. Rep., 2013, 3, 1811 Search PubMed.
- R. H. Sarma, M. H. Sarma, L. Dai and K. Umemoto, FEBS Lett., 1997, 418, 76–82 CrossRef CAS PubMed.
- V. Kocman and J. Plavec, Nat. Commun., 2014, 5, 5831 CrossRef CAS PubMed.
- L. A. Yatsunyk, O. Piétrement, D. Albrecht, P. L. Tran, D. Renčiuk, H. Sugiyama, J. M. Arbona, J. P. Aimé and J. L. Mergny, ACS Nano, 2013, 7, 5701–5710 CrossRef CAS PubMed.
- J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS PubMed.
- A. J. Dingley, R. D. Peterson, S. Grzesiek and J. Feigon, J. Am. Chem. Soc., 2005, 127, 14466–14472 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00611b |
| This journal is © The Royal Society of Chemistry 2016 |